

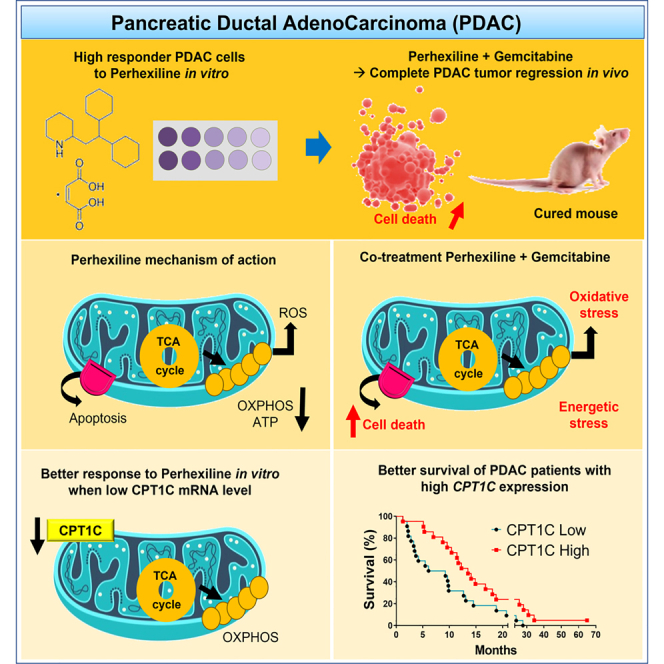
Summary
Pancreatic ductal adenocarcinoma (PDAC) remains one of the human cancers with the poorest prognosis. Interestingly, we found that mitochondrial respiration in primary human PDAC cells depends mainly on the fatty acid oxidation (FAO) to meet basic energy requirements. Therefore, we treated PDAC cells with perhexiline, a well-recognized FAO inhibitor used in cardiac diseases. Some PDAC cells respond efficiently to perhexiline, which acts synergistically with chemotherapy (gemcitabine) in vitro and in two xenografts in vivo. Importantly, perhexiline in combination with gemcitabine induces complete tumor regression in one PDAC xenograft. Mechanistically, this co-treatment causes energy and oxidative stress promoting apoptosis but does not exert inhibition of FAO. Yet, our molecular analysis indicates that the carnitine palmitoyltransferase 1C (CPT1C) isoform is a key player in the response to perhexiline and that patients with high CPT1C expression have better prognosis. Our study reveals that repurposing perhexiline in combination with chemotherapy is a promising approach to treat PDAC.
Subject areas: Biological sciences, Cancer systems biology, Cancer
Graphical abstract
Highlights
-
•
Some PDAC cells respond efficiently to the antianginal drug perhexiline
-
•
Perhexiline synergizes with gemcitabine to induce complete tumor regression
-
•
This co-treatment causes energetic and oxidative stress promoting apoptosis
-
•
The CPT1C isoform is a key actor underlying the response to perhexiline
Biological sciences; Cancer systems biology; Cancer
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is by far the most frequent type of pancreatic cancer. One major concern is that PDAC is predicted to become the second cause of cancer-related death in the United States by 2030, surpassing breast and colorectal cancer.1,2,3 Moreover, the 5-year relative survival rate has only increased from 2.5% in 1970–1977 to 11% in 2022.4,5 This poor progress against PDAC mainly reflects the lack of an early diagnosis, useful biomarkers, and substantial treatment advances. Clinically, only 15%–20% of patients are eligible for surgical tumor resection, and most of them will face chemoresistance and relapse at some point during treatment or after cure.6,7 Moreover, 80–85% of patients present with infiltration of surrounding structures or metastasis at diagnosis,8,9 for which standard chemotherapy only provides a modest increase in survival.10,11 So far, pancreatic cancer remains an incurable disease.
A characteristic of pancreatic cancer is its remarkable therapeutic resistance, mostly due to key features of this cancer, including its constant metabolic reprogramming to survive and proliferate.12 Interestingly, resistant pancreatic cancer cells show reliance on an oxidative phenotype associated with mitochondrial metabolism.13,14 Subsequently, several studies including our own have documented the fundamental role of mitochondria in all facets of PDAC progression and chemotherapeutic resistance.15,16,17,18,19 We demonstrated that inhibiting mitochondrial complex I with phenformin in combination with gemcitabine overcomes chemoresistance in high oxidative phosphorylation (OXPHOS) tumors.18
One of the main substrates that power mitochondria are fatty acids. The fatty acid oxidation (FAO) is responsible for the catabolism of long-chain fatty acids in mitochondria. FAO comprises a cyclical series of reactions that result in the shortening of fatty acid molecules (beta-oxidation) to produce acetyl-coenzyme A (CoA) as well as NADH and FADH2. NADH and FADH2 produced by both FAO and the tricarboxylic acid (TCA) cycle are used by the mitochondrial respiratory chain to produce ATP.20 The carnitine palmitoyltransferase 1 (CPT1) is considered the rate-limiting enzyme of FAO since it is mandatory for fatty acids translocation inside mitochondria, by catalyzing their conjugation with carnitine.
The importance of FAO in cancer has been previously reviewed,20 pointing to the therapeutic potential of its inhibition. The pharmacological blockade of FAO has been pursued for the treatment of heart diseases. Consequently, FAO inhibitors have been approved for human clinical practice. These drugs are used to inhibit degradation of lipids for energy production and thereby to promote more oxygen-efficient utilization of glucose as an energy source in chronic ischemic cardiomyopathy.21 Pharmacological inhibition of FAO in cancer can be achieved with drugs like etomoxir,22,23,24 perhexiline,25,26,27,28 oxfenicine,20 avocatin,29 trimetazidine,30 and ranolazine, among others.20
As most of cancer metabolism researchers are focused on glycolysis, glutaminolysis, and fatty acid synthesis, the importance of FAO in cancer has not been carefully examined, and its relevance has remained obscure, especially in pancreatic cancer. In this work, we investigated the effect of the well-recognized FAO inhibitor perhexiline in combination with chemotherapy and demonstrated that this is a promising therapeutic strategy in the clinic.
Results
Mitochondrial respiration of primary pancreatic cancer cells depends mainly on the FAO pathway
Contrary to the classical view of cancer metabolism that focuses on a high glycolytic dependence, there is an expanding list of molecules besides glucose that fuel cancer cells.31 Therefore, using the Seahorse Agilent oxygraph Mito Fuel Flex assay, we addressed the dependency and flexibility of mitochondria to oxidize glucose, glutamine, and long-chain fatty acids in 21 primary PDAC cells obtained from patient-derived xenografts (PDXs).18 The rate of oxidation of each fuel was determined by measuring the mitochondrial respiration (oxygen consumption rate [OCR]) of cells in the presence or absence of specific inhibitors (Figures 1A and S1A): UK5099, BPTES, and Etomoxir, to inhibit glycolysis, glutaminolysis, and FAO, respectively. In this assay, the concentration of etomoxir (4 μM) specifically inhibits FAO and is much lower than the concentration resulting in off-target effects (200 μM), such as inhibition of mitochondrial complex I.32
Figure 1.

Mitochondrial respiration of primary pancreatic cancer cells depends mainly on the fatty acid oxidation (FAO) pathway
(A) Schematic of Mito Fuel Flex Test carried out on the Seahorse analyzer. The rate of oxidation of glucose, glutamine, and long-chain fatty acids was determined by measuring the mitochondrial respiration (oxygen consumption rate). For this, specific inhibitors were used: UK5099 (2 μM), BPTES (3 μM), and Etomoxir (4 μM), to inhibit the mitochondrial pyruvate carrier (MPC), glutaminase 1 (GLS1), and carnitine palmitoyltransferase 1 (CPT1), respectively, and to determine glycolysis, glutaminolysis, and Fatty Acid Oxidation (FAO), respectively.
(B and C) Percentage of dependency (B) and flexibility (C) of mitochondrial respiration to oxidize three main energetic fuels: glucose, glutamine, and fatty acids, in 21 primary PDAC cells from patients (each bar represents a patient). Data are the mean ± SEM of three independent experiments performed in triplicates. p values were calculated from t test and Mann-Whitney test; ∗∗p < 0.01, ∗∗∗p < 0.001.
Dependency refers to the need for mitochondrial respiration of a specific fuel to meet basic energy needs. Notably, our results show that mitochondrial respiration depends on fatty acids in the 21 primary PDAC cells (Figures 1B and S1B). Indeed, 14 of 21 PDAC cells exhibited a fatty acid dependency higher than 50%, and the remaining showed a moderate dependency degree. Interestingly, a modest correlation between mitochondrial fatty acids’ dependency and the replication rate of the cells was observed (Figure S1C). In contrast, the majority of PDAC cells presented a low or moderate reliance on glucose for mitochondrial respiration. In addition, a very low dependency on glutamine (average of 8%) was observed in all the PDAC cells.
Flexibility means that, when a specific pathway is inhibited, mitochondrial respiration is able to compensate by using other pathways. The results shown in Figure 1C illustrate that mitochondrial respiration in most primary PDAC cells is poorly flexible toward the three tested fuels. Further, the presence of dependency and absence of flexibility for a fuel indicate that mitochondria require that fuel to sustain basal respiration, which is notably observed in the case of fatty acids’ high dependence and low flexibility in our assays.
Thus, in sharp contrast with several studies, our results take the focus away from the glycolysis and glutaminolysis pathways and bring to light the importance of FAO in PDAC. This finding suggests that the FAO pathway could be a novel vulnerability of pancreatic cancer.
The FAO inhibitor perhexiline amplifies the antitumor activity of gemcitabine by decreasing cell viability and increasing apoptosis in primary pancreatic cancer cells
We hypothesized that targeting FAO would be an efficient strategy to promote energetic stress and pancreatic cancer cell elimination. Therefore, we treated primary PDAC cells with well-recognized FAO inhibitors that have been approved for human use to treat cardiac diseases. We used etomoxir and perhexiline, which are inhibitors of the CPT1, a critical fatty acid transporter into mitochondria. In addition, we treated cells with trimetazidine, which targets the 3-ketoacyl-coenzyme A thiolase (3-KAT), the last enzyme that catalyzes the β-oxidation of FAO.20,30
We performed dose-response experiments treating primary PDAC cells for 72 h to determine cell viability with the crystal violet assay. The etomoxir and perhexiline dose-response viability curves (Figure 2A) show that the cells are more sensitive to perhexiline than to etomoxir and display differential sensitivities. The graphs below the curves show the percentage of live cells at a specific concentration of etomoxir or perhexiline (62.5 and 7 μM, respectively) to illustrate the differential sensitivity between cells. The half-maximal inhibitory concentration (IC50) values (Figure S2A) show a similar sensitivity stratification of the primary PDAC cells. We can distinguish PDAC cells that respond efficiently to etomoxir and perhexiline, i.e., showing a percentage of live cells below the mean (75% and 72% for etomoxir and perhexiline, respectively, Figure 2A) and an IC50 below the mean (Figure S2A), such as PDAC084T. Another group of cells show low response with a percentage of live cells and IC50 above the mean, and finally some cells show moderate (intermediate) response with values close to the mean. Only two cases display a different response between the two drugs: the PDAC003T and PDAC012T.
Figure 2.

The FAO inhibitor perhexiline amplifies the antitumor activity of gemcitabine by decreasing cell viability and increasing apoptosis in primary pancreatic cancer cells
(A) Dose-response experiments with etomoxir and perhexiline treatments in selected primary PDAC cells. Live cells were quantified using the crystal violet assay after 72 h of treatment. (Top) Curves are representative of three independent experiments performed in triplicates, and data are the mean ± SD. (Bottom) Percentage of live PDAC cells treated for 72 h with etomoxir (62.5 μM) or perhexiline (7 μM), compared with non-treated controls. The cells showing a percentage of live cells below the mean (75% for etomoxir and 72% for perhexiline [dotted lines]) are considered as responding efficiently to etomoxir and perhexiline (“high responders”). Two other groups of cells show moderate response with values close to the mean (“intermediate responders”) and low response (“low responders”) with a percentage of live cells above the mean. Data are means of triplicates ±SEM and are representative of three independent experiments.
(B) Representative cell death assays in four PDAC cells treated with perhexiline (7 μM) for 72 h compared with controls (vehicle DMSO). Cell survival was measured using an Annexin V apoptosis assay with PI, and the number below each panel is the percentage of viable cells (Annexin V-/PI- cells). The corresponding quantification (right) indicates the percentage of viable cells. Data are presented as mean ± SEM of three independent experiments performed in duplicates.
(C) Dose-response combination assays were performed in four selected PDAC cells according to the sensitivity to Perhexiline. Cells were treated for 72 h with increasing doses of gemcitabine, perhexiline (5 μM), or the combination, and cell viability was measured by crystal violet assay (the curves are shown in Figure S2D). Graphs show the percentage of live cells of each treatment: gemcitabine at one dose (1 nM), perhexiline (5 μM), or the combination. To determine the synergistic effect in the combination treatments, we calculated the predicted values by multiplying the percentage of live cells in the gemcitabine and perhexiline groups, according to the method previously described.28 Then, when the observed value for the combination is lower than the predicted value, we consider it as a synergistic effect. Data are the mean ± SEM of two independent assays performed in triplicates.
(D) Representative cell death assay of PDAC084T cells treated for 24 h with gemcitabine (1 μM), perhexiline (7 μM), or the combination. The corresponding cell survival quantification (%) is shown below. Data are mean ± SEM of two independent assays performed in duplicates. p values from Student’s t test; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Interestingly, no effect in any cell was observed upon trimetazidine treatment compared with the non-treated cells at concentrations lower than 1 mM (Figure S2B). Here is to notice that the concentration of etomoxir used for the Seahorse dependency assay in Figure 1 (4 μM) does not affect cell viability at all in any cell. No correlation was identified between response to perhexiline and mitochondrial fatty acids dependency seen in Figure 1B (not shown) or with the cell replication rate (Figure S2A). By contrast, we found a negative correlation between the basal mitochondrial respiration (basal OCR) previously reported by our group18 and the percentage of live cells with etomoxir at 62.5 μM and perhexiline at 7 μM (Figure S2C), which reached the statistical significance in the case of perhexiline (this significance was lost when the PDAC084T with the highest OCR was removed from the graph, Figure S2C). This suggests that the basal OXPHOS status could predict response to perhexiline.
In our next investigations, we decided to work with perhexiline since this compound was shown to be a potent inhibitor of PDAC cell viability at low concentrations. First, we assessed the mechanism by which perhexiline decreases cell viability. We used four different PDAC cells from Figure 2A that show different sensitivities to perhexiline (high – PDAC084T, intermediate – PDAC012T, and low – PDAC022T, and PDAC032T-responders). We found that perhexiline induces cell death by apoptosis at low concentration (7 μM) in the highly sensitive PDAC084T cells with a significant decrease in cell survival, which was not the case in the other cells (Figure 2B).
Furthermore, we wondered whether treating cells with perhexiline in combination with the standard chemotherapy gemcitabine could increase the efficacy of the latter. Thereby, we treated PDAC cells with perhexiline at low concentration (5 μM that has no impact on cell viability) combined with increasing concentrations of gemcitabine, and we evaluated the synergistic effect by two methods (Figures 2C and S2D). Interestingly, combining perhexiline with a very low gemcitabine concentration (1 nM) sensitized all the cells to cell death, showing a synergistic effect. Finally, we performed apoptosis assays treating the high responder PDAC084T cells with gemcitabine, perhexiline, or the combination, proving that the combination treatment increased the efficacy of the chemotherapy in terms of apoptosis (Figure 2D).
Collectively, these results indicate that the primary PDAC cells show different sensitivities to the FAO inhibitors etomoxir and perhexiline (high/intermediate vs. low responders). More importantly, treatment with perhexiline amplifies the antitumor activity of gemcitabine by decreasing cell viability and increasing apoptosis.
The combination of gemcitabine with perhexiline enhances the energetic and oxidative stress induced by perhexiline in primary pancreatic cancer cells, independently of FAO inhibition
To continue deciphering the synergism between perhexiline and gemcitabine in the high perhexiline responder PDAC084T cells, we assessed the impact of both drugs on mitochondrial respiration using the Seahorse oxygraphy. The Seahorse assay was done 6 h after treatment, when no cell death is observed. We found that mitochondrial respiration was not affected by gemcitabine but that perhexiline decreased basal and maximal mitochondrial respiration of about 50% (Figure 3A). More importantly, the combination of gemcitabine and perhexiline drastically decreased basal and maximal mitochondrial respiration, ATP production by mitochondria, and spare respiratory capacity, leading cells to an energetic crisis (Figure 3A). We performed the same experiment in the low perhexiline responder PDAC032T cells, observing that perhexiline also decreased basal mitochondrial respiration of about 40% (Figure S3A). Interestingly, gemcitabine also decreased basal mitochondrial respiration (35%) compared to controls in the PDAC032T cells (Figure S3A). Concerning the combination treatment, in the same way as with PDAC084T cells, there was a huge decrease in mitochondrial respiration and ATP production.
Figure 3.

The combination of gemcitabine with perhexiline enhances the energetic and oxidative stress induced by perhexiline in primary pancreatic cancer cells
(A) Mitochondrial respiration (oxygen consumption rate, OCR) was measured on a Seahorse oxygraph in PDAC084T cells 6 h after the start of treatment (no cell death was observed at this time point) with gemcitabine (1 μM), perhexiline (10 μM), or combination treatment at same concentrations (DMSO 0.05% was used as vehicle for the controls). The basal and maximal respiration, ATP production by mitochondria, and spare respiratory capacity were calculated (graphics on the right). Values are presented as percentage of the vehicle-treated. Data are mean ± SEM of three independent experiments performed in triplicates. To calculate p values, one-way ANOVA test was used; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(B) PDAC084T cells were treated with gemcitabine (1 μM), perhexiline (7 μM), or the combination for 24 h. Then, total ROS and mitochondrial superoxide anion (O2.-) levels were measured by flow cytometry with the CellROX orange and MitoSOX red probes, respectively. Data are expressed as mean ± SEM and are representative of three independent experiments performed in triplicates (CellROX) or four independent assays done in duplicates (MitoSOX). To calculate p values, one-way ANOVA test was used; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(C) PDAC084T cells were treated with gemcitabine (1 μM), perhexiline (7 μM), or the combination for 24 h; then cells were fixed and stained with BODIPY 493/503 for lipid droplet detection and DAPI. Representative fluorescence microscopy images from three independent experiments performed in duplicates are shown. Scale bar: 50 μM. The graph on the right shows the quantification of BODIPY staining mean area (±SEM) of the three independent experiments. p values calculated from t test; ∗p < 0.05, ∗∗p < 0.01.
(D) PDAC084T cells were treated with gemcitabine (1 μM), perhexiline (7 μM), or the combination for 24 h; then cells were fixed and stained with BODIPY 581/591 C11 for lipid peroxidation detection and DAPI. Representative fluorescence microscopy images from three independent experiments done in duplicates are shown. Scale bar: 50 μM. The graph on the right shows the quantification of BODIPY staining mean area (±SEM) of the three independent experiments. p values calculated from t test; ∗∗p < 0.01, ∗∗∗p < 0.001.
Mitochondria are a major source of reactive oxygen species (ROS), and OXPHOS inhibition could lead to ROS accumulation due to increased leaky electrons partially reducing oxygen into superoxide anion. In addition, several works using perhexiline in different cancers showed that it is cytotoxic by inducing oxidative stress and apoptosis.28,33 Thus, we measured total ROS levels and superoxide anion in PDAC084T cells under treatment. As shown in Figure 3B, gemcitabine and perhexiline alone increased the ROS level, and the combination of both was even more potent in ROS accumulation. Moreover, perhexiline increased mitochondrial ROS (superoxide anion) level compared to control, which was not the case for gemcitabine. However, the combination of the two drugs induced a much greater mitochondrial ROS accumulation than the compounds alone (Figure 3B). These data indicate an oxidative stress state induced by the drugs, through increased mitochondrial superoxide production in the case of perhexiline.
Lipid droplets (LDs) are essential components of the cellular stress response.34 Moreover, FAO inhibition by perhexiline could lead to an excess of free fatty acids diverted toward triglycerides synthesis and LDs formation. Therefore, LDs content was monitored by fluorescence microscopy (BODIPY 493/503 staining) in the high responder PDAC084T cells treated with gemcitabine, perhexiline, or the combination (Figure 3C). Interestingly, the combination of the two drugs induced far more LDs than the drugs alone, whose single effect was not significant compared to control cells. Furthermore, FAO inhibition by perhexiline could increase the level of free unsaturated fatty acids and sensitize cancer cells to lipid peroxidation. Thus, we evaluated lipid peroxidation by fluorescence microscopy and flow cytometry using the BODIPY 581/591 C11 probe (Figures 3D and S3B). Our results show that perhexiline induced lipid peroxidation confirming our hypothesis; however, the combination treatment was not more efficient than perhexiline alone. Thus, we propose that the mechanism behind the synergy is the amplification of energy and oxidative stress induced by gemcitabine and perhexiline, this oxidative stress leading to apoptosis.
Finally, we assessed whether perhexiline effect was due to FAO inhibition in PDAC cells. First, we performed a radioactive labeling assay using 3H-palmitate in the four primary PDAC cells from Figures 2B and 2C, and we observed that perhexiline treatment did not decrease FAO levels except in PDAC022T cells, which was not significant (Figure S3C). Second, we tried the Seahorse Agilent Palmitate assay kit, which requires the removal of serum from the cell culture medium for 6 h before performing the Seahorse assay: this fasting induced the death of the primary cells thus preventing to obtain results. Third, we did octanoate or sodium acetate (acetyl-CoA precursors independently of FAO) supplementation to address the possibility that these metabolites could rescue the perhexiline-induced OCR decrease if this one is due to FAO inhibition. The data obtained by Seahorse did not show rescue by octanoate or acetate supplementation (Figure S3D). Importantly, we must add here that contrary to etomoxir, no OCR decrease was observed upon direct perhexiline addition during Seahorse assay (data not shown), which is another argument pointing to perhexiline-induced energetic stress independently of FAO inhibition.
Altogether, these data provide insight into the cooperative mechanism of perhexiline activity in combination with gemcitabine and demonstrate that the latter enhances the energetic and oxidative stress induced by perhexiline in pancreatic cancer cells.
Perhexiline in combination with gemcitabine induces complete pancreatic cancer regression in vivo in the high responder PDAC084T xenograft model
We then addressed the question of the impact of perhexiline on chemotherapeutic response in vivo. We worked with xenografts using the four primary PDAC cells analyzed above that were selected regarding their in vitro sensitivity to perhexiline (Figure 2A): the high responder PDAC084T cells, the intermediate responder PDAC012T, and the low responder cells PDAC022T and PDAC032T. Tumor-bearing mice were treated during one month using gemcitabine, perhexiline (except PDAC022T), or the combination of both; control mice were vehicle-injected with DMSO and/or PBS. Figure 4A shows the tumor volume upon treatment in the four PDAC xenografts, in which we observed that perhexiline alone did not have any impact on tumor growth compared with control mice. Treatment with gemcitabine alone was arresting the tumor growth in the high and intermediate responder xenografts, as well as in the low responder PDAC022T, suggesting a cytostatic effect. By contrast, the low responder xenograft PDAC032T showed a high sensitivity to gemcitabine alone, which induced tumor regression.
Figure 4.

Perhexiline in combination with gemcitabine induces complete pancreatic cancer regression in vivo in the high responder PDAC084T xenograft
(A) Left. Schematic of experimental protocol. Pieces of tumor from PDX were implanted in the subcutaneous space of recipient female Swiss nude mice. When tumors reached 200 mm3 volume, mice were assigned to treatment groups and treated for one month. Right and below. Tumor volume in four different PDAC PDX (PDAC084T, PDAC012T, PDAC022T, and PDAC032T) treated during one month with gemcitabine (120 mg/kg IP twice a week), perhexiline (5 mg/kg IP every other day), gemcitabine plus perhexiline (with the same indications), and vehicle. PDAC084T (high responder). Photos of representative tumor-bearing mice (left) and tumor volume during one month treatment (right). Perhexiline not only enhances the antitumoral effect of gemcitabine but also results in complete tumor regression. PDAC012T (intermediate responder). The combination treatment shows a better efficiency than gemcitabine alone. PDAC022T and PDAC032T (low responders). No effect was seen upon combination treatment compared to gemcitabine alone. Data are presented as mean ± SEM. ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05 from two-way ANOVA test, compared to gemcitabine treatment alone.
(B) Representative microscopic images from PDAC084T tumor sections obtained in the middle-point of treatment with gemcitabine or gemcitabine plus perhexiline. Tissue sections were stained with hematoxylin phloxine saffron (HPS) and Masson’s Trichrome for histologic examination and fibrosis detection, respectively. Immunohistochemistry was performed staining Ki-67 and cleaved caspase-3 for proliferation and apoptosis detection, respectively. Scale bars: 100 μM for HPS and Ki-67, and 500 μM for Masson’s Trichrome and cleaved caspase-3. Quantification is shown below, and data are the mean ± SEM of the percentage of stained area of tumors from at least two mice per group. p values calculated from t test; ∗p < 0.05.
More importantly, we observed that the co-treatment with perhexiline and gemcitabine significantly potentiated the efficacy of gemcitabine in the high responder xenograft PDAC084T, and excitingly, induced complete tumor regression after one month of treatment. In the intermediate responder xenograft (PDAC012T), the mice treated with the combination showed a higher tumor growth inhibition compared with the chemotherapy alone, even if it did not reach statistical significance. Finally, in tumors from the low responder xenografts PDAC022T and PDAC032T, no impact was observed with the combination treatment compared with gemcitabine alone.
Altogether, we demonstrate that perhexiline enhances the antitumoral activity of gemcitabine in two of our pre-clinical mouse models. Moreover, combining perhexiline with gemcitabine resulted in complete tumor regression in the high responder PDAC084T xenograft. This suggests that applying this therapeutic strategy could notably improve the prognosis of PDAC in a subset of patients.
Furthermore, we addressed the question whether a simultaneous treatment with perhexiline and gemcitabine is necessary to induce the complete tumor regression in the PDAC084T model. For that, we treated a cohort of mice with gemcitabine alone during one month, followed by perhexiline treatment alone for one month (Figure S4A). Interestingly, we observed that this sequential therapeutic strategy did not induce complete tumor regression; however, it seemed to delay the tumor growth in comparison with the sole gemcitabine treatment. These results suggest that the optimal therapeutic sequence to obtain complete regression is simultaneous treatment. Moreover, we wondered what would be the outcome of the tumors that completely regressed. To this purpose, we followed the survival of cured mice after the one-month combination treatment, and we observed that tumors relapsed after several weeks (3 examples are shown in Figure S4B). To address the potential of our combination therapeutic strategy in this context, we treated the relapsed mice with a second cycle of perhexiline and gemcitabine treatment (Figure S4B). Remarkably, we observed that relapsed tumors remained sensitive.
To complete this in vivo study, we investigated the mechanism of cooperation between perhexiline and gemcitabine ex vivo (Figure 4B). Using the mouse model PDAC084T (high responder), we excised the tumors from mice under treatment. Importantly, we analyzed the tumors from controls when they were still small for a better comparison with the treated ones. Next, we performed histologic analysis (hematoxylin phloxine saffron [HPS] staining) and histochemistry with the Masson’s Trichrome staining to detect fibrosis and immunohistochemistry for proliferation (Ki-67) and apoptosis (cleaved caspase-3) markers. First, in the histology examination, we found that the tumors share characteristics of a moderately differentiated adenocarcinoma, showing sheets and groups of epithelial pleomorphic cells. Secondly, we observed that the tumors from mice treated with the combination showed larger fibrotic areas compared with the other groups, which suggest a tissue repair process following damage. Then, for the Ki-67 staining, a huge decrease in proliferation was observed in the gemcitabine and the combination groups in comparison with control mice. However, no difference was seen between gemcitabine and the combination. On the contrary, we found a higher staining for cleaved caspase-3 in the combination therapy tumors in comparison with gemcitabine alone, suggesting that perhexiline permits the gemcitabine-induced apoptosis in vivo, resulting in complete tumor regression.
Collectively, these data show that perhexiline boosts the antitumoral activity of gemcitabine by promoting cell death, resulting in complete tumor regression in the PDAC084T xenograft model. This observation suggests that combining perhexiline with chemotherapy is a promising strategy in some patients.
CPT1C is a key molecular actor behind the response to perhexiline in PDAC
The identification of patients that could respond to perhexiline combined with chemotherapy requires a better understanding of the underlying mechanisms. To decipher the key molecular actors related to this response, we performed transcriptomic (RNA sequencing [RNA-seq] and RT-qPCR) and metabolomic analyses. We first analyzed the RNA-seq data from PDAC cells obtained from PDX of the PaCaOmics cohort.35 Five PDAC cells of the high/intermediate perhexiline responder group and three from the low perhexiline responder group were analyzed. In these data, through a gene set enrichment analysis (GSEA) using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, we determined significantly upregulated or downregulated pathways between the two groups taking as reference the high/intermediate group (Figures 5A and S5). Interestingly, our analysis identified the “OXPHOS” and the “nucleotide and base excision repair” among the top enriched pathways (Figures 5A and S5). Concerning the OXPHOS enriched pathway, these results are in accordance with the correlation between response to perhexiline and the OXPHOS status (Figure S2C). Conversely, several pathways were found downregulated in the high/intermediate responder group. Among these, we were interested in particular in the linoleic acid and α-linolenic acid metabolism (Figures 5A and S5). Accordingly, we analyzed the metabolomic data obtained from the PaCaOmics cohort36 and compared the high/intermediate with the low responder group, showing a lower content of these polyunsaturated fatty acids (PUFAs) in the high responder group (Figure S6). Moreover, the GSEA identified downregulation of the CPT1C isoform, included in the fatty acid degradation pathway (Figures 5A and S5). Given that CPT1 is the rate-limiting enzyme of FAO, we decided to further explore the CPT1C isoform.
Figure 5.

CPT1C is a key molecular actor behind the response to perhexiline in PDAC
(A) Gene set enrichment analysis showing top upregulated and downregulated pathways in the high responder PDAC cells vs. low responder to FAO inhibition with perhexiline. Oxidative phosphorylation is upregulated, and linoleic acid metabolism and fatty acid degradation are downregulated. Specific pathways are described in each box; CPT1C isoform is found in the downregulated fatty acid degradation pathway.
(B) Gene expression values of CPT isoforms using transcriptomic (RNA-seq) data from 43 primary PDAC cells derived from PDX.
(C) The mRNA levels of CPT1 (A, B, C) and CPT2 isoforms were measured by RT-qPCR in three PDAC cells used in the in vivo experiments. The high responder PDAC084T cells exhibit significantly lower mRNA levels of the CPT1A and CPT1C isoforms compared to the intermediate and low responder PDAC cells (PDAC012T and PDAC022T, respectively). Data are presented as mean ± SEM of three independent experiments performed in duplicates. p values from Student’s t test; ∗p < 0.05, ∗∗∗p < 0.001.
(D) CPT1C mRNA levels correlate with response to perhexiline treatment. The plot shows the mean of CPT1C mRNA levels in the previous panel C, compared with the relative cell viability with perhexiline at 7 μM from Figure 2A. Spearman’s correlation coefficient was used.
(E) CPT mRNA levels in PDAC084T cells were measured by RT-qPCR after 24 h treatment with gemcitabine (1 μM), perhexiline (5 μM), or the combination. Higher mRNA levels of CPT1B and CPT1C are observed in the gemcitabine-treated cells in comparison with the other treatments. Moreover, such increase in gene expression upon chemotherapy seems to be counteracted by perhexiline treatment. Data are presented as mean ± SEM of three independent experiments performed in duplicates. p values shown were calculated from the F-test of equality of variances.
(F) Genetic downregulation of CPT1C by transient transfection of CPT1C siRNA or scramble siRNA (negative control) in the PDAC Panc-1 cell line. Perhexiline treatment was started 24 h after the transfection when CPT1C downexpression was observed (Figure S7), and viability was evaluated by counting the cells 72 h later. Data are presented as mean ± SEM of triplicates in two independent experiments. p values from unpaired Student’s t test; ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(G) Kaplan-Meier survival curves using transcriptomic data from 43 primary PDAC cells derived from PDX.18 Patients are divided into high and low CPT1C gene expression groups (n = 21 and n = 22, respectively; the cutpoint was 6). p values from the log rank test and Wilcoxon test.
Next, we examined the expression of the genes encoding the CPT1 (A, B, and C) and CPT2 enzymes using the RNA-seq data from 43 PDAC cells obtained from PDX of the PaCaOmics cohort35 (Figure 5B). Interestingly, the CPT1C isoform showed high heterogeneity among patients, which was not the case for the other isoforms. To further address this point, we assessed the mRNA levels of the four CPT isoforms by RT-qPCR in the PDAC cells used in the in vivo experiments, and we observed that the mRNA levels of the CPT1A and CPT1C isoforms in the PDAC084T cells were significantly lower in comparison with the other cells (Figure 5C). Moreover, we found that the CPT1C mRNA levels correlated with the response to perhexiline treatment (Figure 5D). FAO is activated upon chemotherapy treatment,28 and to address this question in our model, we also measured the CPT mRNA levels in PDAC084T cells upon 24-h treatment with gemcitabine (1 μM), perhexiline (5 μM), or the combination (Figure 5E). Interestingly, we observed significantly higher mRNA levels for the CPT1B and CPT1C isoforms in the gemcitabine-treated cells in comparison with the other treatments. Moreover, such increase in gene expression upon chemotherapy seemed to be counteracted by treatment with perhexiline. We then performed genetic experiments to knock down the CPT1C enzyme followed by cell viability experiments. The low responder primary PDAC cells (PDAC012T and PDAC022T, showing highest expression of CPT1C) are very sensitive to transfection as all the primary cells from the PaCaOmics PDX cohort. For this reason, we used the classical PDAC cell line Panc-1 (Figures 5F and S7), and we found that downregulation of CPT1C decreases cell count, similarly to what was previously reported.37 Of more relevance for this work, we demonstrate that downregulation of CPT1C increases sensitivity to perhexiline. These data show that part of the perhexiline-induced cell death is mediated by CPT1C. Finally, Figure 5G shows that patients with high CPT1C gene expression have a longer survival than patients with low expression, pointing to the relevance of CPT1C as a prognostic biomarker in pancreatic cancer.
Collectively, our molecular analyses point to the CPT1C isoform as a key actor in the response to perhexiline and in the mechanism of cooperation between perhexiline and gemcitabine to induce complete pancreatic cancer regression in the PDAC084T xenograft.
Discussion
This study provides the demonstration that FAO is a novel therapeutic vulnerability in PDAC. Through a functional analysis, we showed a marked dependency of mitochondrial respiration toward fatty acids in the 21 tested primary PDAC cells, which has not been reported so far. By contrast, dependency toward glucose was low or moderate and dependency for glutamine low or absent, which was not expected considering the well-documented “glutamine addiction” in pancreatic cancer.38,39,40,41 The importance of lipid metabolism in PDAC metabolic reprogramming, which was initially demonstrated by our laboratory,42 was reinforced by recent studies.36,43 This study goes one step further by using this knowledge for clinical purpose, developing a new pre-clinical strategy in PDAC using the recognized FAO inhibitor perhexiline to overcome therapeutic resistance.
We used perhexiline in combination with chemotherapy in vitro and in vivo, unveiling antitumoral activity in some patients. We tested drugs inhibiting FAO based on the relevant literature.20,21 Etomoxir, perhexiline, and trimetazidine are compounds commonly used in heart diseases in the clinic, which were repositioned for cancer research. Etomoxir is a drug commonly used to inhibit CPT1 in cancer research. However, even if CPT1 blockade by etomoxir (4 μM) led us to identity FAO dependency in our functional study, an important decrease in PDAC cell viability was seen only at concentrations higher than 60 μM. In triple-negative breast cancer (TNBC) cell lines, etomoxir treatment (200 μM) had a marked effect on cell proliferation but no impact on cell viability.22 Yet, the authors demonstrated both FAO inhibition (metabolite analysis) and reduced tumor growth in vivo. However, Yao and collaborators showed that treatment with high etomoxir concentration (200 μM) inhibited respiratory complex I in isolated mitochondria from TNBC cells and thus presented an off-target effect.32 Nevertheless, in our study, the high responder PDAC cells show important decrease in cell viability at 62.5 μM of etomoxir, suggesting that this decrease could be a consequence of FAO inhibition instead of an off-target effect.
Perhexiline is another FAO inhibitor that targets CPT1 (and to a lesser extent CPT2) and that has gained recent consideration in the cancer research field. In our study, we found that perhexiline notably inhibited pancreatic cancer cells’ viability at concentrations lower than 14 μM, and at only 7 μM in the high responder cells PDAC084T. These results are in accordance with other studies in different cancer subtypes, including gastric and colorectal cancer,28 prostate cancer,25 and leukemia.26 We and others demonstrated that the mechanism of perhexiline-induced cell death is apoptosis, and importantly, this effect is selective in cancer cells without affecting non tumoral cells.26,28
Trimetazidine, another FAO inhibitor tested in our study, is a partial inhibitor of the terminal enzyme in β-oxidation, long-chain 3-ketoacyl coenzyme A thiolase.44 Here, high concentrations of the drug (1 mM) were needed to induce a decrease in PDAC cell viability, in accordance with the range of concentrations used in other cancers.30,43
Given the fact that perhexiline arose to be a potent inhibitor of PDAC viability at low concentrations, we decided to continue our investigation using this FAO inhibitor. We observed a correlation between response to perhexiline and the basal OXPHOS status defined in our previous study of primary PDAC cells;18 high responders to perhexiline correspond to high OXPHOS PDAC cells. Also, PDAC cells with high OXPHOS profile showed resistance to the chemotherapeutic gemcitabine. Hence, we wondered whether treatment with perhexiline would sensitize PDAC cells to gemcitabine. Excitingly, our results showed that perhexiline sensitized all PDAC cells to gemcitabine treatment in vitro with a synergistic effect.
The molecular activity of perhexiline is still a matter of investigation. Whereas it was considered as a CPT1/CPT2 dual inhibitor and described as an FAO inhibitor, some recent reports show that it does not inhibit FAO.33,45 In this work, we were unable to show any cytotoxic activity of perhexiline through FAO inhibition in pancreatic cancer cells. Several works using perhexiline in different cancers showed that it is cytotoxic by inducing oxidative stress and apoptosis.28,33 We showed induction of oxidative stress in perhexiline-treated PDAC cells, which is amplified by co-treatment with gemcitabine (Figure 2), and this suggests that the cytotoxic synergy between gemcitabine and perhexiline relies on pro-apoptotic oxidative stress.
Furthermore, we evaluated the therapeutic potential of perhexiline alone or in combination with gemcitabine in xenograft models of PDAC in vivo. Our findings indicate that perhexiline does not exert antitumor effect as a single agent in vivo, similarly to what was observed in xenografts models of neuroblastoma.27 By contrast, perhexiline alone exhibited antitumoral activity in gastrointestinal cancers and leukemia26,28; however, a higher dose of the drug was used (8 mg/kg versus 5 mg/kg in our study). Another reason for our outcome could be that PDAC cells reside in a rich tumor microenvironment (contrary to in vitro conditions), which could thwart perhexiline-induced cytotoxicity.
Of major importance is that, in one of our xenograft models (PDAC084T), the simultaneous treatment with perhexiline and gemcitabine induced complete pancreatic cancer regression. These findings strongly suggest the potential use of perhexiline in combination with chemotherapy in a clinical context. Furthermore, we evaluated the therapeutic potential of perhexiline combined with gemcitabine comparing a simultaneous versus sequential treatment regimen, showing that the first is necessary to induce complete tumor regression. Moreover, the PDAC084T xenografts that exhibit complete tumor regression showed tumor relapse after some time; however, tumors remained sensitive to a second cycle of combination treatment, thus reinforcing its potential. Importantly, the clinical use of perhexiline is approved in Australia and some Asian countries for the treatment of heart disease since 1970.20 Perhexiline can cause neuro- and hepatoxicity in a small number of patients—“poor-metabolizers”—who have altered activity of CYP2D6 (cytochrome P450 family 2 subfamily D member 6); however, this problem can be overcome by dose reduction and/or genetic testing.25
Our transcriptomic analysis to identify molecular key candidates behind response to perhexiline pointed out to the CPT1C isoform. The CPT1 family of proteins, shuttling long-chain fatty acids from cytosol into mitochondria, constitutes the rate-limiting step of FAO.20,46 This family comprises three tissue-specific isoforms: CPT1A (liver form), CPT1B (muscle form), and CPT1C (brain form, enzymatic inactive) encoded by three paralogous genes.47,48 CPT1A and CPT1B show ubiquitous expression in human organs and demonstrate considerable similarities; however, CPT1A is 30- to 100-fold more resistant to allosteric inhibition by malonyl CoA and thus more likely to be enzymatically active in cancer cells.47 CPT1C was considered as an exclusive brain isoform; however, Zaugg and collaborators demonstrated that cancer cells under metabolic stress express CPT1C and that this isoform could be a regulator of fatty acids homeostasis under this metabolic stress.48 Specifically, these authors showed that CPT1C promoted FAO and ATP production, tumor growth, rescue from metabolic stress, and resistance to mTOR complex 1 (mTORC1) inhibitors. Nevertheless, some publications report that the CPT1C isoform shows differences from the other CPT1A and CPT1B isoforms, with a non-mitochondrial localization and a lack of acyltransferase activity, which both remain controversial.49,50,51,52 Finally, CPT2 is an enzyme that has attracted less attention in the field; however, Wang and collaborators determined its role in chemotherapy resistance and as a target of perhexiline besides CPT1.28
In our study, we observed that the three CPT1 isoforms and CPT2 are constitutively expressed in primary PDAC patients, and we showed a marked heterogeneity of CPT1C expression between patients. This finding, together with the GSEA showing downregulation of CPT1C in the high responder group, prompted us to further explore the role of this isoform in PDAC. We found a correlation between response to perhexiline and CPT1C mRNA levels, the high responder PDAC084T being the one with lowest CPT1C expression. Since FAO is activated upon chemotherapy treatment,28 we evaluated the impact of gemcitabine, perhexiline, and the combination treatment on CPTs expression. Interestingly, we observed significantly higher mRNA levels of CPT1B and CPT1C isoforms in the gemcitabine-treated PDAC084T cells in comparison with the other treatments. Moreover, such increase in gene expression upon chemotherapy seems to be counteracted by the FAO inhibition with perhexiline. Collectively, these data suggest that low expression of CPT1C in PDAC tumors mechanistically underlies the response to perhexiline in combination with chemotherapy. This novel therapeutic approach warrants further investigation in the clinic.
Limitations of the study
This study demonstrates that perhexiline in combination with the chemotherapy gemcitabine is efficient in some PDAC tumors (high responders), showing complete tumor regression in one PDAC xenograft. Even though perhexiline is a well-recognized FAO inhibitor by targeting CPT1 and CPT2, we cannot exclude that it can act on other targets besides CPT. This work demonstrates that perhexiline induces energy stress and cell death independently of FAO inhibition. Its molecular activity and targets have yet to be identified in pancreatic cancer cells.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal [EPR3610] to Ki67 | Abcam | Cat#ab92742; RRID:AB_10562976 |
| Rabbit polyclonal to Cleaved Caspase-3 (Asp175) | Cell Signaling Technology | Cat#9661; RRID:AB_2341188 |
| Goat polyclonal anti-rabbit IgG | Agilent | Cat#E0432; RRID:AB_2313609 |
| Goat polyclonal anti-rabbit IgG | Vector Laboratories | Cat#PK-4001; RRID:AB_2336810 |
| Biological samples | ||
| Primary human PDAC cells from PaCaOmics cohort | Gayet et al.53 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Serum-free ductal media (SFDM) | Gayet et al.53 | N/A |
| Gemcitabine (Gemzar) | Eli Lilly & Co. | N/A |
| Etomoxir sodium salt hydrate | Sigma-Aldrich | Cat#E1905 |
| Perhexiline maleate salt | Sigma-Aldrich | Cat#SML0120 |
| Trimetazidine dihydrochloride | Sigma-Aldrich | Cat#653322 |
| Sodium octanoate | Sigma-Aldrich | Cat#C5038 |
| Sodium acetate | Sigma-Aldrich | Cat#S2889 |
| Critical commercial assays | ||
| Seahorse XF Cell Mito Stress Test Kit | Agilent Technologies | Cat#103015-100 |
| Seahorse XF Mito Fuel Flex Test | Agilent Technologies | Cat#103260-100 |
| Pacific Blue™ Annexin V Apoptosis Detection Kit with PI | BioLegend | Cat#640928 |
| BODIPY™ 493/503 | Invitrogen™ | Cat#D3922 |
| BODIPY™ 581/591 C11 (Lipid Peroxidation Sensor) | Invitrogen™ | Cat#D3861 |
| CellRox Orange | Invitrogen™ | Cat#C10493 |
| MitoSOX Red | Invitrogen™ | Cat#M36008 |
| Trichrome Stain Kit (Connective Tissue Stain) | Abcam | Cat#ab150686 |
| VECTASTAIN® ABC-HRP Kit, Peroxidase (Rabbit IgG) | Vector Laboratories | Cat#PK-4001 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| GoScript™ Reverse Transcription System | Promega | Cat#A5001 |
| Deposited data | ||
| Original and analyzed data | This paper | Mendeley data: https://doi.org/10.17632/8yg3837dsc.1 |
| PaCaOmics RNA-seq data | Nicolle et al.54 | Accession number E-MTAB-5039 |
| PaCaOmics metabolome data | Kaoutari et al.36 | N/A |
| Experimental models: Cell lines | ||
| Panc-1 | ATCC | Cat#CRL-1469 |
| Experimental models: Organisms/strains | ||
| Mouse: Female Swiss nude, SOPF (4 weeks old) | Charles River | Cat#Crl:Nu(lco)-Foxn1nu |
| Oligonucleotides | ||
| CPT1A Forward CCA GAC GAA GAA CGT GGT CA | Brown et al.58 | N/A |
| CPT1A Reverse ATC TTG CCG TGC TCA GTG AA | Brown et al.58 | N/A |
| CPT1B Forward CTG GGC TAT GTG TAT CCG CC | Brown et al.58 | N/A |
| CPT1B Reverse GCA CAG ACT CTA GGT ACC GC | Brown et al.58 | N/A |
| CPT1C Forward TTT GCC TCG TGT TTG TGG G | Zhang et al.59 | N/A |
| CPT1C Reverse CAG CCG TGG TAG GAC AGA A | Zhang et al.59 | N/A |
| CPT2 Forward GTA GCA CTG CCG CAT TCA AG | Brown et al.58 | N/A |
| CPT2 Reverse GCC ATG GTA CTT GGA GCA CT | Brown et al.,58 | N/A |
| CPT1C siRNA, ON-TARGETplus siRNA Reagents | Dharmacon | Cat# L-008824-01-0020 |
| siRNA pool, ON-TARGETplus Non-targeting Pool | Dharmacon | Cat# D-001810-10-20 |
| INTERFERin® transfection reagent | Polyplus-transfection | Cat# POL101000028 |
| Software and algorithms | ||
| Fiji ImageJ | https://imagej.nih.gov/ij | N/A |
| CALOPIX digital software | Tribun Health | N/A |
| FlowJo version 10.0.7 | https://www.flowjo.com/solutions/flowjo | N/A |
| GraphPad Prism | https://www.graphpad.com/ | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alice Carrier (alice.carrier@inserm.fr).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Primary human Pancreatic ductal adenocarcinoma (PDAC) cells
Primary human PDAC cells were acquired from the PaCaOmics cohort.35,53,54,55 The obtaining of these primary PDAC cells has been well described by our group.18 Briefly, PDAC patient samples were used to generate Patient-Derived Xenografts (PDX), and primary cells were obtained from these PDX as reported.53,54 The 21 primary PDAC cells used in this study were previously described by our group.18 Cells were cultured at 37°C with 5% CO2 in a humidified atmosphere in Serum Free Ductal Media (SFDM), which is a complex medium supporting the PDAC primary cell growth and containing DMEM-F12, nicotinamide, glucose, hormones, growth factors and Nu-serum providing a low-protein alternative to fetal bovine serum (FBS). Only the cells PDAC087T and PDAC001T were cultured in a different medium: PDAC087T in DMEM (GIBCO, Life Technologies) + 10% FBS, and PDAC001T in SFDM:DMEM (1:1) + 10% FBS. These 21 PaCaOmics primary PDAC cells are all KRAS mutated except the PDAC012T.18 Cells in exponential growth were harvested using Accutase (Gibco) and passed once per week; all cells were maintained in culture for a maximum of 10 passages. Cell lines were monthly tested for Mycoplasma contamination and found to be negative. Next Generation Sequencing of RNA (RNA-Seq) from these primary PDAC cells was carried out as described and published elsewhere (accession number is E-MTAB5039).54
PDAC cell line
The classical human PDAC cell line Panc-1 was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in DMEM supplemented with 10% FBS. The authentication was performed by the ATCC, and absence of Mycoplasma contamination was tested monthly.
Xenograft mouse models of PDAC
We performed subcutaneous xenografts in immunodeficient mice using four different primary PDAC cells based on in vitro results. Recipient mice were 5- to 6-week-old athymic females, Swiss nude mice, SOPF (Specific and Opportunistic Pathogen Free) health status, strain Crl:Nu(lco)-Foxn1nu (Charles River, France). To obtain the xenografts, subcutaneous tumors from initial mouse donors were removed and finely minced with a scalpel. Then, 150 mg of tumor’s pieces were mixed with 50 μl of Matrigel and implanted with a trocar (10 Gauge) in the subcutaneous space of isoflurane-anesthetized mice. Tumor volume was measured twice per week using a digital caliper and tumor volume was calculated with the formula V = lenght x (width)2/2.
When xenografts reached ∼ 200 mm3 volume, mice were randomly assigned in a treatment group in which the average of all tumors was 200 mm3. Treatments were administered by intraperitoneal injection during one month as follows: Gemcitabine 120 mg/kg twice a week, perhexiline 5 mg/kg every other day, combination of gemcitabine plus perhexiline at the same dose than the drugs alone. Vehicle-injected mice (controls) were injected with PBS in the case of gemcitabine controls or 3% DMSO in PBS for combination treatment controls. Mice whose tumor volume reached 1.5 cm3 were ethically sacrificed and tumors removed. All mice were kept under specific pathogen-free conditions and according to the current European regulation; the experimental protocol was approved by the Institutional Animal Care and Use Committee (#16711).
Method details
Real time metabolic analysis
We used the Seahorse Bioscience XFe24 Extracellular Flux Analyzer (Agilent) for measuring the cellular Oxygen Consumption Rate (OCR, pmoles/min) to determine mitochondrial respiration. The day before the assay, cells at exponential growth were seeded into Seahorse 24-well plates (100 μl volume of SFDM/well) and cultured at 37°C with 5% CO2. One day after seeding, the culture media was replaced by 500 μl of OXPHOS assay media (DMEM without phenol red [Sigma-Aldrich reference D5030], 143 mM NaCl, 2 mM glutamine, 1 mM sodium pyruvate and 10 mM glucose, pH 7.4). Then, the plate was pre-incubated for 1 hour at 37°C in a non-CO2 incubator before carrying out the assay in the Seahorse oxygraph.
XF Mito Fuel Flex Test
The Seahorse XF Mito Fuel Flex Test Kit was used to address the mitochondrial dependency and flexibility of 21 primary PDAC cells to oxidize three main fuels: glucose, glutamine, and long-chain fatty acids (LCFA). One day before the assay, cells were seeded at a specific density ensuring 70-80% confluence the day of analysis (Table S1). The day after, the culture media was replaced by OXPHOS assay media and the plate was pre-incubated for 1 hour at 37°C in a non-CO2 incubator. Then, the oxidation of each fuel was determined by measuring the Oxygen Consumption Rate (OCR) of cells in the presence of specific inhibitors used in different combinations according to manufacturers' instructions: UK5099 at 2 μM, BPTES at 3 μm, and etomoxir at 4 μM, to inhibit the mitochondrial pyruvate carrier (glycolysis), glutaminase (glutaminolysis), and CPT1 (fatty acid oxidation), respectively. Note that etomoxir is not toxic at this low concentration, contrary to high doses inducing cell death through off-target effect (inhibition of mitochondrial complex I). The calculation of dependency and flexibility was done according to Seahorse XF Mito Fuel Flex Test Kit instructions.
XF Cell Mito Stress Test
We addressed the impact of gemcitabine, perhexiline, and the combination treatment on mitochondrial respiration in the primary PDAC cells PDAC084T and PDAC032T. Cells were seeded in the Seahorse plates (30 000 cells/well) and the day after, the media was replaced with media containing vehicle DMSO (0.05%), gemcitabine (1 μM), perhexiline (10 μM), or the combination of gemcitabine and perhexiline at the same concentration that drugs alone. After 6 hours of treatment, the media was replaced with OXPHOS assay media and the plate was pre-incubated for 1 hour at 37°C in a non-CO2 incubator. OCR was measured under basal conditions, and then after sequential injections of oligomycin (1 μM), carbonyl cyanide-p-trifluoromethoxyphenyl-hydrazon (FCCP; 0.5 μM for PDAC084T and 1 μM for PDAC032T cells), and 0.5 μM of rotenone plus antimycin A. Oligomycin is a respiratory Complex V inhibitor that allows to calculate ATP production by mitochondria, and FCCP is an uncoupling agent allowing the determination of the maximal respiration and the spare capacity. Finally, rotenone/antimycin A are Complex I and III inhibitors, respectively, that are injected to stop mitochondrial respiration enabling the calculation of background (i.e., non-mitochondrial respiration driven by processes outside the mitochondria). Supplementation assays with octanoate or sodium acetate metabolites were done with the primary PDAC048T cells. One day after seeding, cells were treated with DMSO (0.05%), octanoate (four concentrations tested from 0.1 to 5 mM), or sodium acetate (three concentrations tested from 1 to 5 mM), perhexiline (10 μM), or the combination of octanoate or sodium acetate and perhexiline at the same concentration that drugs alone. Six hours after treatment, the media was replaced with OXPHOS assay media and the plate was pre-incubated for 1 hour at 37°C in a non-CO2 incubator. OCR was measured under basal conditions, and then after sequential injections of oligomycin (1 μM), FCCP (0.5 μM), and rotenone plus antimycin A (0.5 μM).
Cell viability assays
We tested the sensitivity of PDAC cells to three different compounds targeting the Fatty Acid Oxidation (FAO) pathway: etomoxir, perhexiline, and trimetazidine (all provided by Sigma-Aldrich, Saint-Quentin Fallavier, France), and to the chemotherapeutic gemcitabine (Gemzar, Eli Lilly & Co). Cells were seeded in 96-well plates (5,000 cells per well) and 24 hours later, the medium was supplemented with increasing concentrations of the drugs in triplicates. For the combination treatments, we used perhexiline at 5 μM with increasing doses of gemcitabine. Viability was determined 72 hours later by the Crystal violet assay, which is independent from cell metabolism. For this, cells were fixed in glutaraldehyde (1%), washed twice with PBS, stained with Crystal violet (0.1%) for 10 min, and then washed three times with PBS. Crystals were solubilized in SDS (1%), and absorbance was measured at 600 nm using an Epoch-Biotek spectrophotometer.
We calculated the synergistic effect of the combination treatments as previously reported.28 To calculate the predicted values, the cell viability of gemcitabine-treated cells was multiplied by the cell viability of perhexiline-treated cells. A synergistic effect was considered when the observed value is lower than the predicted value.
Flow cytometry experiments
Cell death assays
Cells were seeded in 6-well plates in duplicate (150 000 cells/2 ml media/well) and the day after, the corresponding treatment was administered. We treated four PDAC cells with 7 μM of perhexiline for 72 h, or for 24 h with perhexiline alone (7 μM) or in combination with gemcitabine (1 μM) for PDAC084T cells. After treatments, cells were detached with pre-warmed accutase (Gibco), resuspended in Annexin V-binding buffer, and stained for 30 min with the BioLegend’s Pacific Blue™ Annexin V Apoptosis Detection Kit with propidium iodide (PI), according to manufacturer’s instructions. Finally, ten thousand events per sample were acquired in a MACSQuant-VYB (Miltenyi Biotec) and data analysis was done with the FlowJo software.
Lipid peroxidation, total ROS, and superoxide anion detection
Cells were seeded in 12-well plates in duplicate or triplicate (60 000 cells/1 ml media/well) and treatments were administered the day after. Cells were treated with perhexiline (7 μM), gemcitabine (1 μM) or the combination for 24 h. For lipid peroxidation detection, cells treated with erastin (2 μM) were used as positive controls. After treatments, the media was supplemented with either the BODIPY 581/591 C11 probe (lipid peroxidation sensor), CellROX Orange (total ROS) or MitoSOX (superoxide anion) at a final concentration of 2, 5 and 10 μM, respectively. Cells were incubated for 30 min (BODIPY and CellROX) or 20 min (MitoSOX) at 37°C, then harvested with pre-warmed accutase (Gibco) and resuspended in PBS for flow cytometry analysis. Five thousand events per sample were acquired in a MACSQuant-VYB (Miltenyi Biotec), and data analysis was done with the FlowJo software.
Fluorescence microscopy
Cells were seeded on coverslips in 12-well plates in duplicates (60 000 cells/1 ml media/well) and treatments were administered the day after. Cells were treated with perhexiline (7 μM), gemcitabine (1 μM) or the combination for 24 hours. Then, cells were washed twice with PBS and fixed with 4% paraformaldehyde (0.5 ml/well) during 20 min at room temperature. Next, cells were stained with the probes BODIPY 493/503 (3 μM) or BODIPY 581/591 C11 (2 μM), for 10 and 30 min, respectively. After incubation, coverslips were mounted on slides using the ProLongTM Gold antifade reagent with DAPI. Fluorescent images were acquired with a ZEISS Axio Imager 2 microscope, under 40x objective lens. For quantification, we used at least two pictures per sample (well) of each independent experiment. The mean area of BODIPY staining per picture was quantified with the Fiji ImageJ software.
FAO measurement
PDAC cells were seeded in 12-well plates in duplicates (50 000 cells/well) and treated with gemcitabine (1 μM), perhexiline (5 μM) or the combination for 16 h. Then, cells were incubated with 3H-labeled palmitate (coupled to BSA) and 1 mM carnitine (Sigma) for 2 h at 37°C. 3H2O produced during FAO was purified on DOWEX columns (Sigma) after TCA extraction and NaOH neutralization. Radioactivity was measured with a wallac reader (Trilux).
Xenografts mouse models of Pancreatic ductal adenocarcinoma (PDAC)
We performed subcutaneous xenografts in immunodeficient mice using four different primary PDAC cells based on in vitro results: PDAC084T, PDAC012T, PDAC022T, and PDAC032T. Recipient mice were 5- to 6-week-old athymic female, Swiss nude mice, SOPF (Specific and Opportunistic Pathogen Free) health status, strain Crl:Nu(lco)-Foxn1nu (Charles River, France). To obtain the xenografts, subcutaneous tumors from initial mouse donors were removed and finely minced with a scalpel. Then, 150 mg of tumor’s pieces were mixed with 50 μl of Matrigel (BD Biosciences) and implanted with a trocar (10 Gauge, Innovative Research of America, Sarasota, FL) in the subcutaneous space (upper dorsal region) of isoflurane-anesthetized mice. Tumor volume was measured twice per week using a digital caliper and tumor volume was calculated with the formula V = length x (width)2/2.
When xenografts reached ∼ 200 mm3 volume, mice were assigned in a treatment group in which the average of all tumors was 200 mm3. Treatments were administered by intraperitoneal injection during one month as follows: gemcitabine 120 mg/kg twice a week (Monday and Thursdays), perhexiline 5 mg/kg every other day (Mondays, Wednesdays, and Fridays), combination of gemcitabine plus perhexiline at the same dose and schedule than the drugs alone. Perhexiline stock solution is dissolved in DMSO, thus before each injection, it was diluted at 3% in PBS. Vehicle-injected mice (controls) were injected with PBS in the case of gemcitabine controls or 3% DMSO in PBS for combination treatment controls.
Mice whose tumor volume reached 1.5 cm3 during the one-month treatment were ethically sacrificed and tumors were removed. All mice were kept under specific pathogen-free conditions and according to the current European regulation; the experimental protocol was approved by the Institutional Animal Care and Use Committee (#16711).
Ex vivo analysis: Histopathology, histochemistry and immunohistochemistry
Tumor-bearing mice were sacrificed under gemcitabine, perhexiline or combination treatments (middle-point of one-month treatment). For control mice, we included small tumors (200 mm3 at Day 0) as well as bigger-size tumors (∼700 mm3 at the middle-point of the one-month treatment) for a better comparison with treated tumors. Tumor samples were fixed in 4% paraformaldehyde, dehydrated and embedded in paraffin. Serial 5 μm sections were cut and stained with Hematoxylin Phloxine Saffron (HPS) and Masson’s Trichrome (Abcam #ab150686) for histopathology and connective tissue detection, respectively.
For proliferation and apoptosis detection, immunohistochemistry was performed using the antibodies anti-Ki-67 (Abcam #ab92742) and anti-cleaved-Caspase 3 (Cell signaling [Asp175] #9661), respectively. Antigen retrieval was done in citrate buffer TRS pH 6 (Dako ref. S1699) in water bath (96°C) for 20 min, before quenching endogenous peroxidase activity (3% H2O2) and blocking solution incubation for 30 min. Tissue sections were then incubated with the primary antibodies anti-Ki-67 (1:200) and anti-cleaved-Caspase 3 (1:50) for one hour at room temperature (RT). Then, an appropriate biotinylated secondary antibody was applied for 30 min at RT (DAKO ref. E0432 or Vectastain Kit Vector ref. pk4001, for Ki-67 and cleaved-Caspase 3, respectively), followed by immunoreactivity visualization using peroxidase-conjugated Streptavidin-HRP (DAKO) for Ki-67 or the Vectastain ABC kit (Vector Laboratories) for cleaved-Caspase 3. Peroxidase activity was revealed using DAB substrate chromogen system (Dako ref. K3468) for 10 min at RT. Counter-staining in Mayer’s Hematoxylin was followed by a sodium bicarbonate (0.1%) clearing before final dehydration and mounting of the sections.
Tumor sections images were captured using the CALOPIX digital software (Tribun Health, France). Quantification data corresponds to the percentage of stained area of the totality of the tumor section of at least two mice per treatment, and it was done with the Fiji ImageJ software.
Differential gene expression analysis
Next Generation Sequencing of RNA (RNA-Seq) from PDAC primary cultures (PaCaOmics cohort) was carried out as described and published elsewhere (accession number is E-MTAB5039).54 We analyzed the RNA-seq data from five PDAC primary cultures (PDAC084T, PDAC082T, PDAC027T, PDAC012T, PDAC021T) of the high and intermediate perhexiline responder group, and three PDAC primary cultures (PDAC003T, PDAC032T, PDAC085T) from the low perhexiline responder group. Differential expression analysis between these two groups was performed using the R package “DESeq2” with default parameters56 to identify differentially expressed genes. Gene Set Enrichment Analysis (GSEA) of KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways was performed using the gseaKEGG function of “ClusterProfiler” R package57 to determine significantly upregulated or downregulated pathways between the two groups taking as reference the high/intermediate responder group.
Real-Time qPCR analysis
We measured the mRNA levels of the CPT1 (A, B, and C) and CPT2 isoforms by RT- qPCR in three PDAC cells: PDAC084T, PDAC012T, PDAC022T. Briefly, cells were seeded in 10 cm2 petri dishes (one million cells per dish), and the day after, cells were subjected to treatments: vehicle DMSO (0.01%), perhexiline 5 μM, gemcitabine 1 μM, and the combination at the same concentration than the drugs alone. Cells were harvested with Accutase (Gibco) 24 hours later, and RNA was extracted from cell pellets using the RNeasy Mini kit (Qiagen) according to manufacturer's instruction. Next, RNA samples were subjected to reverse-transcription using the Go Script reagent (Promega) following manufacturer's instruction. Then, Real-Time quantitative PCR was performed in duplicate using Takara reagents and the Stratagene cycler Mx3005P QPCR System. Raw values were normalized with the housekeeping gene TBP1 for the same cDNA sample. We used the sequence of the human primers for CPT1A, CPT1B, and CPT2 reported elsewhere.58 For CPT1C, human primers sequences were obtained from Zhang et al., 2021.59
Metabolomic analysis
For our metabolic analysis, we used the metabolome dataset from PDX of the PaCaOmics cohort, that has been previously described.36 Briefly, endogenous metabolic profiling experiments were measured using mass spectrometry coupled to ultra-performance liquid chromatography (UPLC-MS). PDX were fractionated into four sections to apply the combination of solvents to extract the specific metabolites group. Methanol and a mix of sodium chloride and chloroform/methanol (2:1) were used to isolate lipids, bile acids, and amino acids. Polar metabolites, including carbon metabolism purification, were done through a mixture of methanol/water (3:2), followed by chloroform and acetonitrile addition. All the measures included three defined quality control samples used to batch normalization.
For our analysis, we extracted the metabolome data corresponding to saturated fatty acids (SFAs), monounsaturated fatty acids (MUFAs) and polyunsaturated fatty acids (PUFAs). Then, using the GraphPad Prism 8 software, we compared the metabolites content between the high/intermediate responder group (PDAC084T, PDAC082T, PDAC027T, PDAC012T, and PDAC021T) and the low responder group (PDAC003T, PDAC022T, PDAC032T, PDAC085T) to Perhexiline treatment.
CPT1C downregulation by siRNA transient transfection
Panc-1 cells (400,000) were plated in 6-well plates in DMEM with 10% FBS. The day after, four CPT1C siRNA (L-008824-01-0010, ON-TARGETplus siRNA Reagents, Dharmacon) were transfected at a final concentration of 20 nM using INTERFERin reagent (Polyplus-transfection) with Opti-MEM (Gibco) medium according to the manufacturer’s protocol. A control siRNA pool (Scramble siRNA) was used as the negative control (D-001810-10-05, ON-TARGETplus Non-targeting Pool, Dharmacon). The medium was replaced with DMEM 10% FBS 6h after the start of transfection. To check the downregulation of CPT1C expression after 24h, cells were detached with pre-warmed accutase (Gibco), triplicates were pooled and processed for RT-qPCR as described above. For the viability test, cells were treated the day after transfection with 7μM of perhexiline in triplicates. Cells were detached 72h later and viability was assessed via Trypan blue exclusion using a cell viability analyzer (Vi-cell XR, Beckman Coulter).
Quantification and statistical analysis
Results are expressed as the mean ± SEM or SD of duplicates or triplicates, and at least two or three independent experiments were done for each analysis. Statistical analysis of data was performed with GraphPad Prism (GraphPad Software) 5 or 8 (only for metabolomic analysis). Significance was calculated as described: two tailed unpaired Student’s t-test, Mann-Whitney U test (Seahorse Mito Fuel Flex Test), One-Way ANOVA test (Seahorse OXPHOS assay, total ROS and mitochondrial superoxide anion), Two-Way ANOVA test (in vivo experiments and FAO assay), and the F- test of equality of variances (RT-qPCR, Figure 5E). The correlation coefficient (r) was calculated using the Pearson coefficient for most of the graphs, except Figure 5D that shows Spearman’s correlation (N too small). The log-rank and Wilcoxon statistic tests were applied to the Kaplan-Meier survival curves. p values < 0.05 were considered statistically significant. Quantification details of experiments can be found in Method details. Statistical details of experiments can be found in the figure legends.
Acknowledgments
We thank the cell culture platform (PCC, TPR2, Marseille, France) for technical assistance and Karim Sari and Régis Vitestelle for assistance with the use of the PSEA animal housing facility. In addition, we thank Martin Bigonnet, Yifan Jiang, and Nicolas Fraunhoffer for their advice in experiments and Eddy Pasquier for the combination index calculation with the Chou and Talalay method. This work was supported by Institut National de la Santé et de la Recherche Médicale, Centre National de la Recherche Scientifique, Institut National Du Cancer, Fondation ARC pour la Recherche sur le Cancer (PJA20181208207), DGOS, Fondation Amidex, and Fondation de France. This work also benefited from grants from Canceropôle Provence-Alpes-Côte d’Azur and Fédération GEFLUC. S-MF acknowledges funding from the European Research Council under the ERC Consolidator Grant Agreement n. 771486–MetaRegulation, FWO Projects, Beug Foundation and Fonds Baillet Latour. GR-C was supported by the CONACYT (Mexico, grant 339091/471717) and La Ligue Nationale contre le Cancer, RM by the Fondation ARC pour la Recherche sur le Cancer, JG and SL by the Fondation pour la Recherche Médicale, NAH by Association AZM et Saade (Lebanon) and Fondation pour la Recherche Médicale, SGA by the CONACYT (Mexico, grant 708538), and TG by the French Agence Nationale pour la Recherche.
Author contributions
Conceptualization and Methodology, GR-C, RM, and AC; Investigation, GR-C, NAH, SGA, RM, JG, SL, TG, MP, and LKL; Formal Analysis, GR-C, NAH, SGA, RM, JG, AEK, FG, S-MF, LKL, and AC; Resources, OG, ND, and JI; Writing – Original Draft, GR-C and AC; Writing – Review & Editing, GR-C, RM, AEK, ND, JI, and AC; Visualization, GR-C, RM, and AC; Supervision, AC; Project Administration and Funding Acquisition, AC.
Declaration of interests
S-MF has received funding from Bayer AG, Merck, Black Belt Therapeutics, and Alesta Therapeutics, has consulted for Fund+, and is in the advisory board of Alesta Therapeutics. This work has led to a patent application (EP20305666.8 18/06/2020; PCT/EP2021/066507 17/06/2021; WO 2021/255204 23/12/2021).
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: May 19, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106899.
Contributor Information
Gabriela Reyes-Castellanos, Email: grc989@hotmail.com.
Alice Carrier, Email: alice.carrier@inserm.fr.
Supplemental information
Data and code availability
-
•
All data analyzed in this study are publicly available. Instructions on how to access each dataset are specified in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Rahib L., Smith B.D., Aizenberg R., Rosenzweig A.B., Fleshman J.M., Matrisian L.M. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 2.Saad A.M., Turk T., Al-Husseini M.J., Abdel-Rahman O. Trends in pancreatic adenocarcinoma incidence and mortality in the United States in the last four decades; a SEER-based study. BMC Cancer. 2018;18:688. doi: 10.1186/s12885-018-4610-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA A Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 4.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022;72:7–33. doi: 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- 5.Roser M., Ritchie H. Cancer. Our World Data. 2018 https://ourworldindata.org/cancer [Google Scholar]
- 6.Conroy T., Hammel P., Hebbar M., Ben Abdelghani M., Wei A.C., Raoul J.-L., Choné L., Francois E., Artru P., Biagi J.J., et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N. Engl. J. Med. 2018;379:2395–2406. doi: 10.1056/NEJMoa1809775. [DOI] [PubMed] [Google Scholar]
- 7.Nevala-Plagemann C., Hidalgo M., Garrido-Laguna I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 2020;17:108–123. doi: 10.1038/s41571-019-0281-6. [DOI] [PubMed] [Google Scholar]
- 8.Haeberle L., Esposito I. Pathology of pancreatic cancer. Transl. Gastroenterol. Hepatol. 2019;4:50. doi: 10.21037/tgh.2019.06.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rawla P., Sunkara T., Gaduputi V. Epidemiology of pancreatic cancer: global trends, etiology and risk factors. World J. Oncol. 2019;10:10–27. doi: 10.14740/wjon1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conroy T., Desseigne F., Ychou M., Bouché O., Guimbaud R., Bécouarn Y., Adenis A., Raoul J.-L., Gourgou-Bourgade S., de la Fouchardière C., et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 11.Von Hoff D.D., Ervin T., Arena F.P., Chiorean E.G., Infante J., Moore M., Seay T., Tjulandin S.A., Ma W.W., Saleh M.N., et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beatty G.L., Werba G., Lyssiotis C.A., Simeone D.M. The biological underpinnings of therapeutic resistance in pancreatic cancer. Genes Dev. 2021;35:940–962. doi: 10.1101/gad.348523.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Viale A., Pettazzoni P., Lyssiotis C.A., Ying H., Sánchez N., Marchesini M., Carugo A., Green T., Seth S., Giuliani V., et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–632. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Viale A., Corti D., Draetta G.F. Tumors and mitochondrial respiration: a neglected connection. Cancer Res. 2015;75:3685–3686. doi: 10.1158/0008-5472.CAN-15-0491. [DOI] [PubMed] [Google Scholar]
- 15.Alistar A., Morris B.B., Desnoyer R., Klepin H.D., Hosseinzadeh K., Clark C., Cameron A., Leyendecker J., D’Agostino R., Topaloglu U., et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: a single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2017;18:770–778. doi: 10.1016/S1470-2045(17)30314-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daemen A., Peterson D., Sahu N., McCord R., Du X., Liu B., Kowanetz K., Hong R., Moffat J., Gao M., et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl. Acad. Sci. USA. 2015;112:E4410–E4417. doi: 10.1073/pnas.1501605112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hollinshead K.E.R., Parker S.J., Eapen V.V., Encarnacion-Rosado J., Sohn A., Oncu T., Cammer M., Mancias J.D., Kimmelman A.C. Respiratory supercomplexes promote mitochondrial efficiency and growth in severely hypoxic pancreatic cancer. Cell Rep. 2020;33:108231. doi: 10.1016/j.celrep.2020.108231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masoud R., Reyes-Castellanos G., Lac S., Garcia J., Dou S., Shintu L., Abdel Hadi N., Gicquel T., El Kaoutari A., Diémé B., et al. Targeting mitochondrial complex I overcomes chemoresistance in high OXPHOS pancreatic cancer. Cell Rep. Med. 2020;1:100143. doi: 10.1016/j.xcrm.2020.100143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sancho P., Burgos-Ramos E., Tavera A., Bou Kheir T., Jagust P., Schoenhals M., Barneda D., Sellers K., Campos-Olivas R., Graña O., et al. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metabol. 2015;22:590–605. doi: 10.1016/j.cmet.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 20.Carracedo A., Cantley L.C., Pandolfi P.P. Cancer metabolism: fatty acid oxidation in the limelight. Nat. Rev. Cancer. 2013;13:227–232. doi: 10.1038/nrc3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lionetti V., Stanley W.C., Recchia F.A. Modulating fatty acid oxidation in heart failure. Cardiovasc. Res. 2011;90:202–209. doi: 10.1093/cvr/cvr038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Camarda R., Zhou A.Y., Kohnz R.A., Balakrishnan S., Mahieu C., Anderton B., Eyob H., Kajimura S., Tward A., Krings G., et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016;22:427–432. doi: 10.1038/nm.4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farge T., Saland E., de Toni F., Aroua N., Hosseini M., Perry R., Bosc C., Sugita M., Stuani L., Fraisse M., et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017;7:716–735. doi: 10.1158/2159-8290.CD-16-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samudio I., Harmancey R., Fiegl M., Kantarjian H., Konopleva M., Korchin B., Kaluarachchi K., Bornmann W., Duvvuri S., Taegtmeyer H., Andreeff M. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Invest. 2010;120:142–156. doi: 10.1172/JCI38942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Itkonen H.M., Brown M., Urbanucci A., Tredwell G., Ho Lau C., Barfeld S., Hart C., Guldvik I.J., Takhar M., Heemers H.V., et al. Lipid degradation promotes prostate cancer cell survival. Oncotarget. 2017;8:38264–38275. doi: 10.18632/oncotarget.16123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu P.-P., Liu J., Jiang W.-Q., Carew J.S., Ogasawara M.A., Pelicano H., Croce C.M., Estrov Z., Xu R.-H., Keating M.J., Huang P. Elimination of chronic lymphocytic leukemia cells in stromal microenvironment by targeting CPT with an antiangina drug perhexiline. Oncogene. 2016;35:5663–5673. doi: 10.1038/onc.2016.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vella S., Penna I., Longo L., Pioggia G., Garbati P., Florio T., Rossi F., Pagano A. Perhexiline maleate enhances antitumor efficacy of cisplatin in neuroblastoma by inducing over-expression of NDM29 ncRNA. Sci. Rep. 2015;5:18144. doi: 10.1038/srep18144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y., Lu J.-H., Wang F., Wang Y.-N., He M.-M., Wu Q.-N., Lu Y.-X., Yu H.-E., Chen Z.-H., Zhao Q., et al. Inhibition of fatty acid catabolism augments the efficacy of oxaliplatin-based chemotherapy in gastrointestinal cancers. Cancer Lett. 2020;473:74–89. doi: 10.1016/j.canlet.2019.12.036. [DOI] [PubMed] [Google Scholar]
- 29.Lee E.A., Angka L., Rota S.-G., Hanlon T., Mitchell A., Hurren R., Wang X.M., Gronda M., Boyaci E., Bojko B., et al. Targeting mitochondria with Avocatin B induces selective leukemia cell death. Cancer Res. 2015;75:2478–2488. doi: 10.1158/0008-5472.CAN-14-2676. [DOI] [PubMed] [Google Scholar]
- 30.Amoedo N.D., Sarlak S., Obre E., Esteves P., Bégueret H., Kieffer Y., Rousseau B., Dupis A., Izotte J., Bellance N., et al. Targeting the mitochondrial trifunctional protein restrains tumor growth in oxidative lung carcinomas. J. Clin. Invest. 2021;131:e133081. doi: 10.1172/JCI133081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reyes-Castellanos G., Masoud R., Carrier A. Mitochondrial metabolism in PDAC: from better knowledge to new targeting strategies. Biomedicines. 2020;8:270. doi: 10.3390/biomedicines8080270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao C.-H., Liu G.-Y., Wang R., Moon S.H., Gross R.W., Patti G.J. Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase I is essential for cancer cell proliferation independent of β-oxidation. PLoS Biol. 2018;16:e2003782. doi: 10.1371/journal.pbio.2003782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kant S., Kesarwani P., Guastella A.R., Kumar P., Graham S.F., Buelow K.L., Nakano I., Chinnaiyan P. Perhexiline demonstrates FYN-mediated antitumor activity in glioblastoma. Mol. Cancer Therapeut. 2020;19:1415–1422. doi: 10.1158/1535-7163.MCT-19-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jarc E., Petan T. Lipid droplets and the management of cellular stress. Yale J. Biol. Med. 2019;92:435–452. [PMC free article] [PubMed] [Google Scholar]
- 35.Bian B., Bigonnet M., Gayet O., Loncle C., Maignan A., Gilabert M., Moutardier V., Garcia S., Turrini O., Delpero J.R., et al. Gene expression profiling of patient-derived pancreatic cancer xenografts predicts sensitivity to the BET bromodomain inhibitor JQ1: implications for individualized medicine efforts. EMBO Mol. Med. 2017;9:482–497. doi: 10.15252/emmm.201606975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaoutari A.E., Fraunhoffer N.A., Hoare O., Teyssedou C., Soubeyran P., Gayet O., Roques J., Lomberk G., Urrutia R., Dusetti N., Iovanna J. Metabolomic profiling of pancreatic adenocarcinoma reveals key features driving clinical outcome and drug resistance. EBioMedicine. 2021;66:103332. doi: 10.1016/j.ebiom.2021.103332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y., Chen Y., Guan L., Zhang H., Huang Y., Johnson C.H., Wu Z., Gonzalez F.J., Yu A., Huang P., et al. Carnitine palmitoyltransferase 1C regulates cancer cell senescence through mitochondria-associated metabolic reprograming. Cell Death Differ. 2018;25:735–748. doi: 10.1038/s41418-017-0013-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeong S.M., Hwang S., Park K., Yang S., Seong R.H. Enhanced mitochondrial glutamine anaplerosis suppresses pancreatic cancer growth through autophagy inhibition. Sci. Rep. 2016;6:30767. doi: 10.1038/srep30767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roux C., Riganti C., Borgogno S.F., Curto R., Curcio C., Catanzaro V., Digilio G., Padovan S., Puccinelli M.P., Isabello M., et al. Endogenous glutamine decrease is associated with pancreatic cancer progression. Oncotarget. 2017;8:95361–95376. doi: 10.18632/oncotarget.20545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Son J., Lyssiotis C.A., Ying H., Wang X., Hua S., Ligorio M., Perera R.M., Ferrone C.R., Mullarky E., Shyh-Chang N., et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang S., Hwang S., Kim M., Seo S.B., Lee J.-H., Jeong S.M. Mitochondrial glutamine metabolism via GOT2 supports pancreatic cancer growth through senescence inhibition. Cell Death Dis. 2018;9:55. doi: 10.1038/s41419-017-0089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guillaumond F., Bidaut G., Ouaissi M., Servais S., Gouirand V., Olivares O., Lac S., Borge L., Roques J., Gayet O., et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA. 2015;112:2473–2478. doi: 10.1073/pnas.1421601112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee J.-S., Oh S.-J., Choi H.-J., Kang J.H., Lee S.-H., Ha J.S., Woo S.M., Jang H., Lee H., Kim S.-Y. ATP production relies on fatty acid oxidation rather than glycolysis in pancreatic ductal adenocarcinoma. Cancers. 2020;12:2477. doi: 10.3390/cancers12092477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kantor P.F., Lucien A., Kozak R., Lopaschuk G.D. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ. Res. 2000;86:580–588. doi: 10.1161/01.RES.86.5.580. [DOI] [PubMed] [Google Scholar]
- 45.Ma Y., Wang W., Devarakonda T., Zhou H., Wang X.-Y., Salloum F.N., Spiegel S., Fang X. Functional analysis of molecular and pharmacological modulators of mitochondrial fatty acid oxidation. Sci. Rep. 2020;10:1450. doi: 10.1038/s41598-020-58334-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qu Q., Zeng F., Liu X., Wang Q.J., Deng F. Fatty acid oxidation and carnitine palmitoyltransferase I: emerging therapeutic targets in cancer. Cell Death Dis. 2016;7:e2226. doi: 10.1038/cddis.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma Y., Temkin S.M., Hawkridge A.M., Guo C., Wang W., Wang X.-Y., Fang X. Fatty acid oxidation: an emerging facet of metabolic transformation in cancer. Cancer Lett. 2018;435:92–100. doi: 10.1016/j.canlet.2018.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zaugg K., Yao Y., Reilly P.T., Kannan K., Kiarash R., Mason J., Huang P., Sawyer S.K., Fuerth B., Faubert B., et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011;25:1041–1051. doi: 10.1101/gad.1987211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Casals N., Zammit V., Herrero L., Fadó R., Rodríguez-Rodríguez R., Serra D. Carnitine palmitoyltransferase 1C: from cognition to cancer. Prog. Lipid Res. 2016;61:134–148. doi: 10.1016/j.plipres.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 50.Price N., van der Leij F., Jackson V., Corstorphine C., Thomson R., Sorensen A., Zammit V. A novel brain-expressed protein related to carnitine palmitoyltransferase I. Genomics. 2002;80:433–442. doi: 10.1006/geno.2002.6845. [DOI] [PubMed] [Google Scholar]
- 51.Sierra A.Y., Gratacós E., Carrasco P., Clotet J., Ureña J., Serra D., Asins G., Hegardt F.G., Casals N. CPT1c is localized in endoplasmic reticulum of neurons and has carnitine palmitoyltransferase activity. J. Biol. Chem. 2008;283:6878–6885. doi: 10.1074/jbc.M707965200. [DOI] [PubMed] [Google Scholar]
- 52.Wolfgang M.J., Kurama T., Dai Y., Suwa A., Asaumi M., Matsumoto S.I., Cha S.H., Shimokawa T., Lane M.D. The brain-specific carnitine palmitoyltransferase-1c regulates energy homeostasis. Proc. Natl. Acad. Sci. USA. 2006;103:7282–7287. doi: 10.1073/pnas.0602205103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gayet O., Loncle C., Duconseil P., Gilabert M., Lopez M.B., Moutardier V., Turrini O., Calvo E., Ewald J., Giovannini M., et al. A subgroup of pancreatic adenocarcinoma is sensitive to the 5-aza-dC DNA methyltransferase inhibitor. Oncotarget. 2015;6:746–754. doi: 10.18632/oncotarget.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nicolle R., Blum Y., Duconseil P., Vanbrugghe C., Brandone N., Poizat F., Roques J., Bigonnet M., Gayet O., Rubis M., et al. Establishment of a pancreatic adenocarcinoma molecular gradient (PAMG) that predicts the clinical outcome of pancreatic cancer. EBioMedicine. 2020;57:102858. doi: 10.1016/j.ebiom.2020.102858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nicolle R., Blum Y., Marisa L., Loncle C., Gayet O., Moutardier V., Turrini O., Giovannini M., Bian B., Bigonnet M., et al. Pancreatic adenocarcinoma therapeutic targets revealed by tumor-stroma cross-talk analyses in patient-derived xenografts. Cell Rep. 2017;21:2458–2470. doi: 10.1016/j.celrep.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu G., Wang L.-G., Han Y., He Q.-Y. clusterProfiler: an R Package for comparing biological themes among gene clusters. OMICS A J. Integr. Biol. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brown Z.J., Fu Q., Ma C., Kruhlak M., Zhang H., Luo J., Heinrich B., Yu S.J., Zhang Q., Wilson A., et al. Carnitine palmitoyltransferase gene upregulation by linoleic acid induces CD4+ T cell apoptosis promoting HCC development. Cell Death Dis. 2018;9:620. doi: 10.1038/s41419-018-0687-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang H., Wang Y., Guan L., Chen Y., Chen P., Sun J., Gonzalez F.J., Huang M., Bi H. Lipidomics reveals carnitine palmitoyltransferase 1C protects cancer cells from lipotoxicity and senescence. J. Pharm. Anal. 2021;11:340–350. doi: 10.1016/j.jpha.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data analyzed in this study are publicly available. Instructions on how to access each dataset are specified in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.