


Abstract
In Trypanosoma brucei the genes are organised into long polycistronic transcription units and only three promoters for protein-encoding genes and a single terminator have been characterised. These promoters recruit a polI-like RNA polymerase for the transcription units encoding the two major stage-specific antigens of the parasite, the variant surface glycoprotein (VSG) of the bloodstream form and procyclin of the insect-specific procyclic form, while the terminator is that of a procyclin transcription unit. By deletional and mutational analysis we defined the two DNA sequences essential for the activity of the VSG promoter from a bloodstream form transcription unit and one of the functional elements of the procyclin terminator. These three short sequences are similar, and their C-rich strand binds the same protein of 40 kDa. In addition, this factor also binds to the C-rich strand of the telomeric repeats, the consensus target sequence being 5′-CCCTNN-3′. The factor-binding sequences are functionally interchangeable in chimeric promoter or terminator constructs, although additional elements are required for full activity.
INTRODUCTION
The protozoan flagellate Trypanosoma brucei is responsible for sleeping sickness in man and nagana in cattle. This parasite is transmitted from mammal to mammal by an insect vector, the tsetse fly. In the mammalian host, T.brucei is covered by a dense and homogeneous coat consisting of 107 molecules of the same antigen termed the variant surface glycoprotein or VSG, which is changed frequently and completely disappears upon cellular differentiation into the insect-specific procyclic form, where it is replaced by another major surface glycoprotein termed procyclin (reviewed in 1,2).
The trypanosomal genome appears to be entirely organised into long polycistronic transcription units and the DNA and protein components involved in transcription initiation and termination are almost totally unknown. Only three promoters for protein-encoding genes and a single terminator have been characterised (1–10). These promoters recruit a polI-like RNA polymerase for the transcription units of the two major stage-specific antigens of the parasite, the VSG and procyclin (5–10), while the terminator is that of a procyclin transcription unit (3). The VSG transcription units are located in telomeres, and contain a battery of about 10 different genes termed expression site-associated genes (ESAGs), in addition to the VSG gene. These units are controlled in the bloodstream form by differential and reversible silencing which underlies antigenic variation of the parasite by allowing the expression of a single unit at a time from a total of about 20 (11,12). The procyclin genes are arranged in two non-telomeric loci termed procyclic acidic repetitive protein (PARP) A and PARP B, each containing two (sometimes three) genes in direct repeats together with a few additional genes termed procyclin-associated genes (PAGs) (1,2). The VSG and procyclin transcription units are developmentally regulated. During the parasite life cycle, or under experimental treatments in vitro, the activities of these units are strikingly and systematically opposite, suggesting inverse regulation by shared transcription factors (13).
We found that the essential elements of the VSG promoter and the procyclin terminator bind a common protein which also binds to the telomeric repeats. Thus, our data provide a possible link between chromatin organisation of telomeres and transcriptional control of the telomeric VSG expression sites, as well as with transcription termination of the procyclin units.
MATERIALS AND METHODS
Trypanosomes
The procyclic form was obtained by in vitro cultivation of isolates from the midgut of flies infected with the EATRO 1125 stock of T.brucei. They were grown in SDM-79 medium (14) supplemented with 15% v/v heat-inactivated foetal calf serum. Bloodstream form AnTat 1.3A were passaged in mice.
Transfections and CAT assays
Procyclic cells (107), harvested at mid-log phase, were resuspended in 500 µl of Zimmerman post-fusion medium (15) and mixed with 50 µg of plasmid DNA. Electroporations were performed with a Bio-Rad gene pulser by two pulses at 1.5 kV and 25 µF. Electroporated cells were inoculated in 5 ml of culture medium and harvested 18 h later for CAT assays which were performed as described by Jefferies et al. (16). All plasmids were assayed between four and 10 times.
Nuclear extracts
Trypanosome nuclei were prepared as described by Murphy et al. (17). The nuclei were washed once in 0.25 M sucrose, 5 mM MgCl2, 10 mM Tris (pH 7.5), resuspended in the same solution containing 0.5% v/v Nonidet P-40 and protease inhibitors (leupeptin, aprotinin, pepstatin A and phenylmethylsulphonyl fluoride), then left at 4°C for 10 min. The suspension was adjusted to 0.3 M NaCl, left for 10 min at 4°C, then centrifuged for 60 min at 30 000 g. The supernatant was harvested and adjusted to 20% v/v glycerol, 0.1% v/v Brij 35, 1 mM dithiothreitol. It was then dialysed overnight against 50 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, 20% v/v glycerol, 1 mM dithiothreitol, 20 mM HEPES (pH 7.9), with protease inhibitors. It was centrifuged for 15 min at 13 000 g and the supernatant was stored at –80°C. One microgram of protein was routinely used for the band shift assays.
Band shift assays and UV crosslinking
Synthetic oligonucleotides (Eurogentec and Life Technologies) were used as probes or competitors. The sequences of the oligonucleotides were (mutations underlined):
1s, 5′-AGAATCATATCCCTATTACCACACCAGTTT-3′;
1as, 5′-AAACTGGTGTGGTAATAGGGATATGATTCT-3′;
MUT1s, 5′-AGAATCATATCAAGCGGACCACACCAGTTT-3′;
2s, 5′-CAGTTTATATTACAGGGGAGGTTATTACAG-3′;
2as, 5′-CTGTAATAACCTCCCCTGTAATATAAACTG-3′;
MUT2as, 5′-CTGTAATAACCGGAGAGGTAATATAAACTG-3′;
Tri1s, 5′-GATCATATCCCTATTACTCATATCCCTATTAC-TCATATCCCTATTA-3′;
Tri1as, 5′-CTAGTAATAGGGATATGAGTAATAGGGATATGAGTAATAGGGATAT-3′;
TriMUT1s, 5′-GATCATATCAAGCGGACTCATATCAAG-CGGACTCATATCAAGCGGA-3′;
TelC, 5′-GATCCCTAACCCTAACCCTAACCCTAACCCTAA-CCCTAACCCTAA-3′;
TelG, 5′-CTAGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGG-3′.
Band shift assays were performed with single-stranded oligonucleotides. When used as probes, the oligonucleotides were end-labelled with T4 polynucleotide kinase then purified on a polyacrylamide gel and eluted. Probes (routinely ~108 c.p.m./µg) were mixed with nuclear extracts, 200 ng of sheared salmon sperm DNA and cold competitor DNA if desired (in increasing molar excesses of 5-, 50- and 500-fold) in a final volume of 50 µl of binding buffer (5% v/v glycerol, 60 mM KCl, 7.5 mM MgCl2, 1 mM dithiothreitol, 45 mM Tris–borate, 1 mM EDTA, 10 mM HEPES, 4 mM Tris, pH 7.9). The mixtures were left for 15 min at room temperature then loaded on a 5% w/v polyacrylamide gel and run at 150 V in 45 mM Tris–borate, 1 mM EDTA for 90 min at room temperature. The gels were dried under vacuum and exposed with intensifying screens.
UV crosslinking was performed by placing the electrophoresis gel on ice at a distance of 5 cm under a UV lamp and irradiating for 20 min at 254 nm before overnight exposure (the energy delivered was not measured). Band II was cut out, boiled for 5 min in SDS–PAGE buffer and submitted to SDS–PAGE.
Preparative band shift
A 4 cm 5% w/v polyacrylamide gel was poured in the 37 mm ID tube of a Model 491 Prep Cell (Bio-Rad). Electrophoresis and elution buffer were 0.5× TBE and 10 mM Tris (pH 7.9), respectively. Nuclear extract (2 ml) from bloodstream forms, equilibrated with binding buffer by dialysis for 1 h at 4°C, was mixed with 60 ng of radiolabelled probe, 6 µg of sheared salmon sperm DNA and 600 µg of non-specific competitor oligonucleotide. The binding reaction was performed for 15 min at room temperature before loading on the preparative gel. The cell was run at 300 V constant. After a 20 min run, fractions of 1 ml were collected using a peristaltic pump set at a flow rate of 0.5 ml/min. The radioactivity of the fractions was evaluated by liquid scintillation counting and the fractions of interest were concentrated to 100 µl by freeze-drying. The concentrated fractions were analysed for protein composition by silver staining after analytical SDS–10% w/v PAGE.
Plasmids
Procyclin terminator plasmids
(i) Plasmids studied in Figure 2. The 2.4 kb EcoRV–XhoI fragment from the procyclin PARP A termination region (3) was inserted between the StuI and XhoI sites of pSL1180 (Pharmacia). The resulting plasmid was cut by NruI and KpnI, digested by exonuclease III, treated with S1 nuclease, then self-ligated. The resulting shortened terminator fragments were cut out of pSL1180 by SalI + HindIII digestion, then ligated between the XhoI and HindIII sites of pCAT-tub2 (3), in which the chloramphenicol acetyltransferase (CAT) gene is inserted downstream from a procyclin promoter. Plasmids 148 and 87 were obtained by cleaving constructs 361 and 297 with BsrGI. (ii) Plasmids studied in Figure 3. Plasmids 1 and 8 were obtained by site-directed mutagenesis using the QuickChange Site-Directed Mutagenesis Kit (Stratagene). Plasmids 2, 3, 7, 9 and 10 were obtained by insertion of annealed complementary oligonucleotides between the Eco47III and SphI sites present just upstream from the terminator fragment in 148, whereas the same strategy, but in plasmid 233, was applied for plasmids 4, 5, 6 and 11. The sequences of the sense oligonucleotides were:
Figure 2.

Defining the key elements of the procyclin terminator. The map of the PARP A locus is shown on the top, with a flag representing the procyclin promoter, PAG3 and GRESAG2 (GRES2) being genes associated with the α and β procyclin genes and the boxes labelled 1, 2 and 3 being three terminator regions identified previously (3). The figure shows the mapping of a minimal terminator by deletion analysis. The numbers indicate the sizes of the fragments assayed and X is for XhoI restriction endonuclease. The small grey box in 297, 148 and 87 contains the strongest inhibitory sequence, which is shown at bottom, with an indication of the stretch conserved in the VSG promoter (boxed).
Figure 3.

The effect of box 1 in transcription termination (explanations of symbols at bottom). Legend as in Figure 2.
2 and 5, ATCCCTATTATCCCTATTATCCCTATTCATG;
3, AATAGGGATAATAGGGATAATAGGGATCATG;
4, ATCCCTATTCATG;
6, ATCCCTATTATCCCTATTATCCCTATTATCCCTATT-ATCCCTATTCATG;
9 and 11, GGATCCCTAACCCTAACCCTAACCCTAACCCT;
10, AGTACTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGCATG.
Plasmids studied in Figure 6. These plasmids were obtained by insertion of annealed oligonucleotides between the SphI and Eco47III sites of pCAT-tub2 (3). The sense oligonucleotides were as follows (mutations underlined):
1, CATATCCCTATTACCACACCAGTTTATATTACAGG-GGAGGTTATTACAGAAATCTCAGATATCAGACTCAC-GGTG;
2, CATATCCCTATTACCACACCAGTTTATATTCCGCTT-GAGGTTATTACAGAAATCTCAGATATCAGACTCACGGTGCATG;
3, CATATAAGCGGTACCACACCAGTTTATATTCCGC-TTGAGGTTATTACAGAAATCTCAGATATCAGACTCA-CGGTGCAT;
4, CCATGGAATATCCCTATTCTTTGCGGCGCATGAGC-CTTCCTCCATTCGTCTTTTGCGTTTGCTTTTATACCCA-CCCGCGTGCATG;
5, CCATGGAATATCCCTATTCTTTGCGGCGCATGAGCC-ACAGGGCATTCGTCTTTTGCGTTTGCTTTTATACCCA-CCCGCGTGCAT.
VSG promoter plasmids. These plasmids were obtained by inserting annealed complementary oligonucleotides between the SphI and StuI sites of pD5 in which the CAT reporter gene is inserted downstream from a VSG promoter (5). The sequences of the sense oligonucleotides used for plasmids 1–6 in Figure 4 were:
Figure 4.

Activity of the VSG promoter after substitution of its crucial boxes with either the minimal procyclin terminator or telomeric repeats (explanations of symbols at bottom).
1, CATATCCCTATTACCACACCAGTTTATATTACAGG-GGAGGTTATTACAGAAATCTCAGATACAGACTCACG-GTG;
2, GAATATCCCTATTCTTTGCGGCGCATGAATTACAG-GGGAGGTTATTACAGAAATCTCAGATATCAGACTCA-CGGTG;
3, GAATATCCCTATTCTTTGCGGCGCATGAGATTACAG-GGGAGGTTATTACAGAAATCTCAGATATCAGACTCA-CGG;
4, GAATATCAAGCGGCTTTGCGGCGCATGAATTACAG-GGGAGGTTATTACAGAAATCTCAGATATCAGACTCA-CGGTG;
5, CATATCAAGCGGACCACACCACTTTATATTACAGG-GGAGGTTATTACAGAAATCTCAGATATCAGACTCACG-GTG;
6, CATATCCCTAATACCACACCACTTTATATTTTAGG-GGAGGTTATTACAGAAATCTCAGATATCAGACTCAC-GGTG.
All plasmids were sequenced before being assayed for CAT activity.
RESULTS
The two essential elements of the VSG promoter bind common factors
Mutagenesis of the VSG promoter allowed the identification of two functionally important boxes, centred on critical stretches at positions –61 to –59 (box 1) and –38 to –35 (box 2) (5) (see the scheme in Fig. 1). The antisense (as) strand of box 2 (2as) was previously reported to specifically bind a 40 kDa protein (5), generating a complex termed band II in band shift assays, in addition to two other non-specific complexes I and III (5; Fig. 1, compare lanes 2 and 1). While all three complexes could be competed out by an excess of cold probe (lanes 3–5), band II selectively resisted competition by non-specific oligonucleotides such as tubulin sequences (5) or the antisense strand of box 1 (1as) (lanes 6–8), and by a mutated box 2 probe (MUT2as) (5).
Figure 1.

The antisense strand of box 2 of the VSG promoter binds the same factor as the sense strand of box 1. Bandshift assays were carried out with the indicated oligonucleotide probes, in the presence of increasing amounts (5-, 50- and 500-fold molar excess) of the indicated cold oligonucleotides. The assays were performed with nuclear extracts from bloodstream forms except in lane 16 (PF, procyclic forms). Complexes are numbered with roman numbers. The specific complex (band II) is arrowed.
A 30mer containing the sense (s) strand of box 1 (1s) (5) was found to compete efficiently for band II (lanes 9–11). This oligonucleotide no longer competed when it was mutated within the short sequence required for promoter activity (MUT1s, CCTATT→AAGCGG; lanes 12–14). These results suggested that the sequence centred on CCTATT is necessary for binding, and that box 1s binds the same protein as box 2as. In order to check these hypotheses oligonucleotides carrying different tandem repeats of box 1s were used as probes in bandshift assays with nuclear extracts of trypanosome bloodstream and procyclic forms. 8mers or 16mers (thus containing one or two repeats) did not generate any shift, as expected from the minimal length believed to meet the steric requirements for factor binding (~25 nt) (Table 1, lines 1and 2). Therefore, a 46mer carrying three repeats centred on box 1 (Tri1s) was used as a probe. Tri1s generated band II in both forms (lanes 15 and 16), while its antisense strand and TriMUT1s did not, even after long exposure of the autoradiograms (lanes 17 and 18). This band exhibited exactly the same competition specificity as band II formed with box 2as (data not shown). Thus, on the basis of (i) formation of band II with both box 1s and box 2as but not with MUT1s, MUT2as or non-specific oligos and (ii) efficient reciprocal competition for formation of band II with box 1s and box 2as but not with mutated or non-specific oligos, we conclude that the two crucial boxes of the VSG promoter bind common factors on opposite strands. The shortest element able to generate band II was defined by using oligomers containing fragments of different sizes from box 1 and box 2 (Table 1). Various base substitution experiments defined the hexamer CCCTNN as the consensus sequence necessary for protein binding. Although the two last nucleotides could vary, pyrimidines led to the strongest binding and guanine was the least efficient (Table 1).
Table 1. Specific band II binding activity to the indicated synthetic oligonucleotides was monitored using bandshift assays.

Oligonucleotides were designed to represent deletions or mutations of DNA segments covering the sense strand of box 1 in the VSG promoter (lines 1–4), the telomeric repeats (line 5), or the antisense strand of box 2 in the VSG promoter (lines 6–18). Boxes 1 and 2 have been previously defined (5) and are underlined in bold in oligonucleotides 1 and 6, respectively. The sequence shared between box 1 in the VSG promoter and the PARP A terminator is indicated by the square bracket. Mutations are framed. The minimal deduced consensus region necessary for band II binding is demarcated by dotted vertical lines.
Box 1 is a key element in the activity of the procyclin terminator
Transcription termination was studied in the 3 kb 3′-end region of the PARP A procyclin unit, which contains three separate fragments each able to inhibit transcription by more than 85% (3; see Fig. 2, top). A deletion analysis was conducted on the third element. As shown in Figure 2, insertion of a 494 bp fragment from the 3′-end of the terminator region in the sense orientation between the procyclin promoter and the CAT gene reduced CAT activity to 8% of the control (no insertion or insertion in the opposite orientation gave 100% activity; see 3). According to our previous work (3), this effect was due to transcription termination. Deletions at either end of this fragment led to a progressive increase in CAT activity, the strongest effect being observed when deleting 22 nt between 297 and 275 bp upstream from the XhoI site, which resulted in an increase from 38 to 78% in CAT activity (Fig. 2). Interestingly, this small fragment contained an 11 bp sequence also present in the VSG promoter and containing box 1 (ATATCCCTATT; Fig. 2, bottom). Therefore, this sequence had the potential to bind the same factor as boxes 1 and 2. To evaluate the significance of this observation, a short terminator fragment (148 bp, ~60% CAT inhibition; see Fig. 2) was altered within this 11mer as performed previously for box 1 (MUT1s, CCTATT→AAGCGG). In the promoter, this change abolished both its activity (5) and binding of the band II factor (Fig. 1). As shown in Figure 3, line 1, it also totally relieved the terminator activity. The role of box 1 was directly assessed by adding three copies of the sequence ATCCCTATT upstream from the 148 bp terminator fragment. As a control, the trimer was also inserted in the opposite orientation. This addition almost totally inhibited CAT activity only when inserted in the same orientation as the terminator (Fig. 3, lines 2 and 3). Moreover, when tested alone, the sequence ATCCCTATT was able to inhibit CAT activity, although several tandem copies were necessary to achieve the full effect (lines 4–6). It was concluded that box 1 is involved in the activity of the procyclin terminator.
We investigated whether other terminator elements contain box 1/2-like sequences. The sequence CCCCCTGCA (positions 1687–1695 in ref. 3) is a candidate for such a box in the first of the three terminator elements (Fig. 2, top). Indeed, this sequence contains the necessary CCCT stretch and resembles the minimal element defined above for box 2. As expected, this sequence generated band II (data not shown). Insertion of three copies of this sequence upstream from the minimal terminator reduced CAT activity (Fig. 3, line 7), while a random trimer was ineffective (not shown). Therefore, different terminator elements may function by recruiting the same factor.
The dual role of box 1 was tested by a functional assay. We constructed a hybrid VSG promoter containing a fragment from the procyclin terminator instead of the normal box 1 region. In the minimal sequence of the VSG promoter (70 bp, see Materials and Methods), the 5′-terminal 28 bp fragment containing box 1 was replaced by a fragment of the same size from the procyclin terminator, the conserved 11mer being placed at exactly the same location with respect to box 2. The hybrid promoter showed 20% of wild-type activity (Fig. 4, lines 1 and 2). Since in the promoter the spacing between boxes 1 and 2 is crucial (5), a control was constructed where the 28 bp terminator fragment was shifted backwards by a single nucleotide. In addition, a hybrid promoter with the correct spacing but including the MUT1s mutation was assayed. Significantly, the activity was strongly reduced when the spacing between boxes 1 and 2 was 1 bp too long (line 3), and, as in the wild-type, it was completely abolished if box 1 was mutated (lines 4 and 5). Therefore, the box 1 sequence shared by the two elements is essential, but full activity of the promoter requires elements not present in the terminator fragment tested here.
The factor which binds to boxes 1 and 2 also binds to the telomeric repeats
The core sequence (CCCTNN) which gives rise to band II in the band shift assay is contained in the C-rich strand of the telomeric repeats (CCCTAA). We evaluated the hypothesis that the same factor may bind to these different sequences. As shown in Figure 5, lanes 2 and 1, an oligo containing seven tandem repeats of sequence CCCTAA (telC) generated band II, whereas its complement (telG) did not, but formed a strong band III. Band II with telC showed the same specificity as observed for boxes 1 and 2 of the promoter. It was efficiently competed out by an excess of telC, box 1s and box 2as (lanes 3–5, 9–11 and 15–17), but not by box 1as, MUT1s and box 2s (lanes 6–8, 12–14 and 18–20), or MUT2as (not shown). Furthermore, band II with box 2as was competed out much better with telC than telG (lanes 21–27). Thus, the factor which binds to boxes 1 and 2 also binds to telC.
Figure 5.

The C-rich strand of telomeric repeats binds the same factor as the crucial elements of the VSG promoter and procyclin terminator. Legend as for Figure 1. UV crosslinking of band II obtained with box 2as, box 1s and telC shows that a common 40 kDa protein is involved. In these experiments the bottom band represents the free probe released from the complex.
The size of the DNA-binding factor contained in band II with box 2as, box 1s and telC was determined by SDS–PAGE after UV crosslinking to DNA. It was found to be the same, ~40 kDa in each case (lanes 28–30).
If the same factor binds to telomeres, the VSG promoter and the procyclin terminator, we speculated that converting the crucial elements of the promoter and terminator into exact telomeric repeats would leave these sequences functional. As shown in Figure 3, line 8, the change of the terminator box 1 (CCCTAT) into a telomeric repeat (CCCTAA) conserved a slightly reduced termination activity. Furthermore, the addition of five telomeric repeats to the 148 bp element strengthened the terminator only when inserted in the sense orientation (lines 9 and 10), and the repeats alone showed a weak termination activity (line 11). Telomeric repeats can thus substitute for box 1 in the terminator, but their efficiency is lower.
Similarly, we converted boxes 1 and 2 of the VSG promoter into telomeric repeats by changing the last nucleotides of the core CCCTPuT hexamer (CCCTAT and CCCTGT, respectively) to CCCTAA. This promoter was still functional, albeit at a very reduced level (Fig. 4, line 6). Therefore, the promoter requires more than the consensus sequence for binding in order to be optimally functional.
Attempts at promoter–terminator interconversions
Given the possibility of relative interchange of boxes between terminator and promoter we attempted interconversion of these elements. Conversion of the promoter into a terminator was assayed by eliminating box 2 (changed to MUT1 as above), thus leaving a single box 1s as in the procyclin terminator. As expected, this totally inactivated the promoter (data not shown), and conferred a slight terminator activity to the mutated promoter (22% CAT inhibition; Fig. 6, line 2). This slight inhibition was reproducible and significant since it disappeared upon conversion of box 1 into MUT1s (Fig. 6, line 3). Conversely, the addition of box 2 to the minimal terminator largely abolished its activity (only 9% CAT inhibition instead of 60%; compare lines 4 and 5). However, this addition did not transform the mutated terminator into a promoter (data not shown).
Figure 6.

Attempts to convert the VSG promoter into a terminator. Line 1, wild-type VSG promoter; line 2, VSG promoter depleted of box 2; line 3, VSG promoter depleted of boxes 1 and 2; line 4, wild-type termination fragment 148; line 5, as line 4, but after insertion of box 2. The flag represents the PARP A procyclin promoter.
Attempts at purifying the factor binding to boxes 1 and 2
In order to purify the factor responsible for formation of band II, a preparative band shift electrophoresis was run using the 32P-labelled box 2 antisense oligonucleotide in the presence of a 100-fold excess of cold non-specific oligonucleotide. As shown in Figure 7A and B, a single radioactive peak, corresponding to band II, was observed. Silver staining of the pooled fractions from this peak still revealed a complex protein pattern (Fig. 7C). The pooled fractions of the peak were checked for the presence of p37, an RNA-binding protein present in the nucleus of bloodstream forms (18,19). As shown in Figure 7D, this protein was absent from the band II complex.
Figure 7.
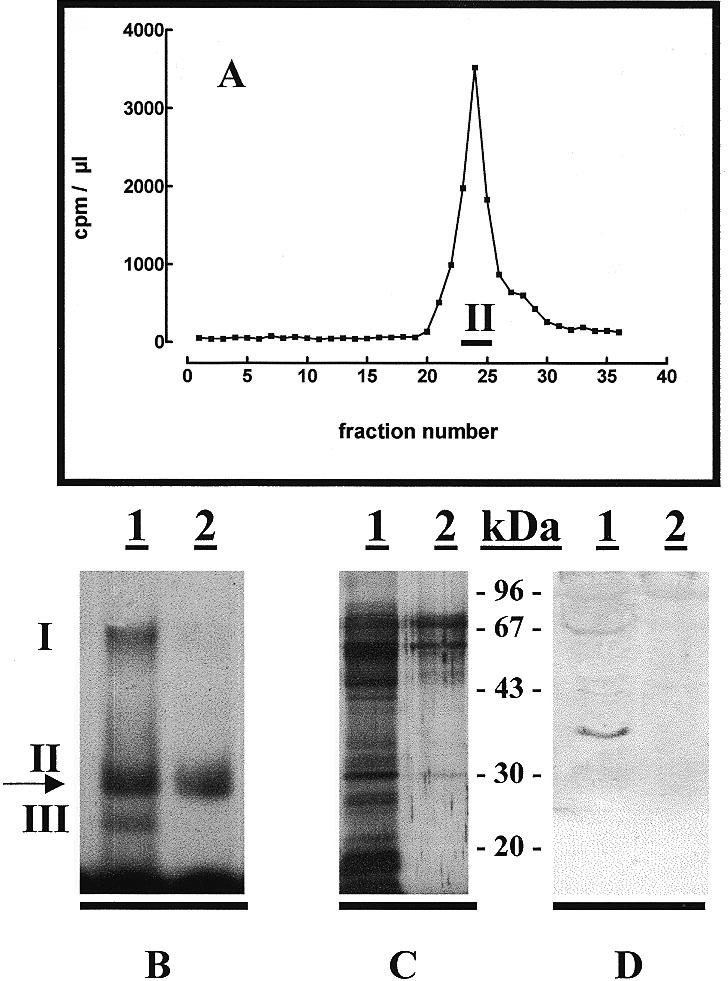
Preparative electrophoresis of the band II complex. (A) Distribution of 32P-labelled box 2 antisense oligonucleotide after incubation with nuclear extracts from T.brucei bloodstream forms in the presence of a 100-fold excess of unlabelled non-specific competitor. Fractions 23–25 were pooled for further analysis.(B–D) The gel retardation, silver staining and western blot patterns, respectively, obtained with the total nuclear extract (lane 1) or the pooled fractions from the peak shown in (A) (lane 2). The western blot was incubated with antibodies directed against the p37 RNA-binding protein (18,19) (dilution 1:2000).
Although some purification was achieved by this method, the isolated fraction lost its ability to perform specific band shifts upon further fractionation (data not shown). This was also true when other approaches were used as the first step of purification (chromatography on Sepharose, Superose, BioRex 70 or heparin), preventing, so far, cloning of the gene for the 40 kDa protein.
DISCUSSION
We found that a common 40 kDa protein, termed band II factor, binds to the essential elements of the VSG promoter and the PARP A procyclin terminator as well as to the telomeric repeats of T.brucei. This binding probably plays a role in the activity of the VSG promoter and procyclin terminator, since the factor binding sites are key elements for these activities. The band II factor clearly exhibited specific binding to single-stranded DNA, suggesting that it either helps to unwind the DNA duplex or makes contact with the C-rich strand alone in double-stranded DNA.
The 3 kb terminator fragment was shown to contain at least three inhibitory regions (3). These elements seem to be redundant as their respective inhibitory activity, taken separately, is almost as high as that of the complete fragment. In this work, we dissected the third region, showing that it contains a box 1 element essential to its activity. Although a similar element present in the first region of the 3 kb terminator fragment was also shown to be inhibitory, at this point it is not known if the PARP A terminator region is made only of box 1-like elements (20 potential elements of this type were found in the 3 kb fragment) or if other terminator motifs are present.
Trypanosoma brucei telomeres were shown to be transcribed by an α-amanitin-resistant polymerase (20). This may result from read-through transcription downstream from the VSG gene in telomeric expression sites, or from the activity of ribosomal promoters that were found upstream from telomeric repeats in some minichromosomes (21). This transcription was shown to be essentially unidirectional, proceeding towards the end of the chromosome (20). This observation does not contradict the transcription termination effect of the telomeric repeats, since the latter effect was observed only when the repeats were used in the antisense orientation with respect to the chromosome end.
In addition to its specificity for the functional elements of the VSG promoter and procyclin terminator, the band II factor may also bind to the procyclin and ribosomal promoters (5,22). Therefore, the latter might be a general transcription factor also involved in termination. However, for both transcription initiation and termination other factors are obviously required, since constructs with substituted binding sites are not optimally active, and changes outside the binding sites also affect the activity (3–5). It is probable that the biological function is influenced by the flanking sequences as well as by the relative affinity of binding, which appears to depend on the nature of the last 2 nt of the target core hexamer.
Numerous attempts to purify the protein responsible for DNA binding have been unsuccessful. All techniques tested, including chromatography on Sepharose blue, Superose, BioRex70 and heparin, led to rapid loss of binding activity. The most useful methods, namely fractionation by differential ammonium sulphate precipitation followed by heparin chromatography or preparative band shift electrophoresis, only allowed partial purification which was insufficient to allow cloning of the gene for the 40 kDa protein. Nevertheless, these methods clearly separated the three band shift complexes, demonstrating that bands I, II and III result from the binding of three different proteins. These data suggest that the complexes are unstable and that binding of the 40 kDa factor to DNA presumably requires the association of other factors.
Although the close matching between the binding sites and the functional elements of DNA sequences suggests that the band II factor is a genuine DNA-binding protein, the possibility that this factor is actually an RNA-binding protein cannot be ruled out. In this respect it is worth noting that different T.brucei proteins able to bind to both single-stranded DNA and RNA in vitro have been analysed in recent reports (18,19). Among these proteins only two exhibited a size close to 40 kDa (18). These proteins, termed p34 and p37, were found to be related and represent major nuclear RNA-binding proteins, only one of which, p37, is present in bloodstream forms (19). Using specific antibodies against p34/37, we detected p37 in total nuclear extracts of bloodstream forms but this protein was clearly absent from the fractions responsible for the band II complex. Moreover, these antibodies did not induce a supershifting of band II (data not shown). Therefore, we conclude that p34/p37 and the 40 kDa band II factor are distinct proteins.
The band II factor also binds to the C-rich strand of telomeric repeats. This characteristic, as well as the size of the protein (40 kDa), are reminiscent of properties described for the 39 kDa T.brucei factor ST-1 which binds to the C-rich strand of double-stranded (sub)telomeric repeats (23). So far we have been unable to determine unequivocally if, like ST-1, the band II factor associates with double-stranded DNA, since in our hands bandshifts with double-stranded oligos consistently resulted in complex patterns of little specificity (data not shown; see also 22,24,25 for a controversial view of results in the field). Moreover, it is presently impossible to determine if ST-1 is indeed the band II factor, since, as is the case for the latter, the ST-1 factor has not been characterised to date. Interestingly, as we postulate for the band II factor, ST-1 was found to cooperate with another factor, termed ST-2, to generate binding complexes with (sub)telomeric repeats (26).
The multifunctional role of the band II factor evokes two yeast factors: RAP1, which binds to both telomeres and promoters, and REB1, which binds to both promoters and the polI terminator (27,28). It is worth mentioning that RAP1 is involved in transcriptional silencing of telomeres, since some reports conclude that the VSG expression sites of T.brucei are controlled by the telomeric position effect (11,12). In this respect, we note that the most important nucleotide of the VSG promoter (T at –61 in the box 1 sequence CCCTAT) (5) might be a target for the unusual telomeric DNA modification which converts T into β-d-glucosylhydroxymethyluracil (29,30). Assuming that this modification prevents binding of the factor, a spreading of the telomeric modification to this region would probably inactivate the promoter.
Acknowledgments
ACKNOWLEDGEMENTS
We thank N. Williams for providing us with the p34/37 antiserum. We also acknowledge A. Pays for help, and D. Perez-Morga and D. Nolan for useful comments on the manuscript. This work was supported by the Belgian FRSM, FRC-IM, a research contract with the Communauté Française de Belgique (ARC) and by the Interuniversity Poles of Attraction Programme, Belgian State, Prime Minister’s Office, Federal Office for Scientific, Technical and Cultural Affairs. M.B. and L.V. are, respectively, chargé de recherche and chercheur qualifié of the Belgian FNRS.
REFERENCES
- 1.Clayton C. (1992) Prog. Nucleic Acid Res. Mol. Biol., 43, 37–66. [DOI] [PubMed] [Google Scholar]
- 2.Vanhamme L. and Pays,E. (1995) Microbiol. Rev., 59, 223–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berberof M., Pays,A., Lips,S., Tebabi,P. and Pays,E. (1996) Mol. Cell. Biol., 16, 914–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pham V.P., Qi,C.C. and Gottesdiener,K.M. (1996) Mol. Biochem. Parasitol., 75, 241–254. [DOI] [PubMed] [Google Scholar]
- 5.Vanhamme L., Pays,A., Tebabi,P., Alexandre,S. and Pays,E. (1995) Mol. Cell. Biol., 15, 5598–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim K.S. and Donelson,J.E. (1997) J. Biol. Chem., 272, 24637–24645. [DOI] [PubMed] [Google Scholar]
- 7.Graham S.V., Wymer,B. and Barry,J.D. (1998) Nucleic Acids Res., 26, 1985–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung H.M., Lee,M.G.S. and Van der Ploeg,L.H.T. (1992) Parasitol. Today, 8, 414–418. [DOI] [PubMed] [Google Scholar]
- 9.Janz L. and Clayton,C. (1994) Mol. Cell. Biol., 14, 5804–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zomerdijk J.C.B.M., Kieft,R., Shiels,P.G. and Borst,P. (1991) Nucleic Acids Res., 19, 5153–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horn D. and Cross,G.A.M. (1995) Cell, 83, 555–561. [DOI] [PubMed] [Google Scholar]
- 12.Rudenko G., Blundell,P.A, Dirks-Mulder,A., Kieft,R. and Borst,P. (1995) Cell, 83, 547–553. [DOI] [PubMed] [Google Scholar]
- 13.Vanhamme L., Berberof,M., Le Ray,D. and Pays,E. (1995) Nucleic Acids Res., 23, 1862–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brun R. and Schoenenberger,M. (1979) Acta Trop., 36, 289–292. [PubMed] [Google Scholar]
- 15.Bellofatto V. and Cross,G.A.M. (1989) Science, 244, 1167–1169. [DOI] [PubMed] [Google Scholar]
- 16.Jefferies D., Tebabi,P. and Pays,E. (1991) Mol. Cell. Biol., 11, 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murphy N., Pays,A.,Tebabi,P., Coquelet,H., Guyaux,M., Steinert,M. and Pays,E. (1987) J. Mol. Biol., 195, 855–871. [DOI] [PubMed] [Google Scholar]
- 18.Zhang J.,R. and Williams,N. (1997) Mol. Biochem. Parasitol., 87, 145–158. [DOI] [PubMed] [Google Scholar]
- 19.Zhang J.,R. and Williams,N. (1998) Mol. Biochem. Parasitol., 92, 79–88. [DOI] [PubMed] [Google Scholar]
- 20.Rudenko G. and Van der Ploeg,L.H. (1989) EMBO J., 8, 2633–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zomerdijk J.C., Kieft,R. and Borst,P. (1992) Nucleic Acids Res., 20, 2725–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown S.D. and Van der Ploeg,L.H.T. (1994) Mol. Biochem. Parasitol., 56, 109–122. [DOI] [PubMed] [Google Scholar]
- 23.Eid J.E. and Sollner-Webb,B. (1995) Mol. Cell. Biol., 15, 389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janz L., Hug,M. and Clayton,C. (1994) Mol. Biochem. Parasitol., 65, 99–108. [DOI] [PubMed] [Google Scholar]
- 25.Pham V.P., Rothman,P.B. and Gottesdiener,K.M. (1997) Mol. Biochem. Parasitol., 89, 11–23. [DOI] [PubMed] [Google Scholar]
- 26.Eid J.E. and Sollner-Webb,B. (1997) J. Biol. Chem., 272, 14927–14936. [DOI] [PubMed] [Google Scholar]
- 27.Lang W.H. and Reeder,R.H. (1993). Mol. Cell. Biol., 13, 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shore D. (1994) Trends Genet., 10, 408–412. [DOI] [PubMed] [Google Scholar]
- 29.Gommers-Ampt J.H., Van Leeuwen,F., de Beer,A.L.J.,. Vliegenthart,J.F.G, Dizdaroglu,M., Kowalak,J.A., Crain,P.F. and Borst,P. (1993) Cell, 75, 1129–1136. [DOI] [PubMed] [Google Scholar]
- 30.Van Leeuwen F., Wijsman,E.R., Kuyl-Yeheskiely,E., Van der Marel,G.A, Van Boom,J.H. and Borst,P. (1996) Nucleic Acids Res., 24, 2476–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]