


Abstract
Phosphorothioate oligodeoxynucleotides (P=S ODNs) are frequently used as antisense agents to specifically interfere with the expression of cellular target genes. However, the cell biological properties of P=S ODNs are poorly understood. Here we show that P=S ODNs were able to continuously shuttle between the nucleus and the cytoplasm and that shuttling P=S ODNs retained their ability to act as antisense agents. The shuttling process shares characteristics with active transport since it was inhibited by chilling and ATP depletion in vivo. Transport was carrier-mediated as it was saturable, and nuclear pore complex-mediated as it was sensitive to treatment with wheatgerm agglutinin. Oligonucleotides without a P=S backbone chemistry were only weakly restricted in their migration by chilling, ATP depletion and wheatgerm agglutinin and thus moved by diffusion. P=S ODN shuttling was only moderately affected by disruption of the Ran/RCC1 system. We propose that P=S ODNs shuttle through their binding to yet unidentified cellular molecules that undergo nucleocytoplasmic transport via a pathway that is not as strongly dependent on the Ran/RCC1 system as nuclear export signal-mediated protein export, U-snRNA, tRNA and mRNA export. The shuttling property of P=S ODNs must be taken into account when considering the mode and site of action of these antisense agents.
INTRODUCTION
Antisense oligonucleotides are short stretches (usually 12–25 nt) of modified DNA or RNA designed to bind through Watson–Crick base pairing to complementary sequences in their target RNA. Their hybridization is thought to interfere with processing, transport and/or translation as well as to elicit degradation of the target RNA. As a final result antisense oligonucleotides lead to inhibition of target gene expression. Since their basis of action is sequence-specific hybridization, antisense molecules are one of the most straightforward examples of ‘rational drug design’ and bear great potential as therapeutic agents (for reviews see 1–3).
Evidence at the molecular level for the mechanisms of action of antisense molecules inside cells is still scarce (4–8). In particular, the interactions of antisense molecules with their targets and other cellular components are poorly characterized in terms of stoichiometry and structure of the complexes, where the interactions take place, and what their fates are. In addition, there have been several reports of antisense oligonucleotides acting by non-antisense mechanisms (9). In order to truly accomplish ‘rational drug design’, an in-depth understanding of the cell biology of antisense molecules must be achieved.
Phosphorothioate oligodeoxynucleotides (P=S ODNs), in which a non-bridging oxygen of the phosphodiester nucleic acid backbone is replaced by sulfur, are the most advanced antisense chemistry in clinical trials, where they have shown great promise (10–12). Recently, the first P=S ODN antisense drug has been approved for medical use in the USA (13). P=S ODNs support, unlike most other modifications, RNase H activity, which in many cases is thought to be a prerequisite for antisense activity (3). Therefore, this chemistry is also being used in the next generation of antisense compounds which employs chimeric molecules with mixed chemistries (14).
Studies using different delivery methods and different cell types have emphasized the nuclear localization of P=S ODNs as important for their potential to exert antisense activity. For example, delivery as free molecules or as complexes with cationic lipids shifted the main distribution of P=S ODN from cytoplasmic vesicles to the nucleus of endothelial cells with a concomitant large increase in antisense activity (15). Using fluorescently tagged antisense P=S ODNs and single cell analysis of antisense activity we recently showed that the P=S ODNs localized to the nucleus under conditions in which they inhibited target gene expression (16). In addition, they induced the formation of nuclear bodies (16). In two other examples the target RNA sequences only existed in the nucleus (intron sequences; 17) or the activity of the antisense oligonucleotide resulted in an aberrant RNA species in the nucleus (7). P=S ODNs are considered to move by diffusion from the cytoplasm into the nucleus as neither chilling, ATP depletion nor wheatgerm agglutinin (WGA), an inhibitor of nuclear pore complex-mediated transport, inhibited their accumulation in the nucleus after cytoplasmic injection of the oligonucleotides (18,19).
Macromolecules such as proteins, RNAs and ribonucleoproteins traffic in a regulated manner through the nuclear pore complexes in a process called nucleocytoplasmic transport. In general, these activities rely on saturable transport receptors that recognize signals on cargo molecules and help feed the cargo into the transport machinery (for reviews see 20–22). The nuclear pore complex lies at the core of the transport machinery (23). Transport receptors, though specific for certain cargoes and transport pathways, thus far all share sequence and functional similarities and fall into one protein superfamily (23). Typical transport signals comprise, for example, the classical basic nuclear localization signal (NLS) and the nuclear export signal (NES) rich in the amino acid leucine. Most nucleocytoplasmic transport processes are strongly dependent on the Ran system: Ran is a small GTPase and exists in a GTP- or GDP-bound form (20,22,24). The nucleotide-bound states of Ran are influenced by several Ran protein partners, among them RCC1, the Ran nucleotide exchange factor, and RanGAP1, a Ran GTPase-activating protein. The functionality of the Ran system is commonly accepted to rely on the assymetric distribution of its components. Ran in its GTP-bound state serves as a sensor to regulate the interactions of transport receptors with their cargo through its interaction with the transport receptors (20,22,24). In contrast to receptor/cargo interactions and their regulation, we know considerably less about the translocation step at the nuclear pores, e.g. transfer of transport complexes within the pore or the extent and source of energy required (23).
The behavior of proteins constantly migrating out of and back into the nucleus is called shuttling. The term shuttling is often meant to implicate mediated transport through the nuclear pores. In fact, in some cases it is clear that signals on the shuttling molecule and specific transport receptors are involved. For example, the M9 sequence in hnRNP-A1 is known to confer nuclear export as well as nuclear import and transportin 1 is required as a nuclear import receptor for hnRNP A1 (25 and references therein). However, in other cases shuttling behavior can be governed by retention through interactions of the migrating molecule with, for example, nuclear sites (26). In the present report we investigated the dynamics of nuclear P=S ODN molecules inside living cells. Here we show that nuclear P=S ODN molecules continuously migrate out of and back into the nucleus, i.e. they shuttle, in a process that is saturable, temperature- and energy-dependent and can be blocked by WGA.
MATERIALS AND METHODS
Oligonucleotides
12182-T is a 20mer, 5′-Texas Red-conjugated, fully modified phosphorothioate oligodeoxynucleotide complementary to a sequence in exon 5 of the rat β-tropomyosin gene. The sequence is 5′-CTTCAGAGCGCTCCAGCTCT-3′. 12183-T is a 5′-Texas Red-conjugated, fully modified 2′-O-propyl phosphodiester oligoribonucleotide of the same sequence as 12182-T where thymidine is replaced by uridine. 10366-X is a 20mer, 5′-XRITC-conjugated, fully modified phosphorothioate oligodeoxynucleotide targeted to porcine E-selectin RNA. Its sequence is 5′-GCTCCTGATTCCTTTGGACT-3′. A second, unlabeled, control 20mer phosphorothioate oligodeoxynucleotide was 8424, with the sequence 5′-GACGC-ATCGCGCCTACATCG-3′. 11068-F is a 5′-fluorescein-conjugated 20mer phosphorothioate oligodeoxynucleotide of sequence 5′-TGCATCCCCCAGGCCACCAT-3′ and targets the 3′-untranslated region of murine ICAM-1 mRNA (27). The oligonucleotides were synthesized utilizing conventional solid phase triester chemistry (28). 2′-Deoxy- and 5′-amino-modified phosphoramidites were obtained from commercial sources (PerSeptive Biosystems, Framingham, MA and Glenn Research, Sterling, VA). 2′-O-propyl amidites were synthesized at ISIS Pharmaceuticals. The fluorophores were attached manually using Texas Red sulfonylchloride, fluorescein isothiocyanate or x-rhodamine isothiocyanate (all from Molecular Probes, Eugene, OR) according to the manufacturer’s protocols. Free fluorophores were separated from oligonucleotides by gel filtration using NAP-25 columns (Pharmacia, Piscataway, NJ)
Expression vectors and recombinant proteins
The expression vectors pCGN-TM1 and pCGN-TM4 encoding HA-tagged forms of rat tropomyosin isoforms TM-1 and TM-4, respectively, were a kind gift of Dr David Helfman (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY) (29). Recombinant export substrate GGNES, which contains glutathione S-transferase fused to green fluorescent protein and the nuclear export signal of RanBP-1, as well as RanT24N, a mutant of the small GTPase Ran which does not stably bind nucleotides and exhibits increased affinity for the guanine nucleotide exchange factor RCC1, were kindly provided by Dr Ian Macara (University of Virginia, Charlottesville, VA) (30). Wild-type Ran protein was a kind gift of Dr Colin Dingwall (SUNY, Stony Brook, NY) (31). In some control experiments purified recombinant glutathione S-transferase fused to the nuclear export sequence of the cAMP-dependent protein kinase inhibitor (GST–PKI–NES) was employed and was kindly provided by Dr Susan Taylor (UCSD, La Jolla, CA) (32).
Cell lines
All cell lines were grown at 5% CO2 and 37°C in high glucose, HEPES-buffered DMEM (Life Technologies, Gaithersburg, MD) supplemented with 10% fetal bovine serum (HyClone, Logan, UT) and antibiotics. The cell lines were obtained from the cell culture core facilities at Cold Spring Harbor Laboratory. Immortalized rat embryo fibroblast Ref52 cells contain an especially high percentage (0.5–1%) of binucleate cells in freshly thawed cultures. Such binucleate cells were also seen in other cell lines, including HeLa, BHK and A549, however with less frequency. In most cases the two nuclei of binucleate cells were very close to each other. However, they seem to be clearly separated by cytoplasm since cytoplasmically injected fluorescently tagged dextrans were detected between the nuclei. In addition, the two nuclei of heterokaryon cells (see below) look similar.
Microinjection and immunostaining for fluorescence microscopy
Cells were injected semi-automatically using an Eppendorf microinjector 5242 and micromanipulator 5170 mounted on a Zeiss Axiovert 10 inverted microscope (33). Microinjection needles were made from glass capillaries (GC120TF-10; Warner Instrument Corp., Hamden, CT) using a Brown-Flaming automatic pipette puller (model P80; Sutter Instruments, San Francisco, CA). Fluorescently tagged oligonucleotides, usually at a concentration of 30 µM in the injection solution, were injected together with the injection marker 70 kDa dextran–Cascade Blue (Molecular Probes, Eugene, OR) at 6 mg/ml. Plasmids were injected into the nucleus at 25 µg/ml along with a 70 kDa fluorescein-conjugated dextran (4 mg/ml) as an injection marker (Molecular Probes, Eugene, OR). 70 kDa dextrans are restricted in their diffusion by the nuclear pores, i.e. they cannot leave the nucleus after injection provided that the nuclear membrane remains intact. One can assume a 1:15–1:20 dilution upon injection into the cell. Molecules to be injected were dissolved in injection buffer (10 mM NaH2PO4, 10 mM K2HPO4, 80 mM KCl, 4 mM NaCl, pH 7.2) and centrifuged for at least 15 min at 10 000 g at 4°C. Injections were performed at room temperature within usually ~10 min. After injection the medium was changed for fresh pre-warmed medium. In the experiments in which binucleate cells received two consecutive injections the location of the cells was retrieved with the help of gridded coverslips (Bellco, Vineland, NJ). Cells were fixed for 15 min with 4% formaldehyde at the indicated times and left unpermeabilized if no immunostaining was involved. For immunofluorescence labeling (33) cells were permeabilized for 5 min in phosphate-buffered saline (PBS) plus 0.5% Triton X-100 and incubated with primary antibodies followed by secondary antibodies diluted in PBS containing 1% normal goat serum for 30 min each. In between each of the above incubations cells were washed three times for 10 min each in PBS. All incubations were at room temperature. Ascites preparations containing anti-SC35 monoclonal antibody (34) or anti-HA-tag monoclonal antibody 12CA5 (35) were used at 1:3000 or 1:500, respectively. As secondary antibodies we used goat anti-mouse IgG preparations conjugated with either fluorescein or Cy-5 (Jackson Immunoresearch Laboratoriess, West Grove, PA). Finally, cells were mounted in 90% glycerol in 0.2 M Tris base (pH 8) with 1 mg/ml p-phenylenediamine.
Image acquisition and quantitation of nucleocytoplasmic movements of P=S ODNs
Images were acquired using a Nikon Microphot-FXA fluorescence microscope equipped with a SenSys cooled CCD camera (1320 × 1035 array, 6.7 µm pixel size; Photometrics, Tucson, AZ) and a Macintosh computer with Oncor Image software (Oncor, Gaithersburg, MD). For quantitation of oligonucleotide shuttling the fluorescence intensities per unit area in the injected and non-injected nuclei of binucleate Ref52 cells were measured using the [find objects/interactive drawing] and [measure objects] functions of the Oncor Image software. Extracellular background was not subtracted as it was negligible. As a measure of transport the ratio between the values of the non-injected and the injected nuclei was calculated. Areas were chosen to exclude nucleoli (which generally did not contain significant amounts of oligonucleotides) but were otherwise random and contained about one-third to half of the total nuclear area. Deviations of the values obtained for different areas within the same nucleus have been found to be smaller than those obtained for different nuclei within the same experimental group. Note that the calculated values reflect two transport processes, nuclear export from the injected nucleus and re-import into either nucleus of binucleate Ref52 cells. Care was taken to only analyze cells with successful injections, i.e. cells which displayed only nuclear fluorescence of the dextran injection marker.
Cell treatments
ATP depletion was performed according to Wen et al. (36). Ref52 cells were incubated at 37°C in Hank’s balanced salt solution containing 6 mM deoxyglucose and 10 mM sodium azide starting 30 min before injection and throughout the experiment. For chilling experiments cells were put on ice for 30 min before the injection and then injected as quickly as possible at room temperature. After injection cells were incubated again on ice in fresh pre-chilled medium for 2 h. To inhibit nuclear pore complex-mediated transport processes binucleate Ref52 cells grown on gridded coverslips were injected into the cytoplasm with 1 mg/ml fluorescein-conjugated WGA (Molecular Probes). After 30 min at 37°C the same cells were injected again into one nucleus with the oligonucleotide to be tested as described above.
Heterokaryon assay
The heterokaryon experiment was done essentially as described (37). Two hours after microinjection of Ref52 fibroblasts, HeLa cells were seeded on the same coverslip. Thirty minutes before fusion, protein synthesis was switched off by addition of 0.1 mg/ml cycloheximide. Cells were fused by immersion of the coverslip containing the cells in 50% polyethyleneglycol 4000 in PBS for 2 min at room temperature. After fusion cells were incubated in medium in the presence of cycloheximide for another 2 h and then fixed.
RESULTS
P=S ODNs undergo nucleocytoplasmic shuttling
We have studied the intracellular dynamics of P=S ODNs by following the movements of molecules tagged with fluorochromes, i.e. Texas Red or fluorescein. Using two different transport assays we have observed that P=S ODNs were able to exit and re-enter the nucleus of tissue culture cells, i.e. they underwent nucleocytoplasmic shuttling (Fig. 1). Both assays reveal nucleocytoplasmic transport events due to the fact that a ‘donor’ nucleus and a ‘recipient’ nucleus share a common cytoplasm. In naturally occurring binucleate Ref52 cells the P=S ODNs were found 2 h post-injection not only in the injected but also in the non-injected nucleus (Fig. 1A–C). No significant P=S ODN fluorescence was detected in the cytoplasm. The injection marker, Cascade Blue-conjugated 70 kDa dextran, too large to diffuse through the nuclear pores, was found only in the injected nucleus (Fig. 1B). Therefore, our observations could only be explained by movement of the P=S ODNs out of the injected nucleus through the cytoplasm and into the non-injected nucleus. This behavior occurred with all tested P=S ODNs, irrespective of sequence and fluorescence tags, and was also observed with a P=S ODN without a tag, which was visualized with a specific monoclonal antibody. In addition, nucleocytoplasmic shuttling was also observed in HeLa, A549 and BHK cells (data not shown).
Figure 1.
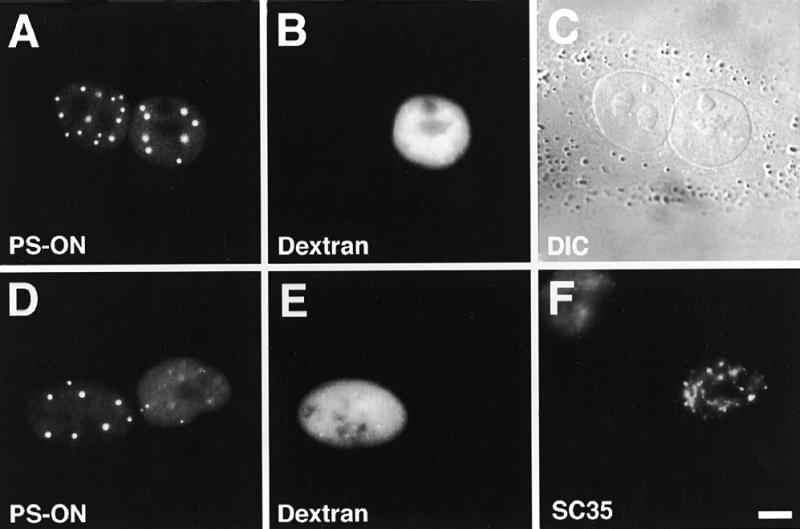
P=S ODNs undergo nucleocytoplasmic shuttling. (A–C) Microinjection of 30 µM P=S ODN 12182–Texas Red (concentration in the injection solution) and injection marker 70 kDa dextran–Cascade Blue into one nucleus of binucleate murine Ref52 fibroblasts. Two hours after injection cells were fixed and the distribution of the P=S ODN examined by fluorescence microscopy. Detection of the P=S ODN in the non-injected nucleus revealed migration across the nuclear envelopes. (D–F) Heterokaryon assay. P=S ODN 12182–Texas Red (30 µM) and injection marker 70 kDa dextran–Cascade Blue were injected into the nucleus of mononucleate Ref52 cells. The Ref52 cells were fused to HeLa cells and fixed 2 h after fusion. To distinguish the two cell types, the cells were labeled with an antibody which only reacted with HeLa but not murine SC35. Detection of the P=S ODN in the HeLa nucleus indicates its nucleocytoplasmic movement. The localizations of the P=S ODN (A and D), the injection marker (B and E), which depicts the injected nucleus, the DIC image (C), which displays the two nuclei within the common cytoplasm, and the anti-SC35 labeling (F) are shown. Bar 10 µm.
To confirm this nucleocytoplasmic movement of P=S ODNs we performed heterokaryon analysis. Mononuclear Ref52 cells were microinjected into the nucleus with the P=S ODNs to be tested. These cells were then fused to HeLa cells using polyethyleneglycol. Two hours after fusion cells were fixed and stained for the splicing factor SC35 (Fig. 1F) with an antibody which preferentially recognizes human SC35 (34). In such a way we were able to distinguish between rat Ref52 and human HeLa cell nuclei. Corroborating the results from the binucleate cells, the P=S ODNs moved from the Ref52 to the HeLa nucleus in heterokaryon cells (Fig. 1D–F). The same result was obtained in HeLa/3T3 heterokaryons and again occurred irrespective of cell type, sequence or the fluorescent tag on the P=S ODNs used (data not shown). It is safe to assume that we characterized the behavior of intact fluorescently labeled P=S ODNs. First, it has been shown that 90% of the fluorescence of a 16mer P=S ODN–fluorescein conjugate persisted even after 6 h inside a nucleus, in contrast to that of the fluorochrome alone or very small P=S ODNs of 4 nt and less (38). Second, stability measurements have shown 80% intact P=S ODNs after 6 h and ~70% intact molecules 1 day after delivery (39). Third, in binucleate cells shuttling molecules induce the formation of PS bodies in the non-injected nucleus (see Fig. 1A), as do P=S ODNs immediately introduced into cells, in a pattern which is stable for at least 6.5 h (16).
Kinetics of nucleocytoplasmic shuttling of P=S ODNs
To measure the kinetics of the movement of a P=S ODN we injected a Texas Red-conjugated molecule together with the injection marker 70 kDa dextran–Cascade Blue into one nucleus of binucleate Ref52 fibroblasts. At different time points after injection the cells were fixed and images of injected cells acquired (Fig. 2A). The increase over time in the accumulation of P=S ODNs in the non-injected nucleus was clearly evident (Fig. 2A, arrowheads). This increase was also reflected in the appearance of bright foci (the PS bodies) in the non-injected nucleus, whose formation was concentration-dependent (16). The movements of the oligonucleotides were quantified by measuring the fluorescence intensities per unit area in both nuclei and calculating the ratio between the values obtained for the non-injected and injected nuclei (Fig. 2B). Quantitation revealed almost linear kinetics up to 2 h after injection, when the processes reached equilibrium. The baseline obtained when cells were fixed immediately after injection (within 5–10 min after injection at room temperature) was ~0.08. The maximum value reached was ~0.8 after 2 h, with no further increase thereafter. It must be stressed that the results reflect two transport processes: first, the movement of the P=S ODNs out of the injected nucleus and, second, their re-migration, which can occur into either of the two nuclei of the binucleate cell. The fact that the oligonucleotide molecules can either re-enter the injected or move into the non-injected nucleus means that the fluorescence intensity in the non-injected nucleus alone only reflected part of the migrating P=S ODN molecules. If there were an equal chance of re-migration into either of the two nuclei, then the maximal ratio that can be obtained would be 1 with all P=S ODN molecules undergoing nucleocytoplasmic shuttling. The maximal value of 0.8 measured in our experiments would then indicate that the majority, but not all, of the injected P=S ODNs were able to shuttle.
Figure 2.
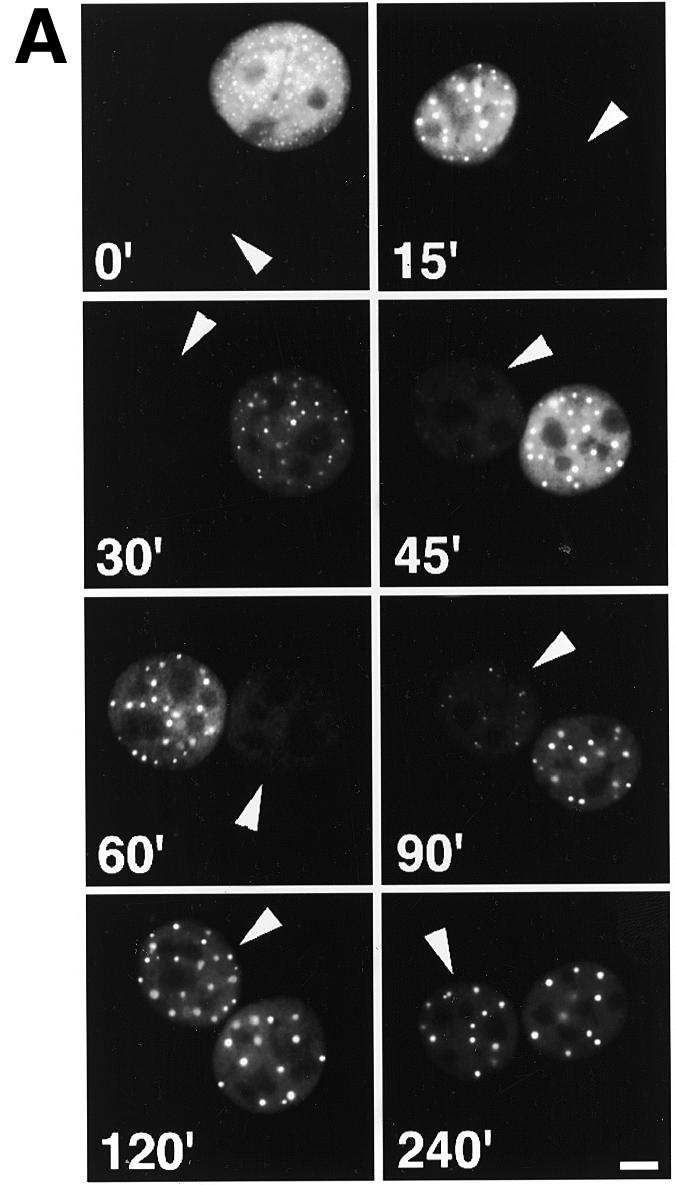

Kinetics of P=S ODN shuttling in binucleate Ref52 cells. P=S ODN 12182–Texas Red (30 µM) and injection marker 70 kDa dextran–Cascade Blue were injected into one nucleus of binucleate Ref52 cells. Cells were incubated at 37°C for various time points and fixed. (A) Examples of cells fixed at different time points. The P=S ODN fluorescence signals are shown. The arrows point to the non-injected nuclei. Bar 10 µm. (B) To quantify the extent of shuttling, the ratio between the fluorescence intensities per unit area of the non-injected and the injected nucleus (see Materials and Methods) were computed and plotted against time. The means of at least 10 cells quantified for each time point ± SD are shown. Note that this quantitation reflects two transport events, nuclear export out of the injected nucleus and re-import into either of the two nuclei.
Shuttling of P=S ODNs shares characteristics of active, nuclear pore-mediated transport
To characterize the shuttling of P=S ODNs we analyzed the influence of chilling, ATP depletion and WGA pre-injection on the movement of fluorescent dye-conjugated P=S ODNs 2 h after their injection into one nucleus of binucleate Ref52 fibroblasts (Fig. 3, quantitation in Fig. 4A). Chilling of cells resulted in a strong inhibition of P=S ODN shuttling as fluorescent P=S ODN molecules were barely detectable in the non-injected nucleus (Fig. 3A). No significant level of fluorescence was observed in the non-injected nucleus even 4 h after injection (data not shown). The block in shuttling by chilling was reversible but it took ~3 h after shifting back to 37°C, in comparison to the usual 2 h, to restore maximal shuttling (data not shown). A similar inhibition with only weak P=S ODN fluorescence in the non-injected nucleus was also observed after inhibition of ATP synthesis (Fig. 3B). Quantitation showed a strong decrease in the ratio between fluorescence intensities per unit area of the non-injected and the injected nucleus to 0.1 at 4°C and 0.24 after ATP depletion, respectively (Fig. 4A). The small differences between the extent of inhibition after chilling in comparison to ATP depletion were reproducible.
Figure 3.
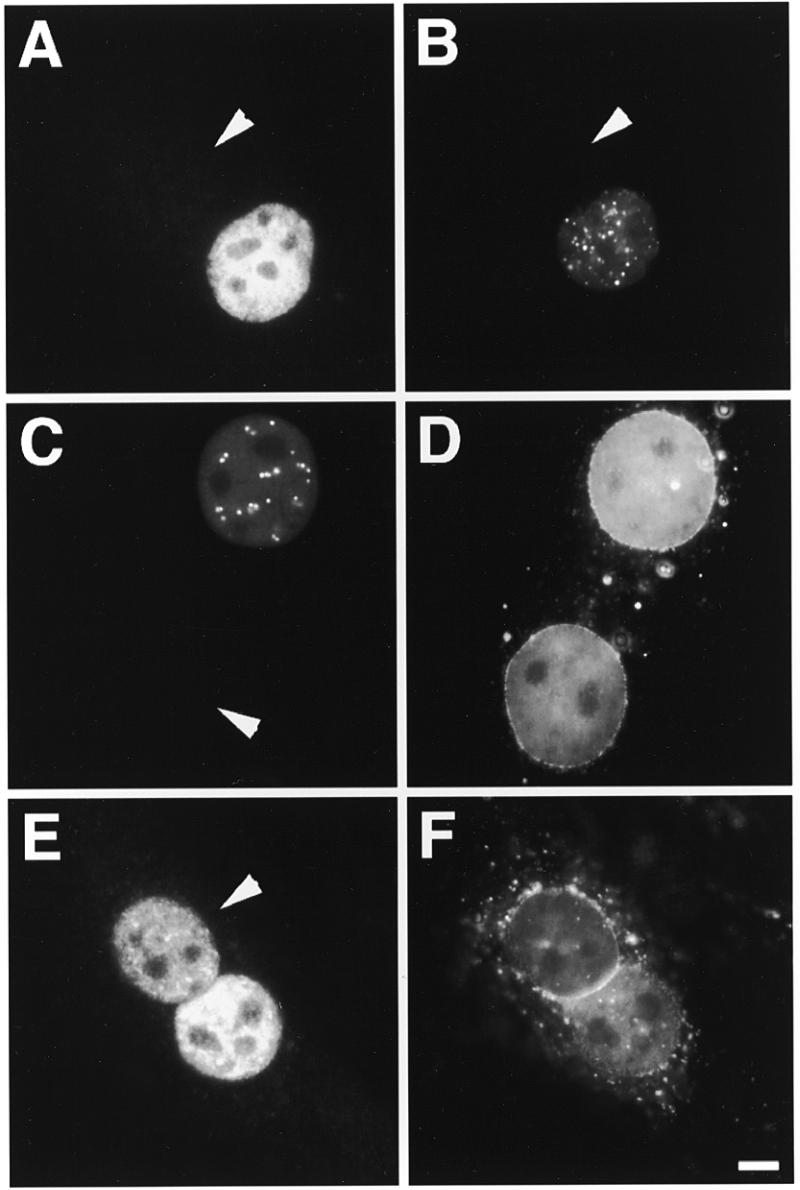
Characteristics of P=S ODN shuttling. Microinjection of 30 µM P=S ODN 12182–Texas Red and 70 kDa dextran–Cascade Blue into one nucleus of binucleate Ref52 cells pretreated as indicated below. After injection cells were incubated for another 2 h before fixation. The arrows point to the non-injected nucleus in each case. (A) Microinjection into chilled cells and incubation on ice after injection. Shuttling was temperature-dependent and strongly inhibited at 4°C. (B) Injection into cells under conditions of ATP depletion showed that nucleocytoplasmic migration of P=S ODN was energy-dependent. (C and D) Cells were pre-injected with 1 mg/ml fluorescein-conjugated WGA into the cytoplasm 30 min prior to oligonucleotide injection. Inhibition of shuttling by WGA suggested a nuclear pore complex-mediated transport process. (E and F) The labeled P=S ODN was injected together with 2 mM unlabeled P=S ODN 8424 into one nucleus of a binucleate cell which had been pre-injected into the cytoplasm with WGA–fluorescein. Detection in the non-injected nucleus despite treatment with WGA indicated a passive diffusion-driven migration of 12182–Texas Red and thus saturation of the active transport process by the excess of unlabeled P=S ODN. The fluorescent images for the P=S ODNs (A–C and E) and WGA (D and F) are shown. Bar 10 µm.
Figure 4.

Quantitative analysis of the characteristics of nucleocytoplasmic shuttling of oligonucleotides. (A and B) Analysis of P=S ODN 12182–Texas Red shuttling. (C) Analysis of phosphodiester 2′-O-propyl-oligoribonucleotide (PO-2′P-ON) 12183–Texas Red migration. Oligonucleotides were injected at a concentration of 30 µM into one nucleus of binucleate Ref52 cells treated as indicated below. Co-injection of 70 kDa dextran–Cascade Blue marked the injected nucleus. Two hours after injection cells were fixed and the fluorescence intensities per unit area in each nucleus were measured. Then the ratio between the values of the non-injected and the injected nuclei was calculated (see Materials and Methods). In each case at least 10 cells of at least two independent experiments were evaluated. The means ± SD are given. The extent of shuttling was compared for untreated cells (w/o), chilled cells (4°C), energy-depleted cells (–ATP), cells pre-injected with wheatgerm agglutinin (WGA), cells pre-injected with WGA and then injected with 30 µM 12182–Texas Red together with 2 mM unlabeled P=S ODN 8424 (WGA+2mM 8424) and cells that received 12182–Texas Red together with 1.7 mg/ml of a Ran mutant protein impairing nuclear protein export (RanT24N).
WGA is known to bind to a number of nuclear pore complex proteins carrying O-linked N-acetylglucosamine units thus blocking transport processes but not diffusion through the nuclear pores (40). To test the effect of WGA on the P=S ODN shuttling we pre-injected WGA–fluorescein into the cytoplasm of binucleate cells before injecting the P=S ODNs into one nucleus. WGA binding to the nuclear pores was indicated by its localization at the nuclear membrane (Fig. 3D). In such cells the injected P=S ODNs were observed almost exclusively in the injected nucleus (Fig. 3C). This was consistent with a nuclear pore complex-dependent transport of the majority of P=S ODNs and was reflected in the drop in the fluorescence ratio of the non-injected and the injected nucleus to 0.15 (Fig. 4A). The effectiveness of our WGA preparation was checked by showing that it abolished the export of a protein export substrate, GST–PKI–NES (data not shown). Preinjection of a control lectin, concanavalin A, instead of WGA did not impair P=S ODN shuttling (data not shown).
None of the above treatments, chilling, ATP depletion or WGA pre-injection, led to an increase in cytoplasmic fluorescence, i.e. accumulation of P=S ODNs in the cytoplasm. Together with the virtual absence of fluorescence in the non-injected nucleus, this suggested that the shuttling process was inhibited at the export step, out of the injected nucleus. Besides the export step, re-import could be inhibited as well. In addition to temperature and energy sensitivity, saturation is another characteristic of a carrier-mediated transport process. When we injected 30 µM fluorescently labeled P=S ODN together with 3 mM unlabeled P=S ODN into one nucleus of binucleate cells, the labeled P=S ODN nevertheless migrated to the non-injected nucleus (data not shown). However, it has been shown that once the carrier-mediated transport mechanism is saturated, passive diffusion through the nuclear pores can provide an alternative pathway for small molecules (41,42). Therefore, it was necessary to determine if at such high concentrations the P=S ODNs moved by diffusion. To do so we observed what happened after injection of a mixture of labeled plus an excess of unlabeled P=S ODN into one nucleus of binucleate cells pre-injected with WGA. Whereas the lectin inhibited the shuttling of P=S ODNs at 30 µM (see above Fig. 3C and D), it no longer inhibited the movement upon co-injection of 3 mM unlabeled P=S ODNs (Fig. 3E and F). The ratio between fluorescence intensities per unit area in the non-injected and the injected nucleus was close to control cells without any treatment (Fig. 4A). This result argues that, after co-injection of high amounts of unlabeled P=S ODN molecules, the active process had been saturated and movement of oligonucleotide out of the injected nucleus was passive.
When RNA polymerse II transcription is inhibited, certain shuttling RNA-binding proteins like hnRNP-A1 (37) and SF2/ASF (43) accumulate in the cytoplasm, probably due to a strongly decreased re-import into the nucleus. In contrast, P=S ODN shuttling was not significantly affected after treatment of cells with RNA polymerase II inhibitors such as actinomycin D (10 µg/ml) or α-amanitin (50 µg/ml) and there was no visible cytoplasmic accumulation (data not shown). Together, our results argue that the nuclear export of the majority of P=S ODNs shows properties of an active process. Similar results were obtained with a different fluorescently tagged P=S ODN (11068-F, different sequence and different fluorochrome; data not shown). Since the various treatments affected the export process of P=S ODNs we could not analyze the characteristics of the re-import process into the nucleus. Cytoplasmic injections of chilled, ATP-depleted or WGA-containing cells did not inhibit movement into and accumulation of the injected P=S ODNs in the nuclei (data not shown). This is in agreement with published results which have suggested that free cytoplasmic P=S ODNs diffuse into nuclei and are bound by retention sites (18,19).
Intracellular movement of a phosphodiester 2′-O-propyl-oligonucleotide primarily occurs by diffusion
Next, we were interested in determining if oligonucleotides without phosphorothioate backbone chemistry show the same intracellular transport behavior. To address this question we chose to characterize the intracellular dynamics of a phosphodiester 2′-O-propyl-oligonucleotide (PO-2′P-ON) of the same sequence as a shuttling P=S ODN. The 2′-O-propyl modification is necessary to render the molecule sufficiently stable against nucleases, in contrast to plain phosphodiester compounds (44,45).
Two hours after injection into one nucleus of binucleate Ref52 cells the PO-2′P-ON was found in the non-injected nucleus as well (Fig. 5A and B). In addition, unlike P=S ODNs, there was some diffuse cytoplasmic localization. Quantitation showed that the ratio between the fluorescence intensities per unit area of the non-injected and the injected nucleus was close to 1 and thus that virtually all of the injected molecules participated in the movement (Fig. 4C). However, in comparison to P=S ODNs there was no strong inhibition of this movement after chilling, ATP depletion or WGA pre-injection (Fig. 5C–F). In all cases the fluorescence ratio of the two nuclei was still ~0.6 (quantitation in Fig. 4C). These results are consistent with a diffusion-driven movement of the majority of PO-2′P-ONs within the cell. Accumulation in the nuclei would be due to the presence of nuclear binding sites.
Figure 5.
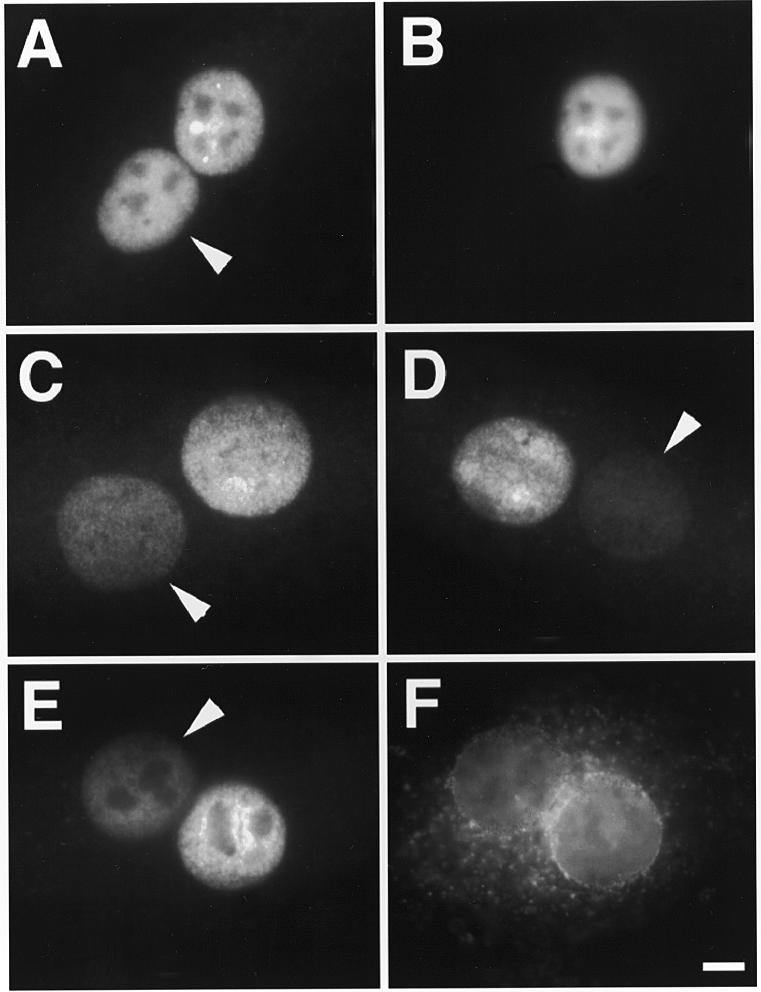
The nucleocytoplasmic migration of a large portion of injected phosphodiester 2′-O-propyl-oligoribonucleotides is a passive process. Microinjection of 30 µM PO-2′P-ON 12183–Texas Red and 70 kDa dextran–Cascade Blue into one nucleus of binucleate Ref52 cells. After injection cells were incubated for 2 h before fixation. (A and B) No further treatment. (C) Injection into chilled cells and incubation on ice after injection. (D) Injection into cells incubated under ATP depletion conditions. (E and F) Pre-injection of 1 mg/ml WGA–fluorescein into the cytoplasm 30 min prior to oligonucleotide injection. In all cases there was a considerable migration into the non-injected nucleus. The fluorescent images for the P=S ODNs (A and C–E), dextran (B) and WGA (F) are shown. The arrows point to the non-injected nucleus in each case. Bar 10 µm.
Relationship of P=S ODN transport and the Ran/RCC1 system
Many nuclear pore-mediated transport processes require the action of the Ran/RCC1 system (22,46). In order to test if this system was also involved in the transport of P=S ODNs, we checked the influence of RanT24N protein injection on shuttling of P=S ODNs in binucleate Ref52 cells. Nuclear RanT24N injection has been shown to efficiently block nuclear export signal-mediated protein export as well as U-snRNA, mRNA and tRNA export (24,30). Co-injection of RanT24N had only a moderate impact on P=S ODN shuttling whereas wild-type Ran protein did not noticeably influence shuttling (Fig. 6A versus B, respectively; note the absence of PS bodies in the non-injected nucleus in Fig. 6A which gives a measure of P=S ODN concentration). Quantitation revealed a decrease in the ratio between fluorescence intensities per unit area of the non-injected and the injected nucleus to 0.42 after RanT24N injection (Fig. 4B). At the same time, in the positive control the same preparation of RanT24N strongly inhibited the export of a reporter protein carrying the NES of RanBP-1 in 98% of injected cells (Fig. 6C). An increase in the concentration of injected RanT24N did not result in stronger inhibition (data not shown). Thus, P=S ODN shuttling, unlike NES-dependent protein export, was only in part inhibited by RanT24N. When a large amount of P=S ODN was co-injected with the protein export substrate, this protein was nevertheless efficiently exported in all injected cells (Fig. 6D). This result indicates that P=S ODNs do not interfere with the cellular protein export machinery or occlude nuclear pores, even at these high concentrations.
Figure 6.
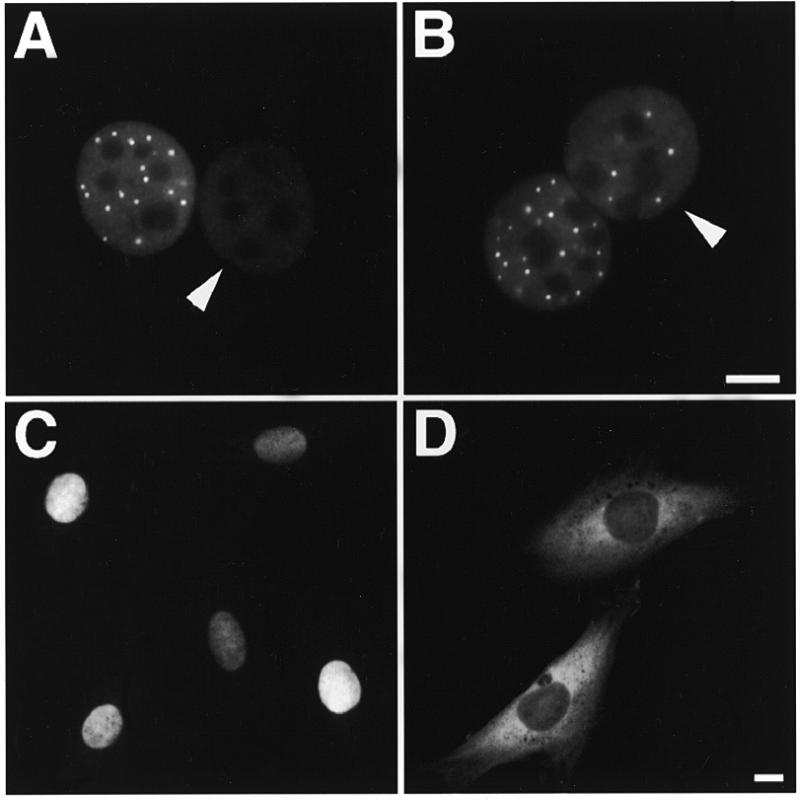
Shuttling of P=S ODNs is less sensitive to nuclear RanGTP depletion than classical protein export. (A) Co-injection of 30 µM P=S ODN 12182–Texas Red and 1.7 mg/ml RanT24N into one nucleus of a binucleate Ref52 cell. Microscopic observation of the fixed cell 2 h after injection indicated that shuttling was only partly inhibited. (B) In a control co-injection of 12182–Texas Red and 1.7 mg/ml wild-type Ran protein shuttling proceeded normally. (C) The same preparation of 1.7 mg/ml RanT24N protein when co-injected with 2 mg/ml export substrate GGNES abolished protein export. The reporter protein was still nuclear 1 h after injection. (D) Co-injection of 2 mg/ml export substrate GGNES with an excess of 2 mM P=S ODN 8424 indicated that GGNES was normally exported within 1 h. The arrowheads in (A) and (B) point to the non-injected nuclei. Bars 10 µm.
Shuttling P=S ODNs display antisense activity
As described above, the majority of the injected P=S ODNs undergo nucleocytoplasmic shuttling. In terms of the use of P=S ODNs as antisense agents it was important to determine if this large percentage was still able to show antisense activity. We employed P=S ODN 12182-T targeted against sequences in exon 5 of rat tropomyosin TM-1, which has been shown to display specific antisense activity when microinjected at 30 µM together with a plasmid encoding the target gene. This antisense molecule is without effect against isoenzyme TM-4 of the tropomyosin family, which does not contain the target sequence (unpublished data and Table 1A). 12182-T was injected along with 70 kDa dextran–Cascade Blue into one nucleus of binucleate Ref52 cells. Two hours after the first injection plasmids encoding the TM-1 target or the TM-4 control gene were injected together with injection marker 70 kDa dextran–fluorescein into the second nucleus. It was possible that the antisense effect would be exerted in the cytoplasm or the second nucleus receiving the expression vectors because these are the two compartments of a binucleate cell where target RNA would be present. We injected the antisense P=S ODN at 60 µM into the first nucleus to ensure that roughly an amount equaling a direct 30 µM injection (which we knew to be effective, see Table 1A) reached the second nucleus. The assumption was that after export from the injected nucleus there would be an equal chance of moving back into either of the two nuclei. Three hours after this second injection cells were fixed and checked for expression of the target or control gene by staining with an antibody recognizing the peptide tag of the newly made protein. As another control, an unrelated P=S ODN was used in the same assay. Expression of the TM-1 target gene injected into the second nucleus, which did not receive oligonucleotide, was abolished in cells which were microinjected into the first nucleus with the antisense (Fig. 7A–D and Table 1B) but not the control P=S ODN (Fig. 7E–H and Table 1B). In addition, the antisense P=S ODN had no effect on TM-4 gene expression. This demonstrates that the antisense P=S ODNs moving out of the first nucleus exerted antisense activity as it was their only possibility to hybridize to their target RNA, either in the cytoplasm or in the second nucleus.
Table 1. Evaluation of antisense activity after co-injection of 30 µM P=S ODNs and expression vectors into the same nucleus of Ref52 cells (A) and evaluation of antisense activity of shuttling P=S ODNs (B).
| Sample | Injected | Positive | Percent |
|---|---|---|---|
| (A) Co-injections into the same nucleus | |||
| 12182-T + TM1 | 74 | 4 | 5 |
| 63 | 13 | 21 | |
| 12182-T + TM4 | 117 | 101 | 86 |
| 70 | 62 | 89 | |
| 10366-X + TM1 | 74 | 59 | 80 |
| 86 | 66 | 77 | |
| (B) Consecutive injections into different nuclei of binucleate cells | |||
| 12182-T/TM1 | 25 | 1 | 4 |
| 10 | 1 | 10 | |
| 12182-T/TM4 | 11 | 9 | 82 |
| 10366-X/TM1 | 32 | 30 | 94 |
| 18 | 13 | 72 |
First P=S ODNs were injected at 60 µM into one nucleus of binucleate Ref52 cells. Two hours later the respective expression vector was injected into the other nucleus that did not receive the P=S ODN. P=S ODN 12182–Texas Red was complementary to sequences in exon 5 of tropomyosin isoform TM-1 but not TM-4. P=S ODN 10366–XRITC was a control P=S ODN without complementary sequences in TM-1. Sites of injections were verified by 70 kDa dextran–Cascade Blue (site of P=S ODN injection) and fluorescein (site of expression vector injection) co-injections. Expression of the tropomyosin isoforms was checked 3 h after vector injection by indirect immunofluorescence with an antibody against the HA tag of the expressed protein and Cy5-conjugated secondary antibodies. The results of independent experiments are shown where the number of successfully injected cells were scored positive or negative for tropomyosin expression and the percentage of expressing cells calculated.
Figure 7.
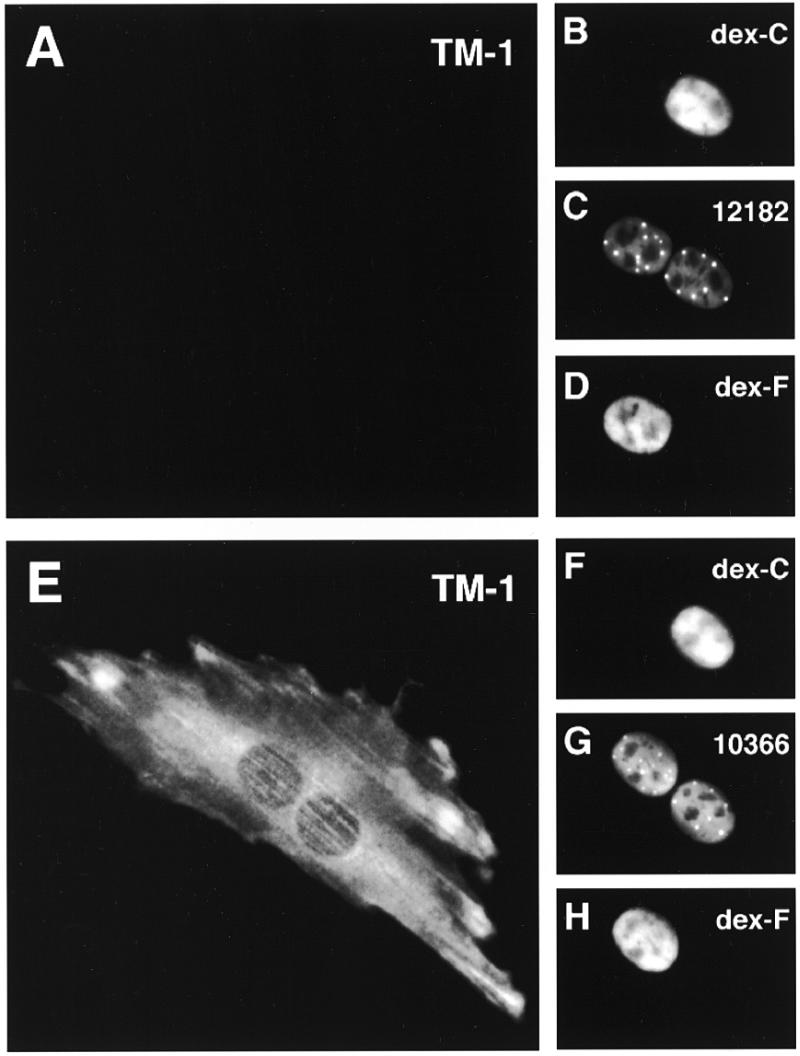
Shuttling P=S ODNs are able to exert their antisense activity. P=S ODN 12182–Texas Red (60 µM) (antisense) or 10366–XRITC (control) and 70 kDa dextran–Cascade Blue (dex-C) were injected into one nucleus of binucleate Ref52 cells. Two hours later the same cells received a second injection into the other nucleus with a vector encoding a tagged form of the tropomyosin TM-1 target gene of 12182–Texas Red together with 70 kDa dextran–fluorescein (dex-F). Cells were fixed 3 h after this second injection and labeled for expression of TM-1 with an antibody against the protein tag and secondary antibodies coupled to Cy5. The antisense P=S ODN abolished TM-1 target gene expression (A). Hybridization was only possible if the P=S ODN moved out of the injected nucleus to contact its target RNA in the cytoplasm or the second nucleus. The control P=S ODN did not interfere with TM-1 expression (E). The fluorescent images for target gene expression TM-1 (A and E), dextran–Cascade Blue (B and F), the P=S ODNs 12182–Texas Red (C) or 10366–XRITC (G) and dextran–fluorescein (D and H) are shown. Bar 10 µm.
DISCUSSION
Characteristics of nucleocytoplasmic shuttling of P=S ODNs
In the present report we describe a novel property of the intracellular behavior of P=S ODNs: nucleocytoplasmic shuttling. We initially observed this shuttling behavior in living cells in which two nuclei share a common cytoplasm. Nucleocytoplasmic shuttling was indicated by migration of the P=S ODNs from an injected nucleus through the cytoplasm into a non-injected nucleus. Heterokaryon assays confirmed this shuttling behavior. P=S ODN shuttling had characteristics of an active transport process as it was temperature-sensitive and ATP-dependent. There were also indications that the process was saturable and therefore carrier-mediated. The inhibition of shuttling by WGA indicated participation of the nuclear pore complex in P=S ODN transport. The observation that the P=S ODNs were largely localized in the injected nucleus with no accumulation in the cytoplasm after inhibition of shuttling suggested that the first step in the shuttling process, nuclear export, was an active nuclear pore complex-mediated process. The properties of the re-import process of P=S ODNs, that were previously exported, back into the nucleus are unclear. Although direct cytoplasmic injection led to passive diffusion into the nucleus (18,19; our unpublished data), it is not certain if that describes the re-import of P=S ODNs properly. It is possible that the exported P=S ODNs were still bound to the presumed export carriers that might also govern their re-import into the nucleus. If these P=S ODN carrier molecules were predominantly nuclear, i.e. their cytoplasmic pools at any given time are small, most free P=S ODNs injected into the cytoplasm would be capable of freely diffusing into the nucleus rather than bind the protein in the cytoplasm. In this scenario, the free P=S ODNs injected into the cytoplasm would not reflect the state of P=S ODNs exported from the nucleus. Thus, both possibilities, active re-import or passive diffusion, remain possible. In this respect, it is noteworthy that in contrast to previous reports, single-stranded phosphodiester oligodeoxynucleotides have recently been found to be actively imported into the nucleus and to even serve as nuclear import signals for otherwise non-imported proteins (47). Interestingly, the extent of import was dependent upon the base composition of the oligodeoxynucleotide. However, P=S ODNs were not tested in that report. The use of the classical in vitro import assay with digitonin-permeabilized cells (48) does not seem to be appropriate as a method to clarify the P=S ODN import mechanism since the distribution after addition of the oligonucleotides to permeabilized cells was not the same as after introduction into intact cells (16).
There is experimental evidence that retention signals can determine the nuclear accumulation of proteins (26,49) and retention of tRNA is rate limiting in its export (50). Nuclear accumulation of P=S ODNs after cytoplasmic injection is also considered to be due to their retention at nuclear binding sites (18,19). It is possible that retention of P=S ODNs in the nucleus might also be altered upon chilling or ATP depletion due to changes in the behavior or properties of nuclear components interacting with the oligonucleotides.
The pathway of P=S ODN shuttling
The easiest explanation for their nucleocytoplasmic transport is the binding of P=S ODNs to molecules, for example proteins, undergoing nuclear pore complex-mediated transport. Hence, shuttling is most likely an indirect property of P=S ODNs conferred on them by binding to molecules undergoing nucleocytoplasmic transport (‘piggy-back’ transport of P=S ODNs). The properties of P=S ODN dynamics described above and discussed below therefore would hint at such carrier molecules. These carrier molecules could be either transport receptors or cargo molecules of transporter systems. Thus far most known transport processes rely on the assymetric distribution of the components of the Ran/RCC1 system (24,30). Ran in its GTP-bound form serves as a sensor to regulate the interaction between the cargo and the members of the superfamily of transport receptors (20,22). Upon nuclear injection the RanT24N mutant protein almost completely abolished protein export, presumably due to depletion of the nuclear RanGTP pool through sequestration and inhibition of the RCC1 exchange factor (30). In contrast, RanT24N injection had only a moderate effect on the extent of P=S ODN shuttling, decreasing it to ~50%. One possibility is that the small amounts of RanGTP that might still be present after RanT24N injection were sufficient to sustain 50% of the transport. Another possibility is that there is a certain Ran-independence of P=S ODN shuttling. It has been shown that different export pathways exhibit distinct sensitivities to RanGTP depletion (24). Besides NES-mediated protein export, U-snRNA, mRNA and tRNA export in Xenopus oocytes is also severely impaired after depletion of the RanGTP pool (24). Thus, all these transport pathways were more sensitive to the depletion of nuclear RanGTP than P=S ODN shuttling and therefore are likely not to be closely related to the P=S ODN/putative P=S ODN carrier molecule pathway. We also employed the tsBN2 cell line which loses most of its functional RCC1 protein at the restrictive temperature (see 30 and references therein), effectively also leading to a decrease in the nuclear RanGTP concentration. We found that P=S ODN shuttling proceeded normally under conditions in which poly(A)+ RNA export and hnRNP-A1 import were severely inhibited (data not shown). This again demonstrated differences between some ‘classical’ transport processes and the process mediating P=S ODN shuttling.
Transport systems not directly linked to the Ran/RCC1 system and nuclear pore complex-mediated transport unassisted by Ran appear to exist and may contribute to P=S ODN shuttling: Yeast cells carrying mutant alleles of PRP20, RNA1 and GSP1 (the Saccharomyces cerevisiae homologs of mammalian RCC1, the GTPase activating protein RanGAP1 and Ran, respectively) upon heat shock still exported the mRNAs encoding heat shock factors (51). There have also been indications for the involvement of a GTPase other than Ran in nucleocytoplasmic transport (52). In addition, it is unclear how the high nuclear accumulation of Ran itself is maintained when Ran is constantly exported during one transport cycle (20). A mechanism different from the known protein import pathways may be a possibility. Further, importin β, part of the nuclear pore targeting complex of NLS-carrying karyophiles, can undergo translocation from the cytoplasm into the nucleus in a Ran-unassisted manner when it does not carry the α-subunit/NLS cargo (53).
Candidates for molecules mediating nucleocytoplasmic P=S ODN transport
It is known that P=S ODNs, which are intended to hybridize very specifically to complementary nucleic acid sequences, nevertheless display a relatively high non-specific binding to a variety of proteins through their highly charged phosphorothioate backbone (3). Since the same sequences made as phosphodiester 2′-O-propyl-oligoribonucleotides did not show a strong dependency of their nucleocytoplasmic migration on temperature and energy and their movement was also not inhibited by WGA, the ability to shuttle is likely due to the relatively high non-specific avidity of P=S ODNs for proteins. After adding P=S ODNs to isolated nuclei a number of proteins have been found to be crosslinked to them (19) and two categories of high and low affinity binding sites totaling ~6 000 000/cell nucleus have been calculated by Scatchard plots (54). Others have shown complex formation between nuclear proteins and P=S ODNs by gel shift assays (55). However, none of the putative binding proteins have been identified. One shuttling protein known to interact in vitro with P=S ODNs is the nucleolar protein nucleolin (56). However, P=S ODNs neither accumulated in the nucleolus nor did the shuttling kinetics resemble those of nucleolin, whose shuttling in heterokaryon assays was maximal ~72 h after fusion (57). We have shown previously that the majority of cellular P=S ODN molecules, under conditions in which they have the potential to display antisense activity, associate with the nuclear matrix fraction of cells (16). Therefore candidates for P=S ODN interacting molecules may be sought in this cellular fraction. Alternatively, it may be that the shuttling behavior was not due to the interaction of P=S ODNs with one or a few particular molecules but to binding to many different classes of molecules. Therefore, our observations may represent a mean result of many different processes. This could explain the small but reproducible differences in the extent of inhibition by low temperature and ATP depletion, e.g. not all of the transport processes involved that were temperature-sensitive were also energy-dependent.
Impact on the use of P=S ODNs as antisense agents
Knowledge about the dynamics and distribution of oligonucleotides in the cell, i.e. their cellular pharmacokinetics, is important to optimally employ them as antisense drugs. One example is the targetting of particular RNA sequences which might exist only in the nucleus (i.e. introns) or through mechanisms like translational arrest which can only take place in the cytoplasm. The question arises in which compartment, the nucleus or the cytoplasm, an antisense oligonucleotide exerts its activity. Hybridization and activity may occur in separate compartments. The translation arrest example indicates activity in the cytoplasm, but not necessarily hybridization in the cytoplasm. We do not know if hybridization and antisense activity towards TM1 transcripts occurred in the cytoplasm and/or the nucleus. However, our observation that P=S ODNs can shuttle between the nucleus and the cytoplasm and that shuttling molecules retain their antisense activity means that a nuclear localization per se cannot be considered a clear cut argument for actual execution of activity in the nucleus. Also, the shuttling of the majority of the injected P=S ODNs in a form capable of inhibiting target gene expression argues that most P=S ODN molecules remained bioavailable in terms of antisense activity and were not inactivated through complex formation with nuclear proteins.
P=S ODNs are predominantly nuclear when introduced into cells under conditions in which they have the potential to display antisense activity (16 and references therein). However, in certain cases one might seek to target cytoplasmic RNAs, e.g. the RNAs of viruses with an exclusively cytoplasmic life cycle. The majority of nuclear P=S ODNs should turn up, at least for a transitory phase, in the cytoplasm by means of shuttling. Though not directly proven, they might possibly be in a state capable of hybridizing and of exerting antisense activity. If that was true an alternative targeting pathway exclusive to cytoplasmic target RNAs would not be necessary as the P=S ODNs that migrated out of the nucleus would provide the cytoplasm with a constant supply of active antisense molecules.
The finding that P=S ODNs actively shuttle between the nucleus and cytoplasm of cells extends our understanding of the cellular behavior of this new class of drugs. This study also identified differences in the cellular pharmacokinetic behavior between P=S ODNs and one class of second generation oligonucleotides. In both cases it was found that oligonucleotides migrated from the injected nucleus to the second nucleus, however movement to the uninjected nucleus appeared to be a passive process for the second-generation oligonucleotide. Interestingly, in a set of preliminary experiments a 2′-methoxyethoxy-oligoribonucleotide with a phosphorothioate backbone, but not the same oligonucleotide with phosphodiester bridges, showed similar characteristics to P=S ODNs in that it migrated from the injected into the non-injected nucleus of binucleate cells in a temperature-dependent manner and its transport was inhibited by WGA (data not shown). Both P=S ODNs and second generation oligonucleotides have been shown to inhibit gene expression by an antisense-dependent mechanism (5,8,44,45). Therefore, it is unlikely that the active shuttling process is required for antisense activity. However, it should be noted that uniformly modified second generation oligonucleotides tend to be more restricted in the number of target sites to which they can effectively bind and inhibit gene expression (8). This fact is very likely related to their inability to support RNase H action on the target RNA, in contrast to P=S ODNs. It is therefore envisaged to employ as third generation antisense agents chimeric molecules with mixed-type chemistries, i.e. with sufficient contiguous P=S ODN units to enable RNase H activity (14). As the cellular localization of potential target transcripts may be highly variable and RNA processing events occur in discrete cellular domains, it is important to more fully understand the dynamics of oligonucleotide localization in cells to broadly apply the antisense technology. In conclusion, these results provide important new insights into the cellular behavior of a potentially significant new class of therapeutic agents.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Colin Dingwall, David Helfman and Ian Macara for kindly providing reagents, Colin Dingwall for valuable discussions and David Goldfarb for valuable discussions and critical reading of the manuscript. We are also grateful for the technical support of Tamara Howard and Jennifer McCann at Cold Spring Harbor Laboratory and for the olignonucleotide synthesis services of John Brugger, Sheri Manalili and Henri Sasmor at ISIS Pharmaceuticals. Thanks are also due to the members of the Spector laboratory for helpful discussions. P.L. was the recipient of a research fellowship from the Deutsche Forschungsgemeinschaft. This work was supported by grants from the National Institutes of Health (NIGMS 42694) and ISIS Pharmaceuticals to D.L.S.
REFERENCES
- 1.Sharma H.W. and Narayanan,R. (1995) Bioessays, 17, 1055–1063. [DOI] [PubMed] [Google Scholar]
- 2.Crooke S.T. (1996) Med. Res. Rev., 16, 319–344. [DOI] [PubMed] [Google Scholar]
- 3.Crooke S.T. (1997) Adv. Pharmacol., 40, 1–49. [DOI] [PubMed] [Google Scholar]
- 4.Shuttleworth J. and Colman,A. (1988) EMBO J., 7, 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiang M.Y., Chan,H., Zounes,M.A., Freier,S.M., Lima,W.F. and Bennett,C.F. (1991) J. Biol. Chem., 266, 18162–18171. [PubMed] [Google Scholar]
- 6.Giles R.V., Spiller,D.G. and Tidd,D.M. (1995) Antisense Res. Dev., 5, 23–31. [DOI] [PubMed] [Google Scholar]
- 7.Condon T.P. and Bennett,C.F. (1996) J. Biol. Chem., 271, 30398–30403. [DOI] [PubMed] [Google Scholar]
- 8.Baker B.F., Lot,S.S., Condon,T.P., Cheng-Flournoy,S., Lesnik,E.A., Sasmor,H.M. and Bennett,C.F. (1997) J. Biol. Chem., 272, 11994–12000. [DOI] [PubMed] [Google Scholar]
- 9.Stein C.A. (1996) Trends Biotechnol., 14, 147–149. [DOI] [PubMed] [Google Scholar]
- 10.Dean N.M., McKay,R., Miraglia,L., Geiger,T., Muller,M., Fabbro,D. and Bennett,C.F. (1996) Soc. Trans., 24, 623–629. [DOI] [PubMed] [Google Scholar]
- 11.Wagner R.W. and Flanagan,W.M. (1997) Mol. Med. Today, 3, 31–38. [DOI] [PubMed] [Google Scholar]
- 12.Webb A., Cunningham,D., Cotter,F., Clarke,P.A., di Stefano,F., Ross,P., Corbo,M. and Dzienwanowska,Z. (1997) Lancet, 349, 1137–1141. [DOI] [PubMed] [Google Scholar]
- 13.Marwick C. (1998) J. Am. Med. Assoc., 280, 871. [Google Scholar]
- 14.Agrawal S. and Iyer,R.P. (1995) Curr. Opin. Biotechnol., 6, 12–19. [DOI] [PubMed] [Google Scholar]
- 15.Bennett C.F., Chiang,M.Y., Chan,H., Shoemaker,J.E. and Mirabelli,C.K. (1992) Mol. Pharmacol., 41, 1023–1033. [PubMed] [Google Scholar]
- 16.Lorenz P., Baker,B.F., Bennett,C.F. and Spector,D.L. (1998) Mol. Biol. Cell, 9, 1007–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagner R.W., Matteucci,M.D., Lewis,J.G., Gutierrez,A.J., Moulds,C. and Froehler,B.C. (1993) Science, 260, 1510–1513. [DOI] [PubMed] [Google Scholar]
- 18.Chin D.J., Green,G.A., Zon,G., Szoka,F.C.,Jr and Straubinger,R.M. (1990) New Biol., 2, 1091–1100. [PubMed] [Google Scholar]
- 19.Leonetti J.P., Mechti,N., Degols,G., Gagnor,C. and Lebleu,B. (1991) Proc. Natl Acad. Sci. USA, 88, 2702–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goerlich D. (1998) EMBO J., 17, 2721–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mattaj I.W. and Englmeier,L. (1998) Annu. Rev. Biochem., 67, 265–306. [DOI] [PubMed] [Google Scholar]
- 22.Weis K. (1998) Trends Biochem. Sci., 23, 185–189. [DOI] [PubMed] [Google Scholar]
- 23.Ohno M., Fornerod,M. and Mattaj,I.W. (1998) Cell, 92, 327–336. [DOI] [PubMed] [Google Scholar]
- 24.Izaurralde E., Kutay,U., von Kobbe,C., Mattaj,I.W. and Goerlich,D. (1997) EMBO J., 16, 6535–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siomi M.C., Eder,P.S., Kataoka,N., Wan,L., Liu,Q. and Dreyfuss,G. (1997) J. Cell Biol., 138, 1181–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmidt-Zachmann M.S., Dargemont,C., Kuhn,L.C. and Nigg,E.A. (1993) Cell, 74, 493–504. [DOI] [PubMed] [Google Scholar]
- 27.Stepkowski S.M., Tu,Y., Condon,T.P. and Bennett,C.F. (1994) J. Immunol., 153, 5336–5346. [PubMed] [Google Scholar]
- 28.Beaucage S.L. and Iyer,R.P. (1992) Tetrahedron, 48, 2223–2311. [Google Scholar]
- 29.Gimona M., Watakabe,A. and Helfman,D.M. (1995) Proc. Natl Acad. Sci. USA, 92, 9776–9780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richards S.A., Carey,K.L. and Macara,I.G. (1997) Science, 276, 1842–1844. [DOI] [PubMed] [Google Scholar]
- 31.Palacios I., Weis,K., Klebe,C., Mattaj,I.W. and Dingwall,C. (1996) J. Cell Biol., 133, 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wen W., Meinkoth,J.L., Tsien,R.Y. and Taylor,S.S. (1995) Cell, 82, 463–473. [DOI] [PubMed] [Google Scholar]
- 33.Spector D.L., Goldman,R.D. and Leinwand,L.A. (1998) Cells: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 34.Fu X.-D. and Maniatis,T. (1990) Nature, 343, 437–441. [DOI] [PubMed] [Google Scholar]
- 35.Niman H.L., Houghten,R.A., Walker,L.E., Reisfeld,R.A., Wilson,I.A., Hogle,J.M. and Lerner,R.A. (1983) Proc. Natl Acad. Sci. USA, 80, 4949–4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wen W., Harootunian,A.T., Adams,S.R., Feramisco,J., Tsien,R.Y., Meinkoth,J.L. and Taylor,S.S. (1994) J. Biol. Chem., 269, 32214–32220. [PubMed] [Google Scholar]
- 37.Piñol-Roma S. and Dreyfuss,G. (1992) Nature, 355, 730–732. [DOI] [PubMed] [Google Scholar]
- 38.Fisher T.L., Terhorst,T., Cao,X. and Wagner,R.W. (1993) Nucleic Acids Res., 21, 3857–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thierry A.R. and Dritschilo,A. (1992) Nucleic Acids Res., 20, 5691–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Forbes D.J. (1992) Annu. Rev. Cell Biol., 8, 495–527. [DOI] [PubMed] [Google Scholar]
- 41.Breeuwer M. and Goldfarb,D.S. (1990) Cell, 60, 999–1008. [DOI] [PubMed] [Google Scholar]
- 42.Pruschy M., Ju,Y., Spitz,L., Carafoli,E. and Goldfarb,D.S. (1994) J. Cell Biol., 127, 1527–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cáceres J.F., Screaton,G.R. and Krainer,A.R. (1998) Genes Dev., 12, 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKay R.A., Cummins,L.L., Graham,M.J., Lesnik,E.A., Owens,S.R., Winniman,M. and Dean,N.M. (1996) Nucleic Acids Res., 24, 411–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Monia B.P., Johnston,J.F., Sasmor,H. and Cummins,L.L. (1996) J. Biol. Chem., 271, 14533–14540. [DOI] [PubMed] [Google Scholar]
- 46.Koepp D.M. and Silver,P.A. (1996) Cell, 87, 1–4. [DOI] [PubMed] [Google Scholar]
- 47.Hartig R., Shoeman,R.L., Janetzko,A., Grueb,S. and Traub,P. (1998) Biol. Cell, 90, 407–426. [PubMed] [Google Scholar]
- 48.Adam S.A., Marr,R.S. and Gerace,L. (1990) J. Cell Biol., 111, 807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakielny S. and Dreyfuss,G. (1996) J. Cell Biol., 134, 1365–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pokrywka N.J. and Goldfarb,D.S. (1995) J. Biol. Chem., 270, 3619–3624. [DOI] [PubMed] [Google Scholar]
- 51.Saavedra C., Tung,K.S., Amberg,D.C., Hopper,A.K. and Cole,C.N. (1996) Genes Dev., 10, 1608–1620. [DOI] [PubMed] [Google Scholar]
- 52.Sweet D.J. and Gerace,L. (1996) J. Cell Biol., 133, 971–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kose S., Imamoto,N., Tachibana,T., Shimamoto,T. and Yoneda,Y. (1997) J. Cell Biol., 139, 841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clarenc J.P., Lebleu,B. and Leonetti,J.P. (1993) J. Biol. Chem., 268, 5600–5604. [PubMed] [Google Scholar]
- 55.Brown D.A., Kang,S.H., Gryaznov,S.M., DeDionisio,L., Heidenreich,O., Sullivan,S., Xu,X. and Nerenberg,M.I. (1994) J. Biol. Chem., 269, 26801–26805. [PubMed] [Google Scholar]
- 56.Weidner D.A., Valdez,B.C., Henning,D., Greenberg,S. and Busch,H. (1995) FEBS Lett., 366, 146–150. [DOI] [PubMed] [Google Scholar]
- 57.Borer R.A., Lehner,C.F., Eppenberger,H.M. and Nigg,E.A. (1989) Cell, 56, 379–390. [DOI] [PubMed] [Google Scholar]