

Summary
Advanced gastric adenocarcinoma (GAC) often leads to peritoneal carcinomatosis (PC) and is associated with very poor outcome. Here we report the comprehensive proteogenomic study of ascites derived cells from a prospective GAC cohort (n = 26 patients with peritoneal carcinomatosis, PC). A total of 16,449 proteins were detected from whole cell extracts (TCEs). Unsupervised hierarchical clustering resulted in three distinct groups that reflected extent of enrichment in tumor cells. Integrated analysis revealed enriched biological pathways and notably, some druggable targets (cancer-testis antigens, kinases, and receptors) that could be exploited to develop effective therapies and/or tumor stratifications. Systematic comparison of expression levels of proteins and mRNAs revealed special expression patterns of key therapeutics target notably high mRNA and low protein expression of HAVCR2 (TIM-3), and low mRNA but high protein expression of cancer-testis antigens CTAGE1 and CTNNA2. These results inform strategies to target GAC vulnerabilities.
Subject areas: Biological sciences, Cancer, Genomics, Proteomics
Graphical abstract
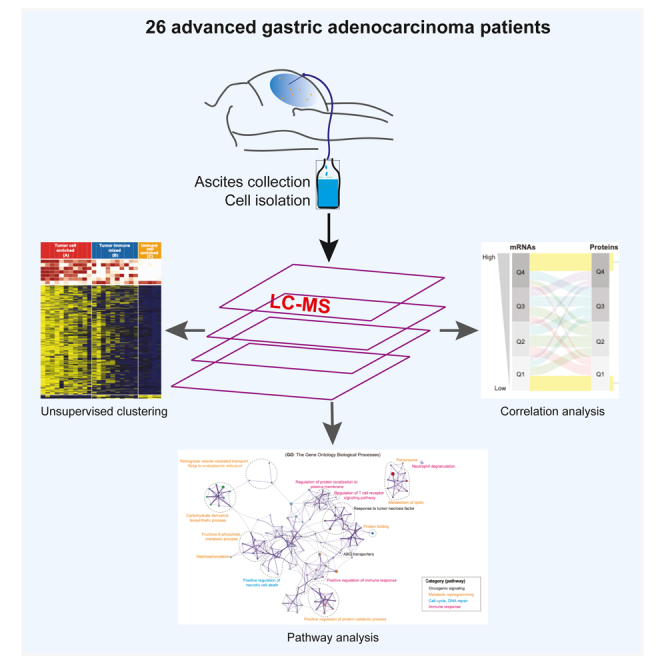
Highlights
-
•
Unsupervised clustering generated three subtypes with distinct enrichment of tumor cells
-
•
Integrated analysis revealed enriched biological pathways and notably, novel druggable targets
-
•
Systematic comparison of proteins and mRNAs expression of key cancer genes
Biological sciences; Cancer; Genomics; Proteomics
Introduction
Gastric adenocarcinoma (GAC) is a common cancer type with more than one million new cases each year.1 GAC is mostly diagnosed at an advanced stage.2 The median survival of patients with advanced GAC remains <12 months mainly because of lack of effective therapies.2 Only one oncogenic driver-based approach (Her2-directed treatment with trastuzumab) has provided modest survival benefit.3 Other therapeutic options (cytotoxic therapy or anti-angiogenic agents) are only transiently effective with the exception of checkpoint inhibitors for patients with microsatellite instable GACs which are infrequent at advanced stage. GAC patients are prone to developing peritoneal carcinomatosis (PC) which can result in accumulation of ascites requiring repeated drainage, bowel/ureteral obstruction(s), cachexia, and rapid decline. PC occurs in ∼45% of GAC patients and often leads to < 6-month survival. Intraperitoneal approaches have produced unsatisfactory results, and there is no accepted standard approach.4
Molecular characterization of GACs in the cancer genome atlas (TCGA) has provided initial insights into GAC biology.5 However, all GACs analyzed were sourced from primary tumor tissues. Proteomics data was limited to ∼300 proteins using reverse phase protein arrays (RPPA), of which 45 were associated with defined molecular subtypes.6 In general, advanced GAC has lagged in molecular investigations compared to other common cancer types.2 We previously reported on whole-exome sequencing (WES), bulk RNA, and single-cell sequencing of PC cells from patients with GAC.7,8 Proteomic profiling of advanced GACs has the potential to provide insights into molecular characteristics of cells that populate the peritoneal cavity. In this study, we present a comprehensive proteomic profiling of PC cells from 26 GACs using liquid chromatography-mass spectrometry (LC-MS) which we compare with their matched transcriptomic profiles to derive distinctive insights into the molecular profiles of GAC.
Results
The proteomics landscape of PC cells
LC-MS was performed on PC specimens from 26 patients (Figure 1A, left, and Figure S1A). Table S1 summarizes the clinical and pathological characteristics of patients. Principal component analysis (PCA) indicated minimal batch effects (Figure S1B), and all samples passed quality filtering were included in subsequent analyses. A total of 16,449 proteins were detected in one or more samples (Figure 1A, middle), of which 3,661 proteins were considered to be highly expressed (Figures 1A and S1C–S1E), of which 637 proteins were identified in all 26 samples. Some of the GAC-associated and functionally well-characterized proteins such as EGFR, ERBB2, YAP1, SOX9, TP53, and CDH1, exhibited variable abundance (Figure 1B). The combined expression values of proteins in each patient sample were similar (Figure 1C), independent of subtypes (NOS or SRC) and histology classifications (diffuse or intestinal). The number of protein IDs was balanced in NOS and SRC samples (Figure 1D) whereas the number of proteins was higher for intestinal compared to diffuse subtype (Figure S1F).
Figure 1.

A landscape of the mass spectrometric-derived cell from 26 advanced gastric cancer patients with peritoneal metastases
From each patient, the ascites was collected and from which the cells were isolated and processed for liquid chromatography-mass spectrometry (LC-MS).
(A) A schematic overview of the protein repertoires from total cell extract (TCE). The total number of proteins detected (total peptides >0) by each approach, the highly expressed proteins (total peptides ≥50) and the proportion of proteins unique to each approach or detected by TCE. The protein categories are annotated in the pie charts based on their molecular functions. TF, transcription factor.
(B) Distribution of protein quantitation measured as mean intensity (Log2 scaled) by the number of samples they are detected in, color coded by the decile. The bar plots on the top display the total counts of proteins quantified across various number of samples. The value 0 stands for the number of proteins that were not detected in any of the samples analyzed. Some representative genes are labeled on the plots.
(C) Scatterplot shows the expression distribution with mean and median values between adenocarcinoma subtypes and histology classifications.
(D) Cumulative number of proteins identified as a function of the patient numbers. Proteins family was shown with pipe plots between NOS and SRC. Statistical method: Wilcox test.
Proteomic profiling classified PC specimens into three groups with differential tumor cellularity
To profile the global proteomic patterns of PC cells, we performed an unsupervised hierarchical clustering analysis of proteins which revealed three subgroups (Figure 2A, S2A and S2B). Analysis based on canonical markers for lineage-specific differentially expressed proteins (DEPs, Table S2) suggested differences in tumor cell compositions between the three groups (Figure 2A). PC specimens in Cluster A displayed the highest expression of epithelial lineage markers EPCAM and KRT8, and GAC driver genes CDH1, YAP1, and ERBB2, and the lowest expression of the immune cell marker PTPRC (CD45), which suggests that Cluster A was predominantly enriched in tumor cells. Similarly, PC specimens in Cluster C displayed the highest expression of the immune cell marker PTPRC and exhibited low or no expression of the epithelial lineage markers or GAC-related oncogenes, indicating a predominance of immune cells. Cluster B, however, exhibited mixed expression features of both epithelial and immune cells and was therefore classified as “tumor-immune mixed”.
Figure 2.

Proteogenomic characterization defined 3 subgroups with differential tumor and immune cell compositions
(A) Proteomic profiling of 3 subgroups based on tumor and immune cell composition. The cell specific markers for tumor (e.g., EPCAM, MUC1, CDH1, YAP1, ERBB2, KRT8) and immune cell (PTPRC) compartments were used for sample classification and expression of these markers are shown in the top tracks. Heatmap shows differentially expressed genes across 3 subgroups.
(B) The OncoPrint plot shows somatic genomic alterations identified by whole-exome sequencing (WES). CNA, copy number alteration.
(C) Copy number gains and losses among cluster A (red), B (blue) and C (orange). The boxplot (black) in the violin represents the interquartile range (IQR) and median value. The points in the violin plot depicts the samples in each group.
(D) Somatic CNAs identified across 22 chromosomes by WES. The frequency of each CNA is labeled on the right axis.
(E) Fraction of histological subtypes in 3 defined subgroups. SRC, signet ring cell carcinoma; NOS, nitric oxide synthase carcinoma. n.s., not statistically significant. N/A, not available.
(F) Kaplan-Meier curve shows the difference in patient survival between patients in subgroup A and the rest of patients from subgroups B and C. The log rank test was used to calculate the p value.
Whole-exome sequencing (WES) data was generated for 22 of 26 samples that had matched germline DNAs available (Table S1). Somatic mutation analysis was then performed, and we identified recurrent mutations in CDH1, TP53, and KMT2C, with frequencies that were similar to those reported in 34 independent GACs in a prior study7 (Figure 2B). We next integrated the mutational profiles and proteomic data which strongly supported the proteomics-based cluster designation. We observed a greater number of somatic mutations in GAC driver genes and a higher mutation burden (TMB) in samples in Cluster A (tumor cell enriched), whereas no somatic mutations were detected in Cluster C (immune cell-enriched) (Figures 2B and 2C). Concordantly Cluster A samples exhibited extensive copy number alterations while no alterations were detected in Cluster C (Figure 2C and Table S3). Overall, the genomic data recapitulated features of the proteomics-based classification. These results are indicative of varied tumor cell enrichment in ascites fluid as would be expected.
We further examined the clinical relevance of proteomics-based classification indicative of varied tumor cell enrichment. Patients’ age was significantly older in clusters A/B than in cluster C (the Fisher exact test p = 0.037, Figure S2C), but it could not independently affect the clusters divided by the significant proteins (Figure S2D). Signet ring cell carcinomas (SRC) were more represented in clusters A and B than C but did not reach statistical significance given the relatively small cohort size (Figures 2E and S2E). We also observed a trend of shortened survival in patients in cluster A (Figure 2F, log rank p = 0.280). These results indicate differences in clinical and histopathological phenotypes between the three clusters based on proteomic profiles.
Dysregulated molecular pathways and potential druggable targets
We next focused on clusters A and C given the enrichment in tumor cells and immune cells, respectively. We performed pathway enrichment analysis to investigate dysregulated molecular processes informed by the proteomics data. A total of 62 upregulated hallmark and KEGG pathways were identified in cluster A which were predominantly composed of oncogenic and metabolic signaling (Figure 3A), such as glycolysis, oxidative phosphorylation (OXPHOS), mTORC1 signaling, MYC, TP53, and, TGF-beta pathways (Figure 3B). Similarly, a total of 35 upregulated pathways were identified which were predominantly immune-related pathways including neutrophil degranulation, neutrophil activation, neutrophil-mediated immunity, and cytokine production (Figure 3A). We next applied a stepwise filtering process to identify proteins that were tumor-specific, highly abundant, and significantly enriched in each cluster (Figure 3C) resulting in 560 proteins, 68 of which were annotated as functionally important (Figure 3D) which included EGFR, ERBB2, CDK4 as known targets with FDA approved inhibitors, PAK4 as an emerging and attractive target involved in cytoskeleton remodeling, invasion, and metastasis9 and two cancer-testis antigens, RQCD1 and CTNNA2 (Figure 3E). The latter exhibited significantly higher expression values of protein and mRNA in the SRC compared to NOS subtype (Figure S3). Meanwhile, a total of 40 proteins including 13 tumor-promoting proteins and 20 tumor-suppressing proteins were annotated as functionally important (Figures 3F and 3G), which included HAVCR2 (TIM-3), CEACAM1, ITGAM, C10orf54, LTF, and S100A12 with relevance to cancer immunotherapy. Three proteins (CAMP, S100A8, and S100A9) were with pro- and anti-tumorigenic functions in different tumor microenvironments.10 Generally, this integrated analysis revealed enriched biological pathways and notably, some druggable targets (cancer-testis antigens, kinases, and receptors) that could be exploited to develop effective therapies and/or tumor stratifications.
Figure 3.

Enriched TCE proteins and associated biological pathways among three clusters and potential targets
(A) Pathway enrichment analysis identified biological pathways enriched in the subgroup A and C. The curated gene sets were downloaded from the Molecular Signature Database (MSigDB). Both the cancer KEGG (left), Hallmark (middle) and GO (right) gene sets are shown. The pathways are colored by their biological functions. FDR q-value, the p value adjusted for the false discovery rate (FDR). A q-value threshold of 0.01 (1% FDR) is applied.
(B) Violin plots of representative pathways from the subgroup A.
(C) Schematic flow chart showing the filtering process of identifying tumor-specific proteins enriched in the subgroups. The filtering conditions were shown during each filtering step.
(D) A heatmap of highly expressed proteins that are enriched in the Subgroup A from the TCE approach. A list of 65 (out of 560) proteins with known functions are shown. The expression values are Z score transformed. Subgroup classification is annotated on the left, sample names are labeled on the right, and protein names are shown at the bottom, with an annotation track color coded by the protein functional category. Top histogram shows for each listed protein, the fraction of samples with detected protein expression by the TCE approach (n = 26 samples). The red filled circle indicates the mean expression level of each protein averaged across 26 samples (y axis on the right, Log2 transformed). The gray dotted horizontal line annotated the expression level of 50 total peptides.
(E) Violin plots of representative proteins from the panels D. The Mann-Whitney U test was used to calculated the p values for data displayed in panels B and E. ∗∗p < 0.01; ∗p < 0.05; n.s., not statistically significant.
(F) Heatmap of highly expressed proteins that were enriched in the subgroup C from the TCE approach. Proteins that were highly expressed and significantly enriched in the subgroup C are shown at the bottom, with an annotation track color coded by the protein functional category. The expression values are Z score transformed. Subgroup classification is annotated on the left, sample names are labeled on the right, and protein names are shown at the bottom. Top histogram each lists protein, the fraction of samples with detected protein expression by TCE (n = 26 samples). The red filled circles indicate the mean expression level of each protein averaged across all samples (y axis on the right, Log2 transformed). The gray dotted horizontal lines annotate the expression levels of 50 total peptides.
(G) Violin plots of representative proteins from the panels C. The Mann-Whitney U test was used to calculated the p values. ∗∗p < 0.01; ∗p < 0.05; n.s., not statistically significant.
Correlation with histological subtypes and clinical features
This cohort included 11 PC samples derived SRC and 13 PC samples derived from NOS. Survival analysis identified no significant difference in these patients’ prognosis between the NOS and SRC subgroups (Log rank p = 0.830, Figure 4A). We then compared their MS expression features, which revealed 131 significantly upregulated proteins in the SRC subtype compared to the NOS subtype including genes involve in response to tumor necrosis factor pathway and ABC transporter pathway (ITGB4, CTNNA2, PAK3, and TAP1), metabolism of lipids (DECR1 and ACSL6), positive regulation of necrotic cell death (PAK1 and CD14), and antigen processing and presentation signaling pathway (TIMP1, CD74, and B2M; Figures 4B and 4C). A total of 6 upregulated GO pathways were identified to be related to ribosomal large subunit biogenesis and regulation of translation (TOP1, STMN1, and IREB2) in the NOS histological subtype (Figure 4D).
Figure 4.

Enriched tumor specific proteins and associated biological pathways between the NOS and SRC subgroups and potential targets
(A) The Kaplan-Meier curve shows the difference in patient survival between patients in NOS and the rest of patients from the SRC histological subtype. The log rank test was used to calculate the p value.
(B) Volcano plot shows the DEPs between two NOS and SRC with a tumor-immune of log2(fold change) > 1 and p value <0.05, different colors indicate the significant genes involving in oncogenic (black), cell cycle (light blue), metabolic (orange) and immune response (pink) pathways.
(C) The pathway enrichment analysis identified biological pathways enriched in the SRC subtype. Pathways are colored by their biological functions.
(D) The pathway enrichment analysis identified biological pathways enriched in the NOS subtype. Pathways are colored by their biological functions.
Next, we compared the proteomics data between the chemotherapy-treated (n = 16) and chemotherapy-naïve (n = 10) groups by cluster but didn’t observe significant changes in their MS profiles (Chi-squared test p = 0.521, Table S4), likely due to the small cohort size. Some significant proteins were upregulated in the chemotherapy-treated group compared to chemotherapy-naïve group, which were involved in cell cycle (RPA1, RFC1, ERCC2, and RAD50) and immune response (MALT1 and DDX5) pathways (Figure S4A). Similarly, a total of 16 samples in the chemotherapy-treated group were identified predominantly in cell cycle and immune response signaling, such as DNA repair, T cell receptor signaling pathway, B cell receptor signaling pathway, E2F targets, and G2M checkpoint (Figure S4B). But in the chemotherapy-naïve group, the oncogenic pathway (PPAR signaling pathway) was upregulated including ACSL1 protein, as well as apical junction pathway in 10 ascites samples. Generally, these results indicated molecular differences in therapy-specific outcomes across the 26 TCE samples. We must acknowledge that our sample size is rather small and further studies are warranted.
Proteomic features associated with somatic mutations of CDH1, TP53, and KMT2C
CDH1, TP53, and KMT2C are frequently mutated in advanced GAC. However, these genomic alterations are not directly druggable. To identify potentially actionable targets in tumor cells with somatic alterations in CDH1, TP53, or KMT2C, we analyzed the proteomic data for statistical associations and for pathway enrichment (Figure 5A).
Figure 5.

Distinct protein expression features and signaling pathways associated with aberrant expression of CDH1, TP53, and KMT2C
(A) Heatmap shows differentially expressed proteins between the “high” and “low” groups for CDH1 (left), TP53 (middle), and KMT2C (right). The expression values are Z score transformed and data from the TCE approach are shown. The names of some presentative genes are listed. The median expression levels of CDH1, TP53, KMT2C (across 26 samples) were used to define the “high” (>median) and “low” (≤median) groups. Other annotation tracks included somatic mutations (of each specific gene) identified from WES, and protein-based subgroup classification from Figure 2. The curved lines in the top plots indicate the protein and mRNA (mRNA sequencing) expression levels of each specific gene across all samples.
(B) Differentially expressed signaling pathways between the “high” and “low” groups for CDH1, TP53, and KMT2C.
Interestingly, proteins that were significantly upregulated in the CDH1-high PC cells included ERBB2, EGFR, CTNNA2, and SOX9, three of which are druggable. Pathway analysis identified upregulated OXPHOS and cell cycle signaling in CDH1-high PC cells (Figure 5B). IGF2R and CREBBP were significantly elevated in tumor cells in the TP53-low group. These data provide potentially druggable targets related to distinct mutation profiles.
Correlation between mRNA and protein expression and key marker genes
To assess the complementary value of proteomic and mRNA data, correlation analysis was carried out that involved 6,638 proteins and mRNAs divided into four quantiles (i.e., Q1, Q2, Q3, and Q4) based on their overall distribution. Protein abundance showed different correlations with their correspondent mRNAs in different quantiles (Figure 6A). Among the consistently highly expressed genes in Q4 (n = 860), we identified enzymes and transporters as the top two families and oncogenic signaling and metabolic signaling as the significantly enriched pathways. Importantly, the oncogene FN1 correlated with the EMT signaling pathway. However, within the consistently low expressed genes in Q1 (n = 744), we identified enzymes and transcriptional factors as the top two families, with a predominance of cell cycle-related processes (Figure 6B). Genes with discordant RNA and protein expression included potentially interesting therapeutic targets notably cancer/testis proteins CTNNA2, LTF, CTAGE1, and immunotherapy targets LAIR1 and HAVCR2 (TIM3) (Figure 6C). These data suggested that the protein data provided complementary information to yield an improved understanding of biological processes involved in PC.
Figure 6.

Correlation between MS protein and mRNA expression and profiling of genes with consistent and distinct expression patterns
(A) Correlation analysis between the protein and mRNA expression. The normalized protein expression data from the TCE approach and the normalized mRNA expression data from the whole transcriptome sequencing (RNA-seq) were used for correlation analysis. The correlation analysis was stratified by the protein and mRNA expression levels (protein low and mRNA low; protein high and mRNA high; protein low and mRNA high; protein high and mRNA low). The Pearson’s correlation is applied and the correlation coefficient (r) and p values are labeled.
(B) Characterization of genes with consistent mRNA and protein expression. The genes were classified into 4 groups (Q1-4) based on their overall distribution of mRNA and protein expression, respectively. The alluvial plot shows the relationship between protein and mRNA expression for genes in each group and the genes that are consistently high (n = 860) or low (n = 744) in their expression from both platforms are highlighted (yellow strips) and taken into subsequent analysis, including protein functional categories and pathway enrichment analysis.
(C) Characterization of genes with inconsistent mRNA and protein expression. The genes were classified into 4 groups (Q1-4) based on their overall distribution of mRNA and protein expression, respectively. The alluvial plot shows the relationship between protein and mRNA expression for genes in each group and the genes that are inconsistently expressed high are highlighted and taken into subsequent analysis, including protein functional categories and pathway enrichment analysis.
Discussion
The ultimate impact of genomic dysregulation is on protein expression.11 Our study was aimed at identifying hallmarks of protein expression in PC ascites cells. Our results provide complementary information to TCGA and Clinical Proteomic Tumor Analysis Consortium (CPTAC) data12,13,14 and a panoramic view of cancer-related proteomics in PC cells. Isolation of tumor cells from biological fluids that contain a multitude of cell types may be accomplished through the enrichment or selection of cells based on the expression of particular markers. However, such an approach may be biased because of heterogeneity among tumor cells with respect to the expression of particular markers. Our profiling did not rely on a tumor cell selection process but rather on unbiased molecular classification which was informative with respect to tumor cell enrichment and correlation with prognosis and biological characteristics.
Molecular subtyping of cancers was aimed to classify patients into distinct subtypes correlated with prognosis, therapy responses, and biological features. Tumors consisted of heterogeneous cells that display varying levels of functional and genetic heterogeneity.15,16 Interestingly, in our study, we discovered three different clusters in the PC ascites cells (tumor-enriched cluster A, tumor-immune mixed cluster B, and immune-enriched cluster C) that could functionally serve as heterogeneity. Cluster A expressed the epithelial lineage markers EPCAM and KRT8, and driver genes of GAC such as CDH1, YAP1, and ERBB2; and cluster C expressed immune-related gene PTPRC. WES analysis showed that a large number of genes were identified with mutations and insertions/deletions in the dataset related to tumor cell enrichment as may be expected. Pathway enrichment analysis displayed that oncogenic and metabolic signaling pathways were predominant in cluster A; whereas immune-related pathways were enriched in cluster C. Thus, cluster A was more representative of mutational and pathway alterations that occur in GAC tumor cells with the occurrence of therapeutic targets including CTAGE1, CTNNA2, ERBB2, and EGFR. GACs with reduced enrichment in tumor cells as in cluster C may benefit from alternative therapies that take advantage of the predominance of immune cells.
As a conserved cellular program, paligenosis undergoes plastic changes respond to damage and maintains homeostasis.17 And in the successful paligenosis program, multiple tissues and cells require stress response proteins to modulate critical cell energy and survival networks centered on MTORC1 and P53.18 Previous study displayed that pre-cancerous metaplasia was a normal, wound-healing, regenerative response produced by paligenosis. However, once the paligenosis was abnormal, the DNA damage were not eliminated or stalled in the paligenotic cells until DNA repair was completed. And then these cells will accumulate DNA damage and undergo malignant transformation, ultimately occurring at the gastric cancer initiation.19,20 In this study, we discovered that PC cells presented a high proliferative state with significant MTORC1, glycolysis, OXPHOS, and TGF-beta signaling pathway in cluster A and B. And it was reported that MTORC1 signaling pathway controls the adaptive transition of quiescent cells in G0 to re-enter the cell cycle in response to injury-induced signals.21 And these proteins analysis within PC ascites cells validated again that paligenosis is a stepwise, specific program with multiple signaling pathways involved. Promisingly, we discovered that targeting paligenosis by MTROC1 inhibitors reduced proliferation of PC cells from ascites by inducing apoptosis.
The immunosuppressive and pro-tumorigenic functions are characterized in tumor infiltrating immune cells that play distinct and complex roles in tumor ecosystems.22,23 Tumorigenesis and metastasis could be propagated by self-renewing the tumor cells with pluripotent and plastic characteristics.24 And immune cells could promote tumor proliferation and metastasis and also reprogram the tumor ecosystem to maintain a suitable niche.25 Our signaling pathway analysis highlighted neutrophil-related pathways in cluster C ascites cells in the peritoneal ecosystem. Therefore, we speculate that neutrophils in the peritoneal cavity maintain tumor cell plasticity via immune-related signaling pathways. In this study, tumor suppressor genes such as C10orf54, ITGAM, LTF, and S100A12 were discovered involved in maintaining homeostasis, and loss tumor suppressor functions will cause cellular plasticity that drives numerous types of cancer,26 including gastric cancer.
While the comparison between the NOS and SRC histotypes is potentially interesting and would be adequately powered for at least preliminary analysis across all samples including 13 NOS samples and 11 SRC samples. It is definitely flawed to compare these two subtypes only within the same cluster with limited number of samples. In our study, we identified the DEPs involved in oncogenic (ITGB4, CTNNA2, PAK3, and TAP1) and immune response (TIMP1, CD74, and B2M) pathways in the SRC group but metabolism (TOP1, STMN1, and IREB2) and cell cycle (KDM6A and SMARCD1) pathways in NOS type. Studying the ecosystems of ascites between NOS and SRC subtypes will be helpful to provide a critical direction for the further investigation of ascites of gastric cancer. A published study indicated that multiple biomarkers of aggressiveness and targets were identified by DEGs analysis and functional studies.7 Ascites from high-grade serous ovarian cancer displayed to be correlated with a poor prognosis and drug resistance,27 which harbored three different cell clusters (immune cells, epithelial cells, and cancer-associated fibroblasts) in the ascites ecosystem.28 Because ascites samples via paracentesis is easier and safer than tissue biopsy which is different from peripheral blood in the immune features. These results provide a direction for the detection of tumor status by sampling ascites instead of biopsy tissue or peripheral blood.
To identify the DEPs among three clusters from the ascites samples also allowed us to more reliably nominate potential drug targets. In this study, we discovered some proteins significantly higher expression in tumor-enriched group belong to cancer/testis antigens, enzyme, ligand, receptor, kinase, or transcription factor, including several “star” molecules including CTAGE1, CTNNA2, CDK4, ERBB2, and EGFR derived from cancer driver gene analysis, their elevated expressions might be associated with poor survival. And it will be interesting to explore these “star” molecules and the other potential drug candidates that we nominated for treatment or developing drugs.
Notably, while we identified a considerable number of genes with DNA mutations, their gene products were sometimes detected in the DEPs analysis. Proteogenomic analysis in relation to CDH1, TP53, and KMT2C revealed a poor correlation between genomic and proteomic data as previously observed in other datasets.12,13,14 We detected frequent occurrences of mutations in CDH1, TP53, and KMT2C genes. Loss-of-function CDH1 mutation was associated with the upregulation of several key genes notably ERBB2, EGFR, SOX9, and CTNNA2 suggesting that loss-of-function of a tumor suppressor gene may yield opportunities to target specific oncoproteins.29 This observation added a cautionary note on nominating treatment candidates based solely on DNA mutations and further strengthen the necessity of considering proteins in the precision medicine.
It is unclear about the relationship between gene expression measured at the mRNA level and the corresponding protein level in human cancer.30 Guoan Chen etc.30 reported that no significant correlation between mRNA and protein expression, which was validated again in our study. The mRNA-protein correlation coefficient varied among proteins with multiple isoforms, indicating potentially separate isoform-specific mechanisms for the regulation of protein abundance.31 So far, the CPTAC, integrating DNA methylation, copy number alterations, and mRNA and protein profiling of TCGA tumor specimens into analysis to portray proteogenomic landscapes of colorectal cancer, breast, and ovarian cancers,12,13,14 discovered that mRNA transcript abundance could not reliably predict protein abundance differences generally, although CNV displayed strong cis- and trans-effects on mRNA abundance but extend to the protein level.12,13 Also, protein level data informs about the extent to which RNA levels are translated into proteins and the extent of protein stability which is needed for the assessment of potential targets. In this study, we discovered some functionally important genes with discordant protein and mRNA expression in PC samples, such as cancer/testis antigens CTNNA2, LTF, CTAGE1, and immunotherapy targets LAIR1 and HAVCR2 (TIM3), which enriched in oncogenic (P53 pathway, TGF-beta signaling, TNFa signaling via NFkb, NOTCH signaling pathway), metabolic (glycolysis, OXPHOS), and immune-related (complement and coagulation cascades, and leukocyte trans-endothelial migration) pathways. And the first four genes were with high protein expression but low in mRNA expression. Inversely, HAVCR2 (TIM3) was with high mRNA expression (Q3/4) but low (Q1) in protein expression. Previous study reported that novel-miR-4885 could promote migration and invasion of esophageal cancer cells by binding to the 3′ untranslated region of CTNNA2,31 and CTNNA2 was a tumor suppressor gene frequently mutated in laryngeal carcinoma.32 LTF (lactotransferrin, also known as lactoferrin) is a critical component of the innate immune defense,33 and its downregulation inhibits the normal immune response in the body, thus making cancer cells escape the immune process.34 For CTAGE1, it is a cutaneous T cell-lymphoma-specific tumor antigen that might lead to further candidate proteins for specific immunotherapy of cutaneous T cell lymphoma and other malignancies.35 LAIR1 (leukocyte associated immunoglobulin like receptor 1) is an immune inhibitory receptor expressed on most immune cell types, and blocking LAIR1 signaling pathway in immune cells could inhibit tumor development.36 LAIR1 has intracellular immunoreceptor tyrosine-based inhibitory motifs in the signaling domains, which decides the different functions of individual proteins. Now, the immuno-oncology therapeutic products based on targeting LAIR1-mediated signaling are anticipated with significant advancement.37 TIM-3 (T cell immunoglobulin mucin 3) is a promising immune checkpoint target in cancer therapy, and the expression level of HAVCR2 was remarkably correlated with cancer immune infiltration or immune checkpoint genes, suggesting that HAVCR2 could participate in immune infiltration in tumorigenesis.38 The discordance of mRNA/protein abundance could be explained by (1) tumor heterogeneity, in which low mRNA abundance in a small number of tumor cells, while their alteration in protein abundance is high; (2) limited number of samples with the same mRNA/protein expression, which diminishes the power of statistical analysis to calculate its correlation. In general, it might be possible to understand the complex mechanisms influencing protein expression in PC from advanced GAC by combining proteomic and transcriptional analysis, and the potential to identify specific protein isoforms correlated with biological behavior in GC would be of quite interest and would promote our realization of the regulation of gene products by transcriptional, translational and post-translational mechanisms.
In summary, our integrative proteogenomic characterization revealed different therapeutic targets in PCs involving signaling proteins, metabolic enzymes, kinases, and cancer antigens. The findings from our study warrant further assessment of the utility of targets and pathways we have uncovered.
Limitations of the study
We acknowledge that the lack of either paired primary GAC samples or an appropriate normal tissue control is a shortcoming of this study. Ideally, we should collect paired tumor and nearby/normal tissue from the same patient to compare with PC cells to identify genetic and proteomic alterations. We agreed with that few normal controls or paired tumor tissue could further complicate proteomics analysis and limit our analysis to identify individually altered cancer proteome and dysregulated-signaling pathways. So, in this study, protein expression data in normal gastric tissues from HPA were used as the control to select tumor-specific proteins. However, to identify tumor-specific proteins, a large number of paired normal gastric tissues are optimal and future investigations are needed to elucidate the consequences of up- or -down-regulated proteins and potential molecular mechanisms involved. Also, some validation experiment is needed to identify the new druggable targets in the future work.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Whole Exome Sequencing Data Of Ascites Samples from TCE |
European Genome-phenome Archive (EGA), the accession number EGAS00001003180 | http://www.nature.com/ng/journal/v47/n7/full/ng.3312.html |
| RNA Sequencing Data Of Ascites Samples from TCE |
European Genome-phenome Archive (EGA), the accession number EGAS00001003180 | http://www.nature.com/ng/journal/v47/n7/full/ng.3312.html |
| Mass Spectrometry Data Of Ascites Samples from TCE |
MD Anderson Cancer Center | The MS data in this study was available from Table S5 |
| Software and algorithms | ||
| R v3.5.3 GSEA |
The R Project for Statistical Computing Gene Set Enrichment Analysis |
https://www.r-project.org/ https://www.gsea-msigdb.org/gsea/index.jsp |
| Other | ||
| Key source codes for this paper | This paper | https://github.com/wpwan/GutAscites |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Jaffer A. Ajani (jajani@mdanderson.org).
Materials availability
All unique/stable reagents generated in the study are available from the lead contact with a completed materials transfer agreement.
Experimental model and study participant details
Ethics approval and consent to participate
Ethical approval was obtained from the institutional review committee. And The design and performance of the study are in accordance with the Declaration of Helsinki. Signed informed consent was obtained from all participants before inclusion, allowing analysis of tumor tissue, blood samples and clinical data.
Patient cohort and sample collection
A total of 26 GAC patients with documented PC were selected for this study. All patients were treated at The University of Texas MD Anderson Cancer Center (Houston, USA). PC specimens were obtained during a therapeutic procedure once patients provided a written approved informed consent document under an Institutional Review Board approved protocol. PC is stage 4 cancer under the American Joint Committee on Cancer Staging Manual (8th edition).2,39 The clinical characteristics including age, sex and race and treatment information of these patients are listed in the Table S1. Information regarding patient treatment is provided in Figure S1A. PC cells were collected through these steps below. Ascites samples ranging from 100 mL to 2000 mL were obtained from GAC patients who underwent a therapeutic and/or diagnostic procedure. Cells were spun down at 2,000g for 20 minutes. Pelleted cells were lysed in red blood cells lysis buffer consisting of 0.079 g of ammonium bicarbonate and 6.1 g of ammonium chloride in 1 L of distilled H2O. Red blood cells were removed to enrich in PC cells for further analysis. The percentage of malignant cells was recorded by a pathologist or cytologist.
Method details
Mass spectrometry based proteomic analysis
Total cell extracts (TCEs) were obtained by sonication of cell lysate pellets in 4M Urea, 3% IsoPropanol, 20 mM Tris containing the detergent 2% octyl-glucoside (OG, Sigma-Aldrich), and protease inhibitors (complete protease inhibitor cocktail, Roche Diagnostics) and phosphatase inhibitor (PhosSTOP, Roche Diagnostics) followed by centrifugation at 20,000 x g at 4 °C for 30 min. TCE proteins were reduced in DTT and alkylated with acrylamide before the fractionation of intact proteins using RP-HPLC. Proteins were desalted and separated using an off-line ACQUITY ultra-performance liquid chromatography (UPLC) Class-H system (WATERS) with reversed-phase column (4.6-mm internal diameter × 150-mm length; Column Technology). Collected fractions were dried by lyophilization followed by in-solution digestion with trypsin (Mass Spectrometry Grade, Thermo Fisher).
A total of 23 fractions were subjected to LC-MS/MS analysis per ascites sample. Tryptic peptides were injected onto reversed phase nanoLC-MS/MS using a nanoAquity LC system coupled online with SYNAPT G2-Si ion-mobility mass spectrometer (WATERS). Separations were performed using 75 μm id × 360 μm od × 25-cm-long fused-silica capillary column (Column Technology) slurry packed with 3 μm, 100 A° pore size C18 silica-bonded stationary phase. Following injection of ∼ 2 μg of protein digest onto a C18 trap column (Waters, 180 μm id × 20 mm), peptides were eluted using a linear gradient of 0.35% mobile phase B (0.1 formic acid in ACN) per minute for 90 min, then to 95% B in an additional 10 min, all at a constant flow rate of 300 nL/min.
LC-HDMSE Data was acquired in high resolution mode using Waters Masslynx (version 4.1, SCN 851). The capillary voltage was set to 2.80 kV, sampling cone voltage to 30 V, source offset to 30 V, and source temperature to 100 °C. Mobility utilized high-purity N2 as the drift gas in the IMS TriWave cell. Pressures in the helium cell, Trap cell, IMS TriWave cell, and Transfer cell were 4.50 mbar, 2.47e-2 mbar, 2.90 mbar, and 2.53e-3 mbar, respectively. IMS wave velocity was 600 m/s, helium cell DC was 50 V, Trap DC bias was 45 V, IMS TriWave DC bias was 3 V, and IMS wave delay was 1,000 μs. The mass spectrometer was operated in V-mode with a typical resolving power of at least 20,000. All analyses were performed using positive mode ESI using a NanoLockSpray source. The lock mass channel was sampled every 60 s. The mass spectrometer was calibrated with a [Glu1] fibrinopeptide solution (300 fmol/μL) delivered through the reference sprayer of the NanoLockSpray source. Accurate mass LC-HDMSE data was collected in an alternating, low energy (MS) and high energy (MSE) mode of acquisition with mass scan range from m/z 50 to 1800. The spectral acquisition time in each mode was 1.0 s with a 0.1-s inter-scan delay. Data were collected in the low energy HDMS mode, at constant collision energy of 2 eV in both Trap cell and Transfer cell. In high energy HDMSE mode, the collision energy was ramped from 25 to 55 eV in the Transfer cell only. The RF applied to the quadrupole mass analyzer was adjusted such that ions from m/z 300 to 2,000 were efficiently transmitted, ensuring that any ions observed in the LC-HDMSE data less than m/z 300 were known to arise from dissociations in the Transfer collision cell.
The acquired LC-HDMSE data were processed and searched against the Uniprot proteome database (Human, January 2017) through ProteinLynx Global Server (PLGS, Waters Company) with two trypsin miss cleavage allowed. The modification search settings included cysteine (Cys) alkylation with acrylamide (71.03714@C) as a fixed modification, and methionine (Met) oxidation (15.99491@M) as a variable modification. The searched data was filtered with False Discovery Rate 4%. The spectral counts for each protein driven by the identified peptides were used as a measure of protein abundance. The normalization was done by each protein spectral counts divided by the total spectral counts of each patient ascites cells multiplied by the factor 50,000.
Protein expression values represented by MS events were generated with the total peptides aggregated based on Ensemble gene symbols (build hg19). Proteins that were not identified in some samples were assigned a value of 0. All values of proteins were then log2-transformed for further downstream processes. Highly expressed proteins were defined using a cut-off of total MS events of > 50 (Figures S1C and S1D).
Quantification and statistical analysis
Sample classification based on proteomic data
A total of 1,980 out of 16,449 proteins detected in ≥ 80% of all samples (n = 26), were selected for principal component analysis (PCA) to assess the distribution of samples based on the global protein expression profiles, explore optimal three clusters (tumor cell enriched, immune cell enriched and tumor immune mixed) and evaluate possible batch effects (Figures 1B–1D, S2A, and S2B). PCA analysis using TCE proteins indicated minimal batch effects and all samples were included for subsequent analyses. Then 2,361 differentially expressed proteins (DEPs) were identified among three clusters defined by PCA and used for the unsupervised hierarchical clustering analysis. Protein expression data in normal gastric tissues were downloaded from the Human Protein Atlas (HPA) and used as controls to select tumor-specific proteins (Figure S1E). Group enriched proteins were selected by applying a cut-off of expression fold change ≥2 and adjusted p-value (FDR q-value) <0.05.
Pathway enrichment analysis
Gene sets of interest were processed using Gene Set Enrichment Analysis (GSEA)40 against the curated gene sets (H and C2 collections) downloaded from Molecular Signature Database (MSigDB)41 to identify significantly enriched signaling pathways ( FDR adjusted p-value < 0.01 and overlapped genes numbers ≥ 3). Results were subsequently visualized in R with ggplot2 package.
DNA and RNA data analysis
Whole-exome sequencing (WES) was performed on 22 of 26 PC specimens, with 11 having matched germline DNA from peripheral blood. We submitted the extracted gDNAs to Sequencing and Microarray Core Facility (SMF) at UT MD Anderson Cancer Center for WES. Then the raw output of Illumina exome sequencing data was processed with Illumina’s Consensus Assessment of sequence And Variation (CASAVA) tool (v1.8.2). Genotyping quality control was completed to rule out any possible contamination or sample swapping. And we called germline SNPs by Platypus (v0.8.1). Finally, we identified somatic mutations and Pindel with MuTect (v1.1.4). We calculated the mutation load by first counting the number of nonsynonymous somatic mutations called in a sample and then normalized the numbers by the sample’s 20x target base coverage. And then we applied the Mutalisk tookit to quality-filtered somatic mutations, and quantified the statistical relative contribution of each of the 30 characterized COSMIC mutational signatures. We conducted the DNA copy number analysis by an in-houses application ExomeLyzer followed by CBS segmentation.
RNA sequencing was performed on 21 specimens by FastQC with RNA-seq FASTQ files to generate a series of RNA-seq related quality control metrics. All samples passed the quality control and generated RNA-seq BAM files with STAR 2-pass alignment (v2.5.3). Finally, the RNA-Seq gene expression raw read counts were produced from HTSeq-count (v0.9.1).
mRNA and protein correlation analysis
To assess the correlation between mRNA and protein abundance, the overlapping genes (n = 1,016) expressed in ≥ 95% samples that were shared between the two platforms for 21 samples were subjected to Pearson correlation analysis.
Statistical analysis
The statistical methods used in this study were performed in the R statistical environment (v3.5.3). The evaluation of normal distribution was checked using the Shapiro-Wilk test. The statistical significance of differences observed among clusters was determined by the ANOVA test or t-test for normal distribution data and the non-parametric Kruskal-Wallis test or Wilcoxon test for non-normal distribution data when comparing continuous variables, and the Fisher’s Exact test when comparing frequencies of clinical factors. To control for false discovery rate (FDR) and correct p-values from multiple testing, the Benjamin-Hochberg method was applied to calculate an FDR adjusted p-value (or q-value). Kaplan-Meier plots and log-rank tests were used for univariate survival analysis. Pearson’s correlation coefficient was calculated to evaluate the association between two continuous variables. Hypothesis testing was performed in a two-sided manner, with p-value or adjusted p-value (if applicable) < 0.05 considered to be statistically significant.
Acknowledgments
The authors would appreciate all the gastric cancer patients for their donating the specimen for this analysis study. This study was supported in part by the National Cancer Institute awards CA129906, CA127672, CA138671 and CA172741 and the DOD grants: CA150334 and CA162445 to JAA and DOD grants CA160433 and CA170906 to SS, and the start-up research funds provided to LW by MD Anderson Cancer Center. LW is a Sabin Fellow. This study was also supported by SMF Core grant CA016672 (SMF). Also supported by Stupid Strong Foundation and V Foundation.
Author contributions
J.A.A. and L.W. conceived and jointly supervised the study. L.W. supervised the bioinformatics analysis and S.Z. conducted the major bioinformatics and biostatistics analysis. R.W., D.H., G.H., J.Z., C.Y.Y., and X.S. assisted with raw data processing and analysis. S.S., M.Z., M.K., B.B., G.C., J.V., and M.P.P. assisted with clinical samples consent, collection, and pathological evaluation and MS data generation. K.H. took charge of ascites cells isolations, DNA/RNA extraction. S.Z., L.W., S.H., H.K., A.F., S.S., and J.A.A. wrote and revised the manuscript.
Declaration of interests
The authors declare that they have no competing interests.
Published: May 19, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106913.
Contributor Information
Samir M. Hanash, Email: shanash@mdanderson.org.
Linghua Wang, Email: lwang22@mdanderson.org.
Jaffer A. Ajani, Email: jajani@mdanderson.org.
Supplemental information
Data and code availability
All WES and RNA-Seq data have been deposited at the European Genome-phenome Archive (EGA). The datasets can be fully accessed under the accession number EGAS00001003180. Further information about EGA can be found on https://ega-archive.org (’the EGA of human data consented for biomedical research’: http://www.nature.com/ng/journal/v47/n7/full/ng.3312.html). The MS data in the current study are available from Table S5.
The key source codes that supported these results and findings can be found on GitHub (https://github.com/wpwan/GutAscites) and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Ajani J.A., Lee J., Sano T., Janjigian Y.Y., Fan D., Song S. Gastric adenocarcinoma. Nat. Rev. Dis. Primers. 2017;3 doi: 10.1038/nrdp.2017.36. [DOI] [PubMed] [Google Scholar]
- 3.Bang Y.J., Van Cutsem E., Feyereislova A., Chung H.C., Shen L., Sawaki A., Lordick F., Ohtsu A., Omuro Y., Satoh T., et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/s0140-6736(10)61121-x. [DOI] [PubMed] [Google Scholar]
- 4.Macrì A., Morabito F. The use of intraperitoneal chemotherapy for gastric malignancies. Expert Rev. Anticancer Ther. 2019;19:879–888. doi: 10.1080/14737140.2019.1671189. [DOI] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Research Network Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Creighton C.J., Huang S. Reverse phase protein arrays in signaling pathways: a data integration perspective. Drug Des. Devel. Ther. 2015;9:3519–3527. doi: 10.2147/dddt.S38375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang R., Song S., Harada K., Ghazanfari Amlashi F., Badgwell B., Pizzi M.P., Xu Y., Zhao W., Dong X., et al. Multiplex profiling of peritoneal metastases from gastric adenocarcinoma identified novel targets and molecular subtypes that predict treatment response. Gut. 2020;69:18–31. doi: 10.1136/gutjnl-2018-318070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang R., Dang M., Harada K., Han G., Wang F., Pool Pizzi M., Zhao M., Tatlonghari G., Zhang S., Hao D., Lu Y., Zhao S., Badgwell B.D., Blum Murphy M., Shanbhag N., Estrella J.S., Roy-Chowdhuri S., Abdelhakeem A.A.F., Wang Y., Peng G., Hanash S., Calin G.A., Song X., Chu Y., Zhang J., Li M., Chen K., Lazar A.J., Futreal A., Song S., Ajani J.A., Wang L. Single-cell dissection of intratumoral heterogeneity and lineage diversity in metastatic gastric adenocarcinoma. Nat. Med. 2021;27:141–151. doi: 10.1038/s41591-020-1125-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murray B.W., Guo C., Piraino J., Westwick J.K., Zhang C., Lamerdin J., Dagostino E., Knighton D., Loi C.M., Zager M., et al. Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proc. Natl. Acad. Sci. USA. 2010;107:9446–9451. doi: 10.1073/pnas.0911863107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srikrishna G. S100A8 and S100A9: new insights into their roles in malignancy. J. Innate Immun. 2012;4:31–40. doi: 10.1159/000330095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 12.Zhang B., Wang J., Wang X., Zhu J., Liu Q., Shi Z., Chambers M.C., Zimmerman L.J., Shaddox K.F., Kim S., et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513:382–387. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang H., Liu T., Zhang Z., Payne S.H., Zhang B., McDermott J.E., Zhou J.Y., Petyuk V.A., Chen L., Ray D., et al. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell. 2016;166:755–765. doi: 10.1016/j.cell.2016.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mertins P., Mani D.R., Ruggles K.V., Gillette M.A., Clauser K.R., Wang P., Wang X., Qiao J.W., Cao S., Petralia F., et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55–62. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bayik D., Lathia J.D. Cancer stem cell-immune cell crosstalk in tumour progression. Nat. Rev. Cancer. 2021;21:526–536. doi: 10.1038/s41568-021-00366-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saygin C., Matei D., Majeti R., Reizes O., Lathia J.D. Targeting cancer stemness in the clinic: from hype to hope. Cell Stem Cell. 2019;24:25–40. doi: 10.1016/j.stem.2018.11.017. [DOI] [PubMed] [Google Scholar]
- 17.Willet S.G., Lewis M.A., Miao Z.F., Liu D., Radyk M.D., Cunningham R.L., Burclaff J., Sibbel G., Lo H.Y.G., Blanc V., et al. Regenerative proliferation of differentiated cells by mTORC1-dependent paligenosis. EMBO J. 2018;37 doi: 10.15252/embj.201798311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miao Z.F., Lewis M.A., Cho C.J., Adkins-Threats M., Park D., Brown J.W., Sun J.X., Burclaff J.R., Kennedy S., Lu J., et al. A dedicated evolutionarily conserved molecular network licenses differentiated cells to return to the cell cycle. Dev. Cell. 2020;55:178–194.e7. doi: 10.1016/j.devcel.2020.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miao Z.F., Cho C.J., Wang Z.N., Mills J.C. Autophagy repurposes cells during paligenosis. Autophagy. 2021;17:588–589. doi: 10.1080/15548627.2020.1857080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miao Z.F., Sun J.X., Adkins-Threats M., Pang M.J., Zhao J.H., Wang X., Tang K.W., Wang Z.N., Mills J.C. DDIT4 licenses only healthy cells to proliferate during injury-induced metaplasia. Gastroenterology. 2021;160:260–271.e10. doi: 10.1053/j.gastro.2020.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodgers J.T., King K.Y., Brett J.O., Cromie M.J., Charville G.W., Maguire K.K., Brunson C., Mastey N., Liu L., Tsai C.R., et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert) Nature. 2014;510:393–396. doi: 10.1038/nature13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DePeaux K., Delgoffe G.M. Metabolic barriers to cancer immunotherapy. Nat. Rev. Immunol. 2021;21:785–797. doi: 10.1038/s41577-021-00541-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thorsson V., Gibbs D.L., Brown S.D., Wolf D., Bortone D.S., Ou Yang T.H., Porta-Pardo E., Gao G.F., Plaisier C.L., Eddy J.A., et al. The immune landscape of cancer. Immunity. 2018;48:812–830.e14. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferguson L.P., Diaz E., Reya T. The role of the microenvironment and immune system in regulating stem cell fate in cancer. Trends Cancer. 2021;7:624–634. doi: 10.1016/j.trecan.2020.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prager B.C., Xie Q., Bao S., Rich J.N. Cancer stem cells: the architects of the tumor ecosystem. Cell Stem Cell. 2019;24:41–53. doi: 10.1016/j.stem.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kirk N.A., Kim K.B., Park K.S. Effect of chromatin modifiers on the plasticity and immunogenicity of small-cell lung cancer. Exp. Mol. Med. 2022;54:2118–2127. doi: 10.1038/s12276-022-00905-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matulonis U.A., Sood A.K., Fallowfield L., Howitt B.E., Sehouli J., Karlan B.Y. Ovarian cancer. Nat. Rev. Dis. Primers. 2016;2 doi: 10.1038/nrdp.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Izar B., Tirosh I., Stover E.H., Wakiro I., Cuoco M.S., Alter I., Rodman C., Leeson R., Su M.J., Shah P., et al. A single-cell landscape of high-grade serous ovarian cancer. Nat. Med. 2020;26:1271–1279. doi: 10.1038/s41591-020-0926-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ge S., Xia X., Ding C., Zhen B., Zhou Q., Feng J., Yuan J., Chen R., Li Y., Ge Z., et al. A proteomic landscape of diffuse-type gastric cancer. Nat. Commun. 2018;9:1012. doi: 10.1038/s41467-018-03121-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen G., Gharib T.G., Huang C.C., Taylor J.M.G., Misek D.E., Kardia S.L.R., Giordano T.J., Iannettoni M.D., Orringer M.B., Hanash S.M., Beer D.G. Discordant protein and mRNA expression in lung adenocarcinomas. Mol. Cell. Proteomics. 2002;1:304–313. doi: 10.1074/mcp.m200008-mcp200. [DOI] [PubMed] [Google Scholar]
- 31.Song J., Zhang P., Liu M., Xie M., Gao Z., Wang X., Wang T., Yin J., Liu R. Novel-miR-4885 promotes migration and invasion of esophageal cancer cells through targeting CTNNA2. DNA Cell Biol. 2019;38:151–161. doi: 10.1089/dna.2018.4377. [DOI] [PubMed] [Google Scholar]
- 32.Fanjul-Fernández M., Quesada V., Cabanillas R., Cadiñanos J., Fontanil T., Obaya A., Ramsay A.J., Llorente J.L., Astudillo A., Cal S., López-Otín C. Cell-cell adhesion genes CTNNA2 and CTNNA3 are tumour suppressors frequently mutated in laryngeal carcinomas. Nat. Commun. 2013;4:2531. doi: 10.1038/ncomms3531. [DOI] [PubMed] [Google Scholar]
- 33.Deng M., Zhang W., Tang H., Ye Q., Liao Q., Zhou Y., Wu M., Xiong W., Zheng Y., Guo X., et al. Lactotransferrin acts as a tumor suppressor in nasopharyngeal carcinoma by repressing AKT through multiple mechanisms. Oncogene. 2013;32:4273–4283. doi: 10.1038/onc.2012.434. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Q., Cheng Y., Xiong Y. LTF regulates the immune microenvironment of prostate cancer through JAK/STAT3 pathway. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.692117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Usener D., Schadendorf D., Koch J., Dübel S., Eichmüller S. cTAGE: a cutaneous T cell lymphoma associated antigen family with tumor-specific splicing. J. Invest. Dermatol. 2003;121:198–206. doi: 10.1046/j.1523-1747.2003.12318.x. [DOI] [PubMed] [Google Scholar]
- 36.Xie J., Gui X., Deng M., Chen H., Chen Y., Liu X., Ku Z., Tan L., Huang R., He Y., et al. Blocking LAIR1 signaling in immune cells inhibits tumor development. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.996026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang C.C. A perspective on LILRBs and LAIR1 as immune checkpoint targets for cancer treatment. Biochem. Biophys. Res. Commun. 2022;633:64–67. doi: 10.1016/j.bbrc.2022.09.019. [DOI] [PubMed] [Google Scholar]
- 38.Li H., Yang D., Hao M., Liu H. Differential expression of HAVCR2 gene in pan-cancer: a potential biomarker for survival and immunotherapy. Front. Genet. 2022;13 doi: 10.3389/fgene.2022.972664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2018. CA. Cancer J. Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 40.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All WES and RNA-Seq data have been deposited at the European Genome-phenome Archive (EGA). The datasets can be fully accessed under the accession number EGAS00001003180. Further information about EGA can be found on https://ega-archive.org (’the EGA of human data consented for biomedical research’: http://www.nature.com/ng/journal/v47/n7/full/ng.3312.html). The MS data in the current study are available from Table S5.
The key source codes that supported these results and findings can be found on GitHub (https://github.com/wpwan/GutAscites) and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.