

Summary
Tau-mediated toxicity is associated with cognitive decline and Alzheimer’s disease (AD) progression. In particular, tau post-translational modifications (PTMs) are thought to generate aberrant tau species resulting in neuronal dysfunction. Despite being well characterized in postmortem AD brain, it is unclear how caspase-mediated C-terminal tau cleavage promotes neurodegeneration, as few studies have developed the models to dissect this pathogenic mechanism. Here, we show that proteasome impairment results in cleaved tau accumulation at the post-synaptic density (PSD), a process that is modulated by neuronal activity. Cleaved tau (at residue D421) impairs neuronal firing and causes inefficient initiation of network bursts, consistent with reduced excitatory drive. We propose that reduced neuronal activity, or silencing, is coupled to proteasome dysfunction, which drives cleaved tau accumulation at the PSD and subsequent synaptotoxicity. Our study connects three common themes in the progression of AD: impaired proteostasis, caspase-mediated tau cleavage, and synapse degeneration.
Subject areas: Molecular neuroscience, Cellular neuroscience
Graphical abstract
Highlights
-
•
Proteasome impairment drives caspase-3 mediated tau cleavage at residue D421
-
•
Cleaved tau is soluble, hypo-phosphorylated, and synaptic-localized
-
•
Activation of the caspase-3/cleaved tau pathway is neuronal-activity regulated
-
•
Cleaved tau is sufficient to drive synaptic alterations and neuronal dysfunction
Molecular neuroscience; Cellular neuroscience
Introduction
Alzheimer’s disease (AD) is characterized by synaptic dysfunction, neuronal loss, cognitive decline, and behavioral/mood disturbances.1,2 Although AD is associated with extracellular amyloid-β plaques and intraneuronal neurofibrillary tau tangles (NFTs), genetic studies indicate that tau correlates with neurodegeneration, as APP mice lacking tau show restored memory and cognition.3 With accelerated disease progression, tau dissociates from microtubules (MTs) and undergoes conformational and structural changes that lead to the formation of paired helical filaments (PHFs) and subsequent NFT formation.4,5,6,7,8 In searching for distinct pathological tau species that mediate toxicity, several studies have identified pools of tau at the post-synaptic density (PSD) interacting with synaptic factors and cytoskeletal components that may drive AD progression.9,10,11,12,13 Given its prominent role in learning and memory,14,15,16 combined with the fact that synaptic degeneration is strongly implicated in the progression of AD,17,18,19 the PSD represents a critical subcellular location of interest to unravel how tau exerts its toxicity.
Tau undergoes many different post-translational modifications (PTMs), with phosphorylation being the most robust marker of NFTs in AD brain.20,21,22,23,24 However, the C-terminal microtubule binding repeat region (MTBR)25 harbors a lysine-rich stretch that is also subject to acetylation,26,27,28,29,30 ubiquitination,31,32,33 methylation,34,35,36 and sumoylation.37,38 These PTMs are known to exert dominant influences on tau’s molecular properties yet remain less well characterized. Beyond covalent PTM conjugation, tau is also abnormally cleaved at the C-terminus in AD brain, generating a series of potentially toxic truncated tau species that may precede, or prime, subsequent PTMs that further drive neurotoxicity.39,40,41,42,43 Given that cleaved tau has been characterized predominantly in postmortem AD brain,39,41,44 details regarding its generation, evolution, and toxicity in vulnerable neurons has not been well studied.
Members of the caspase superfamily, specifically caspases 2, 3, 6, 7, and 8 have been shown to cleave tau at the N- or C-terminus,45,46,47,48,49,50,51 with caspases 3, 7, and 8 showing preference for cleavage at the D421 residue in vitro, leading to the generation of a larger N-terminal tau 1–421 fragment and a smaller C-terminal 20 residue peptide (2N4R tau isoform).39,42,52 Among all caspases capable of cleaving tau, caspase-3 in particular is enriched at the PSD in AD brain and may be highly relevant to the synaptic degeneration induced by the cleaved tau (1–421) fragment.53,54,55,56 Once cleaved, tau is thought to assume a unique conformation that drives neurotoxicity,57,58 however, there are few reliable neuronal systems to examine tau cleavage events and its consequences on neuronal function. Moreover, although a tau 1–421 expressing tau transgenic mouse showed a rapid decline in memory,59 a D421A non-cleavable knock-in mouse also showed cognitive and synaptic plasticity defects,60 creating some debate and inconsistencies as to whether tau cleavage does indeed represent a toxic gain-of-function mechanism.
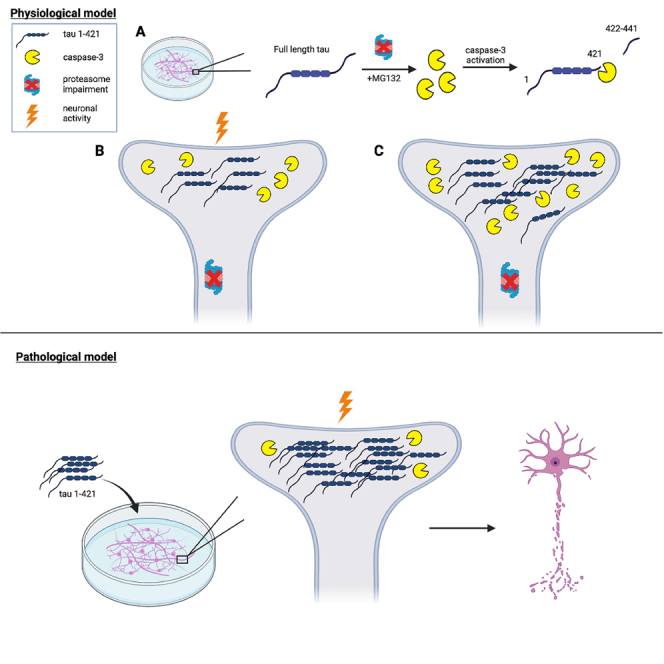
Given that degradative function, including proteasome efficiency, decreases with age and AD progression,61,62,63,64 here we examined whether manipulating the proteasome might provide a reliable approach to interrogate tau cleavage. We found that neuronal silencing, which is coupled to proteasome dysfunction, can modulate caspase-3 activity, resulting in the generation of cleaved tau that is enriched at the PSD. By delivering cleaved tau to neurons, we observed neuronal dysfunction, synaptic degeneration, and a decrease in neuronal activity and network synchrony. Our study highlights an activity-dependent mechanism for the regulation of cleaved tau, which is distinct from other known tau PTMs. We propose that neuronal activity is coupled to proteasome function, which modulates the production of a soluble cleaved tau species and a subsequent feedforward pathogenic cascade leading to synaptotoxicity and cognitive decline.
Results
Developing a neuronal model to characterize tau cleavage
Proteasome defects are strongly implicated in the progression of AD, and we therefore explored proteasome inhibitors as mediators of tau cleavage. We exposed wild-type (WT) mouse cortical neurons to various proteasome inhibitors targeting either the 20S or 26S subunit and found that proteasome inhibition reproducibly led to caspase-3 activation and accumulation of cleaved tau, as assessed with the cleaved tau-specific tauC3 antibody recognizing cleavage at D421 (Figure 1A). MG132, a well characterized 26S subunit inhibitor, produced significantly more cleaved tau than other proteasome inhibitors, providing the most sensitive readout for cleaved tau accumulation. We questioned whether the generation of cleaved tau could similarly be induced by other general proteostasis stressors but detected minimal levels of cleaved tau after exposure to the lysosomal inhibitor chloroquine (CQ) and a complete absence of cleaved tau with the autophagy inhibitor 3-methyladenine (3-MA) (Figure S1A). In addition, we evaluated a known mitochondrial toxin, rotenone, in a dose and time-dependent manner and observed only subtle caspase-3 activation and tau cleavage upon prolonged exposure (Figures S1B and S1C). Thus, our data suggest that tauC3-positive cleaved tau species preferentially accumulate in response to proteasome inhibition.
Figure 1.

Proteasome impairment activates caspase-3 and promotes tau cleavage
(A) Primary cortical neurons (E16) from C57BL/6 WT mice cultured for 10 days in vitro (DIV 10) were treated overnight with proteasome inhibitors MG132 (1 μM), Bortezomib (10 nM), Epoxomicin (20 nM), or Lactacystin (2 μM) and analyzed by immunoblotting. Cleaved tau can migrate as multiple cleavage products ranging from 43 to 50 kDa.
(B) WT or tau knockout (KO) primary neurons were left untreated or treated overnight with MG132 and analyzed by immunoblotting.
(C) WT primary neurons were transduced with either control (GFP expressing) or tau-expressing lentiviruses containing WT tau or the non-cleavable D421 mutant (D421A) at DIV 4, subsequently treated with MG132 at DIV 10, and then analyzed by immunoblotting. We labeled cleaved tau products with an asterisk (∗).
(D and E) Primary neurons treated with MG132 were analyzed by LDH and MTT assays to determine the impact on neuronal viability.
(F and G) Biochemical fractionation of neuronal lysates were used to generate soluble (RIPA) and insoluble (SDS) fractions, which showed cleaved tau was present exclusively in MG132-treated soluble fractions. Statistical tests: p value determined by one-way ANOVA with Tukey’s post hoc test from n > 5 (D-E). Error bars represent means ± SEM (D-E). ∗∗∗∗p< 0.0001, ns not significant.
We next performed a series of control experiments to validate the sensitivity and specificity of the observed cleaved tau immunoreactivity. We employed tau knock-out (KO) neurons as well as a D421A non-cleavable site-specific tau mutant. Even though caspase-3 is still activated by proteasome impairment in tau KO neurons, tauC3 immunoreactivity was lost, confirming the specificity of the cleaved tau antibody (Figure 1B). In addition, the non-cleavable D421A mutant, a substrate no longer recognized by caspases, failed to show any detectable tauC3 immunoreactivity (Figure 1C).
To determine whether cleaved tau was simply generated as a consequence of neuronal damage or cell death, we assessed cell viability by standard lactate dehydrogenase (LDH) release assays in neurons exposed to MG132, which did not reveal apoptotic cell death. However, we did observe an expected ∼20% decrease in metabolic function when assessed by the reduction of the tetrazolium salt 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT assay) (Figures 1D and 1E).
We next investigated whether proteasome impairment showed specificity for the generation of cleaved tau as opposed to other modified types of tau (e.g., phosphorylated tau). Using detergent soluble fractions, we detected AT8 immunoreactive tau in untreated neurons, which was reduced to nearly undetectable levels in response to proteasome inhibition (Figure 1F), consistent with compensatory tau dephosphorylation in response to stress that we and others have previously characterized.65,66,67,68,69 Moreover, analysis of the detergent insoluble fractions did not detect cleaved tau, consistent with the absence of amyloid-like tau aggregation (Figure 1G). Therefore, the generation of cleaved tau in response to proteasome inhibition does not correlate or coincide with the formation of insoluble or hyper-phosphorylated tau, properties typically associated with insoluble NFT formation in AD brain.
To evaluate patterns of cleaved tau localization in neurons, confocal microscopy showed abundant MG132-induced tauC3 immunoreactivity distributed throughout the neuron, with accumulation in neuronal processes and less frequent cleaved tau detected within neuronal soma (Figure 2A). Whereas active caspase-3 accumulated primarily in tauC3-positive nuclei, we also found co-localization of cleaved tau and caspase-3 within neuronal processes, suggesting involvement of a non-nuclear pool of caspase-3.70 In all instances, neurons harboring cleaved tau consistently showed activation of caspase-3, highlighting a strong correlation between caspase-3 and tau cleavage. Lastly, we analyzed cleaved tau and active caspase-3 within plaques and NFTs in human AD brain. Triple labeling immunofluorescence showed that cleaved tau and caspase-3 were found within thioflavin-S (ThS) positive NFTs and neuritic plaques (Figure 2B). Subsequent analysis of AD brain homogenates detected cleaved tau in the soluble tau pool (Figures 2C and 2D), consistent with our in vitro analysis (Figures 1F and 1G). Thus, in two separate systems, a primary neuronal system lacking AD pathology and late-stage AD brain comprised mixed tau/Αβ pathology, we observed co-localization of cleaved tau and active caspase-3.
Figure 2.

Caspase-3-mediated tau cleavage occurs within neuronal processes and in AD brain
(A) Primary mouse neurons were analyzed by immunofluorescence (IF) to detect cleaved tau (green) and cleaved caspase-3 (red), whereas nuclei were marked by DAPI (blue). The white inset highlights a magnified neuronal process depicting co-localization between cleaved tau and cleaved caspase-3 present in the neuronal processes. Scale bar = 50 μm.
(B) Human AD hippocampal sections were analyzed by double-labeling using thioflavin-S (ThS) to mark plaques and NFTs (green), and cleaved tau (red) or cleaved caspase-3 (blue) antibodies to evaluate their co-localization in mature AD pathological lesions. White arrows and the magnified white inset highlight regions of cleaved tau and cleaved caspase-3 co-localization that are associated with ThS-positive plaques and NFTs. Scale bar = 50 μm. Representative images are shown from n ≥ 3 independent experiments (A) and n = AD 5 cases (B).
(C and D) Control and human AD brain tissues were homogenized into either soluble (high salt) or insoluble (urea) fractions and analyzed by immunoblotting with cleaved tau and total tau antibodies.
Molecular and cellular determinants of tau cleavage
Tau proteins in the central nervous system (CNS) are generated as a result of alternative splicing producing six tau isoforms that contain zero (0N), one (1N), or two (2N) N-terminal inserts and either three (3R) or four (4R) C-terminal microtubule-binding repeats within the MTBR. Different neurodegenerative diseases show distinct tau profiles comprised either 3R-tau (e.g., Pick’s disease), 4R-tau (e.g., corticobasal degeneration), or mixed 3R/4R-tau (e.g., AD).71,72 To determine if tau cleavage shows isoform specificity, we asked whether proteasome inhibition preferentially generated cleaved 3R or 4R tau. First, we expressed individual tau isoforms in HEK293A cells and exposed the cultures to MG132. As shown in Figure 3A, tau cleavage was not preferential for either 3R or 4R tau isoforms, though we did observe subtle yet insignificant differences among cleavage of the different tau isoforms. This result is most clearly visualized with a staggered single-lane tau ladder containing all six cleaved tau isoforms derived from individual lysates but spiked into a single sample (Figure 3B).
Figure 3.

Tau cleavage is not dependent on tau isoform or individual MTBR repeat regions
(A) HEK-293A cells were transfected with one of six tau isoforms for 48 h, treated overnight with MG132 (where indicated), and analyzed by immunoblotting.
(B) All independently transfected six-tau isoform expressing HEK-293A cell lysates were combined into one mixed lysate and analyzed by immunoblotting, which detected cleaved tau with no apparent specificity for 3R-tau or 4R-tau.
(C) Immunoprecipitation-western analysis was performed on lysates from WT mouse neurons treated with MG132. Cleaved tau pulldowns revealed that both 3R-tau (RD3-immunoreactive) and 4R-tau (RD4-immnoreactive) were subject to cleavage. Asterisks (∗) denote the cleaved tau product.
(D) HEK-293A cells were transfected with tau mutants lacking individual MTBR repeats or the entire MTBR (ΔR1, Q244-K274; ΔR2, V275-S305; ΔR3, V306-Q336; ΔR4, V337-E372; ΔR1-4, Q244-E372) and treated with MG132 (where indicated). The ΔR1-4 lysate was loaded at twice the volume to provide comparable total tau expression relative to all tau variants. Complete removal of the entire MTBR (ΔR1-4) reduced the ability of tau to undergo caspase-mediated cleavage.
To confirm the lack of tau isoform specificity, we took an alternative biochemistry approach and immunoprecipitated cleaved tau from MG132-treated primary mouse neurons for subsequent detection of the different tau isoforms by immunoblotting with either 3R-tau specific (RD3) or 4R-tau (RD4) specific antibodies. We found that immunopurified cleaved tau pulldowns were both RD3 and RD4-immunoreactive, again reflecting a lack of tau isoform specificity for C-terminal tau cleavage in response to proteasome inhibition (Figure 3C).
We next asked whether internal deletions of the MTBR could impact C-terminal tau cleavage using a panel of MTBR deletion constructs that were expressed in HEK293A cells and exposed to MG132 (Figure 3D). Although deletion of individual MTBR domains (ΔR1, ΔR2, ΔR3, or ΔR4) did not impact C-terminal tau cleavage, we note that the removal of the entire MTBR region (ΔR1-4) abrogated tau cleavage, even when adjusting for slight expression level differences observed with the ΔR1-4 tau variant (Figure 3D, see ΔR1-4). These data suggest that a combination of internal elements across multiple MTBR repeat domains likely contributes to C-terminal cleavage at residue D421.
Since tau cleavage was isoform-independent, we searched for other neuronal properties that might account for the robust MG132-induced caspase-3 activation and subsequent tau cleavage, including neuronal maturity, synaptic activity, and neuronal subtype specificity. First, we exposed neurons to MG132 at different stages of development in vitro (DIV). Beyond DIV 12, our primary neuronal cultures begin to show prominent astrocyte accumulation, based on increasing GFAP immunoreactivity (Figures S2A and S2B). Therefore, to restrict our analysis only to neurons, we analyzed cleaved tau in DIV 4, 8, and 12 cultures, avoiding any potential confounding effects of astrocytes at timepoints beyond DIV 12. We found that immature neurons cultured at DIV 4 showed no detectable cleaved tau even though caspase-3 became activated. Of interest, as neurons matured at DIV 8 and even more so at DIV 12, caspase-3-mediated tau cleavage became more readily apparent (Figures 4A and 4B). These findings suggest that tau cleavage is regulated by neuronal maturity and prompted us to examine the influence of enhanced neuronal activity within this timeframe.
Figure 4.

Tau cleavage is dependent on neuronal maturity and synaptic activity
(A and B) Primary WT neurons were untreated or treated with MG132 overnight, collected at DIV 4, 8, and 12 and analyzed by immunoblotting followed by quantification of cleaved tau normalized to total tau. Cleaved tau accumulates most robustly in mature DIV 8–12 neurons upon proteasome impairment.
(C and D) Primary neurons were either left untreated, pre-treated with Tetrodotoxin (TTX) (1 μM) or Bicuculline (BIC) (20 μM) for 8 h before MG132 treatment (where indicated). Lysates were analyzed by immunoblotting followed by quantification of cleaved tau levels normalized to total tau. BIC-induced hyperexcitability suppressed the formation of cleaved tau while TTX-mediated silencing augmented cleaved tau levels.
(E and F) Primary neurons were either left untreated or treated with TTX (1 μM) for 24 h before being collected at DIV 13 and 15 and analyzed by immunoblotting followed by quantification of cleaved tau normalized to total tau. Representative immunoblotting data are shown from n = 3 biologically independent samples (A-F). Statistical tests: p value determined by one-way ANOVA with Tukey’s post hoc test from n = 3. Error bars represent means ± SEM. ∗p< 0.05, ∗∗p< 0.01, ∗∗∗∗p< 0.0001, ns not significant.
Since neuronal activity increases with differentiation, we asked whether modulating neuronal activity influenced the ability of tau to undergo caspase-dependent cleavage. We employed a pharmacological approach and treated neurons with the sodium channel blocker tetrodotoxin (TTX) to silence neurons or the GABAA receptor antagonist bicuculline (BIC) to increase synaptic firing (Figures 4C and 4D). Strikingly, neurons pre-treated with TTX and then challenged with MG132 resulted in significantly higher cleaved tau levels compared to control neurons. Conversely, pre-treatment with BIC, which disinhibits neurons, completely alleviated the cleaved tau accumulation observed with MG132 (Figure 4C, see BIC+MG lanes). In all instances, cleaved caspase-3 positively correlated with cleaved tau accumulation (Figure 4C). Because neuronal silencing with TTX alone appeared to slightly elevate cleaved tau levels (Figure 4C), we repeated this analysis at two precise timepoints (DIV 13 and 15) and visualized cleaved tau with higher detection sensitivity in the absence of saturating signal from the MG132 exposure. Intriguingly, neurons silenced with TTX alone in the complete absence of proteasome inhibition accumulated cleaved tau (Figures 4E and 4F, see TTX lanes), suggesting silencing is sufficient to generate cleaved tau. These findings support that the cleaved tau cascade is driven by neuronal activity coupled to the regulation of proteasome function.
Although the above results suggest that neuronal activity fine-tunes the levels of cleaved tau, we were still unable to explain the neuron-to-neuron variability in cleaved tau. For example, rather than homogeneous induction of cleaved tau among the entire neuron population, only ∼20% of total neurons treated with MG132 became tauC3-immunoreactive (Figures S3A and S3B), suggesting a cell autonomous property may dictate the propensity to generate cleaved tau. To examine whether tau cleavage shows neuronal subtype differences that could arise from inhibitory versus excitatory neuronal populations in our cultures, we performed confocal imaging of Vglut-positive excitatory and Vgat-positive inhibitory neurons exposed to MG132. We found that cleaved tau preferentially accumulated in excitatory neurons, as tauC3 co-localized with VGlut1 within neuronal soma, whereas Vgat-positive inhibitory neurons were largely devoid of tauC3 immunoreactivity (Figure S3C). Overall, these data support mature excitatory neurons as the major neuronal population that is susceptible to the accumulation of cleaved tau.
Caspase-3 mediated tau cleavage at the post-synaptic density
Prior studies suggest that tau exerts toxicity at the synapse.12,17,18 Our data so far show that cleaved tau is enriched within neuronal processes, and we hypothesized that tau’s subcellular localization may be linked to its responsiveness to neuronal activity. Therefore, we asked whether cleaved tau directly localized to the post-synaptic density (PSD), providing an explanation for its activity responsiveness. Indeed, using isolated PSD preparations from MG132-treated neurons, we detected activated caspase-3 and cleaved tau at the PSD (Figure 5A). Higher magnification of the synapse using high-resolution confocal imaging identified cleaved tau puncta in close proximity to, and in some instances overlapping with the PSD (Figure 5B). Thus, the affinity of cleaved tau for the synapse likely explains its responsiveness to neuronal activity and potential synaptotoxic effects.
Figure 5.

Caspase-3-dependent tau cleavage at the post-synaptic density (PSD)
(A) WT primary neurons cultured at DIV 14 were treated with MG132 and subjected to post-synaptic density (PSD) subcellular fractionation. Total homogenates and PSD fractions were analyzed by immunoblotting using an anti-PSD-95 antibody to validate PSD enrichment during the subcellular fractionation.
(B) Fixed primary neurons treated with MG132 were analyzed by confocal imaging to detect cleaved tau (green) and PSD-95 (red). White arrows highlight areas along neuronal processes where cleaved tau and PSD-95 puncta colocalize. The white inset in the merged panel highlights cleaved tau positive puncta that colocalize with (or are closely adjacent to) PSD-95 puncta. Scale bar = 10 μm. Representative images are shown from n = 3 independent experiments.
(C) WT or caspase-3 KO neurons were left untreated or treated overnight with MG132 and analyzed by immunoblotting using tauC3, cleaved caspase-3 and total tau antibodies.
(D) WT or caspase-3 KO neurons were subjected to subcellular fractionation as in A above and analyzed by immunoblotting.
(E and F) Viability was measured in WT and caspase-3 KO neurons using LDH assay and MTT assays. Statistical tests: p value determined by one-way ANOVA with Tukey’s post hoc test from n = 3 biologically independent experiments. Error bars represent means ± SEM. ∗∗p< 0.01, ns not significant.
Though our findings thus far correlated caspase-3 activation and tau cleavage, other proteases including caspases 2, 3, 6, and 7 have all been shown to generate tau cleavage products. Using breeding-competent caspase-3 KO mice, we generated caspase-3 KO cortical neurons, exposed them to MG132, and evaluated tau cleavage in the complete absence of caspase-3. Cleaved tau was reduced to nearly undetectable levels, suggesting caspase-3 is the dominant caspase generating cleaved tau (at residue D421) in response to proteasome inhibition (Figure 5C). We confirmed this finding using purified isolated PSD preparations and similarly found that caspase-3 KO neurons completely abrogated cleaved tau accumulation at the PSD (Figure 5D). We also note that in the absence of caspase-3, in which cleaved tau levels were significantly reduced, we still observed an MG132-dependent reduction in metabolic function with no apparent impact on neuronal death (Figures 5E and 5F).
Tau cleavage causes synaptic alterations and neuronal dysfunction
To determine the consequences of cleaved tau expression in neurons, we generated a human tau 1–421 lentiviral construct lacking the C-terminal 20 residues of the full-length 2N4R tau isoform. As expected, tau 1–421 was readily detected with the tauC3 antibody, in contrast to neurons expressing WT tau or P301L-tau (PL) (Figure 6A). Tau 1-421-expressing neurons showed moderate activation of caspase-3, indicating that cleaved tau can likely feedback and activate caspase-3. By confocal imaging of a GFP-labeled tau 1–421 construct, ectopically expressed cleaved tau diffusely labeled neuronal processes (Figure 6B). Although caspase-3 activation is associated with apoptosis, we did not observe any overt neuronal death by LDH assay after delivery of tau 1–421 or other tau lentiviruses to neurons, including a non-cleavable D421A mutant (Figure 6C). However, among all tau variants tested, only tau 1–421 showed an ∼ 80% decrease in metabolic activity as assessed by MTT assay, supporting the notion that cleaved tau induces more extensive neuronal dysfunction, but in the absence of overt cell death (Figure 6D).
Figure 6.

Cleaved tau promotes neuronal dysfunction and synaptic degeneration
(A) WT primary neurons were either transduced at DIV 4 with control or tau-expressing lentiviruses (WT tau, P301L tau or tau 1–421), or treated with MG132 overnight at DIV 10, and analyzed by immunoblotting.
(B) Primary neurons were transduced with tau 1-421-GFP and analyzed by confocal imaging to detect ectopically expressed cleaved tau (green) and cleaved caspase-3 (red), while nuclei were marked by DAPI (blue). Scale bar = 50 μm. Representative neurons are shown from n = 3 biologically independent experiments.
(C and D) LDH and MTT assays were performed on WT primary neurons transduced at DIV 4 with control, WT tau, PL tau, tau 1–421 or D421A and analyzed at DIV 10.
(E) Neurons expressing WT tau-GFP or tau 1-421-GFP were analyzed by confocal imaging at DIV 13 and DIV 23 to detect WT tau or cleaved tau (green), PSD-95 (red), and cleaved caspase-3 (cyan). White arrowheads highlight regions where cleaved tau led to reduced PSD-95 signal intensity. In contrast, full arrows depict regions expressing WT tau, or little to no tau 1–421, which retain PSD-95 immunoreactivity, indicating that tau 1–421 is causing synaptic damage. Scale bar = 5 μm.
(F) The percentage of GFP positive puncta found at PSD-95 positive synapse regions of DIV 13 neurons were quantified from n = 7 independently acquired and blinded frames and n = 3 biologically independent experiments.
(G and H) WT neurons transduced with tau 1–421 or treated with MG132 overnight were fractionated into total homogenate and PSD fractions and analyzed by immunoblotting. PSD-enriched lysates were further analyzed by immunoblotting to detect total and phosphorylated glutamate receptors. Statistical tests: p value determined by one-way ANOVA with Tukey’s post hoc test from n = 3 (C-D) or by two-sided unpaired t-test (F) from n = 7. Error bars represent means ± SEM (C,D, F). ∗∗∗p< 0.001, ∗∗∗∗p< 0.0001, ns not significant.
To determine whether ectopically expressed cleaved tau was enriched at the synapse, we performed imaging and PSD fractionation experiments. Neurons expressing tau 1–421, cultured at either DIV 13 or further matured to DIV 23, showed decreased PSD-95-positive puncta that coincided with intensely labeled tau 1–421 puncta, reflecting synapse degeneration and loss of the PSD-95 signal (Figure 6E, see arrowheads highlighting reduced PSD-95 puncta). In contrast, neurons expressing either WT tau-GFP or even those expressing low levels of tau 1–421 at the synapse resulted in more intensely double-labeled PSD-95-positive synapses (Figure 6E, see full arrows highlighting robust PSD-95 puncta). Quantification of these findings showed a ∼ 40% decrease in PSD-95 signal intensity in the presence of tau 1–421 (Figure 6F). To confirm these observations, we analyzed PSD-enriched fractions by immunoblotting and observed diminished levels of PSD-95 in neurons expressing tau 1–421 as well as a decrease in phosphorylated and total levels of glutamate receptors at the PSD (Figures 6G and 6H), again suggesting that cleaved tau promotes synaptic loss.
To assess the effects of cleaved tau on neuronal function, we employed multielectrode arrays (MEAs) to assess neuronal firing and connectivity. We initially employed a 16-electrode array and first evaluated general parameters of neuronal function in WT or tau KO neurons exposed to MG132 to determine whether the presence of tau alters neuronal function in response to proteasome impairment. Indeed, proteasome inhibition led to a time-dependent loss in neuronal firing and synchrony that was partially abrogated by genetic tau depletion, indicating that the synaptic response to proteasome impairment is tau-dependent (Figures S4A and S4B). Next, to evaluate whether the cleaved tau variant phenocopies the reduction in neuronal activity observed with MG132, neurons were transduced with control (GFP), WT tau, and tau 1–421 lentiviruses such that our cultures expressed the tau variants at > 90% infection efficiency (Figure S5A). As in Figure 6C, the tau constructs did not cause overt toxicity in neurons cultured on MEA grids using a viability module (Figure S5B). Electro-physiological changes in firing rates were observed as early as 6 days post-lentiviral transduction (DIV 24) (Figure 7A), coinciding with increasing lentiviral expression at this timepoint. Representative examples of synchronous activity are shown in the raster plots (Figure 7B) depicting activity across all 16 electrodes over a 10-min period, which illustrates a robust decrease in neuronal activity and loss of network spikes (NS, vertical pink bars) in response to tau 1–421 expression, with only a slight loss of activity due to WT tau expression.
Figure 7.

Cleaved tau suppresses neuronal activity
(A) Primary WT neurons cultured for 18 DIV were recorded on Axion Biosystems for 10 min before transduction. After baseline recording, neurons were transduced with control (GFP), WT tau, or tau 1–421, and recordings were performed for 10 min per day until DIV 29 and analyzed for mean firing rate. Statistical tests: p value determined by one-way ANOVA with Tukey’s post hoc test from n = 3 biologically independent experiments. Although tau 1–421 showed a reduction in mean firing rate, there was no statistical significance among groups. Error bars represent means ± SEM.
(B) Raster plots of 10 min recording from primary WT neurons cultured for 24 DIV six days post transduction with either control (GFP), WT tau or tau 1–421 lentivirus.
(C) A histogram averaged overall network spikes (NS) of GFP control (blue), WT tau (dark orange), and tau 1–421 (crimson) networks at DIV 28 (10 days post transduction). Neurons expressing tau 1–421 have nearly double the onset (time to peak) relative to WT (GFP control = 44 ms, WT tau = 59 ms, tau 1–421 = 108 ms, colored dashed lines on excitatory side of NS) with a slightly quicker offset (GFP control = 71 ms, WT tau = 45 ms, tau 1–421 = 57 ms). Tau 1–421 neurons are also further distinguished from control by longer NS durations (GFP control = 115 ms, WT tau = 104 ms, tau 1–421 = 165 ms, blue dashed lines along width of NS).
(D) Instantaneous firing rates in WT neurons illustrating the typical Gaussian distribution of spike rates during the initiation and termination of an NS and instantaneous spike rates for neurons expressing tau WT and 1–421. Note the dips in firing rates during excitation in tau 1–421 expressing neurons, illustrating interference in synaptic communication.
(E) An interspike interval (ISI) histogram with log bins. GFP-expressing control neurons exhibit a peak ISI of 2.5 ms (blue) similar to WT tau (orange) and tau 1–421 (crimson). However, tau 1–421 expressing neurons show a greater dispersion in their ISI distributions with an SD of 0.334 relative to 0.290 for control and 0.026 for WT tau indicating interference in synaptic communication. All high-density MEA results represent an average from n = 4 biologically independent experiments.
To examine the development of network activity more precisely, we utilized a higher density 60-electrode MEA array and studied their average NS profile (Figure 7C, average across all neurons), with representative spike rate histograms from individual neurons shown in Figure 7D. On average, neurons expressing tau 1–421 showed a significantly slower NS onset (Figure 7D, right panel) reflecting weakening of synaptic coupling strength.73 The inefficiency of network communication was also seen in a much higher percentage of aborted NS in the tau 1–421 neurons (67 ± 21%) relative to GFP control neurons (34 + 26%) and WT tau neurons (42 ± 18%). To further probe the synaptic behavior of the networks, interspike interval (ISI) histograms were constructed (Figure 7E), which showed a greater dispersion in the distribution of ISIs for tau 1–421 (SD = 0.334) relative to WT tau (SD = 0.026) or the control (SD = 0.290), supporting a severe lack of synchronization and delay in synaptic communication in tau 1–421 networks.
Next, we probed the network properties and structure in tau expressing neurons. Neuronal network communication, defined as synchronous activity in at least 30% of the electrodes, decreased significantly in neurons expressing tau 1–421 beginning at 6 days post-transduction (DIV 24) (Figure 8A). To further probe this loss of synchronization, an analysis of network properties using the 60-electrode MEAs at DIV 28 revealed a relatively highly connected network in control and WT tau expressing neurons (Figure 8B, left and middle panels) in contrast to 1–421 expressing neurons (Figure 8B, right panel) by several criteria (Figures 8B–8D). Control neurons showed strong probability of nodes with high degrees of connectivity, an average normalized degree of 0.48 ± 0.16, average path length (1 ± 0.50), and average cluster coefficient (0.75 ± 0.16), consistent with strong local communication and strong small world topography. In contrast, neurons expressing tau 1–421 revealed a low and steadily decreasing probability of nodes with high degrees with an average normalized degree of 0.15 ± 0.12, significantly increased average path length (2 ± 0.59) and a decrease in the cluster coefficient (0.32 ± 0.19) consistent with a loss of small world topography (Figures 8C and 8D). Overall, our MEA analysis reveals that ectopic expression of cleaved tau mediates prominent synaptic disruptions that lead to neuronal network and communication defects.
Figure 8.

Cleaved tau causes abnormal synchrony and defects in network communication
(A) Primary WT neurons cultured at DIV 18 were recorded on Axion Biosystems for 10 min before transduction. After baseline recording, neurons were transduced with control (GFP), WT tau, or tau 1–421, and recordings were performed for 10 min per day until DIV 29 and analyzed for synchrony index. Statistical tests: p value determined by one-way ANOVA with Tukey’s post hoc test from n = 3 biologically independent experiments. Error bars represent means ± SEM. ∗p< 0.05.
(B) A representative schematic of efficient communication for a GFP-expressing control network at DIV 28 exhibiting a highly connected network with strong small world topography (strong local communication) evidenced by high cluster coefficients and short path lengths. Each vertex (purple dot) represents a neuron on a 60-electrode array and each green line represents strong synaptic weight with its connecting neuron. A representative schematic of a tau 1–421 network at DIV 28 is shown using the same cross correlation measure and threshold for control networks. Tau 1–421 neurons exhibit loss of small world topography as evidenced by a lower cluster coefficient and longer path lengths between vertices. Occasional tau 1–421 hubs begin to develop (large purple dots).
(C) Complementary Cumulative Distribution Functions (CCDF) of average degree shows that as the average degree (k) increases, tau 1-421-expressing neurons (crimson) show a decreasing probability (pk) of vertices with high degree. In contrast, GFP-expressing control (blue) networks show a high probability of vertices with low to high degrees, while WT tau exhibits a slightly truncated probability of nodes with low to high degrees. The CCDFs are plotted on a log-log plot to show that the tau 1-421s exhibit a mix of a power law and random degree distribution while control and WT tau networks primarily exhibit an exponential degree distribution.
(D) Kernel density estimates of average shortest path length. The average shortest path length of control (blue) networks is approximately 1 illustrating strong small world topography and high clustering and exhibits a bimodal distribution with a significant peak path length of 2 for some networks. In contrast tau 1–421 (crimson) networks exhibit a loss of small world topography and low clustering with a tri-modal distribution of their path lengths and an average path length of 2 (or a path length of 1 and 3 for some networks). WT tau neurons have similar path length distributions to controls. All high-density MEA results represent an average from n = 4 biologically independent experiments.
Discussion
In this study, we unraveled a pathogenic cascade in which caspase-dependent tau cleavage drives neurotoxicity. Although tau cleavage is thought to induce conformational changes in tau's structure that drive NFTs, our study sheds new light on its regulation in vulnerable neurons at earlier stages of disease. We show that proteasome impairment triggers caspase-3-mediated tau cleavage at the PSD, a neuronal activity-regulated process that impacts firing, synaptic communication, and ultimately drives neurotoxicity. Thus, specific modified tau variants (e.g., cleaved tau) may act as putative sensors of neuronal activity which, if perturbed or exacerbated, may drive synaptic and cognitive defects that are characteristic of AD.
A prior study showed rapid memory impairments in a tau transgenic mouse model expressing a 1–421 cleaved tau construct.59 Our data support cleaved tau as a toxic species capable of altering neuronal firing and driving neurodegeneration and network dysfunction. However, the notion that cleaved tau is toxic was not supported by a recent non-cleavable D421A knock-in mouse, which surprisingly also showed cognitive defects.60 One caveat of the non-cleavable D421A point mutation is that it may not solely abrogate tau cleavage at the tauC3 epitope. The point mutation at D421 could potentially alter nearby tau phosphorylation including the immediately adjacent AD-relevant S422 site.74,75 Therefore, future studies are warranted to evaluate the resulting PTM cascade and any downstream consequences of non-cleavable tau mutations.
Caspase-3 is known to control synapse weakening via AMPA receptor trafficking in response to long-term depression (LTD), yet in a non-apoptotic or cell death-related manner.76,77 Although some synaptic proteins are reportedly cleaved by caspase-3, many still remain unidentified. Our data strongly suggest that caspase-3 is the dominant synaptic caspase that targets tau in response to proteasome failure, and whose cleavage leads to the degeneration of post-synaptic structures. The notion that a synaptic pool of caspase-3 may generate D421-cleaved tau is supported by the following observations: 1) caspase-3 activation within neuronal processes parallels cleaved tau accumulation (Figure 2A), 2) synaptic accumulation of D421-cleaved tau was near completely eliminated in caspase-3 KO neurons (Figures 5C and 5D), and 3) prior studies detected caspase-3 at the synapse of AD model mice and human AD postmortem brain.12,17,54,78 Because ectopically expressed tau 1–421 further activated caspase-3 (Figure 6A), we speculate that accumulation of casaspe-3-mediated cleaved tau beyond a critical threshold may initiate a feedforward cycle that further drives the degenerative cascade. Given that caspase-3 normally controls synaptic pruning and is involved in neuronal apoptosis,70,79,80,81 its function may become corrupted under pathological conditions, potentially in response to altered patterns of neuronal activity (see below), generating an abnormally active caspase-3 that preferentially targets tau.
At the moment, we cannot definitively conclude that proteasome inhibition, and the resulting impairments in neuronal activity, results in caspase-3 activation and site-specific tau cleavage at residue D421. Proteasome inhibitors including MG132 are pleiotropic and globally impact neuronal function via activation of redundant caspases that could cleave tau at multiple sites. For example, caspases 2, 6, 7, and 8 cleave tau at multiple N- and C-terminal residues.39 Caspase-2, in particular, cleaves tau at residue D314, a fragment that is implicated in synaptic defects and memory decline.45,47,82 Although our data suggest that caspase-3, rather than caspase-2, is largely responsible for the generation of D421-cleaved tau and the resulting effects on the synapse, we cannot exclude the possibility that other caspases and other tau cleavage sites may impact synaptic activity in response to proteasome dysfunction. Future studies will be required to assess (1) whether redundant and/or sequentially acting caspases coordinately target synaptic tau, and (2) whether site-specific abrogation of tau cleavage at various sites (for example, achieved by inserting a D421A mutation into the endogenous mouse MAPT gene), is sufficient to protect against loss of neuronal activity in response to proteasome inhibition.
We questioned exactly which stressors might initiate caspase-3-dependent tau cleavage. During aging and in AD, proteasome activity decreases and correlates with advancing AD risk.61,62,64 We found that proteasome impairment in particular, more so than other general stressors, preferentially generated cleaved tau. Proteasome impairment may directly stabilize cleaved tau levels because tau is a substrate for the ubiquitin proteasome system (UPS).31,32,33 However, other indirect mechanisms that stabilize cleaved tau are also plausible, as caspase-3 is also itself modified by ubiquitination and targeted in a proteasome-dependent manner,83 which could also result in cleaved tau stabilization on proteasome inhibition. Regardless, once generated in response to proteasome failure, pathological tau may feedback to further block 26S proteasome activity,64 a bidirectional link connecting cleaved tau and the proteasome.
We were intrigued to find that cleaved tau was not expressed in all neurons on proteasome impairment. Instead, cleaved tau was preferentially generated in VGlut1-positive excitatory neurons, consistent with excitatory neurons being more vulnerable to pathological tau accumulation in cortex of AD mice and human AD brain84 and also consistent with our findings that cleaved tau causes defects in excitatory drive (Figures 7C and 7D). In addition to neuronal subtype specificity, we found that neuronal activity itself modulates cleaved tau production. Indeed, we found that silencing neurons pharmacologically was sufficient to induce cleaved tau, whereas enhancing synaptic activity completely abrogated cleaved tau (Figures 4C and 4D), consistent with cleaved tau representing an elusive synaptic activity-regulated tau species. Prior studies have shown that tau is capable of silencing neurons, though the relevant tau species had not been identified. For example, pathological tau expression in the medial entorhinal cortex (MEC) in spatially restricted EC-tau mice led to inhibition of excitatory grid cell firing.85 In addition, Busche et al. showed prominent neuronal silencing in two different tau transgenic mouse models (rTg4510 and rTg21221), which did not depend on tau aggregation or NFT formation.86
Because tau 1–421 is soluble, synaptic-localized, and activity-regulated, we propose that cleaved tau represents a critical tau species that mediates synaptoxicity and network dysfunction in AD, which is supported by the neuronal firing and network properties observed on cleaved tau expression (Figures 7 and 8). Synchronized bursting activity is thought to consolidate memories and underlie cognitive behavior.73,87,88 The longer onset to peak firing in neurons expressing tau 1–421 is consistent with weakening synaptic strength at excitatory synapses. The synaptic changes induced by tau 1–421 thus likely reflect an inability to initiate and maintain effective network communication. Although the specific targets that mediate the effects of cleaved tau at the synapse are not yet entirely clear, synaptic scaffolds such as PSD-9589 and KIBRA,90 AMPA and NMDA receptors,54,89,91 and trafficking factors such as NSF92 are reported candidates that could be explored in future studies.
Finally, how a synaptic caspase-3/tau complex may “sense” neuronal activity is an intriguing and yet to be answered question. The proteasome itself represents a direct target of neuronal silencing and can thereby link neuronal activity to the restructuring, or remodeling, of the synapse. This concept is appealing and supported by studies showing that silencing neurons impairs proteasome activity via direct phosphorylation/dephosphorylation of proteasomal subunits including Rpt6,93 whereas enhancing synaptic activity promotes synaptic homeostasis and activation of pro-survival pathways.94,95,96,97 Based on our current findings, we envision a model in which neuronal silencing directly impairs the proteasome, leading to activation of caspase-3 and subsequent tau cleavage. Once cleaved, tau is sufficient to promote synapse dysfunction, initiating a pathogenic cascade that feeds back to further impair proteasome function and cause additional synapse weakening. Thus, the generation of cleaved tau likely represents a critical event that could be therapeutically targeted to achieve two desirable goals; both to maintain functional synapses and suppress proteasome defects that inevitably accrue with age.
Limitations of the study
Our study demonstrates that proteasome dysfunction can induce cleaved tau accumulation at the synapse, leading to neurotoxicity and synapse loss. Future studies are needed to investigate the exact cellular location in which tau is cleaved. Although cleaved tau is detected at the synapse, our experiments do not address if and how cleaved tau may traffic to and from the synapse. MG132 and other proteasome inhibitors have widespread effects on many cellular processes that may directly or indirectly alter caspase-3 or other caspases that may then, in turn, impact tau cleavage and neuronal activity. This limitation could be addressed using site-specific knock-in models to impede caspase-3 mediated tau cleavage at residue D421, followed by assessment of neuronal activity in response to proteasome impairment. Finally, as with many neuronal-based models, our experiments in primary cultures cannot adequately model late-stage neurons with mature tau pathology, as in human AD brain. There are many tau modifications present in AD brain (e.g., phosphorylation, acetylation, ubiquitination) that may affect the generation, regulation, and/or degradation of cleaved tau.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TauC3 (Cleaved Asp421, Asp422) | Invitrogen | Cat# AHB0061; RRID: AB_2536237 |
| Cleaved caspase-3 (Asp175) | Cell Signaling | Cat# 9661S; RRID: AB_2341188 |
| Total tau K9JA | DAKO | Cat# A0024; RRID: AB_10013724 |
| GAPDH (clone 6C5) | Millipore Sigma | Cat# MAB374; RRID: AB_2107445 |
| Anti-Tau (3-repeat isoform RD3) clone 8E6/C11 | Millipore | Cat# 05–803; RRID: AB_310013 |
| Anti-Tau (4-repeat isoform RD4) clone 1E1/A6 | Millipore | Cat# 05–804; RRID: AB_310014 |
| Anti-Tau-1 Antibody, clone PC1C6 | Millipore | Cat# MAB3420; RRID: AB_94855 |
| Total tau Tau12 | Millipore | Cat# MAB2241; RRID: AB_11211810 |
| Anti-p62 | Progen | Cat# GP62-C; RRID: AB_2687531 |
| LC3A/B | Cell Signaling | Cat# 4108S; RRID: AB_2137703 |
| Tau Monoclonal Antibody (Tau5) | Invitrogen | Cat# AHB0042; RRID: AB_2536235 |
| Phospho-Tau (Ser202, Thr205) Monoclonal Antibody (AT8) | Invitrogen | Cat# MN1020; RRID: AB_223647 |
| VGlut1 | Synaptic Systems | Cat# 135304; RRID: AB_887878 |
| VGAT | Synaptic Systems | Cat# 131008; RRID: AB_2800534 |
| Anti-PSD95 | Abcam | Cat# Ab18258; RRID: AB_444362 |
| Anti-PSD-95 Antibody (K28/43) | NeuroMab/Antibodies Inc | Cat# 75–028; RRID: AB_2292909 |
| Rabbit anti-GFAP | DAKO | Cat# Z0334; RRID: AB_10013382 |
| Homer1 | Synaptic Systems | Cat# 160002; RRID: AB_2120990 |
| Anti-GluA1/GluR1 (N355/1) | NeuroMab/Antibodies Inc | Cat# 75–327; RRID: AB_2315840 |
| Anti-Glutamate Receptor1 Ab, phosphoSer845 | Millipore | Cat# AB5849; RRID: AB_92079 |
| Goat anti-Mouse IgG (H+L) Secondary Antibody, HRP | Invitrogen | 32430 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, HRP | Invitrogen | 32460 |
| Goat anti-Mouse IgG (H+L)- Highly crossed absorbed (HA) Secondary Antibody, Alexa Fluor™ 488 | Invitrogen | A32723 |
| Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor™ 488 | Invitrogen | A11001 |
| Donkey anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor™ 488 | Life technologies | A21202 |
| Goat anti-Guinea pig IgG (H+L) Secondary Antibody, Alexa Fluor™ 568 | Invitrogen | A11075 |
| Donkey anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor™ 568 | Invitrogen | A10037 |
| Donkey anti-Rabbit IgG (H+L) Secondary Antibody, Alexa Fluor™ 594 | Invitrogen | A21207 |
| Goat anti-Rabbit IgG (H+L)-HA Secondary Antibody, Alexa Fluor™ 594 | Invitrogen | A32740 |
| Goat anti-Rabbit IgG (H+L)-HA Secondary Antibody, Alexa Fluor™ 647 | Invitrogen | A32733 |
| Donkey anti-Rabbit IgG (H+L) Secondary Antibody, Alexa Fluor™ 647 | Invitrogen | A31573 |
| Biological samples | ||
| Control 1-5 cortex (IB) | University of Pennsylvania (CNDR) Biorepository | Case 1-5 |
| Alzheimer’s disease 1-6 cortex (IB) | University of Pennsylvania (CNDR) Biorepository | Case 6-11 |
| Alzheimer’s disease 1 hippocampus (IHC) | UNC Biorepository (TPL) | Case 12 |
| Chemicals, peptides, and recombinant proteins | ||
| MG132 | Sigma | M7449 |
| Bortezomib | Millipore | 5.04314.0001 |
| Epoxomicin | Sigma | E3652 |
| Lactacystin | Enzo Life Sciences | BML-PI104-0200 |
| Staurosporine | Enzo Life Sciences | ALX-380-014-M001 |
| Chloroquine diphosphate (CQ) | Sigma | 6628 |
| 3-methyladenine (3-MA) | Sigma | M9281 |
| Rotenone | Sigma | R8875 |
| Bicuculline | Tocris | 0109 |
| Tetrodotoxin citrate | Tocris | 1069 |
| DAPI | ThermoFisher | D1306 |
| Thioflavin S | Sigma | T1892 |
| Critical commercial assays | ||
| CytoTox 96® Non-Radioactive Cytotoxicity Assay (LDH) | Promega | G1780 |
| CellTiter 96® Non-Radioactive Cell Proliferation Assay (MTT) | Promega | G4000 |
| Experimental models: Cell lines | ||
| HEK293A | ThermoFisher | R70507 |
| Experimental models: Organisms/strains | ||
| C57Bl/6 | Charles River | Strain Code: 027 |
| CD1 | Charles River | Strain Code: 022 |
| Tau knockout (KO) | Jackson Laboratory | Strain Code: 7251 |
| Caspase-3 knockout (KO) | Jackson Laboratory | Strain Code: 006233 |
| Oligonucleotides | ||
| Tau KO-F 5′-cattgctgccaagttcaccttag | This paper | N/A |
| Tau KO-R 5′-tgttttgccaagttctaattccatcaga | This paper | N/A |
| Tau KO reporter 1 5′-ctagaggatccccgggtacc | This paper | N/A |
| Caspase-3 common-R 5′-gcgagtgagaatgtgcataaattc | This paper | N/A |
| Caspase-3 WT-F 5′-gggaaaccaacagtagtcagtcct | This paper | N/A |
| Caspase-3 KO-F 5′-tgctaaagcgcatgctccagactg | This paper | N/A |
| Tau-F (33-mer) 5′-ccgaccggtgccaccatggctgagccccgccag | This paper | N/A |
| Tau-R (31-mer) 5′-cgcgtcgactcacaaaccctgcttggccagg | This paper | N/A |
| pUltra tau 1-421-R (33-mer) 5′-cgcgtcgactcagtctaccatgtcgatgctgcc | This paper | N/A |
| D421A-F (26-mer) 5′-gacatggtagcctcgccccagctcgc | This paper | N/A |
| D421A-R (21-mer) 5′-gatgctgccggtggaggagac | This paper | N/A |
| pTX181- control GFP Agel-BsrG1, filled in | This paper | N/A |
| Recombinant DNA | ||
| pTX713 pcDNA5/TO tau T34 (1N4R) | Trzeciakiewicz et al.98 | N/A |
| pTX714 pcDNA5/TO tau T37 (1N3R) | Trzeciakiewicz et al.98 | N/A |
| pTX715 pcDNA5/TO tau T39 (2N3R) | Trzeciakiewicz et al.98 | N/A |
| pTX716 pcDNA5/TO tau T43 (0N4R) | Trzeciakiewicz et al.98 | N/A |
| pTX717 pcDNA5/TO tau T44 (0N3R) | Trzeciakiewicz et al.98 | N/A |
| pTX096 pcDNA5/TO tau T40 (2N4R) | Trzeciakiewicz et al.98 | N/A |
| pTX407 pcDNA5/TO tau T40 ΔR1 | Trzeciakiewicz et al.98 | N/A |
| pTX408 pcDNA5/TO tau T40 ΔR2 | Trzeciakiewicz et al.98 | N/A |
| pTX409 pcDNA5/TO tau T40 ΔR3 | Trzeciakiewicz et al.98 | N/A |
| pTX410 pcDNA5/TO tau T40 ΔR4 | Trzeciakiewicz et al.98 | N/A |
| pTX411 pcDNA5/TO tau T40 ΔR1-4 | Trzeciakiewicz et al.98 | N/A |
| Software and algorithms | ||
| Prism Version 9 | GraphPad Software | https://www.graphpad.com/features |
| ImageJ | Schneider et al.99 | https://imagej.nih.gov/ij/ |
| Zen | Zeiss | https://www.zeiss.com/microscopy/en/products/software/zeiss-zen.html |
| Python (Anaconda 64-bit v3.8.8) code and standard libraries such as numPy, NetworkX, and SciPy. | Python | https://www.python.org |
| Other | ||
| Trypsin | Promega | 25200-056 |
| Dulbecco’s Modified Eagle Media (DMEM) | Gibco | 31-053-028 |
| Fetal bovine serum (FBS) | Sigma | F2442 |
| L-glutamine | Gibco | 25030-081 |
| FuGENE 6 Transfection Reagent | Promega | E2691 |
| Protein A/G PLUS-Agarose beads | Santa Cruz | sc-2003 |
| Penicillin/Streptomycin | Thermo Scientific | 15140-122 |
| BrainPhys Neuronal Medium | StemCell Technologies | 05790 |
| GlutaMAX Supplement | Gibco | 35050061 |
| B-27 Supplement | Gibco | 2450384 |
Resource availability
Lead contact
Further information and any associated requests should be made to the lead contact, Todd Cohen (toddcohen@neurology.unc.edu).
Materials availability
This study did not generate new unique reagents. Pertinent experimental materials are listed in the key resources table and individual methods sections.
Experimental model and study participant details
Human AD brain cortical sections from male and female donors were provided by the University of Pennsylvania, Center for Neurodegenerative Disease Research (CNDR) and the UNC Translational Pathology Laboratory (TPL) (Table S1). Primary cortical neurons were isolated from equal ratios of male and female embryos (C57Bl/6 mice, Charles River) in strict compliance with animal protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the University of North Carolina at Chapel Hill (protocol #21.257). The authors have not observed any influence of sex on the results of this study. Cultured HEK293A cells were confirmed to be negative for mycoplasma contamination.
Method details
Plasmids
All tau expression plasmids for transient transfection were cloned into the pcDNA5/TO vector (Life Technologies). Mutations within the tau protein that were used in this study were generated by site-directed mutagenesis. TauΔR1 is the deletion of 244–274 amino acid region, TauΔR2 is the deletion of 275–305, TauΔR3 is the deletion of 306–336, TauΔR4 is the deletion of 337–372, TauΔR1-4 is the deletion of the whole microtubule binding domain region (MTBR) 244–372. 3R (0N3R,1N3R,2N3R) and 4R (0N4R,1N4R,2N4R) tau plasmids were used to evaluate whether there was cleavage preference between isoforms. WT tau mutations within the MTBR (tauΔR1 (244–274), ΔR2 (275–305), ΔR3 (306–336), ΔR4 (337–372), ΔR1-4 (244–372)) were used to determine which regions of the MTBR impacted tau cleavage compared to the WT full-length tau (2N4R) control.
Cell culture and transfection
Two micrograms of desired DNA plasmid were transfected into HEK-293A cells with Fugene-6 (Promega) at 2.5 μL Fugene per 1 μg DNA plasmid ratio. Twenty-four hours post transfection 5 mM MG132 was added per well overnight. Forty-eight hours post transfection cells were harvested in 250 μL 1X RIPA buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% NP-40, 5 mM EDTA, 0.5% sodium deoxycholate, 0.1% SDS) with protease, phosphatase and deacetylase inhibitors (1 mg/mL pepstatin, leuptin, N-p-Tosyl-L-phenylalanine chloromethyl ketone, Nα-Tosyl-L-lysine chloromethyl ketone hydrochloride, trypsin inhibitor; Sigma, 1 mM NaF, 1 mM sodium orthovanadate, and 1 mM beta-glycerophosphate; Sigma, 2 μM TSA, 10 mM NCA). Samples were sonicated 20 times and then centrifuged at 21,130g for 30 min at 4°C. Supernatant (soluble fraction) was collected and analyzed by immunoblotting. Reaction was finished by adding loading dye solution, boiled for 10 min at 98°C and analyzed by immunoblotting.
Primary mouse neuron culture
Mouse neuron culture protocols were performed in accordance with the University of North Carolina (UNC) Institutional Animal Care and Use Committee (UNC IACUC protocol 21–257). Embryonic neurons from CD1 (Charles River, #022), C57BL/6 (Charles River, #027), tau knockout (KO) (Jackson Laboratory, #7251), and caspase-3 KO (Jackson Laboratory, #006233) mouse strains (C57BL/6 background) were used during this study. Mouse embryos from pregnant females (E15-16) were removed and dissected for primary cortical neurons. Embryos from tau and caspase-3 KO background strains were genotyped after removal. Brains were removed from the embryos where the cortex was isolated and dissected out. The cortex was then minced and digested in a papain solution (Worthington) containing DNase (Promega) for 30 min at 37°C, 5% CO2. After incubation, tissue was dissociated using a P1000 pipette. Single cell suspension was passed through a 40 μm cell strainer, counted, and plated onto poly-D-lysine coated 6, 12 or 24-well culture plates. The next day, plating media was replaced with neuronal media and neurons were differentiated to the indicated timepoints in the absence of mitosis inhibitors, and therefore astrocytes emerge at timepoints greater than DIV 12 (Figure S2A). For lentivirus infected neurons, purified lentivirus was used to infect 6, 12, and 24-well plates (control GFP, wild-type tau, P301L tau, tau 1–421, D421A, WT tau-GFP, tau 1-421-GFP) were added to designated wells at DIV 4. Twenty-four hours post infection, lentivirus was washed out of the well and replaced with half volume of condition media and half volume of fresh neuronal media. To observe early characteristics of cleaved tau, tau 1–421 lentivirus was added into new nontreated wells at DIV 7 with washout on DIV 8. Neurons were harvested on DIV 10, DIV 13, or DIV 23 by scraping 200 μL 1X RIPA buffer with inhibitors per well. Samples were sonicated 20 times and centrifuged at 21,130g for 30 min at 4°C. Supernatant was collected as the soluble fraction and the remaining insoluble pellet was extracted in SDS buffer (1% SDS, 150 mM NaCl, 50 mM Tris, pH7.6), sonicated 10 times and centrifuged at 21,130g for 30 min at room temperature. Lysates were then analyzed by immunoblotting.
Human brain homogenate preparation
Human control and AD brain tissues from Braak V-VI cases were provided by the University of Pennsylvania (Center for Neurodegenerative Disease Research brain bank). Isolated gray matter from frontal cortex was homogenized in 3 vol/g of cold high-salt RAB buffer (0.75 M NaCl, 100 mM Tris, 1 mM EGTA, 0.5 mM MgSO4, 0.02 M NaF, 2 mM DTT, pH 7.4) supplemented with deacetylase, phosphatase, and protease inhibitors as described above. Homogenates were incubated at 4°C for 20 min to depolymerize MTs, then, centrifuged at 100,000g for 30 min at 4°C. The resulting supernatant is labeled as the high-salt fraction. Pellets were re-homogenized and centrifuged in 3 vol/g of cold high-salt RAB buffer. Resultant pellets were homogenized in 5 vol/g of cold RIPA buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% NP-40, 5 mM EDTA, 0.5% sodium deoxycholate, 0.1% SDS) and centrifuged at 100,000g for 30 min at 4°C. Myelin depletion was performed by re-extraction in RIPA buffer supplemented with 20% sucrose to remove the excess myelin in human brain. Finally, resultant insoluble pellets were extracted in 1 vol/g urea extraction buffer (7 M urea, 2 M Thiourea, 4% CHAPS, 30 mM Tris, pH 8.5). High-salt and urea fractions were analyzed by immunoblotting using the indicated antibodies to detect cleaved and total tau proteins.
Drug treatments
All drug treatments, unless otherwise stated in the results, were given between DIV 10-DIV 14 of neuronal cell culture. Proteasome inhibitors targeting the 26S, MG132 (Sigma, #M7449), was treated at a final concentration of 1 μM overnight for around 15 h and Bortezomib (Millipore, #5.04314.0001) was treated at a final concentration at 10 nM overnight. Other proteasome inhibitors targeting the 20S subunit, Epoxomicin (Sigma, #E3652) was treated at 20 nM overnight for 18 h and Lactacystin (Enzo Life Sciences, #BML-PI104-0200) was treated at a final concentration at 2 μM overnight. Staurosporine (Enzo Life Sciences, #ALX-380-014-M001), a non-selective protein kinase inhibitor that induces apoptosis, was treated at 1 μM for 5 h. Lysosomal degradation inhibitor Chloroquine diphosphate (CQ, Sigma, #6628) was treated at 10 μM for 24 h. Autophagy inhibitor 3-methyladenine (3-MA, Sigma, #M9281) was treated at 10 mM overnight for 16 h. Mitochondrial reductase inhibitor, Rotenone (Sigma-Aldrich, #R8875), was treated at final concentrations of either 0.5 nM, 1 nM, 5 nM, 10 nM and 50 nM for 15 h and 36 h. To investigate if neuronal activity effects cleaved tau production, neurons were pre-treated with either GABAA receptor Bicuculline (Tocris, #0109) at a final concentration of 20 μM or sodium channel blocker Tetrodotoxin citrate (TTX, Tocris, #1069) at a final concentration of 1 μM for 8 h prior to MG132 overnight treatment. In HEK-293A cells, MG132 was treated at a final concentration of 5 mM overnight for around 15 h.
Lentivirus generation
To generate lentivirus expression plasmids for full-length WT tau, P301L tau, tau 1–421, and cleaved tau mutant D421A, the mammalian expression vectors were cloned into pcDNA3.1 and amplified by PCR. Fragments were inserted into the pUltra vector using AgeI and SalI restriction endonucleases to replace the GFP cassette. Tau 1-421-GFP, PSD-95-GFP and pUltra-GFP (control) lentivirus were amplified with a GFP-tag at the 5′ end. Empty vector (control) lentivirus (pUltraXGFP) was generated by removing the GFP cassette by digestion with AgeI and BsrGI, and ligation was performed after Klenow blunting. Lentiviral production was performed by cotransfecting 37.5 μg lenti-plasmid with 25 μg psPAX2, 12.5 μg VSVG, and 6.25 μg REV for each 15 cm dish of lenti-X 293T cells (CalPhos Transfection Kit, Takara). Three 15 cm dishes of cells were used for each lentiviral production. Three days post transfection, culture media was collected and centrifuged at 2000g for 10 min. Lentiviral particles were purified using a double-sucrose method. Briefly, the supernatants were loaded onto a 70%-60%-30%–20% sucrose gradient and centrifuged at 70,000g for 2 h at 17°C (using a Beckman Optima LE-80K ultracentrifuge). The 30%–60% fraction containing the lentiviral particles was retrieved, resuspended in 1X PBS, filtered with a 0.45 μm filter flask before loaded onto a 20% sucrose cushion, and centrifuged a second time at 70,000g for 2 h at 17°C. The supernatants were carefully discarded, and the viral particles present in the pellet were resuspended in 1X PBS, aliquoted and stored at −80C. For experiments, purified lentivirus was used to infect 6, 12, and 24-well plates. For infections with smaller volumes, lentivirus was diluted 1:10 and the appropriate amount of lenti was added per well.
Coimmunoprecipitation assay
For coimmunoprecipitation assays, HEK-293A or primary cortical mouse neurons were washed with 1X PBS and lysed in 1X NETN buffer (50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1 mM sodium orthovanadate, 1 mM sodium fluoride, 1 mM PMSF, 0.2 mM leupeptin, protease inhibitor cocktail, 2 μM TSA, 10 mM NCA). Lysate samples were put on wheel at 4°C for 45 min then centrifuged for 15 min at 21,130 g at 4°C. Supernatant was collected and then incubated overnight on the wheel at 4°C with Protein A/G PLUS-Agarose beads (Santa Cruz, #sc2003) and TauC3 (Invitrogren, #AHB0061). Samples were analyzed by immunoblotting. Analyses and quantification of co-IP assays were as described in the immunoblotting and statistical analysis sections.
Immunoblotting
Primary neurons or HEK-293A cells were first washed with 1X PBS and then harvested in 1X RIPA buffer with inhibitors. Samples are sonicated 20 times and centrifuged at 21,130g for 30 min at 4°C. Supernatant was collected as the soluble fraction and the remaining insoluble pellet was extracted in SDS buffer (1% SDS, 150 mM NaCl, 50 mM Tris, pH7.6), sonicated 10 times and centrifuged at room temperature at 21,130g for 30 min. Lysates were prepped in running buffer and boiled for 10 min at 98°C prior to running on gel. Lysates were run using gel electrophoresis SDS-PAGE, transferred onto a nitrocellulose membrane (Biorad), and blocked in 2% milk in 1X TBS for at least 30 min. Membranes were incubated overnight at 4°C in designated primary antibody. The following day, primary antibody was washed with 1X TBST and the incubated in with stabilized peroxidase conjugated Goat Anti-Rabbit or Mouse (H + L) secondary antibody (1:1000 dilution, Invitrogen #32460 and #32430) for 1 h. The following primary antibodies were used in this study: TauC3 (1:500, Invitrogen #AHB0061), Cleaved caspase-3 (1:1000, Cell signaling #9661S), Total tau K9JA (1:5000, DAKO #A0024), Anti-Tau RD3 (1:500, Millipore #05-803), Anti-Tau RD4 (1:500, Millipore #05-804), PSD-95 (1:1000, NeuroMab #75-028), FLAG (1:1000, Sigma #F3165), Total tau Tau12 (1:1000, Millipore #MAB2241), p62 (1:1000, Progen #GP62-C), LC3A/B (1:500, Cell Signaling #4108S), Anti- Tau-1, clone PC1C6 (1:1000, Millipore #MAB3420), Total tau Tau5 (1:1000, Invitrogen #AHB0042), AT8 (1:1000, Invitrogen #MN1020), GFAP (1:2000, Dako #Z0334), Homer1 (1:25,000, Synaptic Systems #160002), Anti-GluA1/GluR1 (N355/1) (1:1000, NeuroMab/Antibodies Inc #75–327), and Anti-Glutamate Receptor 1 Ab, phosphoSer845 (1:1000, Millipore #AB5849). All primary and secondary antibodies used are described in Tables S2 and S3. Membranes were developed with ECL Western Blotting Substrate (ThermoFisher) or ECL Select Western Blotting Detection Reagent (Cytiva, Amersham) and imaged using Image Quant LAS 4000 (Cytiva). Image Quant TL (Cytiva) was used to quantify protein band intensities.
Immunofluorescence microscopy
Primary cortical mouse neurons were plated onto poly-D-lysine coated coverslips. Neurons were either infected with lentivirus and/or treated with MG132 overnight. Coverslips were fixed in 4% paraformaldehyde (PFA) for 10 min and washed in 1X PBS. After fixation, coverslips were permeabilized with 0.1% Triton X-100 in 1X PBS for 8 min at room temperature followed by blocking with 2% milk in 1X TBS for 1 h. Coverslips are double or triple labeled with primary antibodies diluted in 2% milk overnight at 4°C. The following primary antibodies were used in this study: TauC3 (1:500, Invitrogen #AHB0061), cleaved caspase-3 (1:400, Cell signaling #9661S), Total tau K9JA (1:2000, DAKO #A0024), VGlut1 (1:500, Synaptic Systems #135304), VGAT (1:500, Synaptic Systems #131008), PSD-95 (1:1000, Abcam #Ab18258), anti-PSD-95 (K28/43) (NeuroMab/Antibodies Inc. #75-028), Tau5 (1:1000, Invitrogen #AHB0042), and GFAP (1:1000, Dako #Z0334). The next day, coverslips are incubated with Alexa Fluor 488-, 568-,594- or 647-conjugated secondary antibody (Invitrogen or Life Technologies, 1:500 dilutions) staining followed by DAPI. All primary and secondary antibodies used are described in Tables S2 and S3. Coverslips are mounted using Vectashield mounting media (Vector Laboratories) and left to dry overnight before analysis. Images were taken on the confocal Zeiss LSM 780 at either 20X or 63X with oil immersion (for synapse imaging). To quantify GFP-tagged tau puncta that colocalize with the synapse, the percentage of WT tau-GFP or tau 1-421-GFP-positive synapses were counted from 7 independently acquired and blinded frames and plotted as a ratio of the total number of PSD-95-positive puncta within each field. Tau-GFP puncta that colocalized with PSD-95 puncta were considered synaptic, while minimal or no overlay was considered negative.
Thioflavin (ThS) and double labeling of human tissue
Human hippocampal sections on coverslips were acquired from the UNC Translational Pathology Lab (TPL), which maintains a catalogued and archived AD brain repository with neuropathologically confirmed AD cases from UNC donors. Hippocampal brain sections were formalin fixed, paraffin embedded, and sectioned at 5 μm. Tissue on coverslips were deparaffinized by Xylene treatment for a total of 10 min, washed with 100%, 95%, 80% and 70% ethanol for 1 min each and then rehydrated through a series of water and 1X PBS washes. Coverslips are then treated with 0.05% KMnO4/PBS for 20 min, washed in 1X PBS, then distained in 0.2% K2S2O5/0.2% oxalic acid/PBS. Tissues are immersed in freshly made 0.0125% thioflavin-S/40%EtOH/60%PBS for 3 min then differentiated in 50%EtOH/50%PBS for 15 min. For double IF staining, coverslips are washed in 0.1M tris Buffer (pH 7.6), blocked in 0.1M tris Buffer/2% donor bovine serum for 5 min, and incubated overnight with primary antibodies TauC3 (1:500, Invitrogen, #AHB0061) and Cleaved caspase-3 (1:400, Cell signaling, #9661s) in a humidified chamber at 4°C. The following day, primary antibodies are flushed with Tris buffer, tissues are blocked in 0.1M tris Buffer/2% donor bovine serum and Alexa Fluor 568-conjugated and 647-conjugated secondary antibodies (Invitrogen, 1:500 dilutions) are applied for 3 h at room temperature in a humidified chamber. Secondary antibodies are flushed with Tris buffer and then mounted with a coverslip using Vectashield mounting media.
Post-synaptic density (PSD) preparations
Primary cortical mouse neurons were plated onto a poly-D-lysine coated 6-well culture plates and differentiated until DIV 14. Neurons were either infected with lentivirus at DIV 4 and/or treated with MG132 overnight at DIV 13. Prior to harvest, wells were washed in 1X PBS and then six wells of one plate are scraped together in 900 μL of homogenization buffer (320 mM sucrose, 5 mM sodium pyrophosphate, 1 mM EDTA, 10 mM HEPES pH 7.4) with inhibitors (200 nM okadaic acid, 1 mM orthovanadate, 1X protease inhibitor cocktail (Roche)). Lysates were homogenized by passaging through a 26g needle 12 times. Approximately 70 μL of lysate post homogenization was collected as whole cell lysate sample. Homogenate was then centrifuged at 1,000g for 10 min at 4°C to yield P1 (nuclear fraction) and S1 fraction. S1 was further centrifuged at 15,000g for 20 min at 4°C to yield membrane/crude synaptosomes (P2) and cytosol (S2). P2 fraction was resuspended in 500 μL milliQ water (including 200 nM okadaic acid 1 mM orthovanadate, 1X protease inhibitor cocktail) and then suspension was adjusted to 4 mM HEPES pH 7.4 by adding 20 μL of 100 mM HEPES. Samples were put on wheel for agitation and incubated for 30 min at 4°C followed by centrifugation at 25,000g for 20 min at 4°C to yield LP1 and LS2. LP1 was resuspended in 250 μL 50 mM HEPES pH7.4 with inhibitors and then mixed with 250 μL 1% Triton X-100 plus inhibitors, incubated with agitation for 10 min at 4°C and centrifuged at 32,000g for 20 min at 4°C. The final PSD pellet was resuspended in 80–100 μL 50 mM HEPES pH7.4 followed by BCA protein quantification and immunoblotting for analysis. 2 μg of protein was used per sample for immunoblotting.
Cell viability assays (LDH&MTT)
Primary cortical mouse neurons were plated onto a poly-D-lysine coated 24-well culture plates. LDH assay was preformed per the manufacturer’s protocol (Promega, #G1780). All wells were made sure to have 400 μL of media per well. For positive control, 40 μL of 10X lysis solution was added directly into a well with 360 μL of neuronal media. Plates were put in a 37°C, 5% CO2 incubator for 45 min then 50 μL of media was transferred to a new 96-well flat bottom plate. Fifty microliters of reconstituted substrate mix were added to each well, put in a drawer to protect from light and incubated at room temperature for 30 min. Fifty microliters of stop solution was added after 30 min with a final total volume of 150 μL. Immediately after, any large bubbles present in a well were popped using a pipette tip and absorbance was read at 490 or 492 nm using the FLUOstar Omega plate reader. MTT assay was preformed per the manufacturer’s protocol (Promega, #G4000). Remaining 350 μL of media from 24 well plate used in LDH as described above was used for the assay. Fifty-two and a half microliters of Dye Solution were added to each well and incubated at 37°C, 5% CO2 for 4 h. After incubation, 350 μL of Solubilization Solution/Stop Mix was added to each well and put in drawer to protect from light for 1 h. After 1 h, contents of each well were mixed by pipetting up and down 10 times and then 200 μL of each sample were transferred to a new 96-well flat bottom plate. Absorbance wavelengths 570 nm and 650 nm are read using the FLUOstar Omega plate reader.
Multielectrode array (MEA) analysis (16-electrode)
BioCircuit MEA 24 (Axion Biosystems, #M384-BIO-24) plate was coated overnight with poly-D-lysine, washed the next day with sterile DI water and air dried until plating. Primary WT or tau KO cortical mouse neurons were plated over the recording electrode area of each well at a desired concentration of 16 million cells/mL (160,000 neurons in 10 μL of media) and incubated at 37°C, 5% CO2 for 1 h. After the first incubation, 250 μL of media was gently added to each well, put back in the incubator for 1 h, and the remaining 250 μL of media was added to each well for a final volume of 500 μL per well. The following day, media was aspirated and replaced with neuronal media. Neurons were half fed every other day for the remaining of the experiment. At DIV 18, neurons were recorded for 10 min on the Maestro Edge multiwell MEA and Impedance system (Axion Biosystems) for baseline recordings. Prior to recording, MEA plate sat in the system for neurons to acclimate to CO2. After baseline recording, wells were infected with either pUltraXGFP (control), WT tau, or tau 1–421. Twenty-four hours post infection/treatment neural activity was recorded for 10 min and then treatments were washed out and replaced with half new media plus half conditioned media. Neurons continued to be recorded every day for 10 min until DIV 29. For MG132 treated neuron recordings, primary WT and tau KO cortical mouse neurons at DIV 18 were treated with MG132 (1 μM), left in the BioCircuit MEA 24 overnight and recorded for 10 min every hour over 20 h.
Multielectrode array (MEA) analysis (high density)
The methodology for electrophysiological recordings followed the procedures outlined by Pan et al.100 MEA’s from Multichannel Systems (MCS, Reutlingen, Germany) consisted of 60 TiN3 electrodes with diameters of 30 μm and spacing of 200 μm, one of which served as a ground. The electrode area was coated with 0.01% polyethylene imine by adding a 20 μL droplet and incubating for a period of 1 h at 37°C or overnight with 0.1 mg/mL poly-D-lysine in HEPES buffered (pH 8.4) Hanks balanced salt solution. The MEA was then washed 5 times with sterile deionized water and dried in a sterile hood. Neurons from E16 fetal mouse cortex-hippocampus in Neurobasal Plus medium with B27 Plus supplement and 5% FBS (1.5 × 106/mL) were placed on the grid in a volume of 20 μL to achieve a density of ∼150,000 cells/cm2. After attachment, the cells were fed with Neurobasal Plus/B27 Plus medium. The MEA well was covered with a Teflon O-ring containing a permeable Teflon film to prevent evaporation while allowing gas exchange. The Teflon O-ring was designed to fit over the MEA culture well. Cultures were fed by 50% medium exchange three times/week. Recordings were made from 18 to 28 days in culture. The spontaneous activity on the MEA’s was measured using an MCS 1100x amplifier at 25 kHz sampling with a hardware filter of 300–3000Hzat 37°C under continuous flow of sterile 5% CO2/air. The O-ring with a teflon membrane was kept in place during recording to minimize evaporation and contamination, as outlined in Potter and De-Marse.101 MCRack software was used to record up to 4-week old cultures for 40 min of spontaneous activity approximately every weekday beginning on day 18. The action potential spikes were sorted and recorded only if they surpassed the negative threshold of 5 times the standard deviation estimated from the background noise. Our analysis of electrophysiological data was conducted using custom python (Anaconda 64-bit v3.8.8) code and standard libraries such as numPy, NetworkX, and SciPy.
Average network spike and log interspike interval histogram
Average NS were constructed by smoothing each burst with a Gaussian and placing the spikes in a histogram with 1 ms bins. ISI histograms were constructed using 1 μs logbins.
Cross correlation coefficient
For the sake of simplicity, a straightforward Hebbian learning process (wire together fire together) cross correlation coefficient was used. The cross correlation can be formalized by the following equation:
| (Equation 1) |
Let it be a spike time on reference spike train (Si) and jt ± l be a spike time on target spike train (Sj) ± a lag (L) relative to it. If it and jt ± l fall within a certain interval (∫) then they form a synchronized spike pair (Sij(t)). The cross correlation (Cij(t)) is then the sum of Sij(t) normalized over N, with N being the number of spikes in the longest spike train between Si and Sj:
| (Equation 2) |
2 ms bins were used for Cij(t). As the average refractory period of a bursting neuron is approximately 2 msCij(t) can be interpreted as a synaptic weight as a larger bin size would begin to incorporate correlations of firing rates. N was defined as the longest spike train between Si and Sj as when two spike trains are paired faulty coefficients might be obtained when a shorter spike train is paired with a longer spike train.
The Cij(t) were used as edge weights for the graphs (G) and the graphs were thresholded with a value of 0.5. This threshold was used to extrapolate the efficient communication that remains in infected networks while minimizing any weights that might not represent efficient communication.
Network analysis
All basic graph analysis such as node degree, path length, and cluster coefficient were performed with the standard python library NetworkX. The mathematical formulas for average degree, path length and cluster coefficient are briefly:
| (Equation 3) |
Where the average degree of the nth node (<Kn>) of graph G is the sum of the number of edges(E) of the nth node of the graph (n∈G) averaged over N-1 nodes. Since not all electrodes were always active on all arrays during recordings average degrees were normalized out of N-1 electrodes. Since we used 60 electrode MEAs N-1 was 59.
Average shortest path length
| (Equation 4) |
Where the average shortest path length(a) is the sum of the shortest path between two nodes in G (d(s,t)) averaged over the number of nodes (n).
Cluster coefficient
| (Equation 5) |
Where the cluster coefficient (C) is the total number of clusters of vertices (cv) averaged over the number of nodes (n).
Quantification and statistical analysis
GraphPad Prism 9 software was used for all statistical analyses. Results were pooled from a minimum of three independent experiments and presented as average ±SEM. Comparisons between two groups were analyzed using unpaired Student’s t test (where told groups = 2), one-way ANOVA or two-way ANOVA with Tukey’s post hoc test (where total groups n ≥ 3). Significance is presented as ns, ∗p<0.05, ∗∗p< 0.01, ∗∗∗p< 0.001 or ∗∗∗∗p<0.0001.
Acknowledgments
This work was supported by the United States National Institutes of Health (NIH) grants RF1AG068063 (T.J.C and G.H.D), R56AG068063 (T.J.C. and G.H.D.), R01AG061188 (T.J.C), RO1NS108808 (R.B.M), and F32AG072826 (M.R.B). Microscopy was performed at the UNC Neuroscience Microscopy Core (RRID:SCR_019060), supported, in part, by funding from the NIH-NINDS Neuroscience Center Support Grant P30NS045892 and the NIH-NICHD Intellectual and Developmental Disabilities Research Center Support Grant P50HD103573. Michelle S. Itano is also supported by an Imaging Scientist grant from the Chan Zuckerberg Initiative. The content is the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We would like to thank Dr. Mohanish Deshmukh (UNC), Dr. Jonathan Schisler (UNC) and Dr. Shenee Martin (Diering laboratory) for supportive discussions, technical support, and troubleshooting advice.
Author contributions
Conceptualization, C.K.O. and T.J.C.; Investigation, C.K.O., M.R.B., B.H., J.M.M., M.S.I., G.H.D., and R.B.M.; Plasmid construction, X.T.; Mouse colony management and breeding, Y.C.; Writing–original draft, C.K.O, and T.J.C. All authors reviewed, edited, and approved the final version of this manuscript.
Declaration of interests
The authors declare no competing interest with the contents of this article.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. We support inclusive, diverse, and equitable conduct of research.
Published: May 19, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106905.
Supplemental information
Data and code availability
-
•
All data and code supporting the findings of this study are available within the method details of the paper and its supplementary information files and can be obtained from the authors upon request.
-
•
Code availability and all coding requests should be directed to Dr. Rick Meeker (meekerr@neurology.unc.edu).
-
•
Any additional information required to reanalyze the data reported in this paper is available from Dr. Todd Cohen upon request (toddcohen@neurology.unc.edu).
References
- 1.Knopman D.S., Amieva H., Petersen R.C., Chételat G., Holtzman D.M., Hyman B.T., Nixon R.A., Jones D.T. Alzheimer disease. Nat. Rev. Dis. Prim. 2021;7:33. doi: 10.1038/s41572-021-00269-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weintraub S., Wicklund A.H., Salmon D.P. The neuropsychological profile of alzheimer disease. Cold Spring Harb. Perspect. Med. 2012;2:a006171. doi: 10.1101/cshperspect.a006171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roberson E.D., Scearce-Levie K., Palop J.J., Yan F., Cheng I.H., Wu T., Gerstein H., Yu G.-Q., Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 4.Barbier P., Zejneli O., Martinho M., Lasorsa A., Belle V., Smet-Nocca C., Tsvetkov P.O., Devred F., Landrieu I. Role of tau as a microtubule-associated protein: structural and functional aspects. Front. Aging Neurosci. 2019;11:204. doi: 10.3389/fnagi.2019.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo T., Noble W., Hanger D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017;133:665–704. doi: 10.1007/s00401-017-1707-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kopeikina K.J., Hyman B.T., Spires-Jones T.L. Soluble forms of tau are toxic in Alzheimer’s disease. Transl. Neurosci. 2012;3:223–233. doi: 10.2478/s13380-012-0032-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crowther R.A., Olesen O.F., Jakes R., Goedert M. The microtubule binding repeats of tau protein assemble into filaments like those found in Alzheimer’s disease. FEBS Lett. 1992;309:199–202. doi: 10.1016/0014-5793(92)81094-3. [DOI] [PubMed] [Google Scholar]
- 8.Moloney C.M., Lowe V.J., Murray M.E. Visualization of neurofibrillary tangle maturity in Alzheimer’s disease: a clinicopathologic perspective for biomarker research. Alzheimers Dement. 2021;17:1554–1574. doi: 10.1002/alz.12321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu M., Zhang M., Yin X., Chen K., Hu Z., Zhou Q., Cao X., Chen Z., Liu D. The role of pathological tau in synaptic dysfunction in Alzheimer’s diseases. Transl. Neurodegener. 2021;10:45. doi: 10.1186/s40035-021-00270-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoover B.R., Reed M.N., Su J., Penrod R.D., Kotilinek L.A., Grant M.K., Pitstick R., Carlson G.A., Lanier L.M., Yuan L.-L., et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jadhav S., Cubinkova V., Zimova I., Brezovakova V., Madari A., Cigankova V., Zilka N. Tau-mediated synaptic damage in Alzheimer’s disease. Transl. Neurosci. 2015;6:214–226. doi: 10.1515/tnsci-2015-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tracy T.E., Gan L. Tau-mediated synaptic and neuronal dysfunction in neurodegenerative disease. Curr. Opin. Neurobiol. 2018;51:134–138. doi: 10.1016/j.conb.2018.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teravskis P.J., Ashe K.H., Liao D. The accumulation of tau in postsynaptic structures: a common feature in multiple neurodegenerative diseases? Neuroscientist. 2020;26:503–520. doi: 10.1177/1073858420916696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang G., Pan F., Gan W.-B. Stably maintained dendritic spines are associated with lifelong memories. Nature. 2009;462:920–924. doi: 10.1038/nature08577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scannevin R.H., Huganir R.L. Postsynaptic organization and regulation of excitatory synapses. Nat. Rev. Neurosci. 2000;1:133–141. doi: 10.1038/35039075. [DOI] [PubMed] [Google Scholar]
- 16.van der Zee E.A. Synapses, spines and kinases in mammalian learning and memory, and the impact of aging. Neurosci. Biobehav. Rev. 2015;50:77–85. doi: 10.1016/j.neubiorev.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 17.Shao C.Y., Mirra S.S., Sait H.B.R., Sacktor T.C., Sigurdsson E.M. Postsynaptic degeneration as revealed by PSD-95 reduction occurs after advanced Aβ and tau pathology in transgenic mouse models of Alzheimer’s disease. Acta Neuropathol. 2011;122:285–292. doi: 10.1007/s00401-011-0843-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kashyap G., Bapat D., Das D., Gowaikar R., Amritkar R.E., Rangarajan G., Ravindranath V., Ambika G. Synapse loss and progress of Alzheimer’s disease -A network model. Sci. Rep. 2019;9:6555. doi: 10.1038/s41598-019-43076-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Y., Fu A.K.Y., Ip N.Y. Synaptic dysfunction in Alzheimer’s disease: mechanisms and therapeutic strategies. Pharmacol. Ther. 2019;195:186–198. doi: 10.1016/j.pharmthera.2018.11.006. [DOI] [PubMed] [Google Scholar]
- 20.Johnson G.V.W., Stoothoff W.H. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 2004;117:5721–5729. doi: 10.1242/jcs.01558. [DOI] [PubMed] [Google Scholar]
- 21.Liu M., Sui D., Dexheimer T., Hovde S., Deng X., Wang K.-W., Lin H.L., Chien H.-T., Kweon H.K., Kuo N.S., et al. Hyperphosphorylation renders tau prone to aggregate and to cause cell death. Mol. Neurobiol. 2020;57:4704–4719. doi: 10.1007/s12035-020-02034-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wegmann S., Biernat J., Mandelkow E. A current view on Tau protein phosphorylation in Alzheimer’s disease. Curr. Opin. Neurobiol. 2021;69:131–138. doi: 10.1016/j.conb.2021.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Iqbal K., Liu F., Gong C.-X., Grundke-Iqbal I. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010;7:656–664. doi: 10.2174/156720510793611592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alquezar C., Arya S., Kao A.W. Tau post-translational modifications: dynamic transformers of tau function, degradation, and aggregation. Front. Neurol. 2020;11 doi: 10.3389/fneur.2020.595532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee G., Neve R.L., Kosik K.S. The microtubule binding domain of tau protein. Neuron. 1989;2:1615–1624. doi: 10.1016/0896-6273(89)90050-0. [DOI] [PubMed] [Google Scholar]
- 26.Cohen T.J., Guo J.L., Hurtado D.E., Kwong L.K., Mills I.P., Trojanowski J.Q., Lee V.M.Y. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011;2:252. doi: 10.1038/ncomms1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klein H.-U., McCabe C., Gjoneska E., Sullivan S.E., Kaskow B.J., Tang A., Smith R.V., Xu J., Pfenning A.R., Bernstein B.E., et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat. Neurosci. 2019;22:37–46. doi: 10.1038/s41593-018-0291-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caballero B., Bourdenx M., Luengo E., Diaz A., Sohn P.D., Chen X., Wang C., Juste Y.R., Wegmann S., Patel B., et al. Acetylated tau inhibits chaperone-mediated autophagy and promotes tau pathology propagation in mice. Nat. Commun. 2021;12:2238. doi: 10.1038/s41467-021-22501-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim M.-S., Mun Y.-S., Lee S.-E., Cho W.-Y., Han S.-H., Kim D.-H., Yoon S.-Y. Tau acetylation at K280 regulates tau phosphorylation. Int. J. Neurosci. 2022;1:1–5. doi: 10.1080/00207454.2022.2081165. [DOI] [PubMed] [Google Scholar]
- 30.Wang X., Liu E.-J., Liu Q., Li S.-H., Li T., Zhou Q.-Z., Liu Y.-C., Zhang H., Wang J.-Z. Tau acetylation in entorhinal cortex induces its chronic hippocampal propagation and cognitive deficits in mice. J. Alzheimers Dis. 2020;77:241–255. doi: 10.3233/JAD-200529. [DOI] [PubMed] [Google Scholar]
- 31.Cripps D., Thomas S.N., Jeng Y., Yang F., Davies P., Yang A.J. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J. Biol. Chem. 2006;281:10825–10838. doi: 10.1074/jbc.M512786200. [DOI] [PubMed] [Google Scholar]
- 32.Morishima-Kawashima M., Hasegawa M., Takio K., Suzuki M., Titani K., Ihara Y. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron. 1993;10:1151–1160. doi: 10.1016/0896-6273(93)90063-w. [DOI] [PubMed] [Google Scholar]
- 33.Wesseling H., Mair W., Kumar M., Schlaffner C.N., Tang S., Beerepoot P., Fatou B., Guise A.J., Cheng L., Takeda S., et al. Tau PTM profiles identify patient heterogeneity and stages of Alzheimer’s disease. Cell. 2020;183:1699–1713.e13. doi: 10.1016/j.cell.2020.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balmik A.A., Chinnathambi S. Methylation as a key regulator of Tau aggregation and neuronal health in Alzheimer’s disease. Cell Commun. Signal. 2021;19:51. doi: 10.1186/s12964-021-00732-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei X., Du P., Zhao Z. Impacts of DNA methylation on Tau protein related genes in the brains of patients with Alzheimer’s disease. Neurosci. Lett. 2021;763 doi: 10.1016/j.neulet.2021.136196. [DOI] [PubMed] [Google Scholar]
- 36.Huseby C.J., Hoffman C.N., Cooper G.L., Cocuron J.-C., Alonso A.P., Thomas S.N., Yang A.J., Kuret J. Quantification of tau protein lysine methylation in aging and Alzheimer’s disease. J. Alzheimers Dis. 2019;71:979–991. doi: 10.3233/JAD-190604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martins W.C., Tasca C.I., Cimarosti H. Battling Alzheimer’s disease: targeting SUMOylation-mediated pathways. Neurochem. Res. 2016;41:568–578. doi: 10.1007/s11064-015-1681-3. [DOI] [PubMed] [Google Scholar]
- 38.Qin M., Li H., Bao J., Xia Y., Ke D., Wang Q., Liu R., Wang J.-Z., Zhang B., Shu X., Wang X. SET SUMOylation promotes its cytoplasmic retention and induces tau pathology and cognitive impairments. Acta Neuropathol. Commun. 2019;7:21. doi: 10.1186/s40478-019-0663-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rissman R.A., Poon W.W., Blurton-Jones M., Oddo S., Torp R., Vitek M.P., LaFerla F.M., Rohn T.T., Cotman C.W. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J. Clin. Invest. 2004;114:121–130. doi: 10.1172/JCI200420640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Conze C., Rierola M., Trushina N.I., Peters M., Janning D., Holzer M., Heinisch J.J., Arendt T., Bakota L., Brandt R. Caspase-cleaved tau is senescence-associated and induces a toxic gain of function by putting a brake on axonal transport. Mol. Psychiatry. 2022;27:3010–3023. doi: 10.1038/s41380-022-01538-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guillozet-Bongaarts A.L., Garcia-Sierra F., Reynolds M.R., Horowitz P.M., Fu Y., Wang T., Cahill M.E., Bigio E.H., Berry R.W., Binder L.I. Tau truncation during neurofibrillary tangle evolution in Alzheimer’s disease. Neurobiol. Aging. 2005;26:1015–1022. doi: 10.1016/j.neurobiolaging.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 42.Gamblin T.C., Chen F., Zambrano A., Abraha A., Lagalwar S., Guillozet A.L., Lu M., Fu Y., Garcia-Sierra F., LaPointe N., et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA. 2003;100:10032–10037. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fasulo L., Ugolini G., Visintin M., Bradbury A., Brancolini C., Verzillo V., Novak M., Cattaneo A. The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J. Neurochem. 2000;75:624–633. doi: 10.1046/j.1471-4159.2000.0750624.x. [DOI] [PubMed] [Google Scholar]
- 44.Day R.J., Mason M.J., Thomas C., Poon W.W., Rohn T.T. Caspase-cleaved tau Co-localizes with early tangle markers in the human vascular dementia brain. PLoS One. 2015;10 doi: 10.1371/journal.pone.0132637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao X., Kotilinek L.A., Smith B., Hlynialuk C., Zahs K., Ramsden M., Cleary J., Ashe K.H. Caspase-2 cleavage of tau reversibly impairs memory. Nat. Med. 2016;22:1268–1276. doi: 10.1038/nm.4199. [DOI] [PubMed] [Google Scholar]
- 46.Liu P., Smith B.R., Huang E.S., Mahesh A., Vonsattel J.P.G., Petersen A.J., Gomez-Pastor R., Ashe K.H. A soluble truncated tau species related to cognitive dysfunction and caspase-2 is elevated in the brain of Huntington’s disease patients. Acta Neuropathol. Commun. 2019;7:111. doi: 10.1186/s40478-019-0764-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu P., Smith B.R., Montonye M.L., Kemper L.J., Leinonen-Wright K., Nelson K.M., Higgins L., Guerrero C.R., Markowski T.W., Zhao X., et al. A soluble truncated tau species related to cognitive dysfunction is elevated in the brain of cognitively impaired human individuals. Sci. Rep. 2020;10:3869. doi: 10.1038/s41598-020-60777-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Angel A., Volkman R., Royal T.G., Offen D. Caspase-6 knockout in the 5xFAD model of Alzheimer’s disease reveals favorable outcome on memory and neurological hallmarks. Int. J. Mol. Sci. 2020;21:E1144. doi: 10.3390/ijms21031144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Horowitz P.M., Patterson K.R., Guillozet-Bongaarts A.L., Reynolds M.R., Carroll C.A., Weintraub S.T., Bennett D.A., Cryns V.L., Berry R.W., Binder L.I. Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer’s disease. J. Neurosci. 2004;24:7895–7902. doi: 10.1523/JNEUROSCI.1988-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo H., Albrecht S., Bourdeau M., Petzke T., Bergeron C., LeBlanc A.C. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer’s disease. Am. J. Pathol. 2004;165:523–531. doi: 10.1016/S0002-9440(10)63317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Theofilas P., Piergies A.M., Li S.H., Petersen C., Ehrenberg A.J., Eser R.A., Chin B., Yang T., Khan S., Ng R., et al. Caspase-6-cleaved tau is relevant in Alzheimer’s disease but not in other tauopathies: diagnostic and therapeutic implications. Neurology. 2021 doi: 10.1101/2021.01.28.21250322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Means J.C., Gerdes B.C., Kaja S., Sumien N., Payne A.J., Stark D.A., Borden P.K., Price J.L., Koulen P. Caspase-3-Dependent proteolytic cleavage of tau causes neurofibrillary tangles and results in cognitive impairment during normal aging. Neurochem. Res. 2016;41:2278–2288. doi: 10.1007/s11064-016-1942-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.D’Amelio M., Cavallucci V., Middei S., Marchetti C., Pacioni S., Ferri A., Diamantini A., De Zio D., Carrara P., Battistini L., et al. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2011;14:69–76. doi: 10.1038/nn.2709. [DOI] [PubMed] [Google Scholar]
- 54.Louneva N., Cohen J.W., Han L.-Y., Talbot K., Wilson R.S., Bennett D.A., Trojanowski J.Q., Arnold S.E. Caspase-3 is enriched in postsynaptic densities and increased in Alzheimer’s disease. Am. J. Pathol. 2008;173:1488–1495. doi: 10.2353/ajpath.2008.080434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jadhav S., Katina S., Kovac A., Kazmerova Z., Novak M., Zilka N. Truncated tau deregulates synaptic markers in rat model for human tauopathy. Front. Cell. Neurosci. 2015;9:24. doi: 10.3389/fncel.2015.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sokolow S., Henkins K.M., Bilousova T., Gonzalez B., Vinters H.V., Miller C.A., Cornwell L., Poon W.W., Gylys K.H. Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer’s disease. J. Neurochem. 2015;133:368–379. doi: 10.1111/jnc.12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jarero-Basulto J.J., Luna-Muñoz J., Mena R., Kristofikova Z., Ripova D., Perry G., Binder L.I., Garcia-Sierra F. Proteolytic cleavage of polymeric tau protein by caspase-3: implications for Alzheimer disease. J. Neuropathol. Exp. Neurol. 2013;72:1145–1161. doi: 10.1097/NEN.0000000000000013. [DOI] [PubMed] [Google Scholar]
- 58.Zhang Q., Zhang X., Sun A. Truncated tau at D421 is associated with neurodegeneration and tangle formation in the brain of Alzheimer transgenic models. Acta Neuropathol. 2009;117:687–697. doi: 10.1007/s00401-009-0491-6. [DOI] [PubMed] [Google Scholar]
- 59.Kim Y., Choi H., Lee W., Park H., Kam T.-I., Hong S.-H., Nah J., Jung S., Shin B., Lee H., et al. Caspase-cleaved tau exhibits rapid memory impairment associated with tau oligomers in a transgenic mouse model. Neurobiol. Dis. 2016;87:19–28. doi: 10.1016/j.nbd.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 60.Biundo F., d’Abramo C., Tambini M.D., Zhang H., Del Prete D., Vitale F., Giliberto L., Arancio O., D’Adamio L. Abolishing Tau cleavage by caspases at Aspartate421 causes memory/synaptic plasticity deficits and pre-pathological Tau alterations. Transl. Psychiatry. 2017;7:e1198. doi: 10.1038/tp.2017.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Keller J.N., Hanni K.B., Markesbery W.R. Impaired proteasome function in Alzheimer’s disease. J. Neurochem. 2000;75:436–439. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- 62.Keck S., Nitsch R., Grune T., Ullrich O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem. 2003;85:115–122. doi: 10.1046/j.1471-4159.2003.01642.x. [DOI] [PubMed] [Google Scholar]
- 63.Tang M., Harrison J., Deaton C.A., Johnson G.V.W. Tau clearance mechanisms. Adv. Exp. Med. Biol. 2019;1184:57–68. doi: 10.1007/978-981-32-9358-8_5. [DOI] [PubMed] [Google Scholar]
- 64.Myeku N., Clelland C.L., Emrani S., Kukushkin N.V., Yu W.H., Goldberg A.L., Duff K.E. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 2016;22:46–53. doi: 10.1038/nm.4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kátai E., Pál J., Poór V.S., Purewal R., Miseta A., Nagy T. Oxidative stress induces transient O-GlcNAc elevation and tau dephosphorylation in SH-SY5Y cells. J. Cell Mol. Med. 2016;20:2269–2277. doi: 10.1111/jcmm.12910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takahashi M., Chin Y., Nonaka T., Hasegawa M., Watanabe N., Arai T. Prolonged nitric oxide treatment induces tau aggregation in SH-SY5Y cells. Neurosci. Lett. 2012;510:48–52. doi: 10.1016/j.neulet.2011.12.067. [DOI] [PubMed] [Google Scholar]
- 67.Davis D.R., Anderton B.H., Brion J.P., Reynolds C.H., Hanger D.P. Oxidative stress induces dephosphorylation of tau in rat brain primary neuronal cultures. J. Neurochem. 1997;68:1590–1597. doi: 10.1046/j.1471-4159.1997.68041590.x. [DOI] [PubMed] [Google Scholar]
- 68.Galas M.-C., Dourlen P., Bégard S., Ando K., Blum D., Hamdane M., Buée L. The peptidylprolyl cis/trans-isomerase Pin1 modulates stress-induced dephosphorylation of Tau in neurons. Implication in a pathological mechanism related to Alzheimer disease. J. Biol. Chem. 2006;281:19296–19304. doi: 10.1074/jbc.M601849200. [DOI] [PubMed] [Google Scholar]
- 69.Tseng J.-H., Xie L., Song S., Xie Y., Allen L., Ajit D., Hong J.-S., Chen X., Meeker R.B., Cohen T.J. The deacetylase HDAC6 mediates endogenous neuritic tau pathology. Cell Rep. 2017;20:2169–2183. doi: 10.1016/j.celrep.2017.07.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ertürk A., Wang Y., Sheng M. Local pruning of dendrites and spines by caspase-3-dependent and proteasome-limited mechanisms. J. Neurosci. 2014;34:1672–1688. doi: 10.1523/JNEUROSCI.3121-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Irwin D.J. Tauopathies as clinicopathological entities. Parkinsonism Relat. Disord. 2016;22 Suppl 1(Suppl 1):S29–S33. doi: 10.1016/j.parkreldis.2015.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Irwin D.J., Brettschneider J., McMillan C.T., Cooper F., Olm C., Arnold S.E., Van Deerlin V.M., Seeley W.W., Miller B.L., Lee E.B., et al. Deep clinical and neuropathological phenotyping of Pick disease. Ann. Neurol. 2016;79:272–287. doi: 10.1002/ana.24559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eytan D., Marom S. Dynamics and effective topology underlying synchronization in networks of cortical neurons. J. Neurosci. 2006;26:8465–8476. doi: 10.1523/JNEUROSCI.1627-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cao L., Liang Y., Liu Y., Xu Y., Wan W., Zhu C. Pseudo-phosphorylation at AT8 epitopes regulates the tau truncation at aspartate 421. Exp. Cell Res. 2018;370:103–115. doi: 10.1016/j.yexcr.2018.06.010. [DOI] [PubMed] [Google Scholar]
- 75.Guillozet-Bongaarts A.L., Cahill M.E., Cryns V.L., Reynolds M.R., Berry R.W., Binder L.I. Pseudophosphorylation of tau at serine 422 inhibits caspase cleavage: in vitro evidence and implications for tangle formation in vivo. J. Neurochem. 2006;97:1005–1014. doi: 10.1111/j.1471-4159.2006.03784.x. [DOI] [PubMed] [Google Scholar]
- 76.Li Z., Jo J., Jia J.-M., Lo S.-C., Whitcomb D.J., Jiao S., Cho K., Sheng M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell. 2010;141:859–871. doi: 10.1016/j.cell.2010.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lo S.-C., Wang Y., Weber M., Larson J.L., Scearce-Levie K., Sheng M. Caspase-3 deficiency results in disrupted synaptic homeostasis and impaired attention control. J. Neurosci. 2015;35:2118–2132. doi: 10.1523/JNEUROSCI.3280-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dejanovic B., Huntley M.A., De Mazière A., Meilandt W.J., Wu T., Srinivasan K., Jiang Z., Gandham V., Friedman B.A., Ngu H., et al. Changes in the synaptic proteome in tauopathy and rescue of tau-induced synapse loss by C1q antibodies. Neuron. 2018;100:1322–1336.e7. doi: 10.1016/j.neuron.2018.10.014. [DOI] [PubMed] [Google Scholar]
- 79.Nikolaev A., McLaughlin T., O’Leary D.D.M., Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 80.Imbriani P., Tassone A., Meringolo M., Ponterio G., Madeo G., Pisani A., Bonsi P., Martella G. Loss of non-apoptotic role of caspase-3 in the PINK1 mouse model of Parkinson’s disease. Int. J. Mol. Sci. 2019;20:E3407. doi: 10.3390/ijms20143407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fieblinger T., Li C., Espa E., Cenci M.A. Non-apoptotic caspase-3 activation mediates early synaptic dysfunction of indirect pathway neurons in the parkinsonian striatum. Int. J. Mol. Sci. 2022;23:5470. doi: 10.3390/ijms23105470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Steuer E.L., Kemper L.J., Hlynialuk C.J.W., Leinonen-Wright K., Montonye M.L., Lapcinski I.P., Forster C.L., Ashe K.H., Liu P. Blocking site-specific cleavage of human tau delays progression of disease-related phenotypes in genetically matched tau-transgenic mice modeling frontotemporal dementia. J. Neurosci. 2022;42:4737–4754. doi: 10.1523/JNEUROSCI.0543-22.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Choi Y.E., Butterworth M., Malladi S., Duckett C.S., Cohen G.M., Bratton S.B. The E3 ubiquitin ligase cIAP1 binds and ubiquitinates caspase-3 and -7 via unique mechanisms at distinct steps in their processing. J. Biol. Chem. 2009;284:12772–12782. doi: 10.1074/jbc.M807550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fu H., Possenti A., Freer R., Nakano Y., Hernandez Villegas N.C., Tang M., Cauhy P.V.M., Lassus B.A., Chen S., Fowler S.L., et al. A tau homeostasis signature is linked with the cellular and regional vulnerability of excitatory neurons to tau pathology. Nat. Neurosci. 2019;22:47–56. doi: 10.1038/s41593-018-0298-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen Y., Liu L., Li M., Yao E., Hao J., Dong Y., Zheng X., Liu X. Expression of human Tau40 in the medial entorhinal cortex impairs synaptic plasticity and associated cognitive functions in mice. Biochem. Biophys. Res. Commun. 2018;496:1006–1012. doi: 10.1016/j.bbrc.2017.04.153. [DOI] [PubMed] [Google Scholar]
- 86.Busche M.A., Wegmann S., Dujardin S., Commins C., Schiantarelli J., Klickstein N., Kamath T.V., Carlson G.A., Nelken I., Hyman B.T. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat. Neurosci. 2019;22:57–64. doi: 10.1038/s41593-018-0289-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Buzsáki G. Neural syntax: cell assemblies, synapsembles, and readers. Neuron. 2010;68:362–385. doi: 10.1016/j.neuron.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zeldenrust F., Wadman W.J., Englitz B. Neural coding with bursts-current state and future perspectives. Front. Comput. Neurosci. 2018;12:48. doi: 10.3389/fncom.2018.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ittner L.M., Ke Y.D., Delerue F., Bi M., Gladbach A., van Eersel J., Wölfing H., Chieng B.C., Christie M.J., Napier I.A., et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 90.Tracy T.E., Sohn P.D., Minami S.S., Wang C., Min S.-W., Li Y., Zhou Y., Le D., Lo I., Ponnusamy R., et al. Acetylated tau obstructs KIBRA-mediated signaling in synaptic plasticity and promotes tauopathy-related memory loss. Neuron. 2016;90:245–260. doi: 10.1016/j.neuron.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kobayashi S., Tanaka T., Soeda Y., Almeida O.F.X., Takashima A. Local somatodendritic translation and hyperphosphorylation of tau protein triggered by AMPA and NMDA receptor stimulation. EBioMedicine. 2017;20:120–126. doi: 10.1016/j.ebiom.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Prikas E., Paric E., Asih P.R., Stefanoska K., Stefen H., Fath T., Poljak A., Ittner A. Tau target identification reveals NSF-dependent effects on AMPA receptor trafficking and memory formation. EMBO J. 2022;41 doi: 10.15252/embj.2021110242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Djakovic S.N., Schwarz L.A., Barylko B., DeMartino G.N., Patrick G.N. Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 2009;284:26655–26665. doi: 10.1074/jbc.M109.021956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Warmus B.A., Sekar D.R., McCutchen E., Schellenberg G.D., Roberts R.C., McMahon L.L., Roberson E.D. Tau-mediated NMDA receptor impairment underlies dysfunction of a selectively vulnerable network in a mouse model of frontotemporal dementia. J. Neurosci. 2014;34:16482–16495. doi: 10.1523/JNEUROSCI.3418-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Styr B., Slutsky I. Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer’s disease. Nat. Neurosci. 2018;21:463–473. doi: 10.1038/s41593-018-0080-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chakroborty S., Hill E.S., Christian D.T., Helfrich R., Riley S., Schneider C., Kapecki N., Mustaly-Kalimi S., Seiler F.A., Peterson D.A., et al. Reduced presynaptic vesicle stores mediate cellular and network plasticity defects in an early-stage mouse model of Alzheimer’s disease. Mol. Neurodegener. 2019;14:7. doi: 10.1186/s13024-019-0307-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Frere S., Slutsky I. Alzheimer’s disease: from firing instability to homeostasis network collapse. Neuron. 2018;97:32–58. doi: 10.1016/j.neuron.2017.11.028. [DOI] [PubMed] [Google Scholar]
- 98.Trzeciakiewicz H., Ajit D., Tseng J.-H., Chen Y., Ajit A., Tabassum Z., Lobrovich R., Peterson C., Riddick N.V., Itano M.S., et al. An HDAC6-dependent surveillance mechanism suppresses tau-mediated neurodegeneration and cognitive decline. Nat. Commun. 2020;11:5522. doi: 10.1038/s41467-020-19317-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pan L., Alagapan S., Franca E., Leondopulos S.S., DeMarse T.B., Brewer G.J., Wheeler B.C. An in vitro method to manipulate the direction and functional strength between neural populations. Front. Neural Circuits. 2015;9:32. doi: 10.3389/fncir.2015.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Potter S.M., DeMarse T.B. A new approach to neural cell culture for long-term studies. J. Neurosci. Methods. 2001;110:17–24. doi: 10.1016/s0165-0270(01)00412-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data and code supporting the findings of this study are available within the method details of the paper and its supplementary information files and can be obtained from the authors upon request.
-
•
Code availability and all coding requests should be directed to Dr. Rick Meeker (meekerr@neurology.unc.edu).
-
•
Any additional information required to reanalyze the data reported in this paper is available from Dr. Todd Cohen upon request (toddcohen@neurology.unc.edu).