

Abstract
The use of protein sequence to inform enzymology in terms of structure, mechanism, and function has burgeoned over the past two decades. Referred to as genomic enzymology, the utilization of bioinformatic tools such as sequence similarity networks and phylogenetic analyses has allowed the identification of new substrates and metabolites, novel pathways, and unexpected reaction mechanisms. The holistic examination of superfamilies can yield insight into the origins and paths of evolution of enzymes and the range of their substrates and mechanisms. Herein, we highlight advances in the use of genomic enzymology to address problems which the in-depth analyses of a single enzyme alone could not enable.
Keywords: genomic enzymology, sequence similarity network, enzyme evolution, genome neighborhood, metabolic pathways
Graphical Abstract

Introduction
Over the past decade, the definitive study of individual enzymes in terms of specificity and catalytic and chemical mechanism has been leveraged using bioinformatic tools to extrapolate that knowledge to numerous homologues. Subsequently, deviations from the original classification are employed to define those enzymes with different structure-function and mechanism for further study. These innovations stemmed from the initial insight in 1996, when Babbitt et al. demonstrated how identifying common ancestry of mechanistically diverse superfamilies can allow for function and sometimes, specificity predictions [1]. The study focused on annotation of members of the enolase superfamily. Extensive mechanistic characterization and key structural knowledge of only a few family members allowed the authors to make the structure-function linkage as related to mechanistically diverse enzymes within the superfamily [1, 2]. The biochemical insight gained through this study gave rise to the concept of genomic enzymology, wherein the sequence-function space of a protein family can be analyzed within its genomic context to allow for the prediction of enzymatic activity and metabolic pathways [3]. Genomic enzymology also can be greatly enhanced by the inclusion of structural information, though the sequence-structure-function linkage is still being explored. The implementation of genomic enzymology, supported by biochemical and biophysical experimentation, has enabled the identification of novel pathways and functions (Figure 1).
Figure 1.

Overview of applications of genomic enzymology in recent years: determining pathways (PDB ID: 6P78), defining a superfamily or family (SSN generated from sequences from Knox et al. and visualized in Cytoscape [4]), exploring substrate specificity (PDB ID: 5V1V), understanding enzyme evolution, and predicting mechanism (mechanism adapted from Braffman et al. [5])
Bioinformatic tools have been developed that facilitate the sequence- and genomic- based study of enzymes [6–12]. A key development has been the ability to visualize the sequence-function space in sequence similarity networks (SSNs) paired with genome neighborhood networks and/or diagrams (GNNs or GNDs) [3, 6, 8, 11]. The SSN shows how homologous sequences relate through grouping into clusters defined by pairwise sequence alignments, whereas a GNN/GND shows the occurrence of genes found in common in the operons encoding homologues of the gene of interest [6, 8, 9]. The colocalization of genes in a GNN/GND is often taken as evidence of participation of the genes in a common pathway (guilt by association) and/or possible protein-protein interactions [13]. In mechanistically diverse superfamilies with overall low sequence identity, the evolutionary drift and resulting noise make it difficult to differentiate signatures for distinct families and subfamilies [14]. To test and quantify relationships within an SSN, Hidden Markov Models (HMMs) can be useful in probing whether subgroupings of enzymes are likely to be isofunctional [15]. HMMs are statistical models that can be used to identify hidden patterns or relationships within protein sequences. Likewise, the convergence ratios (CRs) when used within a superfamily can yield information about the similarity between sequences and aid in distinguishing paralogues from orthologues [16]. CRs measure sequence similarity at a specific alignment score by calculating the ratio of the number of edges of a cluster to the maximum possible number of edges [16]. Taxonomic information can be integrated into SSNs by attributing known properties to the nodes. This information together with phylogenetic analyses, can be powerful tools in making conclusions about relatedness and ancestral origins. In this review, we highlight examples of the conjoined use of primary and tertiary structure information together with genome context, which through these and other tools, reveal mechanisms of substrate specificity, find new chemistries and new pathways, and reveal evolutionary paths.
Benefits of superfamily analysis
As of May 2022, the UniProtKB/Swiss-Prot database contains more than 500,000 unique and annotated sequences and is still expanding [17, 18]. With this wealth of information, comparative analyses of superfamilies can be utilized to aid in the annotation of uncharacterized enzymes as well as determine mechanisms, assign functions, and correct misannotations. A recent application of this “comparative anatomy” approach is the work by Tararina and Allen, which deconvoluted the flavin-dependent amine oxidase (FAO) superfamily, organizing it into eight subgroups based on substrate type (Figure 2) [19]. A notable feature of the FAO superfamily is the flexibility in the multi-domain scaffold, which allows for facile evolution of new reactivity through insertions or deletions to the substrate-binding domains with retention of the critical flavin-binding domain, creating new functions amongst the subfamilies [19]. The evolvability of the superfamily, through use of modular binding domains, was further revealed by the reinvention of functions amongst the eight subfamilies [19].
Figure 2.

Abbreviated SSN of the FAO superfamily adapted from Tararina and Allen [16] and visualized in Cytoscape. The eight enzyme functional subgroups are labeled with representative structures for illustrative purposes. PDB IDs are 2IID (L-amino acid oxidase), 3NKS (protoporphyrinogen IX oxidase), 5KRQ (renalase), 6C71 (nicotine oxidoreductase), 1OJ9 (monoamine oxidase), 2UXN (lysine-specific demethylase), 5MOG (phytoene desaturase), and 5MBX (N1-acetylpolyamine oxidase).
Combining structural information with bioinformatic analysis can aid greatly in surveying a family or superfamily [20]. In Knox et al., the authors structurally characterized an enzyme within the cobalamin-dependent radical S-adenosylmethionine (SAM) methylase class, the largest and most diverse radical SAM methylase class [4]. Radical SAM methylases are able to append methyl groups to inert sp2- and sp3-hybridized carbonas well as phosphinate phosphorus. The low sequence conservation and mechanistic divergence amongst these enzymes make their study challenging. The authors discuss the diversity of this class of enzymes through structural comparisons of three different cobalamin-dependent radical SAM enzymes [4]. Although the three enzymes have the same three structural domains (a Rossmann fold that binds cobalamin, a shortened TIM barrel that ligates the Fe/S cluster, and a structurally variable C-terminal domain), the mechanistic determinants are dictated largely by the cofactor interactions and the secondary structural information [4]. In a second example, Sun et al. enlisted a SSN and phylogenetic tree to draw links between evolutionary and functional diversification of a phosphodiesterase subfamily, allowing for prediction of the enzyme metallocofactor and reactivity [21]. The study focused on metal-dependent phosphodiesterases involved in regulating cyclic dinucleotides [21]. Within this study, Sun et al. were able to functionally categorize the metal-dependent phosphodiesterases and surmise evolutionary paths by incorporating information on metallocofactors and coordination spheres, secondary structural differences, and specific activities [21]. Generally, the identification of sequence patterns and divergences from those patterns have been very successful in flagging possible new substrates and reactivities [22–26]. The survey of a superfamily, labeled with known reactivities and reaction types, can guide selection of new targets to cover unexplored areas or find additional examples of known reactions, increasing project success.
Sequence analysis is a boon to target selection [27–30]. For instance, identifying homologs can support the study of intractable enzymes. In 2019, Guo et al. completed the experimental mechanistic and structural characterization of an imine reductase (Bsp5) which, unlike its homologues (Ind5 and Pel5), does not require an additional enzyme (Ind4 and Pel4 respectively) for stability and activity [31]. Bsp5 is capable of stand-alone activity and crystallized readily. Moreover, its simplification allowed for a better understanding of the structural constraints for activity [31]. The selection of this more divergent member (Bsp5 has 50% sequence identity to Pel5) also allowed the authors to observe evidence that Bsp5 can accept alternative substrates, which suggests the expansion of the scope of its ability to synthesize D-amino acids. Describing the scope of a superfamily through sequence analysis can also aid in target selection for applications in structural biology [32–34]. Where X-ray crystallographic analysis may be facilitated by using the minimal core catalytic fold, cryo-electron microscopy necessitates a larger macromolecule. The divergence of the folds within a superfamily can be defined through SSNs to identify the minimal, common catalytic fold versus the structurally elaborated folds [35].
In 2021, Taujale et al. designed a convolution neural network with an attention-based deep learning model to predict folds within the glycosyltransferase superfamily by combining secondary structure representations with primary sequence analysis [36]. The authors further delineated the individual folds into distinct clusters using a probabilistic mathematical model to segregate the secondary structures of evolutionarily divergent families [36]. Their approach worked best for the GT-A fold type glycosyltransferases but is also able to predict several unknown fold types [36]. The same approach worked well for the kinase superfamily, leading the authors to suggest that this method is adaptable for exploring fold diversity within large protein superfamilies [36]. The ability to accurately predict structures with no homologous protein fold is a powerful innovation, showing the strength of using deep learning to complement experimental biochemical approaches [37]. As such, machine learning can greatly enhance the studies in which structural information is lacking.
Defining structure-function links within a superfamily
A recent insight is that SSNs can inform not only the relationship, but the evolutionary path between mechanistically divergent enzymes. Bioinformatic studies of the tautomerase superfamily identify linker proteins- enzymes connecting two different clusters within the SSN (see example in Figure 3)- shedding light on the structural changes in the active sites yielding diversification of activity [38, 39]. Structural and kinetic characterization of two linker proteins revealed amino-acid substitutions that allow the switch in mechanism between tautomerase and dehalogenase activities [39]. The identification of linker proteins can uncover sequence variation driving diversity and suggest the evolutionary trajectory within a superfamily, although evolutionary relationships require further support from phylogenetic tree reconstruction [15, 40].
Figure 3.
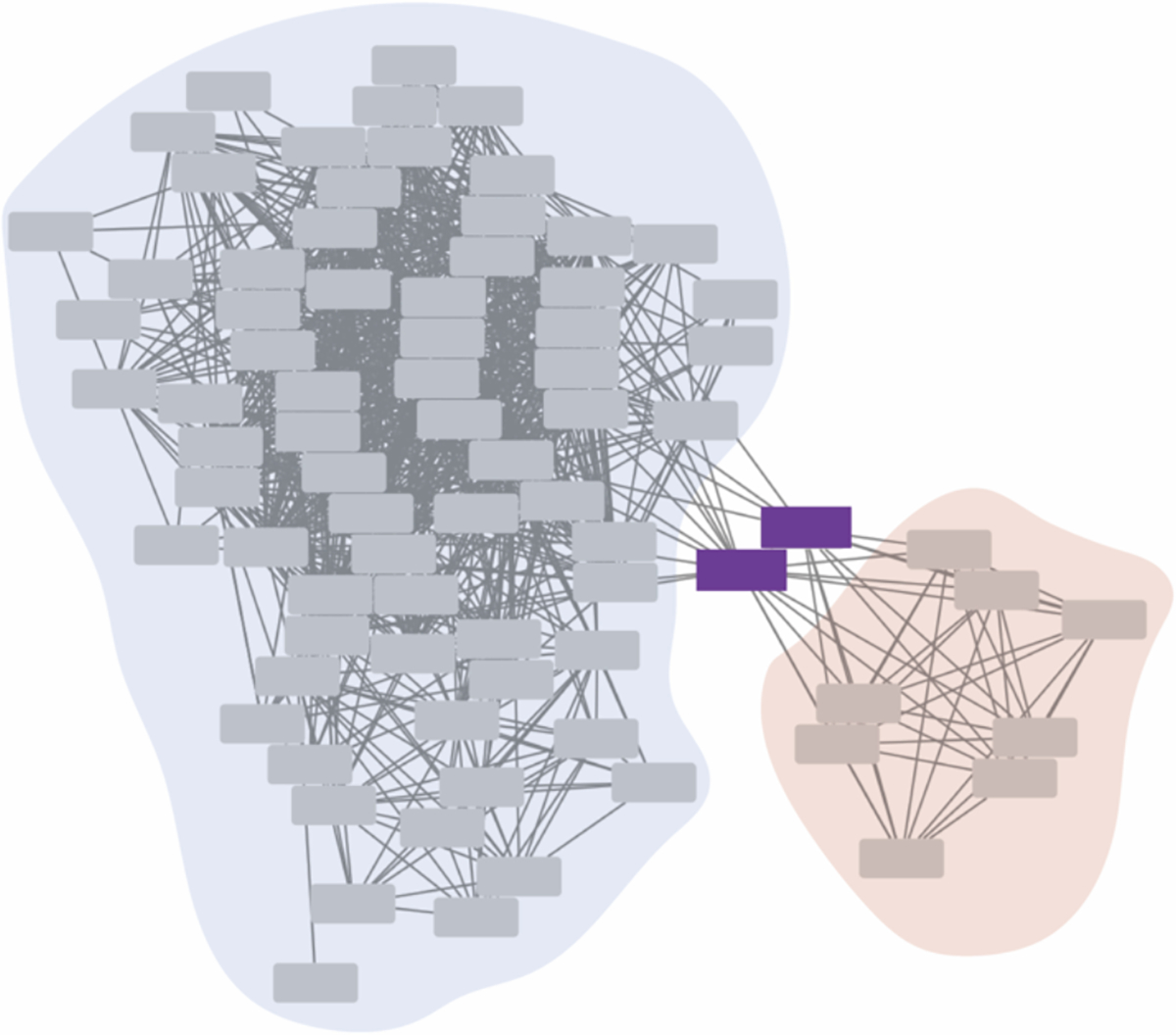
An example of linker proteins found within a SSN, shown as purple boxes. The enzymes act as a “link” between the two clusters, illustrated in blue and red. SSN generated from sequences in O’Toole et al. [41] and visualized in Cytoscape.
Phylogenetic evaluation can be used to assess evolutionary origins, as highlighted in the recent study of DNA ligases by Pan et al. [42]. Using genomic information gathered in a bioinformatics survey combined with the mechanistic and crystallographic data, the authors identify a probable entry point for horizontal acquisition of ATP-dependent DNA ligases by bacteria [42]. In 2021, O’Toole et al. performed bioinformatic analyses of the monotopic phosphoglycosyl transferase (monoPGT) superfamily, identifying numerous fusion proteins within the SSN [41]. The fusions were grouped into sugar-modifying, glycosyltransferase, and regulatory domains. Intriguingly, a number of fusions with an isofunctional, but distinct superfamily, the polytopic PGTs (polyPGT), were also identified, suggesting an evolutionary link through the shared substrate [41]. Phylogenetic reconstruction revealed a “radial burst of functionalization, with a minority of members comprising only the minimal phosphoglycosyl transferase catalytic domain” [41].
Discovery of new pathways
Genomic enzymology has enabled the identification of novel or unique metabolites and pathways [43–47]. Macdonald et al. surveyed the glycoside phosphorylase family, specifically CAZy family GH94 [48]. The Carbohydrate-Active enZYmes (CAZy) database includes a number of sequences of glycoside phosphorylases, but the classification and characterization of this class are limited [49]. The phylogenetic exploration by the authors allowed for the identification and then characterization of a previously unclassified glycoside phosphorylase and its substrate, a new biopolymer referred to as acholetin [48]. In Yuan et al., the authors utilized a comparative genomic approach to identify two queuosine salvage pathways in pathogenic, commensal bacteria [50]. Many bacteria are capable of de novo queuosine biosynthesis, but the identification of the salvage pathway in bacteria suggests that there may be direct competition for the queuine-base precursor [50]. The study also uncovered the first example of a radical SAM enzyme catalyzing this queuine-lyase reaction mechanism, which itself had never been previously described in the literature [50].
The catabolic pathway of 1-deoxy-D-sorbitol has remained unknown, but Li et al. in 2022 were able to identify an operon that contained genes involved in catabolism of D-sorbitol and 1-deoxy-D-sorbitol [51]. A phylogenetic distribution analyses suggested the conservation of the gene cluster in the Firmicutes phylum (recently suggested to be renamed Bacillota) [51]. The authors propose that their integrated strategy will be applicable to the discovery of other novel metabolites [51]. Genomic enzymology can also aid in organizing complex families, enabling the dissection of sequence markers for each subfamily or chemical activity. In 2021, Beal et al. performed bioinformatic analysis of aminotransferases, dehydrogenases, and aminopolyol dehydrogenases (a three gene cluster of these is typically indicative of bacterial azasugar biosynthesis), which allowed for determination of a consensus sequence for aminotransferases involved in azasugar biosynthesis. This, in turn, identified more than 400 microorganisms that putatively produce azasugar [52]. The utilization of a pan-taxonomic survey allowed for a broader perspective of the genomic context of azasugar biosynthesis [52].
Often, examining the crystal structures within a bioinformatic study reveals information about the substrate specificity or mechanism [53–55]. Li et al. identified a unique ArsR-SmtB transcriptional regulator that was proposed to use disulfide-bond formation to upregulate the expression of thioredoxin as a way to maintain cellular redox homeostasis [56]. From the structural work, the authors were able to identify an entrance channel and key catalytic residues involved in peroxide activation [56]. Sequence analysis found a conserved Cys residue signature which allowed tracking of this divergent function through the superfamily. Their findings suggest that the vicinal disulfide redox switch is a feature of cyanobacteria within the Nostocales order, identifying a distinct cluster within the ArsR-SmtB regulator family [56]. This provides an excellent example of how evolution of novel pathways often depends on the recruitment and tailoring of existing enzymes to adopt a new substrate [57].
Exploring substrate specificity
Bioinformatic study can help inform the factors determining substrate specificity [58–60]. In the study of ribosomally-synthesized and post-translationally modified peptides (RiPPs), the recent focus is on identifying features that control substrate recognition. RiPPs comprise a large number of natural products, many with favorable pharmacokinetic properties, but the specificity of the enzymes that biosynthesize these peptides is still unclear. Generally, enzymes in RiPP biosynthetic pathways are believed to utilize sequence-specific recognition of the leader peptide sequence, which is removed after post-translation modifications on the core peptide (Figure 4). Typically, there is some conservation of the leader peptide sequence within a RiPP class, which suggests strict leader-sequence specificity [61]. However, enzymatic study guided by bioinformatics has found flexibility in the recognition of the leader peptide [61, 62]. Chekan et al. found in their enzymatic study of the lasso peptide class using alternative leader peptides, that the minimal binding pocket allows for recognition and binding of sequence-divergent leader peptides. The authors conclude that the binding of the leader peptide is controlled by hydrophobic packing rather than a specific amino-acid sequence [61]. Moreover, by curating the extensive SSN for the occurrence of the signature sequence, the authors conclude that it is likely that in many other RiPP classes leader peptide recognition does not dictate activity [61]. Likewise, in a recent study of class II lanthipeptide synthetases, Zhang et al. found that the enzyme’s recognition of the leader peptide is promiscuous whereas the enzyme catalyzes the same reaction on the core peptide, despite significant diversity of the leader peptide [62]. From detailed biochemical analysis and extensive sequence alignment, the authors suggest that the precursor leader peptides coevolve with the cognate enzyme [62]. There is much interest in bioengineering RiPPs and one such point of interest is in the usage of chimeric leader peptides, which can be recognized by enzymes in unrelated RiPP pathways, to allow for biosynthesis of novel RiPPs [63].
Figure 4.

Schematic example of RiPP processing. Amino acids and modified residues are represented by colored spheres.
In 2021, Lachowicz et al. explored the range of substrates accepted by viperins [64]. The radical SAM enzyme viperin (virus inhibitory protein, endoplasmic reticulum associated, interferon inducible) is able to restrict replication of several different RNA and DNA viruses, making it of interest for use in combatting pathogens including HIV, West Nile virus, and Dengue virus [64]. Human viperin catalyzes biosynthesis of 3’-deoxy-3’,4’-didehydro-CTP (ddhCTP) from CTP (making viperin a ddh-synthase), which functions as a chain terminator for RNA polymerases from a variety of flaviviruses [64]. From the structural and kinetic study of viperin homologues, Lachowicz et al. were able to decipher the catalytic mechanism of viperin as well as define the physical and chemical determinants of ddh-synthase activity and substrate selectivity [64]. The SSN revealed discrete clusters delineated by substrate selectivity and this information was used to identify residues that control the specificity [64].
Discovery of novel reaction mechanisms
Enzyme genomic context can aid in identifying unique reactions, mechanisms, or enzymatic properties [65–68]. In 2021, Braffman et al. characterized a cyanobacterial enzyme that performs two unusual alkylation reactions between aromatic rings and secondary alkyl halide substrates in the biosynthesis of cylindrocyclophane natural products [5]. Their bioinformatic study of homologues established that the key catalytic residues likely associate with divergent reactivity and altered secondary metabolism [5]. Phylogenetic study indicated that this specific family of enzymes likely catalyzes other reactions that do not involve alkyl halides or resorcinols [5]. Uncovering the molecular mechanism of this unprecedented biosynthetic reaction allows for a better understanding of the activation of alkyl halides. Similarly, Chekan et al. were able to utilize bioinformatics to aid in identifying key catalytic residues that dictate divergent functionalities in their study of enzymatic dehalogenation [69]. From their crystallographic and computational study of Bmp8, an enzyme that replaces a halogen with a hydrogen in the biosynthesis of the natural product and antibiotic pentabromopseudilin, the authors propose a mechanism similar to that of human deiodinases [69]. To further demonstrate the debromination of Bmp8, the authors characterized a homologue with the same activity found in a distinct genomic context [69]. From this observation, the authors conclude that there are conserved enzymes with the capacity to dehalogenate tetrabromopyrroles outside of the typical biosynthetic pathway [69].
The usage of comparative genomic approaches is a powerful way to discover novel enzymatic reaction mechanisms. In 2019, Rizzolo et al. identified a previously undiscovered class of diheme enzymes that exhibits mechanistically distinct peroxidase activity through stabilization of a bis-Fe(IV) state [70]. Together, the crystallographic, spectroscopic, and bioinformatic study identified essential catalytic residues and distinguished this enzyme from other superfamily members [70]. Another example is found in metalloenzymes, where the metal coordination sphere plays a key role in controlling reactivity. In 2022, Cheng et al. studied a non-heme iron enzyme with two distinct activities- acting as a thiol oxygenase and a sulfoxide synthase [71]. The authors identified homologs that enabled further study of the variation in the coordination sphere of the Fe(II) ion and ultimately concluded that both the substrates and the residues comprising the secondary coordination shell of the Fe(II) cofactor control the dual activity of the enzyme [71].
Enzyme engineering has long been of interest in the biochemical community, especially towards designing catalysts with altered substrate specificity, increased thermostability, and enhanced catalytic activity [72]. Zetzsche et al. explored biocatalytic cross-coupling with cytochrome P450 enzymes [73]. The authors developed a strategy for engineering these biocatalysts for improved activity and selectivity using the structural information available and bioinformatic approaches [73]. The goal of the research was to explore the cytochrome P450 sequence space for biocatalysts with complementary reactivity [73]. The authors identified candidates with the ability to catalyze oxidative cross-coupling reactions with naphthols and were able to use these candidates to construct fusion enzymes with cytochrome P450 to perform complementary reactions [73]. In addition to providing a framework for bioengineering, genomic enzymology provides a starting point for directed evolution or rational design. The assessment of structure-function space can empower the choice of target, pushing design towards the optimal evolutionary path.
Conclusion
With the wealth of genomic information available, structural and kinetic characterization of superfamilies is vital for probing the paths to evolve different families and the factors that distinguish their cognate activities. Looking forward, four main activities will significantly enhance the outcomes from the methodologies discussed above. First, continued enzyme functional assignment and mechanistic work is needed to provide the prototypes for each isofunctional cluster within SSNs. Second, the greater reliability of structure prediction (as implemented in AlphaFold [37]) will allow for integration of structure as additional data in these analyses. Third, the increased use of machine learning approaches to extract information from the evolutionary drift present in the sequence data of divergent superfamilies will facilitate the connection of primary and tertiary structure to new functionalities. Finally, despite the abundance of sequences, the addition of newly sequenced genomes will continue to accelerate study. As sequence information is added, enhancing the understanding of the more divergent members, better links can be made within and between families from both a functional and an evolutionary perspective.
Acknowledgements:
Thank you to Christian Whitman and Katherine O’Toole for reading and commenting on our manuscript.
Funding:
This work was supported by the National Institutes of Health [R01 GM039334 and R01 GM131627 to KNA].
Abbreviations:
- ddhCTP
3’-deoxy-3’,4’-didehydro-CTP
- CAZy
Carbohydrate-Active enZYmes
- CRs
convergence ratios
- FAO
flavin-dependent amine oxidase
- GNDs
genome neighborhood diagrams
- GNN
genome neighborhood networks
- HMMs
Hidden Markov Models
- monoPGT
monotopic phosphoglycosyl transferase
- polyPGT
polytopic phosphoglycosyl transferase
- RiPPs
ribosomally-synthesized and post-translationally modified peptides
- SAM
S-adenosylmethionine
- SSNs
sequence similarity networks
- virus inhibitory protein, endoplasmic reticulum associated, interferon inducible
viperin
Footnotes
Declarations of interest: none.
References
- 1.Babbitt PC, Hasson MS, Wedekind JE, Palmer DR, Barrett WC, Reed GH, Rayment I, Ringe D, Kenyon GL, and Gerlt JA (1996). The enolase superfamily: a general strategy for enzyme-catalyzed abstraction of the alpha-protons of carboxylic acids. Biochemistry-Us 35, 16489–16501. [DOI] [PubMed] [Google Scholar]
- 2.Allen KN, and Whitman CP (2021). The Birth of Genomic Enzymology: Discovery of the Mechanistically Diverse Enolase Superfamily. Biochemistry-Us 60, 3515–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zallot R, Oberg N, and Gerlt JA (2021). Discovery of new enzymatic functions and metabolic pathways using genomic enzymology web tools. Curr Opin Biotechnol 69, 77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Knox HL, Sinner EK, Townsend CA, Boal AK, and Booker SJ (2022). Structure of a B12-dependent radical SAM enzyme in carbapenem biosynthesis. Nature 602, 343–348. * The structures of a cobalamin-dependent radical SAM enzyme display the shared structural folds and identifies the features that determine the reactivity of the family of enzymes.
- 5.Braffman NR, Ruskoski TB, Davis KM, Glasser NR, Johnson C, Okafor CD, Boal AK, and Balskus EP (2022). Structural basis for an unprecedented enzymatic alkylation in cylindrocyclophane biosynthesis. Elife 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zallot R, Oberg N, and Gerlt JA (2019). The EFI Web Resource for Genomic Enzymology Tools: Leveraging Protein, Genome, and Metagenome Databases to Discover Novel Enzymes and Metabolic Pathways. Biochemistry-Us 58, 4169–4182. ** This paper describes the resources providing genomic enzymology webtools.
- 7.Gerlt JA, Allen KN, Almo SC, Armstrong RN, Babbitt PC, Cronan JE, Dunaway-Mariano D, Imker HJ, Jacobson MP, Minor W, et al. (2011). The Enzyme Function Initiative. Biochemistry-Us 50, 9950–9962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerlt JA, Bouvier JT, Davidson DB, Imker HJ, Sadkhin B, Slater DR, and Whalen KL (2015). Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim Biophys Acta 1854, 1019–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerlt JA (2017). Genomic Enzymology: Web Tools for Leveraging Protein Family Sequence-Function Space and Genome Context to Discover Novel Functions. Biochemistry-Us 56, 4293–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pereira J, and Alva V (2021). How do I get the most out of my protein sequence using bioinformatics tools? Acta Crystallogr D Struct Biol 77, 1116–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akiva E, Brown S, Almonacid DE, Barber AE, Custer AF, Hicks MA, Huang CC, Lauck F, Mashiyama ST, Meng EC, et al. (2014). The Structure-Function Linkage Database. Nucleic Acids Res 42, D521–D530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao S, Kumar R, Sakai A, Vetting MW, Wood BM, Brown S, Bonanno JB, Hillerich BS, Seidel RD, Babbitt PC, et al. (2013). Discovery of new enzymes and metabolic pathways by using structure and genome context. Nature 502, 698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Esch R, and Merkl R (2020). Conserved genomic neighborhood is a strong but no perfect indicator for a direct interaction of microbial gene products. BMC Bioinformatics 21, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akiva E, Copp JN, Tokuriki N, and Babbitt PC (2017). Evolutionary and molecular foundations of multiple contemporary functions of the nitroreductase superfamily. Proc Natl Acad Sci U S A 114, E9549–E9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Copp JN, Anderson DW, Akiva E, Babbitt PC, and Tokuriki N (2019). Exploring the sequence, function, and evolutionary space of protein superfamilies using sequence similarity networks and phylogenetic reconstructions. Methods Enzymol 620, 315–347. [DOI] [PubMed] [Google Scholar]
- 16.Oberg N, Precord TW, Mitchell DA, and and Gerlt JA (2022). RadicalSAM.org: A Resource to Interpret Sequence-Function Space and Discover New Radical SAM Enzyme Chemistry. ACS Bio & Med Chem Au 2, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.UniProt C (2021). UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49, D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, et al. (2005). The Universal Protein Resource (UniProt). Nucleic Acids Res 33, D154–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tararina MA, and Allen KN (2020). Bioinformatic Analysis of the Flavin-Dependent Amine Oxidase Superfamily: Adaptations for Substrate Specificity and Catalytic Diversity. J Mol Biol 432, 3269–3288. * This work describes the flavin-dependent amine oxidase family and shows mapping of the clusters to specific substrate types.
- 20.Quaye JA, and Gadda G (2020). Kinetic and Bioinformatic Characterization of d-2-Hydroxyglutarate Dehydrogenase from Pseudomonas aeruginosa PAO1. Biochemistry-Us 59, 4833–4844. [DOI] [PubMed] [Google Scholar]
- 21.Sun S, and Pandelia ME (2020). HD-[HD-GYP] Phosphodiesterases: Activities and Evolutionary Diversification within the HD-GYP Family. Biochemistry-Us 59, 2340–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cleveland ME, Mathieu Y, Ribeaucourt D, Haon M, Mulyk P, Hein JE, Lafond M, Berrin JG, and Brumer H (2021). A survey of substrate specificity among Auxiliary Activity Family 5 copper radical oxidases. Cell Mol Life Sci 78, 8187–8208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lescallette AR, Dunn ZD, Manning VA, Trippe KM, and Li B (2022). Biosynthetic Origin of Formylaminooxyvinylglycine and Characterization of the Formyltransferase GvgI. Biochemistry-Us 61, 2159–2164. [DOI] [PubMed] [Google Scholar]
- 24.Schupfner M, Busch F, Wysocki VH, and Sterner R (2019). Generation of a Stand-Alone Tryptophan Synthase alpha-Subunit by Mimicking an Evolutionary Blueprint. Chembiochem 20, 2747–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lloyd CT, Iwig DF, Wang B, Cossu M, Metcalf WW, Boal AK, and Booker SJ (2022). Discovery, structure and mechanism of a tetraether lipid synthase. Nature 609, 197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ervin SM, Simpson JB, Gibbs ME, Creekmore BC, Lim L, Walton WG, Gharaibeh RZ, and Redinbo MR (2020). Structural Insights into Endobiotic Reactivation by Human Gut Microbiome-Encoded Sulfatases. Biochemistry-Us 59, 3939–3950. [DOI] [PubMed] [Google Scholar]
- 27.Russell AH, Vior NM, Hems ES, Lacret R, and Truman AW (2021). Discovery and characterisation of an amidine-containing ribosomally-synthesised peptide that is widely distributed in nature. Chem Sci 12, 11769–11778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kostenko A, Lien Y, Mendauletova A, Ngendahimana T, Novitskiy IM, Eaton SS, and Latham JA (2022). Identification of a poly-cyclopropylglycine-containing peptide via bioinformatic mapping of radical S-adenosylmethionine enzymes. J Biol Chem 298, 101881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chow JY, Choo KLS, Lim YP, Ling LH, Nguyen GKT, Xue B, Chua NH, and Yew WS (2021). Scalable Workflow for Green Manufacturing: Discovery of Bacterial Lipases for Biodiesel Production. Acs Sustain Chem Eng 9, 13450–13459. [Google Scholar]
- 30.Santos CR, Costa P, Vieira PS, Gonzalez SET, Correa TLR, Lima EA, Mandelli F, Pirolla RAS, Domingues MN, Cabral L, et al. (2020). Structural insights into beta-1,3-glucan cleavage by a glycoside hydrolase family. Nat Chem Biol 16, 920–929. [DOI] [PubMed] [Google Scholar]
- 31.Guo J, Higgins MA, Daniel-Ivad P, and Ryan KS (2019). An Asymmetric Reductase That Intercepts Acyclic Imino Acids Produced in Situ by a Partner Oxidase. J Am Chem Soc 141, 12258–12267. [DOI] [PubMed] [Google Scholar]
- 32.Yang G, Hong S, Yang P, Sun Y, Wang Y, Zhang P, Jiang W, and Gu Y (2021). Discovery of an ene-reductase for initiating flavone and flavonol catabolism in gut bacteria. Nat Commun 12, 790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lancaster EB, Yang W, Johnson WH Jr., Baas BJ, Zhang YJ, and Whitman CP (2022). Kinetic, Inhibition, and Structural Characterization of a Malonate Semialdehyde Decarboxylase-like Protein from Calothrix sp. PCC 6303: A Gateway to the non-Pro1 Tautomerase Superfamily Members. Biochemistry-Us. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vogt MS, Ngouoko Nguepbeu RR, Mohr MKF, Albers SV, Essen LO, and Banerjee A (2021). The archaeal triphosphate tunnel metalloenzyme SaTTM defines structural determinants for the diverse activities in the CYTH protein family. J Biol Chem 297, 100820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ray LC, Das D, Entova S, Lukose V, Lynch AJ, Imperiali B, and Allen KN (2018). Membrane association of monotopic phosphoglycosyl transferase underpins function. Nat Chem Biol 14, 538–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taujale R, Zhou Z, Yeung W, Moremen KW, Li S, and Kannan N (2021). Mapping the glycosyltransferase fold landscape using interpretable deep learning. Nat Commun 12, 5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davidson R, Baas BJ, Akiva E, Holliday GL, Polacco BJ, LeVieux JA, Pullara CR, Zhang YJ, Whitman CP, and Babbitt PC (2018). A global view of structure-function relationships in the tautomerase superfamily. J Biol Chem 293, 2342–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baas BJ, Medellin BP, LeVieux JA, Erwin K, Lancaster EB, Johnson WH Jr., Kaoud TS, Moreno RY, de Ruijter M, Babbitt PC, et al. (2021). Kinetic and Structural Analysis of Two Linkers in the Tautomerase Superfamily: Analysis and Implications. Biochemistry-Us 60, 1776–1786. ** This works highlights the identification and chemical validation of linkers in a superfamily.
- 40.Conte JV, and Frantom PA (2020). Biochemical characterization of 2-phosphinomethylmalate synthase from Streptomyces hygroscopicus: A member of the DRE-TIM metallolyase superfamily. Arch Biochem Biophys 691, 108489. [DOI] [PubMed] [Google Scholar]
- 41. O’Toole KH, Imperiali B, and Allen KN (2021). Glycoconjugate pathway connections revealed by sequence similarity network analysis of the monotopic phosphoglycosyl transferases. Proc Natl Acad Sci U S A 118. ** Through detailed SSN and phylogenetic analyses, the authors suggest evolutionary paths in a superfamily.
- 42.Pan J, Lian K, Sarre A, Leiros HS, and Williamson A (2021). Bacteriophage origin of some minimal ATP-dependent DNA ligases: a new structure from Burkholderia pseudomallei with striking similarity to Chlorella virus ligase. Sci Rep 11, 18693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Riegert AS, Narindoshvili T, Coricello A, Richards NGJ, and Raushel FM (2021). Functional Characterization of Two PLP-Dependent Enzymes Involved in Capsular Polysaccharide Biosynthesis from Campylobacter jejuni. Biochemistry-Us 60, 2836–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patteson JB, Putz AT, Tao L, Simke WC, Bryant LH 3rd, Britt RD, and Li B (2021). Biosynthesis of fluopsin C, a copper-containing antibiotic from Pseudomonas aeruginosa. Science 374, 1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morgan KT, Zheng J, and McCafferty DG (2021). Discovery of Six Ramoplanin Family Gene Clusters and the Lipoglycodepsipeptide Chersinamycin. Chembiochem 22, 176–185. [DOI] [PubMed] [Google Scholar]
- 46.Shin I, Wang Y, and Liu A (2021). A new regime of heme-dependent aromatic oxygenase superfamily. Proc Natl Acad Sci U S A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stack TMM, Morrison KN, Dettmer TM, Wille B, Kim C, Joyce R, Jermain M, Naing YT, Bhatti K, Francisco BS, et al. (2020). Characterization of an l-Ascorbate Catabolic Pathway with Unprecedented Enzymatic Transformations. J Am Chem Soc 142, 1657–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Macdonald SS, Pereira JH, Liu F, Tegl G, DeGiovanni A, Wardman JF, Deutsch S, Yoshikuni Y, Adams PD, and Withers SG (2022). A Synthetic Gene Library Yields a Previously Unknown Glycoside Phosphorylase That Degrades and Assembles Poly-beta-1,3-GlcNAc, Completing the Suite of beta-Linked GlcNAc Polysaccharides. Acs Central Sci 8, 430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Drula E, Garron ML, Dogan S, Lombard V, Henrissat B, and Terrapon N (2022). The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res 50, D571–D577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yuan Y, Zallot R, Grove TL, Payan DJ, Martin-Verstraete I, Sepic S, Balamkundu S, Neelakandan R, Gadi VK, Liu CF, et al. (2019). Discovery of novel bacterial queuine salvage enzymes and pathways in human pathogens. Proc Natl Acad Sci U S A 116, 19126–19135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Y, Huang H, and Zhang X (2022). Identification of catabolic pathway for 1-deoxy-D-sorbitol in Bacillus licheniformis. Biochem Biophys Res Commun 586, 81–86. [DOI] [PubMed] [Google Scholar]
- 52.Beal HE, and Horenstein NA (2021). Comparative genomic analysis of azasugar biosynthesis. AMB Express 11, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abraham N, Schroeter KL, Zhu Y, Chan J, Evans N, Kimber MS, Carere J, Zhou T, and Seah SYK (2022). Structure-function characterization of an aldo-keto reductase involved in detoxification of the mycotoxin, deoxynivalenol. Sci Rep 12, 14737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kincannon WM, Zahn M, Clare R, Lusty Beech J, Romberg A, Larson J, Bothner B, Beckham GT, McGeehan JE, and DuBois JL (2022). Biochemical and structural characterization of an aromatic ring-hydroxylating dioxygenase for terephthalic acid catabolism. Proc Natl Acad Sci U S A 119, e2121426119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shi Q, Wang H, Liu J, Li S, Guo J, Li H, Jia X, Huo H, Zheng Z, You S, et al. (2020). Old yellow enzymes: structures and structure-guided engineering for stereocomplementary bioreduction. Appl Microbiol Biotechnol 104, 8155–8170. [DOI] [PubMed] [Google Scholar]
- 56.Li B, Jo M, Liu J, Tian J, Canfield R, and Bridwell-Rabb J (2022). Structural and mechanistic basis for redox sensing by the cyanobacterial transcription regulator RexT. Commun Biol 5, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Noda-Garcia L, Liebermeister W, and Tawfik DS (2018). Metabolite-Enzyme Coevolution: From Single Enzymes to Metabolic Pathways and Networks. Annu Rev Biochem 87, 187–216. [DOI] [PubMed] [Google Scholar]
- 58.De Doncker M, De Graeve C, Franceus J, Beerens K, Kren V, Pelantova H, Vercauteren R, and Desmet T (2021). Exploration of GH94 Sequence Space for Enzyme Discovery Reveals a Novel Glucosylgalactose Phosphorylase Specificity. Chembiochem 22, 3319–3325. [DOI] [PubMed] [Google Scholar]
- 59.Zheng CJ, Kalkreuter E, Fan BY, Liu YC, Dong LB, and Shen B (2021). PtmC Catalyzes the Final Step of Thioplatensimycin, Thioplatencin, and Thioplatensilin Biosynthesis and Expands the Scope of Arylamine N-Acetyltransferases. Acs Chem Biol 16, 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li A, Laville E, Tarquis L, Lombard V, Ropartz D, Terrapon N, Henrissat B, Guieysse D, Esque J, Durand J, et al. (2020). Analysis of the diversity of the glycoside hydrolase family 130 in mammal gut microbiomes reveals a novel mannoside-phosphorylase function. Microb Genom 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chekan JR, Ongpipattanakul C, and Nair SK (2019). Steric complementarity directs sequence promiscuous leader binding in RiPP biosynthesis. Proc Natl Acad Sci U S A 116, 24049–24055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang SS, Xiong J, Cui JJ, Ma KL, Wu WL, Li Y, Luo S, Gao K, and Dong SH (2022). Lanthipeptides from the Same Core Sequence: Characterization of a Class II Lanthipeptide Synthetase from Microcystis aeruginosa NIES-88. Org Lett 24, 2226–2231. [DOI] [PubMed] [Google Scholar]
- 63.Burkhart BJ, Kakkar N, Hudson GA, van der Donk WA, and Mitchell DA (2017). Chimeric Leader Peptides for the Generation of Non-Natural Hybrid RiPP Products. ACS Cent Sci 3, 629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lachowicz JC, Gizzi AS, Almo SC, and Grove TL (2021). Structural Insight into the Substrate Scope of Viperin and Viperin-like Enzymes from Three Domains of Life. Biochemistry-Us 60, 2116–2129. * This study uses structure and sequence analysis to delineate markers for substrate specificity.
- 65.Schneider NO, Tassoulas LJ, Zeng D, Laseke AJ, Reiter NJ, Wackett LP, and Maurice MS (2020). Solving the Conundrum: Widespread Proteins Annotated for Urea Metabolism in Bacteria Are Carboxyguanidine Deiminases Mediating Nitrogen Assimilation from Guanidine. Biochemistry-Us 59, 3258–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wullich SC, Arranz San Martin A, and Fetzner S (2020). An alpha/beta-Hydrolase Fold Subfamily Comprising Pseudomonas Quinolone Signal-Cleaving Dioxygenases. Appl Environ Microbiol 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu J, Wei Y, Lin L, Teng L, Yin J, Lu Q, Chen J, Zheng Y, Li Y, Xu R, et al. (2020). Two radical-dependent mechanisms for anaerobic degradation of the globally abundant organosulfur compound dihydroxypropanesulfonate. Proc Natl Acad Sci U S A 117, 15599–15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang JQ, Fang X, Tian X, Chen P, Lin JL, Guo XX, Li JX, Fan Z, Song WM, Chen FY, et al. (2020). Aromatization of natural products by a specialized detoxification enzyme. Nat Chem Biol 16, 250–256. [DOI] [PubMed] [Google Scholar]
- 69.Chekan JR, Lee GY, El Gamal A, Purdy TN, Houk KN, and Moore BS (2019). Bacterial Tetrabromopyrrole Debrominase Shares a Reductive Dehalogenation Strategy with Human Thyroid Deiodinase. Biochemistry-Us 58, 5329–5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rizzolo K, Cohen SE, Weitz AC, Lopez Munoz MM, Hendrich MP, Drennan CL, and Elliott SJ (2019). A widely distributed diheme enzyme from Burkholderia that displays an atypically stable bis-Fe(IV) state. Nat Commun 10, 1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cheng R, Weitz AC, Paris J, Tang Y, Zhang J, Song H, Naowarojna N, Li K, Qiao L, Lopez J, et al. (2022). OvoAMtht from Methyloversatilis thermotolerans ovothiol biosynthesis is a bifunction enzyme: thiol oxygenase and sulfoxide synthase activities. Chem Sci 13, 3589–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marques WL, Anderson LA, Sandoval L, Hicks MA, and Prather KLJ (2020). Sequence-based bioprospecting of myo-inositol oxygenase (Miox) reveals new homologues that increase glucaric acid production in Saccharomyces cerevisiae. Enzyme Microb Technol 140, 109623. [DOI] [PubMed] [Google Scholar]
- 73.Zetzsche LE, Yazarians JA, Chakrabarty S, Hinze ME, Murray LAM, Lukowski AL, Joyce LA, and Narayan ARH (2022). Biocatalytic oxidative cross-coupling reactions for biaryl bond formation. Nature 603, 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]