


Abstract
A 2′-O-methylribooligonucleotide containing a G1·U·G3 triad modified by trans-diamminedichloro-platinum(II) was targeted to the RNA region responsible for the gag–pol frameshifting during translation of the HIV-1 mRNA. The binding of the platinated oligonucleotide to its target RNA induced a rearrangement of the (G1,G3)-intrastrand crosslink, leading to the formation of an intermolecular oligonucleotide–RNA G–A crosslink. This resulted in the selective arrest of translation of a luciferase gene placed downstream of the HIV-1 frameshift signal both in a cell-free extract (rabbit reticulocyte lysate) and in RNA-transfected cells. A specific inhibition of luciferase activity was still observed when the oligonucleotide–RNA complex was not pre-formed prior to either translation or transfection. Moreover, a selective inhibition was also observed when the oligonucleotide and the plasmid DNA encoding the luciferase and bearing the RNA gag–pol frameshifting signal were co-transfected in NIH 3T3 cultured cells. Therefore the intrastrand→interstrand conversion of the platinum crosslink kinetically competes with the translation machinery and blocks the polypeptide elongation. These transplatin-modified oligonucleotides which operate within a live cell on a ‘real-time’ basis and do not need an external triggering signal constitute a promising new class of selective reactive probes.
INTRODUCTION
A large number of mammalian retroviruses use –1 ribosomal frameshifting for controlling the expression of their pol genes [see (1,2) for reviews]. Ribosomal frameshifting which occurs at a specific site and at a defined frequency, allows the ribosome to avoid the gag stop codon and to read the pol gene, leading to the synthesis of a Gag–Pol fusion protein subsequently matured by proteolytic cleavage. In HIV-1 the Gag to Pol ratio is critical for the viral production. The overproduction of the Gag–Pol polyprotein and the increase of the protease amount was shown to inhibit virus assembly and budding (3,4). The key role played by the balance between the Gag and Pol products in the development of the virus was further demonstrated by the variants that accumulate upon treatment by protease inhibitors: the mutations responsible for resistance not only improved polyprotein processing but also increased the frameshifting efficiency thus leading to a higher level of protease expression (5). Therefore, altering the frameshift process would unbalance the production of structural proteins and enzymes and might allow the control of virus production.
In HIV-1 the gag–pol frameshifting is prompted by an RNA signal comprising two different elements: a slippery heptanucleotide and a hairpin structure located 7 nt downstream (6). Even if the slippery sequence mediates per se a low level of frameshifting in vivo, the stem–loop structure enhances this basic level (7,8). The presence of the secondary structure is generally thought to make the ribosome stalling on the heptanucleotide, thus increasing the probability of frameshifting. Therefore one may think that any ligand that would perturb the movement/stalling of the ribosome on the slippery sequence would interfere with the translation of the pol gene (9).
Antisense oligonucleotides have been used to this end (10). In particular, it was recently reported that oligopyrimidines were able to bind to the stem of the hairpin responsible for the gag–pol frameshifting (11). However, the resulting three-stranded RNA–RNA–DNA complex had no effect on the in vitro translation of a construct in which the luciferase gene was fused downstream of the gag–pol frameshifting signal of HIV-1. We used such constructs to investigate the properties of platinated oligonucleotides which were reported to crosslink complementary RNA targets (12).
The reaction of an oligonucleotide carrying two guanines with trans-diamminedichloro-platinum(II) results in the formation of an intramolecular crosslink which is stable as far as the oligomer remains single-stranded. The intramolecular crosslink rearranges into an intermolecular one upon binding of the oligonucleotide to a target RNA sequence. A fast intrastrand→interstrand crosslink rearrangement was observed using an oligo(2′-O-methylribonucleotide) containing a G1·N·G3 triad as a reactive site for platination. In addition the half-life of the (G1,G3) intrastrand adduct was strongly reduced when the platinum-bridged GUG motif was facing a 5′ pyrimidine–adenine doublet. This specific crosslink rearrangement makes platinated oligonucleotides attractive antisense molecules. Indeed, such an oligomer was shown to block in vitro translation of HaRas and to inhibit the proliferation of HBL100 ras1 cells but the mechanism responsible for this inhibition was not firmly established (13).
We targeted the gag–pol frameshifting signal of HIV-1 by OMeFSPt, a platinated oligomer comprising 19 2′-O-methylribonucleotides, including a GUG triad. We demonstrated that the selective crosslinking of this oligomer to a luciferase mRNA bearing the gag–pol frameshifting motif resulted in the specific inhibition of translation both in vitro in a cell free assay and ex vivo in cultured cells.
MATERIALS AND METHODS
Oligonucleotides
Oligoribonucleotides listed in Figure 1 were synthesised on a Perkin-Elmer Expedite synthesiser as previously described (11). Oligodeoxynucleotides were purchased from Eurogentec. RNA and DNA oligomers were purified by gel electrophoresis on a denaturing polyacrylamide gel. Oligo(2′-O-methylribonucleotide) was from Eurogentec. The reaction between trans-diamminedichloro-platinum(II) (transplatin) and the oligonucleotide as well as the purification of the platinated oligonucleotide were performed as previously described (13).
Figure 1.

RNA and oligonucleotide sequences. The sequence of the transcript used throughout this study is given at the top; the A residue of the initiator AUG corresponds to the +1 position. The slippery sequence UUUUUUA is underlined. The G(70) residue is present only in the pGFs(0) construct, not in pGFS(–1) for which the translation of luciferase requires a –1 frameshift. The actual luciferase coding region starts at position +84. Bold face characters [from A(13) to U(66)] indicate the 53FS RNA sequence used for crosslinking experiments. The stem–loop structure extending from U(29) to G(56), termed 28FS RNA, is italicised. The oligonucleotide OMeFSPt and OMeFS are the antisense 2′-O-methylribo analogues in which the GUG triad faces the U(20), A(21) doublet of the target RNA. The transplatin intramolecular crosslink in OMeFSPt is indicated by ‘∧’. DNAFS is the sense DNA sequence corresponding to the 12–28 frameshift region of the target RNA. The sequence of the gag–pro frameshifting signal of HTLV-I inserted upstream of the luciferase gene in the control construct pAC 1515 is given at the bottom (HTLV-I RNA). The slippery sequence is underlined.
Oligonucleotide crosslinking
The platinated oligonucleotide OMeFSPt (2 µM) was incubated at 37°C with 32P 5′ end-labelled 53FS RNA adjusted at 2 µM with unlabelled RNA, in a 20 mM HEPES buffer pH 7.4 containing 20 mM sodium acetate, 140 mM potassium acetate and 3 mM magnesium acetate. At the indicated time samples were collected and placed on dry ice after addition of the loading buffer. Samples were then heated for 5 min at 70°C, prior to electrophoresis, on a 10% polyacrylamide denaturing gel in Tris–borate EDTA (TBE) buffer. The ratio of bound over free oligonucleotide was determined by INSTANTIMAGER analysis.
Alkaline hydrolysis
For alkaline hydrolysis, 5′ end-labelled 53FS RNA (0.02 pmol) was mixed with 20 µM of the indicated oligonucleotide in 10 µl of a 5 mM phosphate buffer adjusted at pH 7.5, containing 50 mM NaCl. After 10 min incubation at 37°C, 5 µl were collected and were mixed with 5 µl of 100 mM NaHCO3 adjusted at pH 9.4. The mixture was then placed for 4.5 min in boiling water and the reaction was stopped on dry ice. Samples were then analysed by polyacrylamide gel electrophoresis.
Plasmid construction
Two plasmids derived from a commercially available luciferase construct (pGEM-luc; Promega) were designed. Both plasmids contained the gag–pol frameshift sequence at the 5′-end of the gene coding for luciferase. pGFS(–1) presents the luciferase coding region in the –1 frame with respect to the initiation AUG codon (Table 1). As a consequence the luciferase sequence is extended by 28 amino acids at the NH2 terminus. Therefore the synthesis of luciferase from this construct requires ribosome frameshifting. In contrast the luciferase was inserted in frame ‘0’ in the plasmid pGFS(0) (11). Plasmid pRL-SV40, from which the Renilla luciferase is expressed, was from Promega. pAC1789 was constructed as previously described (14). Plasmid pAC1515 was prepared from pAC74 (14): the sequence 5′-CAACGACATTCCACATCCCAAAA-AACTCC-3′ which contains the slippery heptanucleotide (underlined) responsible for gag–pro frameshifting on the HTLV-I mRNA, was inserted upstream of the luciferase gene at BclI and NhcI sites of pAC74 (Table 1).
Table 1. Luciferase constructs. The promoter, frameshift signal and the reading frame of the luciferase gene are indicated.
| Plasmid |
Promoter |
Frameshift signal |
Luc reading frame |
| pGFS(0) |
SP6 |
+ (HIV-1) |
0 |
| pGFS(–1) |
SP6 |
+ (HIV-1) |
–1 |
| pAC 1789 |
SV40 |
+ (HIV-1) |
–1 |
| pAC 1515 |
SV40 |
+ (HTLV-I) |
0 |
| pGEM luc |
SP6 |
– |
0 |
| pRL-SV40 | T7 and SV40 | – | 0 |
HIV-1, the gag–pol RNA region shown in Figure 1; HTLV-I, the slippery part of the HTLV-I gag–pro motif (see Fig. 1).
Transcription
The three plasmids pGEM-luc, pGFS(–1) and pGFS(0) were cut at the SalI site located downstream of the luciferase open reading frame. Transcription was then performed with the Sp6 RiboMAXΤΜ large-scale RNA production system (Promega) according to the manufacturer’s instructions. For translation in cell-free mixtures, RNAs were synthesised uncapped, whereas 3 mM of m7G(5′)ppp(5′)G (Boehringer) were added to the transcription mix, together with GTP adjusted at 0.6 mM for the synthesis of the RNA which had to be transfected in cells. pRL-SV40 was cut with XbaI (Gibco) and transcription was performed under similar capping conditions with the T7 transcription kit (Tebu).
In vitro translation
RNA (5 nM final concentration) was preincubated for 10 min at 37°C in a 5 mM phosphate buffer containing 25 mM NaCl in the absence or in the presence of the desired oligonucleotide. Then rabbit reticulocyte lysate and amino acids (Promega) were added following the manufacturer’s instructions except that the final volume was adjusted at 15 µl. Translation was then allowed to proceed for 1 h at 30°C. Luciferase activity was determined using a luminometer (Lumat Berthold) with the luciferase assay reagent (Promega). The reaction, without preforming the RNA–oligonucleotide complex, was performed exactly under the same conditions except for the 10 min preincubation period at 37°C.
Reverse transcription
RNA obtained from in vitro transcription of pGFS(–1), adjusted at a final concentration of 25 nM, was mixed with 500 nM of the primer 5′-TCCAGCGGTTCCATCCTCTA complementary to the 5′-end of luc and with the desired antisense oligonucleotide, in a 50 mM Tris–HCl pH 8.3 buffer containing 75 mM KCl, 3 mM MgCl2 and 10 mM DTT. The mix was incubated for 10 min at 37°C prior to the addition of 3 U of HIV-1 reverse transcriptase (Amersham), 1 µl of dCTP α32P (111 Tbq/mmol) and 2.5 mM of each of the dNTPs except dCTP which was 0.25 mM. After the reaction the samples were extracted with a phenol/chloroform/isoamyl alcohol (50:49:1) mix prior to ethanol precipitation. Samples were then loaded on a 10% polyacrylamide gel containing 7 M urea in TBE buffer. Electrophoresis was run at 50 V/cm and the gel was analysed by autoradiography. The band intensity was evaluated by INSTANTIMAGER measurement.
Tissue culture—RNA and DNA transfection
NIH 3T3 cells were grown at 37°C under 5% CO2 in DM7, a Dulbecco’s modified Eagle’s medium containing 2 mM glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin, 0.25 µg/ml amphotericin B and 7% of heat-inactivated foetal calf serum. Vero cells were grown in DM10 which is identical to the DM7 medium except that the foetal calf serum was adjusted at 10%. NIH 3T3 or Vero cells were seeded in 24 well plates at 8 × 104 cells per well in 500 µl of DM7 or DM10 respectively, 24 h prior to transfection with RNA or plasmid DNA.
RNA derived from pGEM-luc, pGFS(–1) or pGFS(0), coding for the firefly luciferase, were mixed with RNA derived from pRL-SV40 (3 nM each), coding for the Renilla luciferase, in the presence or the absence of the oligonucleotide. This mixture was then added to 200 µl of OptiMEM medium (Gibco) containing 6 µl of DMRIE-C reagent (Gibco). The lipid–RNA complex was then added to NIH 3T3 or Vero cells, previously rinsed two times with the OptiMEM medium, and incubated for 5 h at 37°C. Firefly or Renilla luciferase activity was then determined by using the Dual-LuciferaseΤΜ reporter assay system (Promega). The percentage of inhibition induced by the antisense oligonucleotide was calculated as [(UFx/URx)/(UF0/UR0)] × 100 where UF and UR correspond to the luciferase activity of the firefly and Renilla variant respectively, U(F,R)x and U(F,R)0 are the activity measured in the presence and in the absence of the oligonucleotide, respectively.
For DNA transfection 1 µg of plasmid pAC1789 or pAC1515 was added to 5 ng of pRL-SV40 in the presence of various concentrations of oligonucleotide in 400 µl of OptiMEM containing 6 µl of DMRIE-C. The mixture was left for 30 min at room temperature before addition to NIH 3T3 cells previously washed two times with OptiMEM. After 5 h incubation at 37°C under 5% CO2, the medium was replaced by DM7 and the cells were then incubated for a further 24 h. The luciferase activity was then determined by the luciferase assay described above for RNA transfection.
RESULTS
We studied the properties of a platinated antisense oligonucleotide targeted to the gag–pol frameshift sequence of HIV-1. The characterisation of the complex was achieved using a 53 nt long RNA fragment (53FS RNA) which included the binding site of the oligonucleotide of interest. This RNA species also carried the key determinants for ribosome frameshifting during HIV-1 mRNA translation, i.e. the slippery heptanucleotide 5′-U6A followed by a stem–loop structure seven bases downstream (Fig. 1).
The transplatin-modified antisense 19mer was synthesised in the 2′-O-methylribonucleotide series. This oligomer (OMeFSPt) contained a (G1,G3) intrastrand crosslink at the GUG motif facing 5′-UA in the oligonucleotide–RNA complex (Fig. 1). According to previous studies this modified nucleoside backbone combined with this particular mismatched association ensured the fastest intrastrand→interstrand crosslink rearrangement (12,13). The non-platinated homologue OMeFS of the reactive antisense oligomer was also available as a control.
Binding of the oligonucleotide and crosslinking reaction
We first monitored the binding of the control unplatinated oligonucleotide OMeFS to its target RNA by band-shift assay on a non-denaturing polyacrylamide gel. In spite of the central mismatched positions, a retarded band was observed (not shown) indicating the formation of a 53FS RNA–OMeFS complex characterised by a Kd of ~10 nM under our conditions (50 mM Tris–acetate buffer pH 7.0, 10 mM Mg2+; 4°C) determined as previously described (11).
Hybridisation of OMeFSPt with 53FS RNA resulted in the (G1,G3) intrastrand→interstrand crosslink rearrangement. This reaction was monitored by polyacrylamide gel electrophoresis under denaturing conditions. Upon incubation the band corresponding to the free RNA disappeared at the expense of a low mobility species corresponding to the RNA–oligonucleotide conjugate (Fig. 2a). About 50% of RNA was crosslinked to the platinated oligonucleotide after 2 min incubation (Fig. 2b). The reaction was much slower when a ‘complementary’ DNA sequence DNAFS was substituted for 53FS RNA (not shown): a half-life of ~45 min was determined in the case of the DNA target in good agreement with a previous study (13). In order to assess the sequence specificity of the crosslinking reaction we used 28FS RNA, a truncated target, corresponding to the stem–loop part of the frameshift RNA signal; i.e. devoid of the binding sequence of the antisense oligonucleotide (Fig. 1). No crosslinked product was detected upon incubation of 28FS RNA with OMeFSPt underlining the specificity of the oligonucleotide conjugation (Fig. 2).
Figure 2.
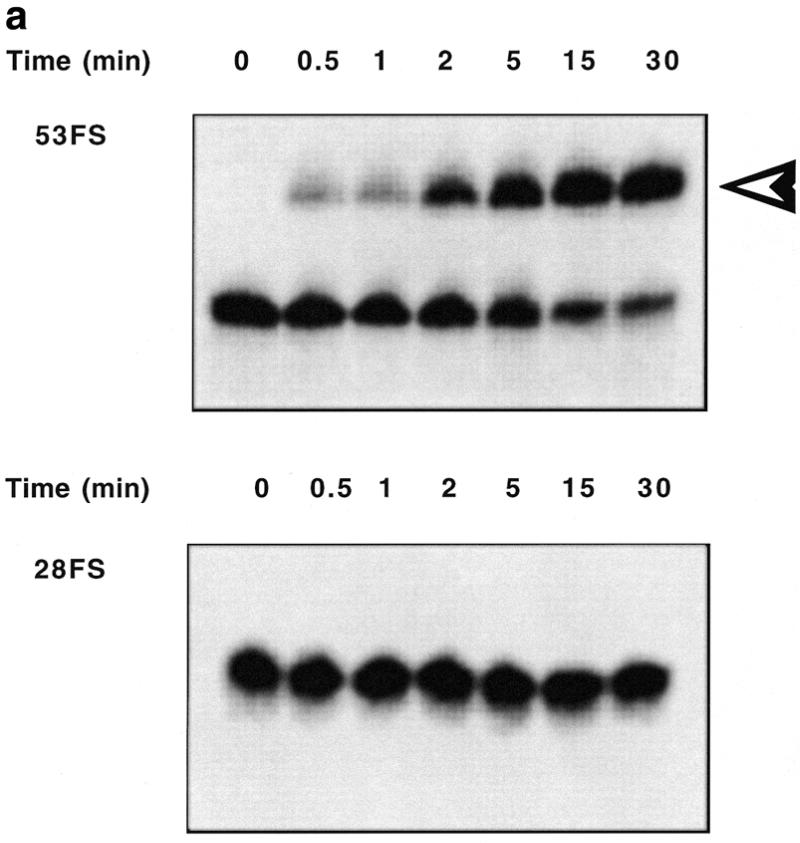

Conversion of the intramolecular transplatin intrastrand crosslink within OMeFSPt. The platinated oligonucleotide has been incubated under the conditions described in Materials and Methods either with 53FS or 28FS RNA. (a) Kinetic of the cross-linking reaction between OMeFSPt and 53FS (top) or 28FS (bottom) monitored by electrophoresis on a denaturing polyacrylamide gel (RNAs were 32P 5′ end-labelled). The OMeFSPt–53FS RNA adduct is indicated by an arrowhead to the right. The incubation time is given at the top of each lane. (b) Autoradiographs shown in (a) were quantitated by INSTANTIMAGER analysis and the crosslinked RNA was plotted versus time for 53FS (circle) and 28FS RNA (square).
Alkaline hydrolysis of the RNA–oligonucleotide conjugate was performed in order to determine the position of the platinum interstrand crosslink between OMeFSPt and 53 FS RNA. Whereas a ladder of bands was observed with both 53FS RNA and the 53FS RNA–OMeFS oligonucleotide complex, only the short fragments up to U(20) were detected for the 53FS RNA–OMeFSPt mixture, indicating that interstrand crosslinking took place at the A(21) position (Fig. 3). Breakdown products resulting from hydrolysis at positions downstream of U(20), i.e. beyond A(21), migrated slowly as a consequence of their conjugation to the oligonucleotide. This result was confirmed by diethylpyrocarbonate footprinting: the signal corresponding to acylation of the A(21) residue was missing following piperidine treatment of the 53FS RNA–OMeFSPt product (not shown).
Figure 3.
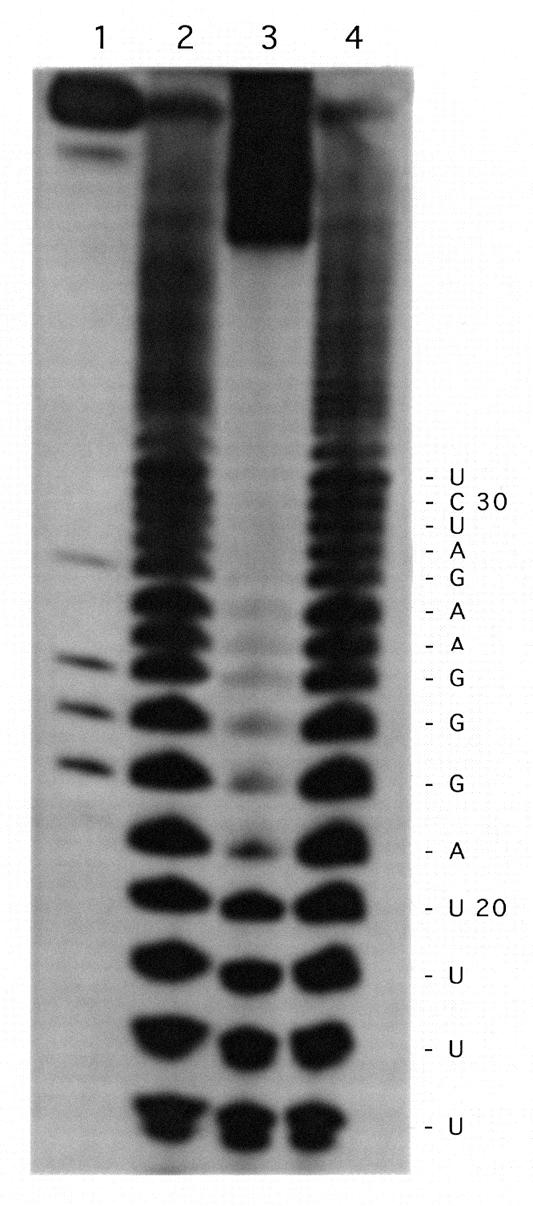
Determination of the Pt-crosslinked residues on the target RNA. 32P 5′ end-labelled 53FS RNA alone (lane 2), mixed with OMeFSPt (lane 3) or with OMeFS (lane 4) was submitted to alkaline hydrolysis and run on a denaturing polyacrylamide gel. Lane 1 corresponds to the RNase T1 digest of 53FS RNA which was used to determine the RNA sequence given to the right.
Inhibition of in vitro translation
The effect of the oligonucleotide OMeFSPt on translation was assessed using the different constructs presented in Table 1. In the first series of experiments the RNA of interest was pre-incubated with OMeFSPt prior to being added to the rabbit reticulocyte lysate. Under these conditions a strong and specific inhibition of protein synthesis was observed: luciferase activity from pGFS(0) was reduced by >80% in the presence of 0.5 µM OMeFSPt whereas a reduction of <10% was seen using pGEM luc control RNA which does not contain the frameshift signal (not shown). The inhibition of luciferase was lower when the oligonucleotide was added directly to the translation mixture containing the RNA (i.e. without pre-incubation). But the reduction of luciferase activity remained concentration-dependent and specific of the added oligonucleotide indicating that it was merely due to the association of the platinated-oligomer with its RNA target (Fig. 4). The luciferase activity was still reduced (20% in the presence of 5 µM OMeFSPt) when the oligonucleotide was added to a reticulocyte lysate in which the translation of pGFS(0) mRNA was running for 5 min or more, showing that the oligonucleotide was able to compete with the ongoing translation reaction.
Figure 4.

Effect of OMeFSPt on the in vitro translation of luciferase transcripts prepared from pGEM (triangle), pGFS(–1) (square) or pGFS(0) (circle). The platinated oligonucleotide was added to the rabbit reticulocyte lysate without pre-incubation with the mRNA (see text). Inhibition was calculated as indicated in Materials and Methods. Each data point is the average of three independent experiments.
Despite a lower yield of translation due to the frameshift efficiency (from 10 to 20% depending on the batch of reticulocyte lysate) the effect of OMeFSPt on pGFS(–1) was similar to that on pGFS(0) under the different conditions described above (Fig. 4 and data not shown). Therefore the oligomer was acting on translation elongation. The non-platinated control oligomer OMeFS did not induce any effect on the translation of the various constructs, whatever the conditions used, demonstrating that the reduction of the luciferase activity observed with OMeFSPt resulted from the covalent crosslink of the oligonucleotide to the target RNA.
Inhibition of in vitro reverse transcription
As the target sequence is available on the viral RNA, the oligomer can potentially interfere with reverse transcription. We investigated the effect of the platinated antisense oligomer on in vitro reverse transcription by the HIV-1 enzyme. The experiment shown in Figure 5 was performed using a transcript prepared from pGFS(–1) as a template. Increasing the OMeFSPt concentration from 0 to 2 µM decreased the synthesis of full-lengh cDNA to ~15%. This effect was specific as no abortive cDNA could be observed when OMeFS was added, or when RNA issued from pGEM luc, which did not contain the frameshift signal, was used as template (not shown).
Figure 5.
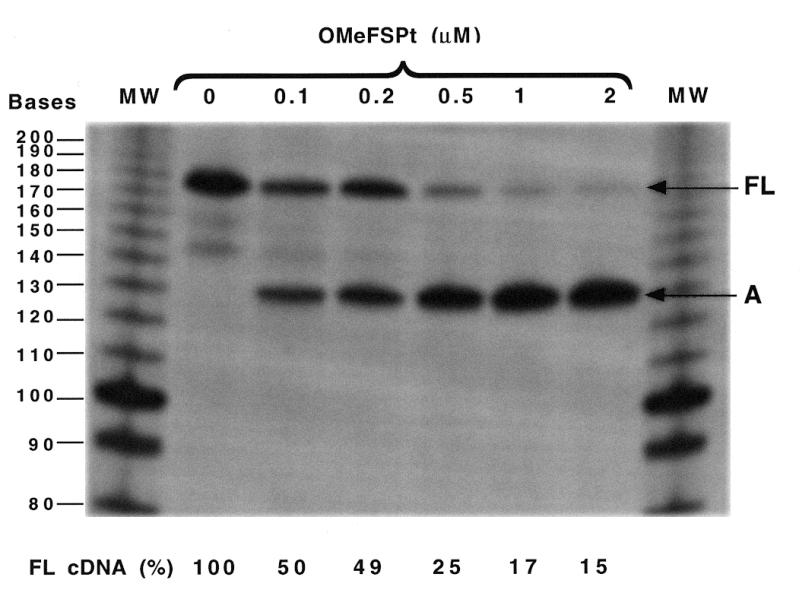
Effect of OMeFSPt on the in vitro reverse transcription of pGFS(–1) RNA by the HIV-1 reverse transcriptase. The 174 nt long full-length cDNA (FL) and the abortive cDNA fragment (A) obtained at different oligonucleotide concentrations (indicated at the top) are marked to the right. The relative intensity of the band corresponding to the full-length product (FLcDNA), determined by INSTANTIMAGER analysis, is given at the bottom for each oligonucleotide concentration.
Inhibition of translation in cultured cells
To evaluate the potency of OMeFSPt in live cells, RNAs obtained by in vitro transcription of pGFS(–1), pGFS(0) or pGEM were transfected in NIH 3T3 and in Vero cells with various amounts of OMeFSPt. For standardisation the transfection efficiency cells were simultaneously transfected with a transcript derived from pRL-SV40 which encodes the Renilla luciferase and does not contain the frameshift signal. Specific inhibition was obtained in NIH 3T3 cells at very low oligonucleotide concentration, 50% inhibition being reached with ~5 nM of oligonucleotide (Fig. 6a). Similar results were obtained with Vero cells: the extent of inhibition was even higher than with NIH 3T3 cells (Fig. 6b). Such an effect, although of lower amplitude, was still observed when OMeFSPt was transfected 1 h prior to the target RNA. The recipient cells were first transfected by a mixture containing the oligonucleotide at the desired concentration in the presence of DMRIE-C reagent (6 µl for 200 µl of oligonucleotide dissolved in 200 µl OPTIMEM). The cells were secondarily rinsed by 500 µl of OPTIMEM medium prior to RNA transfection as described in Materials and Methods. About 1 µM oligonucleotide was needed to induce a 50% reduction of luciferase activity in this case (not shown). Therefore OMeFSPt–RNA crosslink did not need to be pre-formed; the intrastrand→interstrand crosslink conversion took place in the cell. No difference was observed between the two frameshifting signal-containing constructs [pGFS(0) and pGFS(–1)] but no effect was seen with the control RNA derived from pGEM, underlining the specificity of the inhibition.
Figure 6.


Effect of OMeFSPt on the translation of luciferase RNA-transfected cells. (a) NIH 3T3 cells, (b) Vero cells. OMeFSPt was co-transfected with RNA obtained from pGEM (triangle), pGFS(0) (circle) or pGFS(–1) (square) under the conditions described in Materials and Methods. The data points are the average of three independent experiments.
In order to evaluate the ability of OMeFSPt to inhibit the in situ translation of a neo-synthesised RNA, OMeFSPt was co-transfected with plasmids pRL-SV40 and either pAC1789 or pAC1515. The former was a control Renilla reporter. The latter contained the firefly luciferase, downstream of a frameshift signal of either HIV-1 (pAC1789) or of HTLV-I (pAC1515) under the control of a eukaryotic promotor (Table 1). The transplatin oligonucleotide induced a modest but significant decrease of luciferase activity: 50% inhibition was observed at ~1 µM of oligonucleotide with the HIV-1 construct whereas no decrease (or even a moderate increase) was detected when the firefly luciferase was placed downstream of the slippery sequence of the gag–pro frameshifting signal of the virus HTLV-I (Fig. 7).
Figure 7.

Effect of OMeFSPt on the expression of luciferase from plasmid DNA-transfected cells. NIH 3T3 cells were co-transfected with the platinated oligonucleotide and with a luciferase construct under the control of the HIV gag–pol frameshifting motif (pAC1789; black bars) or of the HTLV-1 gag–pro slippery sequence (pAC1515; hatched bars). The results are the average of three independent experiments.
DISCUSSION
The platinated oligo(2′-O-methyl ribonucleotide) OMeFSPt crosslinked very efficiently the RNA motif responsible for translation frameshifting on the HIV-1 mRNA. The particular conformation of the oligonucleotide–RNA hybrid, in the region containing the transplatin-modified triad GUG of the oligomer, allowed the interconversion of the intrastrand crosslink with a half-life of ~2 min. As a consequence a specific oligonucleotide–RNA conjugate was formed which involved on the RNA side the mismatched adenine facing the GUG motif (Figs 1 and 3). These results are in agreement with previous studies according to which the sequences in the reactive centre of both the oligonucleotide and the RNA were optimised (12,13). The above results demonstrate that the rearrangement reaction was not sensitive to the surrounding sequences: in the present study the crosslinked target residue was located downstream of a U6 stretch and upstream of a G3 motif which lead to helical portions of different stability and geometry.
The chosen RNA target which folds into a stem–loop structure is a key element for retroviral gene expression. In a previous study it was demonstrated that this RNA structure might extend beyond the 12 bp stem of the perfect hairpin (11). Alternatively a pseudoknotted structure was also suggested (15). In any case the intramolecular interactions in which the target sequence of the antisense molecules might be involved were efficiently competed out by the 2′-O-methyl oligomers: both the control and the platinated oligonucleotides were indeed able to invade the putative RNA structure as demonstrated by the band shift assay and by the half-life of the intrastrand crosslink, respectively.
In vitro inhibition of reverse transcription by oligo(2′-O-methyl ribonucleotide) has been reported on different RNA templates read by avian (AMV) or human (HIV) reverse transcriptases (16,17). At variance with these reports, the 19mer used in this study (OMeFS) did not prevent cDNA synthesis by the HIV reverse transcriptase. These contradictory results might be due to the relative stability of RNA–oligonucleotide hybrids, in particular the central mismatches in the template RNA–OMeFS complex which weaken the duplex. The modification of the oligo(2′-O-methyl ribonucleotide) by transplatin converted the inactive OMeFS into an efficient inhibitor of reverse transcription as 50% inhibition was achieved at ~100 nM oligonucleotide; i.e. at a 1/4 RNA/OMeFSPt ratio. Reverse transcriptase could not bypass template RNA adducts as shown for other types of reagents (18,19).
The unplatinated oligonucleotide OMeFS did not prevent in vitro translation either. This was not unexpected as chemically modified antisense sequences such as alpha or 2′-O-methyl analogues, targeted to the coding region did not inhibit protein synthesis (20,21). Even strong binders like PNA and N3′→5′ phosphoramidates do not arrest the elongating ribosome (22,23). Previous studies have shown that unless the antisense oligonucleotide-sense RNA hybrid is a substrate for RNaseH (24), crosslinking is the only way to block protein synthesis with an oligomer targeted downstream the AUG initiation codon (25,26). Compared to these compounds the rearrangement of transplatin intrastrand crosslinks displays properties of interest in the frame of antisense studies. On the one hand it does not need to be triggered by an external agent such as UV light. On the other hand, it is stable until the platinated oligonucleotide binds to the target sequence. This also accounts for the specificity of the observed effects.
Indeed, the oligomer OMeFSPt selectively inhibited the translation of a reporter gene under the control of the HIV frameshift RNA signal. This was demonstrated both in a cell-free medium (rabbit reticulocyte lysate) and in live NIH 3T3 (or Vero) cells. Interestingly, the oligonucleotide–RNA intermolecular adduct did not need to be pre-formed. The inhibition of luciferase activity was still observed, albeit with a reduced efficiency, when the addition of the oligonucleotide was delayed with respect to that of the mRNA in the reticulocyte lysate (Fig. 4). This indicates that the binding of the oligomer and the crosslinking reaction can kinetically compete with the translation process. This was also true in intact cells: transfection of the oligomer prior to that of the luciferase transcript or co-transfection with the plasmid DNA led to a selective and significant reduction of luciferase activity. This also means that both partners move to the same compartment and that oligonucleotides are not trapped by proteins.
In cultured cells an increased inhibition efficiency of the OMeFSPt was observed upon co-transfection with the RNA compared to the cell-free experiment. We might speculate that the association between RNA and the oligonucleotide was promoted by the cationic lipid used for transfection.
Results of in vitro and ex vivo translation experiments demonstrate that OMeFSPt does not induce any change in the frameshift efficiency. The result obtained with the oligomer OMeFS confirms that 2′-O-methyl oligoribonucleotides which bind 5′ to the stem of the frameshift hairpin have no effect on frameshift ratio (10) suggesting an all or nothing mode of action of the transplatin-modified 2′-O-methyl oligoribonucleotide.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Justine Michel for oligodeoxynucleotide and RNA synthesis. Karine Aupeix-Scheidler was the recipient of a fellowship from the Fondation pour la Recherche Médicale. This work was supported in part by the ‘Agence Nationale de Recherche contre le SIDA’ (M.L. and J.J.T.) and by the ‘Association pour la Recherche contre le Cancer’ (contract 9873 to J.P.R).
REFERENCES
- 1.Jacks T. (1990) Curr. Top. Microbiol. Immunol., 157, 93–124. [DOI] [PubMed] [Google Scholar]
- 2.Levin J.G., Hattfield,D.L., Oroszian,S. and Rein,A. (1993) Reverse Transcriptase. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, pp. 5–31.
- 3.Park J. and Morrow,C.D. (1991) J. Virol., 65, 5111–5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karacostas V., Wolfe,E.J., Nagashima,K., Gonda,M.A. and Moss,B. (1993) Virology, 193, 661–671. [DOI] [PubMed] [Google Scholar]
- 5.Doyon L., Payant,C., Brakier-Gingras,L. and Lamarre,D. (1998) Virology, 193, 661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacks T., Power,M.D., Masiarz,F.R., Luciw,P.A., Barr,P.J. and Varmus,H.E. (1988) Nature, 331, 280–283. [DOI] [PubMed] [Google Scholar]
- 7.Reil H., Kollmus,H., Weidle,U.H. and Hauser,H. (1993) J. Virol., 67, 5579–5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kollmus H., Honigman,A., Panet,A. and Hauser,H. (1993) J. Virol., 68, 6087–6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toulmé J.J., Le Tinévez,R., Boiziau,C. and Dausse,E. (1997) Nucleic Acids Symp. Ser., 36, 39–41. [PubMed] [Google Scholar]
- 10.Vickers T. and Ecker,D.J. (1992) Nucleic Acids Res., 20, 3945–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aupeix K., Le Tinévez,R. and Toulmé,J.J. (1999) FEBS Lett., 449, 169–174. [DOI] [PubMed] [Google Scholar]
- 12.Colombier C., Boudvillain,M. and Leng,M. (1997) Antisense Nucleic Acid Drug Dev., 7, 397–402. [DOI] [PubMed] [Google Scholar]
- 13.Boudvillain M., Guerin,M., Dalbies,R., SaisonBehmoaras,T. and Leng,M. (1997) Biochemistry, 36, 2925–2931. [DOI] [PubMed] [Google Scholar]
- 14.Stahl G., Bidou,L., Rousset,J.P. and Cassan,M. (1995) Nucleic Acids Res., 23, 1557–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du Z., Giedroc,D.P. and Hoffman,D.W. (1996) Biochemistry, 35, 4197–4198. [Google Scholar]
- 16.Boiziau C., Larrouy,B., Sproat,B. and Toulmé,J.J. (1995) Nucleic Acids Res., 23, 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boulmé F., Freund,F., Moreau,S., Nielsen,P., Gryaznov,S., Toulmé,J.J. and Litvak,S. (1998) Nucleic Acids Res., 26, 5492–5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ericson G. and Wollenzien,P. (1988) Anal. Biochem., 174, 215–223. [DOI] [PubMed] [Google Scholar]
- 19.Chu B.C.F. and Orgel,L.E. (1989) Nucleic Acids Res., 17, 4783–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boiziau C., Kurfurst,R., Cazenave,C., Roig,V., Thuong,N.T. and Toulmé,J.J. (1991) Nucleic Acids Res., 19, 1113–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johansson H.E., Belsham,G.J., Sproat,B.S. and Hentze,M.W. (1994) Nucleic Acids Res., 22, 4591–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knudsen H. and Nielsen,P.E. (1996) Nucleic Acids Res., 24, 494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gryaznov S., Skorski,T., Cucco,C., Skorska,M.N., Chiu,C.Y., Lloyd,D., Chen,J.K., Koziolkiewicz,M. and Calabretta,B. (1996) Nucleic Acids Res., 24, 1508–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toulmé J.J. and Tidd,D. (1998) In Crouch,R.J. and Toulmé,J.J. (eds), Ribonucleases H. John Libbey, Paris, pp. 225–250.
- 25.Boiziau C., Boutorine,A.S., Loreau,N., Verspieren,P., Thuong,N.T. and Toulmé,J.J. (1991) Nucl. Nucl., 10, 239–244. [Google Scholar]
- 26.Gee J.E., Robbins,I., van der Laan,A.C., van Boom,J.H., Colombier,C., Leng,M., Raible,A.M., Nelson,J.S. and Lebleu,B. (1998) Antisense Nucleic Acid Drug Dev., 8, 103–111. [DOI] [PubMed] [Google Scholar]