

Abstract
Glycogen synthase kinase 3β (GSK-3β) is a serine/threonine kinase and an attractive therapeutic target for Alzheimer’s disease. Based on proteolysis-targeting chimera (PROTAC) technology, a small set of novel GSK-3β degraders was designed and synthesized by linking two different GSK-3β inhibitors, SB-216763 and tideglusib, to pomalidomide, as E3 recruiting element, through linkers of different lengths. Compound 1 emerged as the most effective PROTAC being nontoxic up to 20 μM to neuronal cells and already able to degrade GSK-3β starting from 0.5 μM in a dose-dependent manner. PROTAC 1 significantly reduced the neurotoxicity induced by Aβ25–35 peptide and CuSO4 in SH-SY5Y cells in a dose-dependent manner. Based on its encouraging features, PROTAC 1 may serve as a starting point to develop new GSK-3β degraders as potential therapeutic agents.
Keywords: proteolysis targeting chimeras, glycogen synthase kinase 3β, Alzheimer’s disease, chemical knockdown, protein degradation
Introduction
Glycogen synthase kinase 3β (GSK-3β) is a highly conserved serine/threonine kinase ubiquitously expressed and constitutively active.1 Being involved in crucial signaling pathways (such as PI3K, Wnt, Hedgehog, Notch) and regulating a wide spectrum of cellular functions, it is highly implicated in a series of diseases such as cancer, diabetes, and inflammatory, immune, and neurological disorders.1 Regarding neurological conditions, GSK-3β controls a multitude of central nervous system (CNS)-specific signaling pathways related to development, metabolic homeostasis, neuronal growth, and differentiation,2 and it is found to be hyperactivated in the brain of Alzheimer’s disease (AD) patients.3 Compelling evidence supports GSK-3β as the main kinase involved in AD pathology, being implicated in tau- and Aβ-mediated toxicities as well as in oxidative stress, inflammation, memory formation, and synaptic plasticity.4 Furthermore, GSK-3β is networked with several other factors involved in AD.5 In light of this, GSK-3β represents a promising drug target and a multitude of inhibitors have been developed, some of which have reached clinical studies.6 Based on the mechanism of action, such inhibitors can be categorized into ATP competitive and non-ATP competitive inhibitors.7
Recently, beyond classical target inhibition, a new paradigm based on so-called proteolysis targeting chimeras (PROTACs) is in the spotlight.8 This revolutionary modality uses small-molecule PROTACs to control protein levels rather than modulating its function. Indeed, PROTACs do not inhibit a given protein of interest (POI) but instead induce its removal by binding to it and by harnessing the cell disposal ubiquitin–proteasome system (UPS). Based on this mechanism of action, PROTACs catalytically remove different quantities of proteins through multiple rounds of activity and trigger potent effects even at low doses. As such, many issues associated with classical small molecule inhibitors, such as drug resistance and adverse effects, could be avoided.9,10 Since the first report almost 20 years ago, more than 1000 different PROTACs have been described, and some of them have entered clinical trials.11
Last year, two research groups independently reported about PROTACs targeting GSK-3β.12,13 These two PROTACs turned out to be able to induce GSK-3β degradation in cells, and one of them was also effective in an AD mouse model.12
Owing to the availability of an arsenal of GSK-3β inhibitors and our interest in GSK-3β for AD,5,14,15 we sought to develop new PROTACs characterized by different and previously unexplored GSK-3β recruiting elements. Particularly, we were interested to evaluate whether any difference could be observed between an ATP competitive and non-ATP competitive GSK-3β engagement. Considering this insight, in this Letter, we report the preliminary design, synthesis, and evaluation of these novel GSK-3β-directed PROTACs (Figure 1a).
Figure 1.

(a) Design strategy leading to GSK-3β-directed PROTAC 1–4. Black circles represent tethering points. (b) MD-extracted structure of the 1–GSK-3β (left) and 3–GSK-3β (right) complexes showing H-bond contacts with kinase residues.
Results and Discussion
Drug Design
From a medicinal chemistry point of view, PROTACs are heterobifunctional molecules consisting of a POI-binding ligand, connected via a linker to a recruitment moiety for an E3 ubiquitin ligase, leading to polyubiquitination and degradation of the POI.16 To develop such PROTACs we focused our attention on compounds SB-216763 and tideglusib as GSK-3β recruiting elements (Figure 1). We selected these compounds because they are characterized by two different mechanisms of inhibition. SB-216763 is the prototype of reversible maleimide-based ATP-competitive inhibitors able to block GSK-3β with high selectivity,17 while tideglusib selectively inhibits GSK-3β, with a non-competitive inhibition pattern with respect to ATP.18 Analyzing the structure–activity relationships (SAR) of SB-216763, it turned out that the indolyl nitrogen may be substituted with groups bulkier than methyl without significant activity reduction.7 Similarly, we observed that a chain may be introduced at the phenyl group para-position of tideglusib, by means of amide-bond connection, without significant loss of the inhibitory activity (unpublished results).
Therefore, we identified these two positions, i.e., the indolyl nitrogen of SB-216763 and the para-position of the phenyl ring of tideglusib, as the tethering site to the E3 ligase recruiting element. As cereblon (CRBN) E3 ligase recruiter, pomalidomide was selected. Since the degrading potential of PROTACs depends on their ability to form a ternary complex with the POI and the ligase, we hypothesized the initial use of two linkers of different length, as 3–4–3 and 2–2–2-poly(ethylene glycol), designing compounds 1–4 (Figure 1a). We selected PEG linkers since they are by far the most common motifs incorporated into PROTACs due to their favorable properties, in terms of synthetic accessibility, flexibility, availability, and physicochemical profile.19 To support such choice, we performed computational analysis to assess whether the chosen linkers were able to project the E3 ligase-binding element outside the POI binding site.
First, to have indications about the possible conformations that the designed PROTACs would assume in solution, we submitted 1–4 to 200 ns long MD simulations in replicate.20,21 Given the presence of several aromatic rings in pomalidomide, SB-216763, and tideglusib, it is likely that the formation of extended stacking interactions leads to bent PROTAC conformations, less able to interact with the corresponding targets. The analysis of intramolecular H-bonds, solvent accessible surface area (SASA; Figure 1–3SI), and radius of gyration (Rg) values (Figure 4SI) pointed out that PROTACs 1 and 2, featuring a longer linker, might adopt a more open and extended conformation compared to 3 and 4, carrying a shorter linker. Next, to investigate the orientation of the designed PROTACs at GSK-3β, molecular modeling studies were performed. SB-216763 is known to bind the protein orthosteric site,17 and more importantly, the X-ray structures of GSK-3β complexed with maleimide-based inhibitors similar to SB-216763 (PDB codes 1r0e and 1q4l) are available.22 Thus, we docked SB-216763-based PROTACs 1 and 3 at GSK-3β, observing that the compound core was able to properly fit the binding site, forming a bidentate H-bond with the Asp133 and Val135 backbone, plus hydrophobic contacts with Ile62, Val70, Lys85, Val110, Leu188, and Cys199. The linker and the pomalidomide moiety can assume different poses, spanning the protein outside surface. The best obtained 1–GSK-3β and 3–GSK-3β complexes (in terms of number of formed interactions and docking scores) were then submitted to 200 ns long plain molecular dynamics (MD) simulations to verify and compare the ligand behavior, according to the different linker lengths. As shown in Figure 1b, the SB-216763 core is able to form H-bonds and hydrophobic contacts in the binding site. The linker gains additional contacts with Tyr134 for compound 1 and with Pro136 and Arg141 for 3 (Figure 1b). The results of the in silico studies might suggest a higher propensity for compound 1, because of the longer linker, to orient the pomalidomide moiety toward the kinase β-hairpin loop, interacting with Lys74 and with Gly79 also during MD simulations (Figure 1b, left panel). In contrast, compound 3’s shorter linker does not allow the pomalidomide moiety to reach the β-hairpin loop, remaining for most of the simulation trapped in a cavity lined by Arg141, His145, and Arg148 (Figure 1b, right panel). Furthermore, the proximity of the pomalidomide moiety to the β-hairpin loop in 1–GSK-3β complex might suggest 1 to be more likely to bind CRBN, stabilizing a ternary complex. Similarly to the β-hairpin loop of CK-1α reported to bind the carbon terminal domain of CRBN (PDB ID: 5fqd)23 leading to CK-1α degradation,23,24 we might suppose the β-hairpin loop could play a similar role for GSK-3β, which shares a high 3D structure similarity with CK-1α. Unfortunately, we could not perform the same investigation on tideglusib-derived PROTACs 2 and 4, as no experimental data or models have unequivocally defined the binding mode of tideglusib with GSK-3β25 and no crystal structures have been solved.
Synthesis
Designed compounds 1–4 have been synthesized following the sequences reported in Schemes 1 and 2, starting with the synthesis of fragments 8 and 14. For the synthesis of SB-216763-like fragment 8, commercially available indole 5 was alkylated with tert-butyl bromoacetate in potassium carbonate to give 6 that was then treated with ethyl chlorooxoacetate to give glyoxalate 7. Perkin-type condensation between 7 and 2-(2,4-dichlorophenyl) acetamide generated the desired deprotected maleimide 8. To obtain tideglusib-like fragment 14, we started from trifluoroacetamide derivative 9(26) that was protected at the carboxylic acidic functionality by treatment with tert-butanol to obtain 10. Basic hydrolysis of the trifluoroacetamide protecting group generated the free primary amino ester 11 that was transformed in isothiocyanate 12 following treatment with 1,1′-thiocarbonyl-di-2(1H)-pyridone. The thiadiazolidinone 13 was obtained via a straightforward cyclization/oxidation sequence involving first a cyclization between isothiocyanate 12 and the commercially available naphthyl isocyanate in the presence of sulfuryl chloride and then the oxidation of the obtained product under atmospheric oxygen. The thiadiazolidinone 13 was then deprotected in trifluoroacetic acid to obtain fragment 14 ready to be coupled via linker to the E3 ligase recruitment element.
Scheme 1. Synthesis of the Precursors 8 and 14.
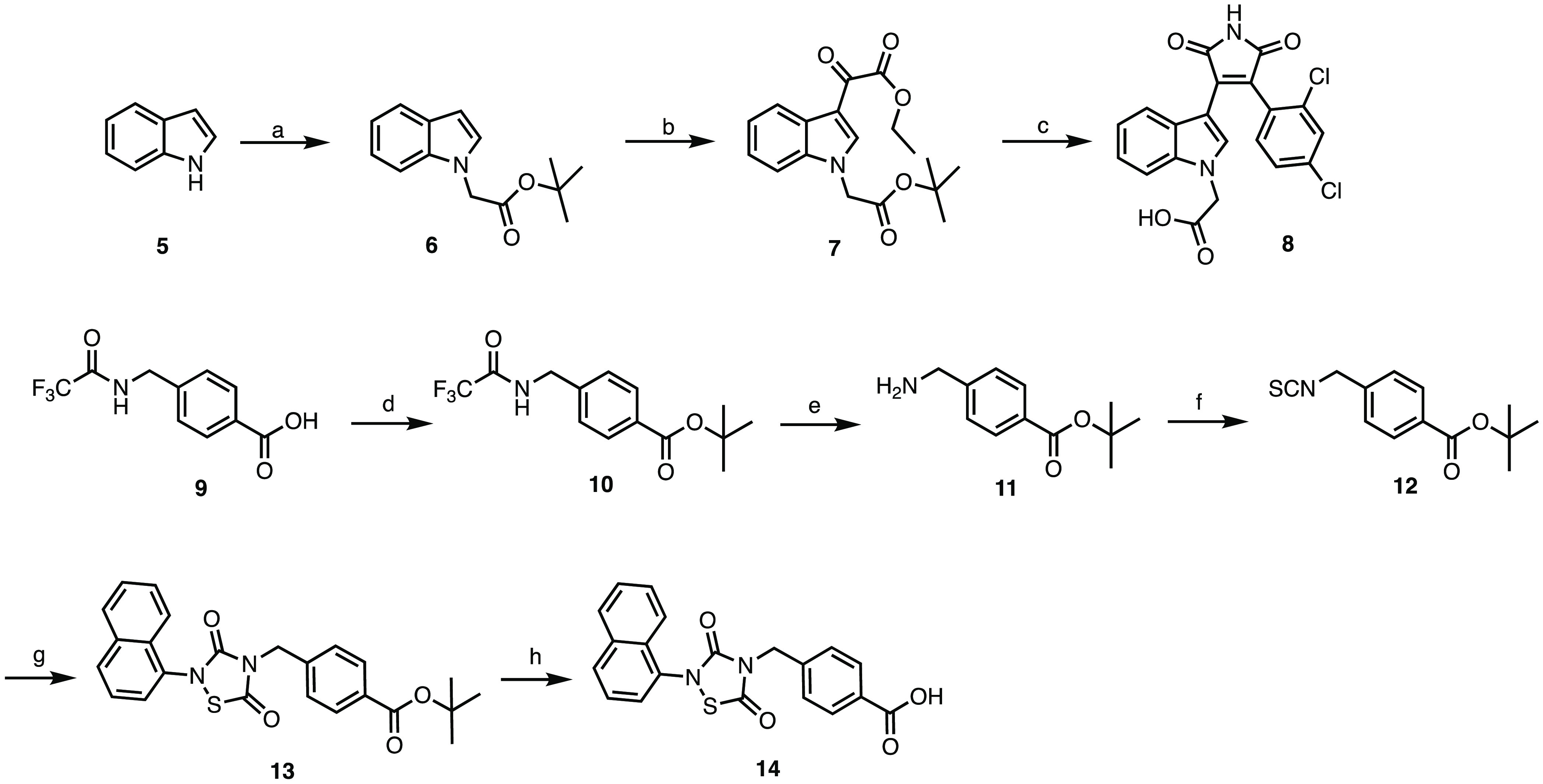
Reagents and conditions. (a) tert-butyl bromoacetate, K2CO3, acetone, reflux, 12 h, 50% yield; (b) ethyl chlorooxoacetate, diethyl ether, rt, 12 h, N2, 42% yield; (c) 2-(2,4-dichlorophenyl)acetamide, KOtBu, DMF, rt, 12 h, N2, 39% yield; (d) tert-butanol, EDCI, DMAP, THF, rt, 12 h, 77% yield; (e) K2CO3, H2O/MeOH, rt, 12 h, 79% yield; (f) 1,1′-thiocarbonyldi-2(1H)-pyridone, DCM, rt, 12 h, 55% yield; (g) naphthyl isocyanate, sulfuryl dichloride, THF, rt, 12 h, N2 then air, THF, rt, 30 min, 57% yield; (h) TFA, DCM, rt, 12 h, 81% yield.
Scheme 2. Synthesis of the Target Compounds 1–4.

Reagents and conditions. (a) 8, EDCI, HOBT, DIPEA, DMF, rt, 12 h, 32% yield for 1, 25% yield for 3; (b) 14, EDCI, HOBT, DIPEA, DMF, rt, 12 h, 48% yield for 2, 44% yield for 4.
EDCI/HOBT assisted amide formation between the common intermediates 15(27) and 16(27) and the GSK-3β recruiting elements 8 and 14 led to the target compounds 1–4 (Scheme 2).
Biological Evaluation
To check whether the structural modification generated by introducing the ligase recruiting element on both inhibitor scaffolds would lead to impaired target recognition, we evaluated the ability of 1–4 and parent inhibitors (SB-216763 and tideglusib) to block GSK-3β activity (Table 1).
Table 1. Effects of Compounds 1–4, SB-216763, and Tideglusib on GSK-3β Activity.

IC50 value is defined as the drug concentration that reduces by 50% the target activity. The results are reported as the mean value of at least two determinations each carried out in duplicate.
Compounds displayed anti-GSK-3β activity spanning from micromolar to nanomolar scale. Activity was clearly influenced by GSK-3β warhead and linkers. The structural modification introduced on the maleimide scaffold is highly detrimental for GSK-3β inhibitory activity: the PROTACs 1 and 3 are characterized by IC50 values (1: 833 nM; 3: 457 nM) which are <1 order of magnitude higher than the starting inhibitor SB-21676 (20 nM). Interestingly, the length of the linker has an impact on the interaction with GSK-3β, since compound 1, characterized by the 3–4–3 PEG linker, is less active than compound 3, characterized by the shorter 2–2–2 linker. On the other hand, the introduction of the linker–E3 ligase recruiting moiety on tideglusib significantly increases the inhibitory activity in the case of 3–4–3 PROTAC (2), when tested in the same assay conditions (2: 79.26 nM; tideglusib: 200 nM). This does not apply to 2–2–2 PROTAC 4, where the structural modification has no significant impact on the inhibitory activity (IC504: 177.56 nM). Overall, the inhibitory profiles displayed by the four PROTACs seem sufficient to engage GSK-3β and allow the formation of the ternary GSK-3β–PROTAC–E3 ligase complex, which is necessary to trigger the degradation process. Indeed, one of the advantages of PROTACs over classical inhibition is that, while traditional small molecule inhibitors need to form strong interactions with their biological counterparts, PROTACs may only require moderate binding to the POI to catalytically induce its degradation.28
Considering that (neuro)toxicity of AD drug candidates has been a drawback for clinical translation, cytotoxicity of compounds 1–4, SB-216763, and tideglusib was evaluated in neuronal SH-SY5Y cells (Figure 5SI). Viability was measured using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) reduction assay after 24 h of treatment with various concentrations of compounds 1–4, SB-216763 and tideglusib (1.25–40 μM). Compounds 1, 3, 4, and SB-216763 showed cytotoxicity at concentrations higher than 20 μM, while for compound 2 cytotoxicity was detected already at 20 and 40 μM. Before performing the degradation assay, we confirmed that 48 h treatment of SH-SY5Y cells with 1–4 at 10 μM concentration did not significantly modify cell viability (data not shown). Then, the ability of compounds 1–4, SB-216763, and tideglusib to induce the degradation of GSK-3β was evaluated in terms of total GSK-3β protein level decrease by Western blotting (Figure 2a). Inhibitors SB-216763 and tideglusib did not modify the basal level of GSK-3β protein. Among the PROTACs, only 1 and 3 (and not 2 and 4) significantly decreased the total level of GSK-3β protein, suggesting the specific ability of the two SB-216763-derived compounds to degrade GSK-3β protein. Compound 1 showed a higher activity in comparison to 3, maybe due to the formation of a more stable GSK-3β–1–E3 ligase complex (see Computational Analysis). There are many potential reasons why tideglusib-derived 2 and 4 did not induce GSK-3β degradation. The one related to their binding to an allosteric site should be ruled out, since PROTAC efficacy depends on POI recruitment, irrespective of the binding site. In fact, there are several positive examples of allosteric PROTACs, such as the allosteric EGFR degrader reported by Gray et al. in 2020.29 A plausible explanation could rely on the fact that the selected linkers are not the suitable ones, considering the critical role the linker plays on the formation of the ternary complex. Furthermore, the availability (lacking for tideglusib) of an X-ray structure that characterizes the binding mode of the recruiting element in complex with its target is a fundamental prerequisite for PROTAC design and assembly. As a general remark, these data support the view that the selection of a suitable kinase inhibitor, as well as ample structural variations on the linkers, including length, flexibility, and attachment points, are crucial for the development of effective degraders.30 Clearly, a means to answer these questions is to experimentally assess ternary complex formation.31
Figure 2.

Degradation GSK-3β protein by compounds 1–4, SB-216763, and tideglusib in SH-SY5Y cells. (a) GSK-3β protein level after 48 h of treatment with all compounds (10 μM); (b) GSK-3β protein level after 48 h of treatment with compound 1 (0.5–10 μM); (c) GSK-3β protein level after 24 h of treatment with compound 1 (10 μM) and lactacystin (1.25–5 μM). GSK-3β protein level was measured by Western blotting. Data are expressed as mean ± SEM of three independent experiments (**p < 0.01 and ***p < 0.001 vs untreated cells, §§§p < 0.001 vs cells treated with compound 1, at one-way ANOVA with Dunnett or Bonferroni post hoc test).
We further investigated the dose-dependent GSK-3β degradation capacity of 1, by treating SH-SY5Y cells for 48 h at 0.5, 1, 5, and 10 μM concentrations. As shown in the Western blot of Figure 2b, compound 1 significantly decreased GSK-3β protein level at all tested concentrations in a dose-dependent manner, and with a half-maximal degradation (DC50) of 6.22 μM. This seems to support a consistent degradation effectiveness.
To confirm the involvement of the UPS in the GSK-3β degradation, SH-SY5Y cells were treated for 24 h with compound 1 (10 μM) in the presence of increased concentration of lactacystin (1.25–2.5–5 μM), a potent and selective irreversible 20S proteasome inhibitor. As shown in Figure 2c, the treatment with compound 1 in the presence of lactacystin at a concentration of 5 μM had no effect on the total GSK-3β protein level, indicating that the GSK-3β degradation induced by compound 1 involves the UPS. Interestingly, being that GSK-3β degradation induced by 1 is directly proportional to the dose of lactacystin, the involvement of the UPS in the mechanism of degradation seems confirmed.
Several studies have reported that GSK-3β is involved in tau protein phosphorylation and neuronal death in AD.3,4 In this regard, in vitro and in vivo studies have demonstrated the role of copper to exacerbate tau hyperphosphorylation, ultimately contributing to both synaptic failure and neuronal death.32 Based on this evidence, to assess the effect of compound 1 on neuronal death induced by copper, SH-SY5Y cells were incubated for 24 h with compound 1 (0.5–1 μM), SB-216763, and tideglusib (1 μM) in the presence of copper sulfate (CuSO4, 150 μM), and cell viability was measured by MTT assay.14 As shown in Figure 3a, compound 1, SB-216763, and tideglusib significantly counteracted the neurotoxicity induced by CuSO4 at 1 μM. Although we did not check whether copper sulfate induced GSK-3β upregulation, it is encouraging that, remarkably, the neuroprotective effect of 1 was abolished by lactacystin (5 μM) (Figure 6SI). In parallel, the neuroprotective activity of compound 1 was also evaluated against insult from Aβ25–35 peptide, the neurotoxic fragment of Aβ involved in the neuropathology of AD. GSK-3β is aberrantly activated by the presence of Aβ and contributes to neural damage.33 Thus, SH-SY5Y cells were incubated for 2 h with compound 1 (0.5 and 1 μM) and further 3 h with Aβ25–35 peptide (10 μM). At the end of incubation, cell viability was evaluated by MTT assay. The treatment with both concentrations of 1 markedly reduced the neurotoxicity induced by Aβ25–35 peptide, in a dose-dependent manner (Figure 3b).
Figure 3.

Compound 1 reduced the neurotoxicity induced by CuSO4 and Aβ25–35 in SH-SY5Y cells. (a) Cells were incubated for 24 h with compound 1 (0.5–1 μM), SB-216763, and tideglusib (1 μM) in the presence of CuSO4 (150 μM); (b) cells were incubated for 2 h with compound 1 (0.5–1 μM) and further 3 h in the presence of Aβ25–35 (10 μM). At the end of incubation, the cell viability was measured by MTT assay as described in the Methods section. Data are reported as mean ± SEM of three independent experiments (*p < 0.05 and ***p < 0.001 vs cells treated with CuSO4; *p < 0.05 and **p < 0.01 vs cells treated with Aβ25–35 at one-way ANOVA with Dunnett post hoc test).
For AD-directed drugs, the ability to cross the blood–brain barrier (BBB) is a fundamental prerequisite. We tested SB-216763-derived PROTACs 1 and 3 in BBB specific parallel artificial membrane permeability assay (PAMPA-BBB) (Supporting Information Table 1 and Figure 7SI). Compound 1 had an effective permeability (Pe) of 15.33 ± 1.12, while compound 3 had a Pe of 20.68 ± 3.93. Based on these results, we can classify compound 1 as CNS± permeable approaching CNS+ permeability values (Figure 8SI), while 3 is predicted to cross the BBB.
Conclusions
GSK-3β has become one of the most investigated AD targets by both companies and academia, due to its critical roles in cellular homeostasis and in a multitude of neurodegeneration-specific signaling pathways. Despite this, up to now, no GSK-3β inhibitor has been approved for clinical practice. In recent years, the PROTAC paradigm has emerged as a compelling strategy for modulating challenging or traditionally considered “undruggable” targets. Based on this approach, a POI is degraded rather than being simply blocked, leading to a number of advantages over classical inhibition. Following the explosion of the PROTAC paradigm,34 we applied this strategy to the development of GSK-3β-directed degraders based on the structure of two chemically and mechanistically different GSK-3β inhibitors, i.e., SB-216763 and tideglusib. The obtained compounds 1–4, which employ pomalidomide as CRBN E3 ligase targeting element, show a good level of POI engagement (as indirectly evaluated via enzymatic assay). Compound 1, characterized by the SB-216763 recruiting moiety and the 3–4–3 PEG linker, is not toxic and is the most potent degrader of the set, able to induce significant GSK-3β degradation already at 0.5 μM and in a dose-dependent manner. Moreover, by using a specific proteasome inhibitor, we demonstrated that GSK-3β degradation is mediated by the UPS. Finally, PROTAC 1 is effective in two disease cell models: in SH-SY5Y cells, it is able to counteract toxic insults induced by Cu2+ and Aβ25–35 at the low concentrations of 1 and 0.5 μM, respectively.
Above all, these results demonstrated that SB-216763-based PROTACs are potent GSK-3β degraders, and further SAR optimization, together with development of PK–PD relationships, is underway to obtain GSK-3β degraders for preclinical development.
Methods
Procedures for the synthesis of targets compounds 1–4 and their characterization, in vitro and in cell assays, PAMPA-BBB assay, and computational studies are included in the Supporting Information.
Acknowledgments
This work is dedicated to Prof. Carlo Melchiorre, on the occasion of his 80th birthday. This research was supported by the University of Bologna (Grant RFO), University of Turin (SPY_RILO_20_01, SPY_RILO_21_01), and partially by MIUR-FISR2019_03796 “Proteolysis targeting chimeras (PROTACs) to treat leishmaniasis”- PROLEISH.
Glossary
Abbreviations
- Aβ
amyloid β
- AD
Alzheimer’s disease
- CK-1α
casein kinase 1α
- CRBN
cereblon
- GSK-3β
glycogen synthase kinase 3β
- MD
molecular dynamics
- MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide
- PROTAC
proteolysis-targeting chimera
- POI
protein of interest
- UPS
ubiquitin–proteasome system
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.3c00096.
Experimental procedures for chemistry, biology and computational analysis, compounds synthesis and characterization, NMR spectra, and supplementary figures (PDF)
Author Contributions
∇ M.G. and L.P. contributed equally. M.G., A.S., and E.U. performed chemical synthesis, L.P., B.P., F.F., and A.T. designed and performed biological assays and analyzed the data, A.D.S., V.P., P.F., and V.A. designed and performed in vitro assays and analyzed the data, M.B. and F.S. designed and performed computational studies and analyzed the data, A.M. and M.L.B. conceived the idea, supervised the project, analyzed the data, and wrote the manuscript with the contribution of all coauthors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Beurel E.; Grieco S. F.; Jope R. S. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther 2015, 148, 114–131. 10.1016/j.pharmthera.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo-Tello P.; Ortiz-Matamoros A.; Arias C. GSK3 Function in the Brain during Development, Neuronal Plasticity, and Neurodegeneration. Int. J. Alzheimers Dis 2011, 2011, 189728. 10.4061/2011/189728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy K.; Yilmaz Z.; Brion J. P. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol Appl. Neurobiol 2007, 33 (1), 43–55. 10.1111/j.1365-2990.2006.00795.x. [DOI] [PubMed] [Google Scholar]
- Lauretti E.; Dincer O.; Praticò D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim Biophys Acta Mol. Cell Res. 2020, 1867 (5), 118664. 10.1016/j.bbamcr.2020.118664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simone A.; Tumiatti V.; Andrisano V.; Milelli A. Glycogen Synthase Kinase 3β: A New Gold Rush in Anti-Alzheimer’s Disease Multitarget Drug Discovery?. J. Med. Chem. 2021, 64 (1), 26–41. 10.1021/acs.jmedchem.0c00931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arciniegas Ruiz S. M.; Eldar-Finkelman H. Glycogen Synthase Kinase-3 Inhibitors: Preclinical and Clinical Focus on CNS-A Decade Onward. Front Mol. Neurosci 2022, 14, 792364. 10.3389/fnmol.2021.792364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M.; Wang S. L.; Zhu L.; Wu P. Y.; Dai W. B.; Rakesh K. P. Structure-activity relationship (SAR) studies of synthetic glycogen synthase kinase-3β inhibitors: A critical review. Eur. J. Med. Chem. 2019, 164, 448–470. 10.1016/j.ejmech.2018.12.073. [DOI] [PubMed] [Google Scholar]
- Burslem G. M.; Crews C. M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181 (1), 102–114. 10.1016/j.cell.2019.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churcher I. Protac-Induced Protein Degradation in Drug Discovery: Breaking the Rules or Just Making New Ones?. J. Med. Chem. 2018, 61 (2), 444–452. 10.1021/acs.jmedchem.7b01272. [DOI] [PubMed] [Google Scholar]
- Burslem G. M.; Smith B. E.; Lai A. C.; Jaime-Figueroa S.; McQuaid D. C.; Bondeson D. P.; Toure M.; Dong H.; Qian Y.; Wang J.; et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol. 2018, 25 (1), 67–77. 10.1016/j.chembiol.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Békés M.; Langley D. R.; Crews C. M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov 2022, 21 (3), 181–200. 10.1038/s41573-021-00371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu L.; Li S.; Ji L.; Luo S.; Ding M.; Yin F.; Wang C.; Luo H.; Lu D.; Liu X.; et al. Discovery of PT-65 as a highly potent and selective Proteolysis-targeting chimera degrader of GSK3 for treating Alzheimer’s disease. Eur. J. Med. Chem. 2021, 226, 113889. 10.1016/j.ejmech.2021.113889. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Zhou J.; Wang Y.; Liu X.; Xu K.; Xu J.; Feng F.; Sun H. PROTACs suppression of GSK-3β, a crucial kinase in neurodegenerative diseases. Eur. J. Med. Chem. 2021, 210, 112949. 10.1016/j.ejmech.2020.112949. [DOI] [PubMed] [Google Scholar]
- De Simone A.; La Pietra V.; Betari N.; Petragnani N.; Conte M.; Daniele S.; Pietrobono D.; Martini C.; Petralla S.; Casadei R.; et al. Discovery of the First-in-Class GSK-3β/HDAC Dual Inhibitor as Disease-Modifying Agent To Combat Alzheimer’s Disease. ACS Med. Chem. Lett. 2019, 10 (4), 469–474. 10.1021/acsmedchemlett.8b00507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandini A.; Bartolini M.; Tedesco D.; Martinez-Gonzalez L.; Roca C.; Campillo N. E.; Zaldivar-Diez J.; Perez C.; Zuccheri G.; Miti A.; et al. Tau-Centric Multitarget Approach for Alzheimer’s Disease: Development of First-in-Class Dual Glycogen Synthase Kinase 3β and Tau-Aggregation Inhibitors. J. Med. Chem. 2018, 61 (17), 7640–7656. 10.1021/acs.jmedchem.8b00610. [DOI] [PubMed] [Google Scholar]
- Paiva S. L.; Crews C. M. Targeted protein degradation: elements of PROTAC design. Curr. Opin Chem. Biol. 2019, 50, 111–119. 10.1016/j.cbpa.2019.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan M. P.; Culbert A. A.; Cross D. A.; Corcoran S. L.; Yates J. W.; Pearce N. J.; Rausch O. L.; Murphy G. J.; Carter P. S.; Roxbee Cox L.; et al. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 2000, 7 (10), 793–803. 10.1016/S1074-5521(00)00025-9. [DOI] [PubMed] [Google Scholar]
- Domínguez J. M.; Fuertes A.; Orozco L.; del Monte-Millán M.; Delgado E.; Medina M. Evidence for irreversible inhibition of glycogen synthase kinase-3β by tideglusib. J. Biol. Chem. 2012, 287 (2), 893–904. 10.1074/jbc.M111.306472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troup R. I.; Fallan C.; Baud M. G. J. Current strategies for the design of PROTAC linkers: a critical review. Explor Target Antitumor Ther 2020, 1 (5), 273–312. 10.37349/etat.2020.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poongavanam V.; Atilaw Y.; Siegel S.; Giese A.; Lehmann L.; Meibom D.; Erdelyi M.; Kihlberg J. Linker-Dependent Folding Rationalizes PROTAC Cell Permeability. J. Med. Chem. 2022, 65 (19), 13029–13040. 10.1021/acs.jmedchem.2c00877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerakoon D.; Carbajo R. J.; De Maria L.; Tyrchan C.; Zhao H. Impact of PROTAC Linker Plasticity on the Solution Conformations and Dissociation of the Ternary Complex. J. Chem. Inf Model 2022, 62 (2), 340–349. 10.1021/acs.jcim.1c01036. [DOI] [PubMed] [Google Scholar]
- Bertrand J. A.; Thieffine S.; Vulpetti A.; Cristiani C.; Valsasina B.; Knapp S.; Kalisz H. M.; Flocco M. Structural characterization of the GSK-3beta active site using selective and non-selective ATP-mimetic inhibitors. J. Mol. Biol. 2003, 333 (2), 393–407. 10.1016/j.jmb.2003.08.031. [DOI] [PubMed] [Google Scholar]
- Petzold G.; Fischer E. S.; Thomä N. H. Structural basis of lenalidomide-induced CK1α degradation by the CRL4(CRBN) ubiquitin ligase. Nature 2016, 532 (7597), 127–130. 10.1038/nature16979. [DOI] [PubMed] [Google Scholar]
- Asatsuma-Okumura T.; Ito T.; Handa H. Molecular mechanisms of cereblon-based drugs. Pharmacol Ther 2019, 202, 132–139. 10.1016/j.pharmthera.2019.06.004. [DOI] [PubMed] [Google Scholar]
- Balasubramaniam M.; Mainali N.; Bowroju S. K.; Atluri P.; Penthala N. R.; Ayyadevera S.; Crooks P. A.; Shmookler Reis R. J. Structural modeling of GSK3β implicates the inactive (DFG-out) conformation as the target bound by TDZD analogs. Sci. Rep 2020, 10 (1), 18326. 10.1038/s41598-020-75020-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S.; Zang J.; Gao Q.; Liang X.; Ding Q.; Li X.; Xu W.; Chou C. J.; Zhang Y. Design, synthesis and anti-tumor activity study of novel histone deacetylase inhibitors containing isatin-based caps and o-phenylenediamine-based zinc binding groups. Bioorg. Med. Chem. 2017, 25 (12), 2981–2994. 10.1016/j.bmc.2017.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinebach C.; Sosič I.; Lindner S.; Bricelj A.; Kohl F.; Ng Y. L. D.; Monschke M.; Wagner K. G.; Krönke J.; Gütschow M. A MedChem toolbox for cereblon-directed PROTACs. Medchemcomm 2019, 10 (6), 1037–1041. 10.1039/C9MD00185A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng G.; Li D.; Kang Y.; Hou T. Integrative Modeling of PROTAC-Mediated Ternary Complexes. J. Med. Chem. 2021, 64 (21), 16271–16281. 10.1021/acs.jmedchem.1c01576. [DOI] [PubMed] [Google Scholar]
- Jang J.; To C.; De Clercq D. J. H.; Park E.; Ponthier C. M.; Shin B. H.; Mushajiang M.; Nowak R. P.; Fischer E. S.; Eck M. J.; et al. Mutant-Selective Allosteric EGFR Degraders are Effective Against a Broad Range of Drug-Resistant Mutations. Angew. Chem., Int. Ed. Engl. 2020, 59 (34), 14481–14489. 10.1002/anie.202003500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidou M.; Oun A.; Pathak P.; Zhang B.; Wang Z.; Ter Brake F.; Dolga A. M.; Kortholt A.; Dömling A. The tale of proteolysis targeting chimeras (PROTACs) for Leucine-Rich Repeat Kinase 2 (LRRK2). ChemMedChem. 2021, 16 (6), 959–965. 10.1002/cmdc.202000872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casement R.; Bond A.; Craigon C.; Ciulli A. Mechanistic and Structural Features of PROTAC Ternary Complexes. Methods Mol. Biol. 2021, 2365, 79–113. 10.1007/978-1-0716-1665-9_5. [DOI] [PubMed] [Google Scholar]
- Zubčić K.; Hof P. R.; Šimić G.; Jazvinšćak Jembrek M. The Role of Copper in Tau-Related Pathology in Alzheimer’s Disease. Front Mol. Neurosci 2020, 13, 572308. 10.3389/fnmol.2020.572308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo S.; Rivière C.; Piazzi L.; Bisi A.; Gobbi S.; Bartolini M.; Andrisano V.; Morroni F.; Tarozzi A.; Monti J. P.; et al. Benzofuran-based hybrid compounds for the inhibition of cholinesterase activity, beta amyloid aggregation, and abeta neurotoxicity. J. Med. Chem. 2008, 51 (10), 2883–2886. 10.1021/jm8002747. [DOI] [PubMed] [Google Scholar]
- Salerno A.; Seghetti F.; Caciolla J.; Uliassi E.; Testi E.; Guardigni M.; Roberti M.; Milelli A.; Bolognesi M. L. Enriching Proteolysis Targeting Chimeras with a Second Modality: When Two Are Better Than One. J. Med. Chem. 2022, 65 (14), 9507–9530. 10.1021/acs.jmedchem.2c00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.