

Abstract
Chronic alcohol consumption, Alzheimer’s disease (AD), and vascular dementia are all associated with cognitive decline later in life, raising questions about whether their underlying neuropathology may share some common features. Indeed, recent evidence suggests that ethanol exposure during adolescence or intermittent drinking in young adulthood increased neuropathological markers of AD, including both tau phosphorylation and beta-amyloid (Aβ) accumulation. The goal of the present study was to determine whether alcohol consumption later in life, a time when microglia and other neuroimmune processes tend to become overactive, would influence microglial clearance of Aβ(1–42), focusing specifically on microglia in close proximity to the neurovasculature. To do this, male and female Fischer 344 rats were exposed to a combination of voluntary and involuntary ethanol consumption from ~10 months of age through ~14 months of age. Immunofluorescence revealed profound sex differences in microglial co-localization, with Aβ(1–42) showing that aged female rats with a history of ethanol consumption had a higher number of iba1+ cells and marginally reduced expression of Aβ(1–42), suggesting greater phagocytic activity of Aβ(1–42) among females after chronic ethanol consumption later in life. Interestingly, these effects were most prominent in Iba1+ cells near neurovasculature that was stained with tomato lectin. In contrast, no significant effects of ethanol consumption were observed on any markers in males. These findings are among the first reports of a sex-specific increase in microglia-mediated phagocytosis of Aβ(1–42) by perivascular microglia in aged, ethanol-consuming rats, and may have important implications for understanding mechanisms of cognitive decline associated with chronic drinking.
Keywords: aging, amyloid beta, chronic ethanol, microglia, sex differences
Introduction
Alcohol consumption among older individuals is on the rise, particularly in women (Ahlner et al., 2018; Keyes et al., 2019). Some reports suggest that low to moderate alcohol consumption may have a neuroprotective effect, characterized by a decreased risk of cognitive decline, dementia, and increased cardiovascular benefits, whereas chronic and heavy alcohol consumption is detrimental (Kim et al., 2012; Lao et al., 2021; Rehm, Hasan, Black, Shield, & Schwarzinger, 2019). Forty-three% of individuals over the age of 65 report engaging in alcohol consumption, with 10% of individuals over the age of 65 reporting engaging in binge drinking (Blazer & Wu, 2009; Han, Jones, Einstein, Powell, & Compton, 2019). Between 1997 and 2014, prevalence of drinking and binge drinking among elderly women has increased at rates of 1.6% and 3.7% per year, respectively, suggesting elderly women may be increasingly vulnerable to the deleterious effects on neuropathology and cognitive decline relative to men (Breslow, Castle, Chen, & Graubard, 2017). These statistics may not simply reflect generational differences in alcohol use; evidence suggests that alcohol misuse rates are increasing across most sociodemographic groups in the United States, and that the increases were greatest in subgroups including women and older adults within a 12-month period from 2012 to 2013, indicating that these populations are initiating increased alcohol use later in life (Grant et al., 2017).
Alzheimer’s disease (AD) and vascular dementia account for the largest percentage of dementia cases (Iadecola, 2013). AD is an age-related neurodegenerative disorder characterized by aggregation of amyloid-beta (Aβ) and phosphorylated tau, which progressively affects cognition, social and executive function, and memory (Venkataraman, Kalk, Sewell, Ritchie, & Lingford-Hughes, 2017). Though no universally accepted disease origin has been identified, the Aβ hypothesis posits that Aβ plaques develop from insoluble Aβ fragments that accumulate over time, ultimately leading to neuronal death. These plaques arise from deleterious cleavage of the amyloid precursor protein (APP), a transmembrane protein involved in metabolic functions, by β-secretase one (BACE1) and gamma-secretase, which release fragments that are 39–43 residues in length (Nunan & Small, 2000). In support of the Aβ hypothesis, elevated levels of Aβ and APP have been associated with cognitive dysfunction (Mokhtar, Bakhuraysah, Cram, & Petratos, 2013; Näslund et al., 2000).
Vascular dementia is a diagnostic term used to identify a wide array of brain disorders associated with dementia and pathological changes in brain vascular function. The mechanisms of these disorders can be varied; however, they share altered NVU function through factors such as reduced cerebral perfusion volumes (Jellinger, 2013), altered immune cell trafficking (Rossi, Angiari, Zenaro, Budui, & Constantin, 2011), cytokine release (Zuliani et al., 2007), and BBB permeability (Ueno et al., 2016). A shared characteristic of vascular cognitive impairment shared between AD and vascular dementia is the accumulation of Aβ in cerebral blood vessels (Iadecola, 2013). The accumulation of Aβ(1–40) and Aβ(1–42) are both associated with cognitive decline in both AD and vascular dementia, with Aβ(1–42) being expressed at higher levels in vascular dementia patients (Lewis et al., 2006). This accumulation of Aβ has been associated with BBB compromise and subsequent increases in BBB permeability that may then permit increased Aβ access to the brain (Donahue & Johanson, 2008; Hartz et al., 2012). Within the parenchyma of the CNS, Aβ plaques are believed to drive microglial activation (likely to mitigate Aβ infiltration), but ultimately this shift in inflammatory tone may contribute to cytotoxicity and disease pathology (Nelson, Soma, & Lavi, 2002).
Amyloid peptides interact with microglia in the parenchyma, leading to a proinflammatory response that attracts other microglia to the area of damage, as well as contributes to the phagocytosis of Aβ fragments, likely through the activation of TREM2 receptors (Fassbender et al., 2004; Liu et al., 2020; Wang et al., 2015; Zheng et al., 2018). Post mortem brains from AD patients indicate that microglia are reactive in the cortex, and that the stage of reactivity is proportionate to the distance from the Aβ plaque (McGeer, Itagaki, Tago, & McGeer, 1987). This has also been observed in the temporal association cortex, where both microglia and astrocytes co-localized with amyloid plaques in a distance-dependent manner. Interestingly, a larger number of microglia were associated with larger plaques, while number of astrocytes did not change when compared to plaque size, indicating that microglia may have a more active role in Aβ plaque management (Serrano-Pozo et al., 2013).
Phenotypic changes in microglial function induced by alcohol may contribute to the self-perpetuating cycle of inflammatory responses that ultimately moderate Aβ aggregation and disease progression (Venkataraman et al., 2017). Some studies have indicated that following ethanol binge exposure, microglia increase in quantity, phenotypically appear to be partially activated, and increase the release of pro-inflammatory cytokines such as IL-1β and TNF-alpha, though these changes may be species-dependent and require large amounts of ethanol to manifest (Marshall et al., 2013; Marshall, McClain, Wooden, & Nixon, 2020; Zhao et al., 2013). Mice are more sensitive to inflammatory responses compared with rats, and a commonly used mouse model, C57BL/6J, appears to show hyper-inflammatory responses compared with other mouse strains, which may account for species differences in ethanol-induced neuroimmune responses observed across experiments (Boschen et al., 2021).
Partial microglial activation is thought to reflect microglial priming (i.e., maintaining a persistent state of microglial activation in the absence of challenge), resulting in an exaggerated inflammatory response to secondary insult (Norden, Trojanowski, Villanueva, Navarro, & Godbout, 2016). Primary rat microglia cultures treated with 78 mM ethanol had a decreased ability to phagocytose Aβ peptides, suggesting that microglial clearance of Aβ is directly inhibited by the presence of ethanol (Kalinin et al., 2018). Chronic ethanol intake increased APP and BACE1 in the hippocampus and striatum of rats; similarly, chronic exposure to ethanol increased both 1–40 and 1–42 isoforms of Aβ in male mice (Gong et al., 2017; Kim et al., 2011). Taken together, these data suggest that ethanol may increase amyloid deposition load, regardless of animal model. A 3x Tg-AD mouse model demonstrated that heavy ethanol consumption in adulthood increased the onset and magnitude of AD-like pathology, as well as increased Aβ accumulation in the lateral entorhinal and prefrontal cortex, whereas tau pathology was found in the lateral entorhinal cortex, amygdala, and hippocampus (Hoffman et al., 2019). Thus, microglia play an important role in the clearance of amyloid and consequent disease progression, and these effects seem to depend on the level of ethanol exposure. However, less is known about the effect of chronic moderate ethanol consumption on microglia in aging in typical rodent models that lack a transgenic AD phenotype.
The present study had two primary goals. First, we sought to determine whether late-life initiation of ethanol consumption would lead to an escalation of voluntary drinking. To test this, rats were exposed to multiple phases of voluntary and single-bottle ethanol consumption periods, with a final assessment of voluntary ethanol intake in a 2-bottle choice test. Using this varied history of ethanol consumption to mimic more naturalistic, human drinking patterns, our second goal was to investigate whether chronic drinking would alter microglial dynamics and Aβ(1–42) protein in the aging brain in both males and females. This work is especially timely given the potential role of aging-related changes in neuroimmune function that may render the aged brain vulnerable or resistant to the effects of chronic ethanol consumption (Deak & Savage, 2019; Perkins, Vore, Lovelock, Varlinskaya, & Deak, 2019). Immunofluorescence on hippocampal sections was therefore used to assess microglia and Aβ(1–42), with a specific focus on perivascular microglial populations.
Materials and methods
Subjects
Male and female Fischer 344 rats were purchased from Envigo at ~2 months of age, maintained in our colony, and were ~10 months of age at the beginning of testing (n = 10/group, N = 40). All rats were pair-housed in a standard Plexiglas cage with chew blocks provided for enrichment. Rats received ad libitum access to food, and colony conditions were maintained on a 12:12-hour light/dark cycle (lights on 7:00 AM) at 22 ± 1° C. They were handled for 2–3 minutes for 2 days prior to experimentation to acclimate to experimenter handling. Rats housed together were assigned the same experimental condition. Due to the nature of the longitudinal, aging experiment, a few rats were required to be euthanized due to the emergence of late-life illness (n = 5). All experimental subjects were maintained in accordance with PHS policy and the Institutional Animal Care and Use Committee (IACUC) at Binghamton University, and experiments were approved prior to procedures.
Experimental design and procedures
Male and female middle-aged F344 rats were given intermittent access to two bottles (ethanol and water) followed by a single-bottle ethanol consumption procedure. The present experiment took place between approximately 10–18 months of age, equivalent to about 30–45 years old in humans (Sengupta, 2013). For the intermittent 2-bottle choice (IA2BC) procedure, at 10 months of age, experimental subjects were given 4-hour access to either two water bottles or one water bottle and one bottle containing 5% ethanol solution (v/v), three times weekly (2:30 PM–6:30 PM, Mondays, Wednesdays, and Fridays) for a total of 3 weeks (9 exposures) as in our previous work (Doremus-Fitzwater et al., 2014). Ethanol and water were provided in volumetric bottles with metal sipper tubes. Ethanol bottles were prepared from 95% stock ethanol diluted in tap water to a final concentration of 5% (v/v). During the IA2BC phase, both cage mates were present in the cage, therefore total amount consumed by a pair of rats and a sum of their body weights was used for calculation of g/kg ethanol intake or mL/kg water intake.
Next, we moved the rats to an intermittent, single-bottle procedure to increase the level of ethanol intake/exposure and establish a history of ethanol consumption with intermittent periods of abstinence, which are critical for producing long-lasting deficits associated with chronic ethanol intake (Spear, 2020). Rats that received only water in the earlier phase continued to receive water during all phases of experimentation (water group). Each cycle of single bottle access in the ethanol condition consisted of 2 days of ethanol exposure followed by 2 days of water access (see Figure 1A). Rats were weighed prior to presentation of ethanol on the first day of each cycle and ethanol was delivered to the home cage. This model was chosen in order to keep animals pair-housed due to the long-term nature of the studies; isolation stress has been shown to alter drinking patterns in rats, as well as alter neuropathology of Alzheimer’s disease (Becker, Lopez, & Doremus-Fitzwater, 2011; Dong & Csernansky, 2009). Ethanol was given in a concentration of 10% (v/v) for the first 9 cycles of forced consumption. From the 10th cycle, the concentration of ethanol was increased to 20% for an additional 10 weeks. Tail blood samples for assessment of blood ethanol concentration (BEC) were taken on the 2nd day (11:00 PM) of cycle 8 (10% ethanol) and cycle 18 (20% ethanol), approximately 5 hours after lights-off on the second day of the 2-day cycle (~32 hours after ethanol onset). Intermittent single-bottle ethanol exposures continued for a total of 32 cycles (19 weeks). Although consumption was not recorded during the single-bottle 10% ethanol access portion of this experiment, other studies from our lab using the same model indicate that adult males reliably consumed an average of ~13.3 g/kg ethanol per day, whereas adult females consumed an average of ~16.7 g/kg ethanol per day (data not shown), with peak BECs observed within ~1 hour after lights-off (60–90 mg/dL).
Figure 1. Timeline and behavioral characterization of drinking model.
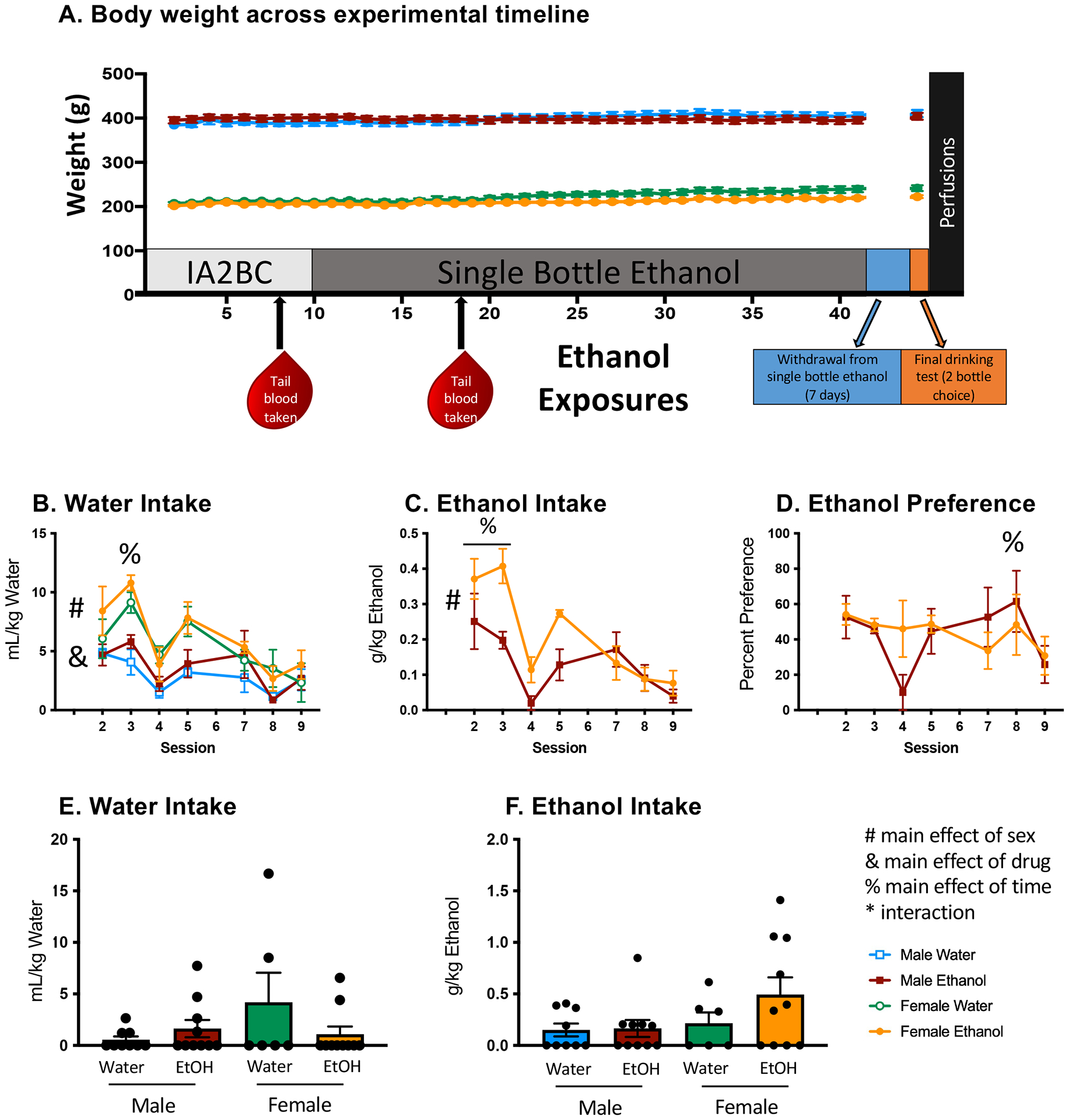
(A) Body weight in rats across experimental timeline; no weight differences were observed as a result of ethanol consumption. (B) Females consumed more water in mL/kg than males (#), with data collapsed across sessions and exposure condition, and ethanol-exposed animals consumed more water than their water-exposed counterparts (&), with data collapsed across sex and session. Water consumption was highest (%) during session 3. (C) Females consumed more g/kg ethanol than males (#), with data collapsed across sessions, and the highest amounts of ethanol (see %) were consumed during sessions 2 and 3 (D) Preference for ethanol was highest during session 8 (%), but lowest during session 9. (E & F) A final, 30-minute acute drinking test was performed to determine whether ethanol preference had developed. No differences in water intake or ethanol intake were observed during the final drinking test. BECs reported in Table 1. All data expressed as mean ± SEM, alpha set to p = 0.05.
Following the intermittent single-bottle procedure, rats underwent a 1-week abstinence period from ethanol. At the end of this abstinence period, all rats were given a 2-bottle choice test to assess voluntary ethanol intake. They were taken from their home cages, placed alone in an opaque-sided Plexiglas bin under low-light conditions, and given access to two volumetric bottles containing ethanol (20% v/v) and water to determine whether a history of ethanol intake in a forced consumption model would result in ethanol preference. Immediately following the drinking test, tail blood samples were taken to determine BECs. On the next day (24 hours following the final drinking test), rats were perfused with 4 °C 0.01 M phosphate-buffered saline (PBS; 0.01 M phosphate buffer, 120 mM NaCl; 2.7 mM KCl), followed by 4 °C paraformaldehyde (4% PFA in 0.1 M PBS), and brains were collected and post-fixed for 4 hours in 4% PFA. Brains were then transferred to sucrose (30% sucrose in 0.01 M PBS) for 3 days to cryoprotect. Brains were then flash-frozen in 2-methylbutane at −20 °C for 2 minutes, then stored at −20 °C until time of sectioning.
Blood ethanol concentration
Tail blood samples were collected for assessment of blood ethanol concentrations (BECs). Plasma was collected from whole blood following refrigerated centrifugation and stored at −20° C until plasma analysis was performed. BECs were measured using Analox AM-1 alcohol analyzer (Analox Instruments, Lunenburg, Massachusetts, United States). Analox was calibrated to a 50 mg% industry standard and quality control was checked every 15 samples against a known concentration (Analox Instruments).
Immunofluorescence
Sections were washed in 0.01 M phosphate-buffered saline three times for 10 minutes each, quenched in 0.6% hydrogen peroxide for 30 minutes and washed again for 5 minutes. Antigen retrieval was completed by bathing sections in 10 mM sodium citrate (pH 8.5) at 80 °C for 30 minutes, followed again by three, 5-minute 0.01 M PBS washes. Sections were then permeabilized for 1 hour at room temperature using a normal donkey serum-based block (5% NDS; 1% Bovine Serum Albumin, 22.52 mg/mL glycine; 0.2% Triton-X; in 1x PBS). Tissue sections were incubated with primary antibody goat anti-rat IBA-1 (Abcam, AB5076, 1:300) and rabbit anti-human beta amyloid (1–42) (ThermoFisher Scientific, 44344, [1:1000]) diluted in blocking buffer (described above) at 4 °C overnight. Sections were washed in PBS before being incubated with secondary antibody donkey anti-goat AF488 (705–545-147, Jackson Immunoresearch, 1:500), donkey anti-rabbit AF680 (A10043, ThermoFisher Scientific, 1:500) diluted in blocking buffer, and stained with lycopersicon esculentum (tomato) lectin conjugated to DyLight 594 (L32471, ThermoFisher Scientific, 1:500) at room temperature for 1 hour. Excess secondary was removed via three 10-minute PBS washes and tissue-mounted to gelatin-coated slides using ProLong Diamond Anti-Fade Mountant with DAPI (P3691, ThermoFisher Scientific).
Micrographs of the hippocampus were collected using Olympus VS200 Slide Scanner (Olympus, United States) at 20× magnification. A DAPI, FITC (AF488), mCherry (Dylight594), and Cy5.5 (AF680) cube were used to capture signal for each secondary antibody. Capture settings were automatically determined using Olympus software algorithm for optimal range, with exposure time manually set to 500 msec maximum. A Z-stack of 20 ± 10 microns was collected for each image with a 1.18-micron interval. Olympus software was used to represent the data across the Z-stack in a single two-dimensional “enhanced focus image” allowing pixel data from across the Z-range to be reflected in one image. Micrographs were imported into HALO FISH v3.1.3 software (Indica Labs) where the amygdala, entorhinal cortex, and CA1, CA2, CA3, and dentate gyrus subregions of the hippocampus were investigated for Iba-1+ microglia, tomato lectin, and amyloid signal (Figure 2). Cells were identified by the presence of DAPI signal and a three-micron radius was artificially placed around each cell. Cells were considered microglia- or amyloid-positive based on the presence of Iba1 and Aβ(1–42) signal, respectively. To determine whether effects of ethanol history specifically influenced perivascular microglia, Iba1+/Aβ(1–42)+ cells within 3 μm of tomato lectin were used to identify microglia in close proximity to the vasculature as a distinct population from microglia more deeply embedded in the CNS parenchyma. All slides were analyzed using the same settings determined by optimizing the capture of all fluorophore signals. Immunofluorescence of tomato lectin was reported as RFU/total area, and percentage of microglia and amyloid beta were reported as percentage of positive cells co-stained with Dapi nuclei.
Figure 2. Assessment of microglia and Aβ(1–42) after chronic ethanol.
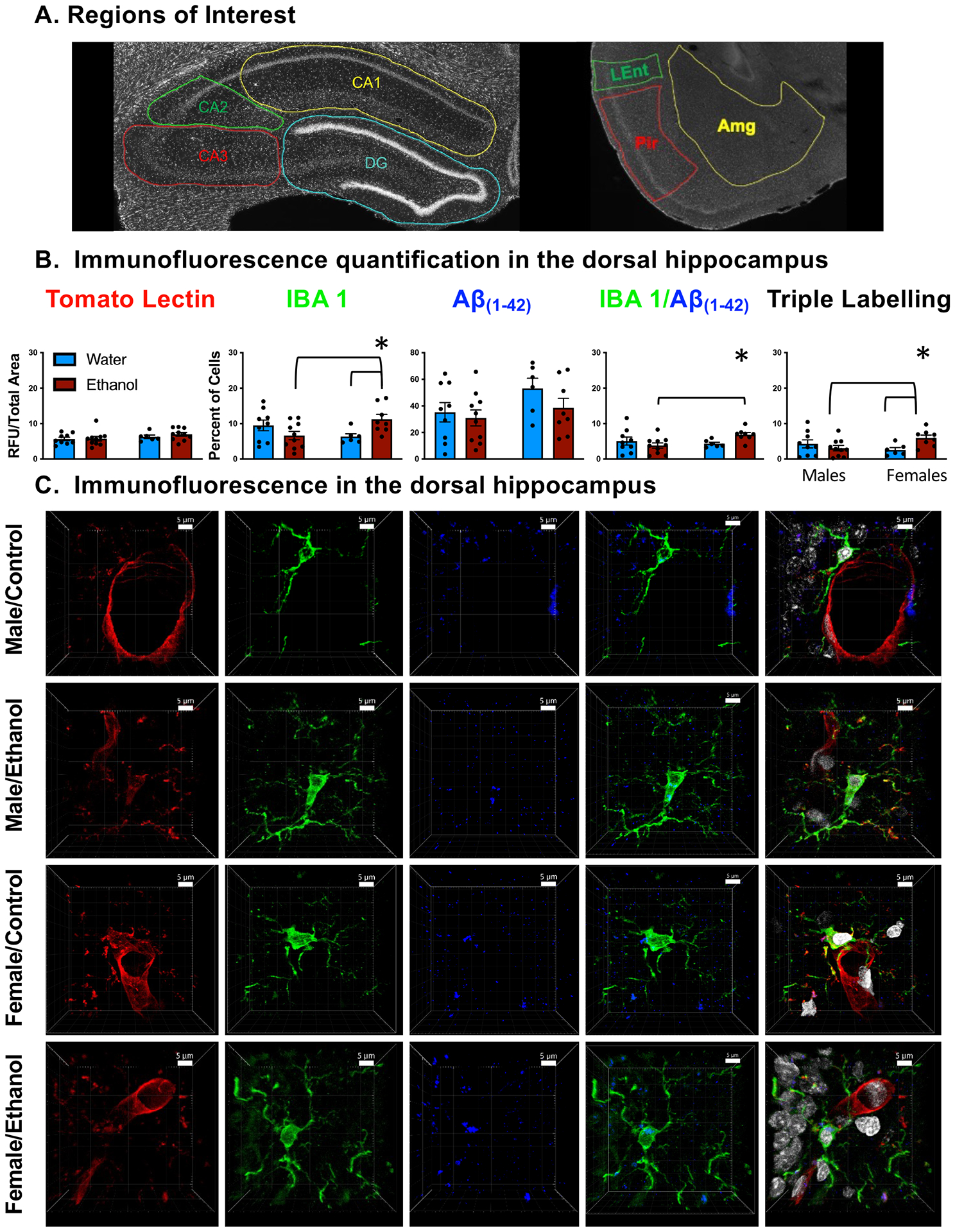
(A) Regions of interest denoted in the dorsal hippocampus (CA1, CA2, CA3, and DG) as well as amygdala (AMG), piriform cortex (Pir), and lateral entorhinal cortex (Lent) (B) Quantification of labeling in the dorsal hippocampus. Females with a history of ethanol had increased Iba-1+ positive cells, Iba-1 / Aβ(1–42) co-localization, and Iba-1 / Aβ(1–42) / tomato lectin triple localization. Asterisk (*) denotes a significant interaction (p < 0.05). (C) Representative images of immunofluorescence in the dorsal hippocampus. All representative images were collected at 40× for better clarity and include a 5-micron scale bar for reference.
Statistical analysis
All behavioral data were analyzed by Statistica (TIBCO Data Science; Palo Alto, California, United States). A repeated-measures ANOVA was used to assess body weight and water consumption with session as a within-subject factor and sex and exposure condition as between-subject factors. For the experimental group, a repeated-measures ANOVA was used to assess ethanol consumption during the IA2BC portion as within-subject and sex as a between-subject factor. Immunofluorescence data were analyzed using GraphPad Prism (GraphPad Software Inc.; California). Separate two-way two-exposure (ethanol, water) × Sex (male, female) ANOVAs were used to assess changes following ethanol exposure, with Tukey’s HSD test used to clarify group differences when a significant interaction was observed. Alpha level was set at 0.05 for all statistical tests.
Results
Mixed ethanol history
As expected, an ANOVA of body weight revealed a main effect of sex (F(1,31) = 802.662, p < 0.01), with males having higher body weights than females. Additionally, animal weight increased with time (F(40,1240) = 45.099, p < 0.01) (Figure 1A). No changes in body weight were observed due to ethanol consumption at any phase of the experiment, suggesting the ethanol was well-tolerated in aged rats (p values > 0.05). When examining consumption of water, we observed a main effect of sex (F(1,15) = 43.604, p < 0.01), with females consuming more water than males. We also observed a main effect of exposure condition (F(1,15) = 5.679, p < 0.05), in that ethanol-exposed rats consumed more water than their water-exposed counterparts (Figure 1B). In the ethanol-consuming animals, we observed a main effect of sex (F(1,8) = 39.6349, p < 0.01), such that females consumed significantly more ethanol than males (Figure 1C). A main effect of session (F(6,48) = 11.538, p < 0.01], was revealed for ethanol intake, with the highest ethanol consumption evident during sessions 2 and 3 (Figure 1C). Finally, ethanol preference differed as a function of session (F(6,90) = 2.453, p < 0.05): percent ethanol preference was highest during session 8 and lowest during session 9 (Figure 1D).
As expected, tail blood samples collected after the 8th cycle revealed a main effect of ethanol exposure (F(1,32) = 10.757, p < 0.01), where rats receiving ethanol had elevated blood ethanol concentrations during the IA2BC procedure (Table 1). Similarly, blood samples collected after the 18th cycle indicated a main effect of exposure (F(1,32) = 8.441, p < 0.01), with elevated BECs during the single-bottle consumption procedure evident in ethanol-exposed rats relative to water-exposed controls (Table 1). In the final drinking test, all rats were given access to both an ethanol bottle and a water bottle. No differences were observed in BECs following the final 2-bottle choice test regardless of ethanol history (Table 1). Similarly, no differences in BECs were noted between groups, suggesting surprisingly limited intake in this brief 2-bottle choice test in aged rats. This seems to indicate that rats with a history of forced ethanol consumption did not develop a preference for ethanol consumption following a period of abstinence (Table 1).
Table 1.
Blood ethanol concentrations (BECs) assessed at various phases of experimentation.
| Males | Females | ||||
|---|---|---|---|---|---|
| Time of Sample | Water | Ethanol | Water | Ethanol | Interaction Term |
| 8th Cycle BEC | 6.52 ± 1.40 | 19.00 ± 4.07 # | 6.55 ± 2.32 | 15.28 ± 3.40 # |
F(1,32) = 10.757, p < 0.01 |
| 18th Cycle BEC | 8.80 ± 0.83 | 17.85 ± 4.35 # | 5.90 ± 1.11 | 14.82 ± 3.02 # |
F(1,32) = 8.441, p < 0.01 |
| Final Drinking Test BEC | 5.84 ± 0.47 | 5.56 ± 0.40 | 6.07 ± 0.90 | 4.75 ± 0.60 |
F(1,31) = 0.788, p > 0.05 |
Values represent mean ± SEM and are provided in mg/dL. A main effect (#) was observed, such that animals consuming ethanol, regardless of sex, had higher BECs.
Immunofluorescence analysis
Immunofluorescence of tomato lectin was reported as RFU/total area, and percentage of microglia were reported as percentage of IBA-1+ cells co-stained with Dapi nuclei. When examining the dorsal hippocampus without subregional distinctions (Figure 2B), we observed a sex by exposure condition interaction (F(1,29) = 8.374, p < 0.01) for percentage of Iba-1+ microglia: female rats with a history of ethanol had a higher percentage of Iba-1+ microglia compared with both control females and males with a history of ethanol. We also found a sex by exposure condition interaction (F(1,29) = 5.206, p < 0.05) for percentage of Iba-1+ microglia associated with Aβ(1–42), in that a higher percentage was evident in females exposed to ethanol compared with ethanol-exposed males. Similarly, a sex by experimental condition interaction (F(1,29) = 6.083, p < 0.05) was evident for Iba-1+ microglia associated with both Aβ(1–42) and tomato lectin-staining cells, with ethanol-exposed females demonstrating significant increases in this immunofluorescent measure relative to both control females as well as males with a history of ethanol. In males, all three immunofluorescent measures did not differ as a function of exposure condition. No differences were observed in the dHPC of tomato lectin, measured as immunoreactivity divided by total area measured (p > 0.05). It is noteworthy to mention that immunofluorescence for Aβ(1–42) appeared visually as localized fragment staining, rather than as large plaque formations (Figure 3A, B). Sporadic plaques were evident visually in the hippocampus (see Figure 3E, F), but the frequency of Aβ(1–42) plaques was quite low (i.e., not more than a few per rat/structure), did not vary as a function of sex or ethanol exposure condition, and were not quantified due to the low incidence rate. No significant differences were observed when examining for Iba-1+ microglia, Aβ(1–42, Iba-1+ microglia / Aβ(1–42 co-localization, and Iba-1+ microglia / Aβ(1–42 / tomato lectin triple labeling in the amygdala, piriform cortex, or entorhinal cortex (Table 2).
Figure 3. Immunofluorescence and 3D rendering of amyloid beta plaques.
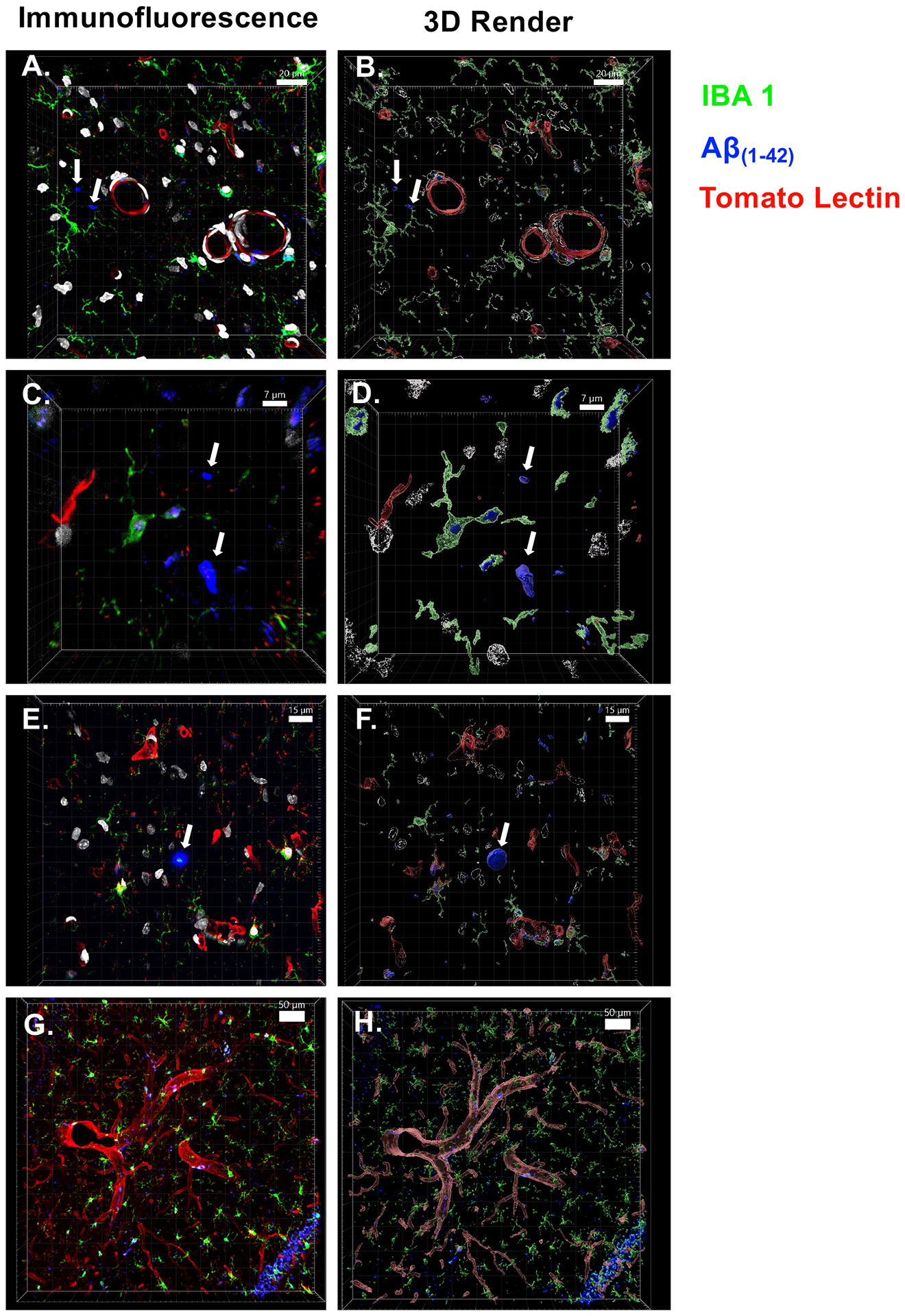
Images to the left are immunofluorescent images taken with a confocal microscope, images to the right are rendered using IMARIS software for improved visual clarity. Arrows denote Aβ fragments and plaques. (A & B) Prodromal Aβ fragments, relatively small (<75 micron2), Aβ primarily found in vascular-bound microglia, captured at 40×. (C & D) Plaque development beginning, roughly the size of a microglial soma (350 micron2), captured at 40×. (E & F) Traditional Aβ plaque, larger than a microglial soma (519 micron2), with high density of Aβ internalized in surrounding microglia, captured at 40×. (G & H) Representative image of microglia-vasculature interaction, captured at 20×. Scale bar is included for size reference in each image.
Table 2.
Quantification of immunofluorescence signals in CNS sites outside the hippocampus.
| Males | Females | |||
|---|---|---|---|---|
| Amygdala | ||||
| Water | Ethanol | Water | Ethanol | |
| IBA 1 | 12.64 ± 1.67 | 13.92 ± 1.78 | 15.06 ± 4.22 | 12.34 ± 3.73 |
| Aβ(1–42) | 29.16 ± 5.98 | 31.59 ± 6.21 | 45.73 ± 6.80 | 37.66 ± 3.61 |
| IBA 1 / Aβ(1–42) | 8.63 ± 1.44 | 8.56 ± 0.94 | 10.50 ± 2.71 | 8.46 ± 2.27 |
| IBA 1 / Aβ(1–42) / Tomato Lectin | 6.99 ± 1.44 | 6.36 ± 1.00 | 7.94 ± 3.38 | 5.96 ± 2.85 |
| Piriform Cortex | ||||
| IBA 1 | 15.67 ± 2.27 | 15.4 ± 2.17 | 17.03 ± 5.66 | 13.52 ± 4.54 |
| Aβ(1–42) | 35.72 ± 7.15 | 28.47 ± 5.82 | 48.45 ± 5.01 | 35.50 ± 3.66 |
| IBA 1 / Aβ(1–42) | 11.13 ± 2.03 | 8.72 ± 1.50 | 12.55 ± 3.87 | 9.57 ± 2.94 |
| IBA 1 / Aβ(1–42) / Tomato Lectin | 9.30 ± 2.04 | 6.32 ± 1.43 | 9.56 ± 4.48 | 6.56 ± 3.71 |
| Lateral Entorhinal Cortex | ||||
| IBA 1 | 17.38 ± 2.49 | 15.35 ± 2.11 | 18.20 ± 5.39 | 14.79 ± 4.90 |
| Aβ(1–42) | 39.64 ± 7.26 | 30.30 ± 5.86 | 49.64 ± 3.50 | 39.83 ± 3.72 |
| IBA 1 / Aβ(1–42) | 12.15 ± 2.10 | 8.91 ± 1.37 | 12.79 ± 3.46 | 10.36 ± 3.11 |
| IBA 1 / Aβ(1–42) / Tomato Lectin | 10.30 ± 2.22 | 6.72 ± 1.42 | 9.88 ± 4.17 | 7.11 ± 3.94 |
Values represent mean ± SEM and are provided in mg/dL. No significant differences were observed.
In the CA1, we observed significant interactions of sex and exposure condition (F(1,29) = 5.58, p < 0.05) for percentage of Iba-1+ microglia associated with Aβ(1–42), with ethanol exposure significantly increasing these immunofluorescent measures in ethanol-exposed females relative to all other groups. We also observed a significant interaction, where percentage of Iba-1+ microglia associated with Aβ(1–42) and cells positively stained with tomato lectin (triple labeling) (F(1,29) = 5.94, p < 0.05) increased in females with a history of ethanol, compared with both female-controls as well as males with a history of ethanol (see Figure 4A). In the CA2, no significant differences were observed between groups (Figure 4B). When investigating the CA3, we observed significant sex by ethanol exposure condition interactions for percentage Iba-1+ microglia associated with Aβ(1–42) (F(1,29) = 5.129, p < 0.05) and percentage of Iba-1+ microglia associated with both Aβ(1–42) and tomato-lectin+ cells (F(1,29) = 6.074, p < 0.05). However, post hoc analysis revealed no significant loci of interest (p > 0.05) (Figure 4C). In the dentate gyrus, the percentage of cells expressing Aβ(1–42) was lower in animals with a history of ethanol exposure than in water-exposed controls, as evidenced by a main effect of exposure condition (F(1,29) = 4.508, p < 0.05). Additionally, a history of ethanol exposure increased percentage of Iba-1+ microglia associated with Aβ(1–42), in females with a history of ethanol consumption, relative to ethanol-exposed males. Iba-1+ microglia were associated with both Aβ(1–42) and tomato lectin in females when compared with water-exposed females, with no ethanol exposure effects evident in males, as evidenced by significant sex by ethanol exposure condition interactions (F(1,29) = 7.707, p < 0.05 and F(1,29) = 7.633, p < 0.05) (Figure 4D).
Figure 4. Quantification of immunofluorescence in subregions of the hippocampus.
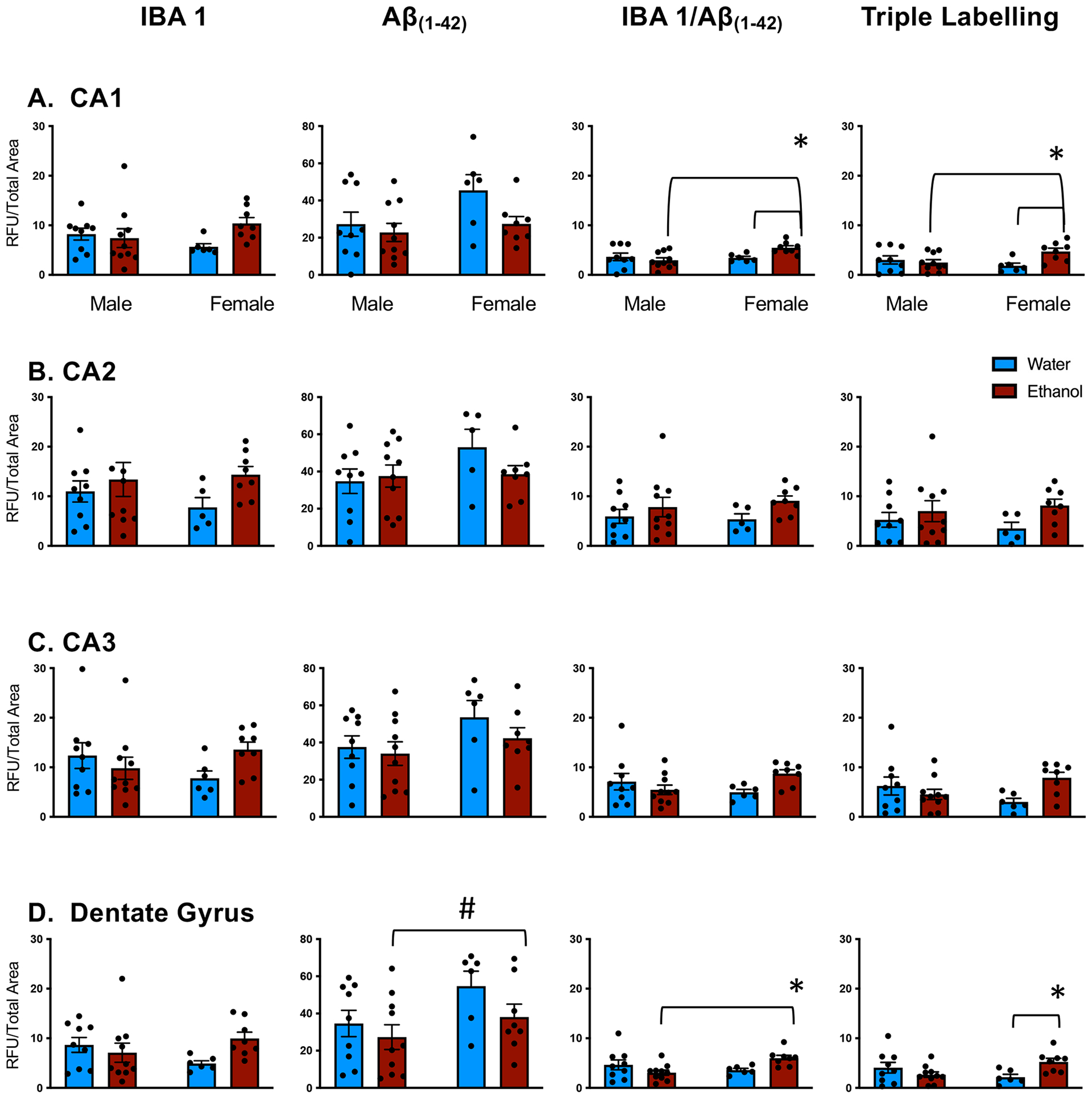
(A) Females with a history of ethanol had increased Iba-1 / Aβ co-localization Iba-1 / Aβ / tomato lectin triple localization in the CA1 region of the dorsal hippocampus. (B) No differences were observed across groups in the CA2 region of the dorsal hippocampus. (C) No differences were observed across groups in the CA3 region of the dorsal hippocampus. (D) Ethanol history resulted in lowered Aβ(1–42) in both males and females, while females with a history of ethanol had increased Iba-1 / Aβ co-localization Iba-1 / Aβ / tomato lectin triple localization in the dentate gyrus. Asterisk (*) denotes a significant interaction (p < 0.05). All data expressed as mean ± SEM.
Discussion
The present study observed an increase in IBA1 expression in the dorsal hippocampus in females only following chronic ethanol in middle-aged rats. An increase in co-localization of Iba1/Aβ(1–42) was also observed in females only, in the overall dorsal hippocampus, as well as in the CA1, CA2, and dentate gyrus. Finally, an increase in triple-labeled co-localization between IBA1, Aβ(1–42), and tomato lectin was observed in the overall dorsal hippocampus, as well as in the CA1. These results indicate that females may have higher phagocytic activity of microglia interacting with Aβ(1–42), and that this interaction is happening primarily near the vasculature in the CA1 region of the hippocampus.
A unique feature of the present study was the assessment of chronic ethanol consumption on Aβ(1–42) deposition in both male and female mid-late adult rats, aged 10–14 months. The approach in this study was to introduce and maintain a long-term ethanol exposure procedure comparable to what humans may consume across their lifespan, which tends to fluctuate between periods of consumption and abstinence, and in which drinking periods and amounts of ethanol are both variable and intermittent. During the varied history of ethanol exposure, rats showed relatively low blood ethanol concentrations at the time points examined. Despite the low BECs reported, it should be noted that only a single, probative time point was assessed across the diurnal cycle, which may not have captured peak BECs for all rats. Furthermore, samples were taken on the second day of continuous access to ethanol, which may have been impacted by metabolic tolerance. Supporting the low-moderate levels of ethanol exposure, no changes in body weight gain occurred due to ethanol consumption, even during the prolonged single-bottle exposure period, suggesting that the chronic ethanol consumption was well-tolerated by both sexes. Thus, there is translational value of this model of ethanol consumption, which may shed light on the relationship between Aβ plaque development and a varied ethanol history throughout adulthood.
Regions of interest were chosen due to their relevance in dementia-related decline and propensity for Aβ accumulation. The hippocampus is heavily implicated in memory deficits relating to AD, appears to be particularly vulnerable to Aβ accumulation, and is one of the first brain areas affected during early stages of AD (Braak, Braak, & Bohl, 1993; Fjell et al., 2014; Mu & Gage, 2011). Subregional analysis of the dorsal hippocampus revealed several sex-specific effects wherein microglia of ethanol-exposed females were co-localized with Aβ(1–42), as well as Aβ(1–42) and neurovasculature, more often in the CA1 and the dentate gyrus. No differences in Aβ(1–42) accumulations were observed in CA1, CA3, and DG. However, microglia were increased and more readily interacted with Aβ(1–42) at these areas. Surprisingly, chronic ethanol consumption had no effect on Aβ(1–42) in the entorhinal cortex and amygdala. In this way, the highly localized effects within the hippocampus support the conclusion that the effects of low-to-moderate drinking, initiated later in life, had only modest effects on Aβ(1–42). Although there is a tendency to assume that microglial activation and clearance of Aβ(1–42) may be neuroprotective, accumulation of microglia specifically in the perivascular regions may also be associated with other elements of mild inflammation, including expression of cytokines, chemokines, or other inflammatory mediators. Examination of inflammation markers was beyond the scope of what could be accomplished here and could be a fruitful path for future studies.
A lesser discussed symptom of dementia is an increase in anxiety, fear, and aggression, likely caused by damage to the amygdala during neurodegeneration. Amygdala atrophy occurs in early stages of AD and is related to severity of both neuropsychiatric as well as motor symptoms in patients (Poulin et al., 2011). Post mortem analyses of patients with symptoms of mild cognitive impairment (MCI) reveal increased Aβ load in the entorhinal cortex (EC) and piriform cortex (PiRC), suggesting these cortices are areas of vulnerability in early dementia (Mufson et al., 1999; Saiz-Sanchez, De la Rosa-Prieto, Ubeda-Banon, & Martinez-Marcos, 2015). Animal models of AD report increased Aβ deposition in both the hippocampus as well as the entorhinal cortex, and exogenous Aβ injection into the entorhinal cortex results in AD symptoms (Frontiñán-Rubio et al., 2018; Sipos et al., 2007). For these reasons, the present study focused on the hippocampus, lateral entorhinal cortex, piriform cortex, and amygdala as areas of vulnerability in early pathological disease states of AD and dementia. Contrary to the findings in the hippocampus, no changes were observed following chronic ethanol exposure in the amygdala, piriform cortex, or lateral entorhinal cortex as a result of chronic ethanol exposure in either males or females.
Women are more susceptible to dementia than men for reasons that are not fully understood. Nearly two-thirds of AD patients are women who also suffer more severe cognitive deterioration than men (Hebert, Weuve, Scherr, & Evans, 2013; Laws, Irvine, & Gale, 2018). While both males and females display behavioral dysfunction after chronic ethanol exposure, females may be especially vulnerable to alcohol-related brain damage and cognitive impairments (Nixon & Glenn, 1995; Wilsnack, Wilsnack, & Kantor, 2013). Females also showed greater microglial activation than males following binge ethanol exposure, particularly in the medial prefrontal cortex and the hippocampus (Barton, Baker, & Leasure, 2017). Consistent with this, effects of chronic ethanol consumption on Aβ(1–42) in the dorsal hippocampus were sex-specific: female rats showed increased Iba1+ microglia, consistent with human post mortem reports, which indicates that a history of alcohol misuse increases microglial populations as well as inflammatory markers (He & Crews, 2008). Perhaps paradoxically, ethanol decreased overall Aβ(1–42) expression, yet microglia showed greater co-localization with Aβ(1–42), especially in perivascular regions. Since microglia are known to phagocytize Aβ(1–42), these data may reflect more effective clearance of Aβ(1–42) after chronic ethanol exposure. Whether this might be indicative of prodromal vascular dementia and/or AD remains to be determined.
While adults accrue amyloid with age, many do not manifest symptoms related to dementia, indicating that Aβ is not the only factor involved in cognitive impairment. Despite this, PET imaging of Aβ differentiates between healthy controls and those with AD, as well as correlating increased amyloid load with decreased performance on cognitive tests (Okamura & Yanai, 2010; Sperling et al., 2020). Presently, we used immunofluorescence to examine emerging pathology and possible associations with clearance by microglia, with particular interest in perivascular microglia. Considering that both Aβ and alcohol can elicit an inflammatory response from microglia, examining specific cytokines, chemokines, and their cognates may be an important next step in understanding the connection between alcohol intake and AD. Pro-inflammatory cytokines such as IL-6 and SOCS3 have both been expressed at high levels in post mortem brains with AD, and reduction of IL-6 signaling via inhibition of STAT3 signaling reduced cognitive impairment in an AD mouse model (Lyra e Silva et al., 2021). Thus, while increased microglial responding may be initially helpful in Aβ clearance in the aging brain, sustained inflammation via microglial signaling may be detrimental over time.
The present study tested the hypothesis that an increased Aβ load would be observed following chronic ethanol exposure, and that females would be particularly vulnerable to Aβ deposition. However, results from this study instead suggest that ethanol increased Aβ-associated phagocytosis by microglia, perhaps as a result of low to moderate ethanol intake levels. Results from the IA2BC data indicate that ethanol intake declined across time. Evidence suggests that adults are more sensitive to the aversive effects of ethanol, and that this sensitivity increases from adulthood throughout aging, which may explain the decrease in ethanol consumption observed (Perkins et al., 2018). Alternatively, the tail sampling procedure during the IA2BC procedure may have contributed to the decline in ethanol preference observed over time. The present study did not evaluate the amount of ethanol consumed during the forced consumption portion of the procedure, which is a major limitation. This model was chosen in order to keep animals pair-housed throughout the majority of their lives because separation and isolation stress have been shown to alter both drinking patterns, as well as neuropathology of Alzheimer’s disease (Becker, Lopez, & Doremus-Fitzwater, 2011; Dong & Csernansky, 2009). Ongoing studies from our lab using this model are characterizing the amounts and patterns of ethanol consumed using this procedure.
Other studies have found that chronic, high ethanol intake results in increased expression of APP and Aβ-producing enzymes such as BACE1, indicating that chronic ethanol likely leads to increased Aβ fragment deposition (Kim et al., 2011). Ethanol has also been shown to increase microglial activation; these results may indicate that ethanol exposure resulted in increased Aβ production, but that microglia have been primed to increase phagocytic activity to compensate. Purinergic receptors on microglia, particularly P2X4, are sensitive to ethanol exposure (Popova, Trudell, Alkana, Davies, & Asatryan, 2013). Ethanol exposure increases expression of P2X4 receptors on microglia, as well as increases gene expression of complement proteins that contribute to phagocytosis and clearance from apoptotic cells including C1qa, C1qb, C1qc, C3, and C3aR1 (Galvan, Greenlee-Wacker, & Bohlson, 2012; Kalinin et al., 2018). Consistent with the reduced amyloid beta load observed following chronic ethanol in this paper, low levels of ethanol may increase phagocytic activity in microglia. Other studies have observed that ethanol does not increase phagocytic microglia in the CNS following a 4-day, high-dose ethanol procedure. However, lower-dose ethanol exposure for 3 weeks resulted in microglial activation and higher phagocytic activity in microglia (Marshall et al., 2013; Ward et al., 2009). It is possible that a dose-dependent curve of ethanol-microglia interaction is at play here: at low to moderate ethanol consumption, Aβ fragments are increased, with microglia increasing phagocytosis to compensate; however, at high doses of ethanol, Aβ fragments will overwhelm the capacity for microglia to clear Aβ, resulting in Aβ plaque deposition and pathology. Taken together, these data support the growing literature that alcohol likely acts in a dose-dependent manner, where low-dose ethanol can be anti-inflammatory while high-dose ethanol can be pro-inflammatory and lead to deleterious consequences such as decreased cognition and increased risk of neurodegenerative diseases (Sinforiani et al., 2011).
Overall, the present findings support two important conclusions. First, initiation of ethanol consumption later in life (~10 months of age) through a progressive series of voluntary, then involuntary, consumption procedures did not increase ethanol intake in a subsequent test of voluntary consumption. Although BECs were low, clear effects on microglial dynamics were observed exclusively in females, indicating a sex-specific increase in microglia-mediated phagocytosis by perivascular microglia in aged, ethanol-consuming rats. As there are no current treatments or cures for AD, identifying risk factors and prevention of AD is crucial. These findings indicate that ethanol consumption produces a female-specific increase in microglia-mediated phagocytosis by perivascular microglia in aging rats, which may have important implications for understanding the mechanisms of cognitive decline associated with chronic alcohol consumption.
Highlights.
The present experiment used a novel, moderate intermittent ethanol consumption procedure across middle to late aging.
Aged females with a history of ethanol consumption have increased co-localization of microglia and Aβ(1–42) in the dorsal hippocampus.
This paper offers insight into microglia-Aβ(1–42) co-localization around neurovasculature after moderate chronic ethanol exposure.
Acknowledgments
Supported in part by NIH grants P50AA017823 (T.D.), T32AA025606 (ASV, AAB), and F31AA027959 (ASV), as well as the Center for Development and Behavioral Neuroscience at Binghamton University. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the above-stated funding agencies. The authors have no conflicts of interest to declare.
References
- Ahlner F, Sigström R, Rydberg Sterner T, Mellqvist Fässberg M, Kern S, Östling S, et al. (2018). Increased Alcohol Consumption Among Swedish 70-Year-Olds 1976 to 2016: Analysis of Data from The Gothenburg H70 Birth Cohort Studies, Sweden. Alcoholism: Clinical and Experimental Research, 42(12), 2403–2412. [DOI] [PubMed] [Google Scholar]
- Barton EA, Baker C, & Leasure JL (2017). Investigation of Sex Differences in the Microglial Response to Binge Ethanol and Exercise. Brain Sciences, 7(10), 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker HC, Lopez MF, & Doremus-Fitzwater TL (2011). Effects of stress on alcohol drinking: a review of animal studies. Psychopharmacology, 218, 131–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazer DG, & Wu L-T (2009). The epidemiology of at-risk and binge drinking among middle-aged and elderly community adults: National Survey on Drug Use and Health. The American Journal of Psychiatry, 166(10), 1162–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boschen KE, Ptacek TS, Berginski ME, Simon JM, & Parnell SE (2021). Transcriptomic analyses of gastrulation-stage mouse embryos with differential susceptibility to alcohol. Disease models & mechanisms, 14(6), dmm049012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E, & Bohl J (1993). Staging of Alzheimer-Related Cortical Destruction. European Neurology, 33(6), 403–408. [DOI] [PubMed] [Google Scholar]
- Breslow RA, Castle I-JP, Chen CM, & Graubard BI (2017). Trends in Alcohol Consumption Among Older Americans: National Health Interview Surveys, 1997 to 2014. Alcoholism: Clinical and Experimental Research, 41(5), 976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue JE, & Johanson CE (2008). Apolipoprotein E, Amyloid-β, and Blood-Brain Barrier Permeability in Alzheimer Disease. Journal of Neuropathology and Experimental Neurology, 67(4), 261–270. [DOI] [PubMed] [Google Scholar]
- Deak T, & Savage LM (2019). Preface: Setting the stage for understanding alcohol effects in late aging: A special issue including both human and rodent studies. International review of neurobiology, 148, xiii–xxv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doremus-Fitzwater TL, Buck HM, Bordner K, Richey L, Jones ME, & Deak T (2014). Intoxication‐and withdrawal‐dependent expression of central and peripheral cytokines following initial ethanol exposure. Alcoholism: Clinical and Experimental Research, 38(8), 2186–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, & Csernansky JG (2009). Effects of stress and stress hormones on amyloid-β protein and plaque deposition. Journal of Alzheimer’s Disease, 18(2), 459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassbender K, Walter S, Kühl S, Landmann R, Ishii K, Bertsch T, et al. (2004). The LPS receptor (CD14) links innate immunity with Alzheimer’s disease. FASEB Journal, 18(1), 203–205. [DOI] [PubMed] [Google Scholar]
- Fjell AM, McEvoy L, Holland D, Dale AM, Walhovd KB, & Alzheimer’s Disease Neuroimaging Initiative. (2014). What is normal in normal aging? Effects of aging, amyloid and Alzheimer’s disease on the cerebral cortex and the hippocampus. Progress in Neurobiology, 117, 20–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frontiñán-Rubio J, Sancho-Bielsa FJ, Peinado JR, LaFerla FM, Giménez-Llort L, Durán-Prado M, et al. (2018). Sex-dependent co-occurrence of hypoxia and β-amyloid plaques in hippocampus and entorhinal cortex is reversed by long-term treatment with ubiquinol and ascorbic acid in the 3 × Tg-AD mouse model of Alzheimer’s disease. Molecular and Cellular Neuroscience, 92, 67–81. [DOI] [PubMed] [Google Scholar]
- Galvan MD, Greenlee‐Wacker MC, & Bohlson SS (2012). C1q and phagocytosis: the perfect complement to a good meal. Journal of Leukocyte Biology, 92(3), 489–497. [DOI] [PubMed] [Google Scholar]
- Gong Y-S, Guo J, Hu K, Gao Y-Q, Hou F-L, Song F-L, et al. (2017). Chronic Ethanol Consumption and Thiamine Deficiency Modulate β-Amyloid Peptide Level and Oxidative Stress in the Brain. Alcohol and Alcoholism, 52(2), 159–164. [DOI] [PubMed] [Google Scholar]
- Grant BF, Chou SP, Saha TD, Pickering RP, Kerridge BT, Ruan WJ, et al. (2017). Prevalence of 12-month alcohol use, high-risk drinking, and DSM-IV alcohol use disorder in the United States, 2001–2002 to 2012–2013: results from the National Epidemiologic Survey on Alcohol and Related Conditions. JAMA Psychiatry, 74(9), 911–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Jones CM, Einstein EB, Powell PA, & Compton WM (2021). Use of Medications for Alcohol Use Disorder in the US: Results From the 2019 National Survey on Drug Use and Health. JAMA Psychiatry, 78(8), 922–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartz AMS, Bauer B, Soldner ELB, Wolf A, Boy S, Backhaus R, et al. (2012). Amyloid-β contributes to blood-brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke, 43(2), 514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, & Crews FT (2008). Increased MCP-1 and microglia in various regions of the human alcoholic brain. Experimental Neurology, 210(2), 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert LE, Weuve J, Scherr PA, & Evans DA (2013). Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology, 80(19), 1778–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman JL, Faccidomo S, Kim M, Taylor SM, Agoglia AE, May AM, et al. (2019). Alcohol drinking exacerbates neural and behavioral pathology in the 3xTg-AD mouse model of Alzheimer’s disease. International Review of Neurobiology, 148, 169–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C (2013). The pathobiology of vascular dementia. Neuron, 80(4), 844–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA (2013). Pathology and pathogenesis of vascular cognitive impairment – a critical update. Frontiers in Aging Neuroscience, 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinin S, González-Prieto M, Scheiblich H, Lisi L, Kusumo H, Heneka MT, et al. (2018). Transcriptome analysis of alcohol-treated microglia reveals downregulation of beta amyloid phagocytosis. Journal of Neuroinflammation, 15(1), 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Lee DY, Lee BC, Jung MH, Kim H, Choi YS, et al. (2012). Alcohol and cognition in the elderly: a review. Psychiatry Investigation, 9(1), 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S-R, Jeong H-Y, Yang S, Choi S-P, Seo MY, Yun Y-K, et al. (2011). Effects of chronic alcohol consumption on expression levels of APP and Aβ-producing enzymes. BMB Reports, 44(2), 135–139. [DOI] [PubMed] [Google Scholar]
- Keyes KM, Jager J, Mal‐Sarkar T, Patrick ME, Rutherford C, & Hasin D (2019). Is there a recent epidemic of women’s drinking? A critical review of national studies. Alcoholism: Clinical and Experimental Research, 43(7), 1344–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao Y, Hou L, Li J, Hui X, Yan P, & Yang K (2021). Association between alcohol intake, mild cognitive impairment and progression to dementia: a dose–response meta-analysis. Aging: Clinical and Experimental Research, 33(5), 1175–1185. [DOI] [PubMed] [Google Scholar]
- Laws KR, Irvine K, & Gale TM (2018). Sex differences in Alzheimer’s disease. Current Opinion in Psychiatry, 31(2), 133–139. [DOI] [PubMed] [Google Scholar]
- Lewis H, Beher D, Cookson N, Oakley A, Piggott M, Morris CM, et al. (2006). Quantification of Alzheimer pathology in ageing and dementia: age-related accumulation of amyloid-β(42) peptide in vascular dementia. Neuropathology and Applied Neurobiology, 32(2), 103–118. [DOI] [PubMed] [Google Scholar]
- Liu W, Taso O, Wang R, Bayram S, Graham AC, Garcia-Reitboeck P, et al. (2020). Trem2 promotes anti-inflammatory responses in microglia and is suppressed under pro-inflammatory conditions. Human Molecular Genetics, 29(19), 3224–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyra e Silva NM, Gonçalves RA, Pascoal TA, Lima-Filho RAS, Resende E.de P. F. , Vieira ELM , et al. (2021). Pro-inflammatory interleukin-6 signaling links cognitive impairments and peripheral metabolic alterations in Alzheimer’s disease. Translational Psychiatry, 11(1), 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall SA, McClain JA, Kelso ML, Hopkins DM, Pauly JR, & Nixon K (2013). Microglial activation is not equivalent to neuroinflammation in alcohol-induced neurodegeneration: The importance of microglia phenotype. Neurobiology of Disease, 54, 239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall SA, McClain JA, Wooden JI, & Nixon K (2020). Microglia Dystrophy Following Binge-Like Alcohol Exposure in Adolescent and Adult Male Rats. Frontiers in Neuroanatomy, 14, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Tago H, & McGeer EG (1987). Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neuroscience Letters, 79(1–2), 195–200. [DOI] [PubMed] [Google Scholar]
- Mokhtar SH, Bakhuraysah MM, Cram DS, & Petratos S (2013). The Beta-amyloid protein of Alzheimer’s disease: communication breakdown by modifying the neuronal cytoskeleton. International Journal of Alzheimer’s Disease, 2013, 910502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Y, & Gage FH (2011). Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Molecular Neurodegeneration, 6, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, Chen E-Y, Cochran EJ, Beckett LA, Bennett DA, & Kordower JH (1999). Entorhinal Cortex β-Amyloid Load in Individuals with Mild Cognitive Impairment. Experimental Neurology, 158(2), 469–490. [DOI] [PubMed] [Google Scholar]
- Näslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, et al. (2000). Correlation Between Elevated Levels of amyloid beta-peptide in the Brain and Cognitive Decline. JAMA, 283(12), 1571–1577. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Soma LA, & Lavi E (2002). Microglia in diseases of the central nervous system. Annals of Medicine, 34(7–8), 491–500. [DOI] [PubMed] [Google Scholar]
- Nixon SJ, & Glenn SW (1995). Cognitive Psychosocial Performance and Recovery in Female Alcoholics. In Galanter M, Begleiter H, Deitrich R, Gallant D, Goodwin D, Gottheil E, Paredes A, Rothschild M, Van Thiel D, & Edwards H (Eds.), Recent Developments in Alcoholism: Alcoholism and Women (pp. 287–307). Springer. [DOI] [PubMed] [Google Scholar]
- Norden DM, Trojanowski PJ, Villanueva E, Navarro E, & Godbout JP (2016). Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia, 64(2), 300–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunan J, & Small DH (2000). Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Letters, 483(1), 6–10. [DOI] [PubMed] [Google Scholar]
- Okamura N, & Yanai K (2010). Florbetapir (18F), a PET imaging agent that binds to amyloid plaques for the potential detection of Alzheimer’s disease. IDrugs, 13(12), 890–899. [PubMed] [Google Scholar]
- Perkins AE, Vore AS, Lovelock D, Varlinskaya E, & Deak T (2018). Late aging alters behavioral sensitivity to ethanol in a sex-specific manner in Fischer 344 rats. Pharmacology, Biochemistry, and Behavior, 175, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova M, Trudell J, Li K, Alkana R, Davies D, & Asatryan L (2013). Tryptophan 46 is a site for ethanol and ivermectin action in P2X4 receptors. Purinergic Signalling, 9(4), 621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin SP, Dautoff R, Morris JC, Barrett LF, Dickerson BC, & Alzheimer’s Disease, Neuroimaging Initiative. (2011). Amygdala atrophy is prominent in early Alzheimer’s disease and relates to symptom severity. Psychiatry Research, 194(1), 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehm J, Hasan OSM, Black SE, Shield KD, & Schwarzinger M (2019). Alcohol use and dementia: a systematic scoping review. Alzheimer’s Research & Therapy, 11(1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi B, Angiari S, Zenaro E, Budui SL, & Constantin G (2011). Vascular inflammation in central nervous system diseases: adhesion receptors controlling leukocyte–endothelial interactions. Journal of Leukocyte Biology, 89(4), 539–556. [DOI] [PubMed] [Google Scholar]
- Saiz-Sanchez D, De la Rosa-Prieto C, Ubeda-Banon I, & Martinez-Marcos A (2015). Interneurons, tau and amyloid-β in the piriform cortex in Alzheimer’s disease. Brain Structure & Function, 220(4), 2011–2025. [DOI] [PubMed] [Google Scholar]
- Sengupta P (2013). The Laboratory Rat: Relating Its Age With Human’s. International Journal of Preventive Medicine, 4(6), 624–630. [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A, Muzikansky A, Gómez-Isla T, Growdon JH, Betensky RA, Frosch MP, et al. (2013). Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. Journal of Neuropathology and Experimental Neurology, 72(6), 462–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinforiani E, Zucchella C, Pasotti C, Casoni F, Bini P, & Costa A (2011). The effects of alcohol on cognition in the elderly: from protection to neurodegeneration. Functional Neurology, 26(2), 103–106. [PMC free article] [PubMed] [Google Scholar]
- Sipos E, Kurunczi A, Kasza A, Horváth J, Felszeghy K, Laroche S, et al. (2007). β-Amyloid pathology in the entorhinal cortex of rats induces memory deficits: implications for Alzheimer’s disease. Neuroscience, 147(1), 28–36. [DOI] [PubMed] [Google Scholar]
- Spear LP (2020). Timing Eclipses Amount: The Critical Importance of Intermittency in Alcohol Exposure Effects. Alcoholism: Clinical and Experimental Research, 44(4), 806–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Donohue MC, Raman R, Sun C-K, Yaari R, Holdridge K, et al. (2020). Association of Factors With Elevated Amyloid Burden in Clinically Normal Older Individuals. JAMA Neurology, 77(6), 735–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Chiba Y, Matsumoto K, Murakami R, Fujihara R, Kawauchi M, et al. (2016). Blood-brain barrier damage in vascular dementia. Neuropathology, 36(2), 115–124. [DOI] [PubMed] [Google Scholar]
- Venkataraman A, Kalk N, Sewell G, Ritchie CW, & Lingford-Hughes A (2017). Alcohol and Alzheimer’s Disease – Does Alcohol Dependence Contribute to Beta-Amyloid Deposition, Neuroinflammation and Neurodegeneration in Alzheimer’s Disease? Alcohol and Alcoholism, 52(2), 151–158. [DOI] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell, 160(6), 1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward RJ, Colivicchi MA, Allen R, Schol F, Lallemand F, De Witte P, et al. (2009). Neuro‐inflammation induced in the hippocampus of ‘binge drinking’ rats may be mediated by elevated extracellular glutamate content. Journal of Neurochemistry, 111(5), 1119–1128. [DOI] [PubMed] [Google Scholar]
- Wilsnack SC, Wilsnack RW, & Kantor LW (2013). Focus on: women and the costs of alcohol use. Alcohol Research: Current Reviews, 35(2), 219–228. [PMC free article] [PubMed] [Google Scholar]
- Zhao Y-N, Wang F, Fan Y-X, Ping G-F, Yang J-Y, & Wu C-F (2013). Activated microglia are implicated in cognitive deficits, neuronal death, and successful recovery following intermittent ethanol exposure. Behavioural Brain Research, 236(1), 270–282. [DOI] [PubMed] [Google Scholar]
- Zheng H, Cheng B, Li Y, Li X, Chen X, & Zhang Y (2018). TREM2 in Alzheimer’s Disease: Microglial Survival and Energy Metabolism. Frontiers in Aging Neuroscience, 10, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuliani G, Ranzini M, Guerra G, Rossi L, Munari MR, Zurlo A, et al. (2007). Plasma cytokines profile in older subjects with late onset Alzheimer’s disease or vascular dementia. Journal of Psychiatric Research, 41(8), 686–693. [DOI] [PubMed] [Google Scholar]