


Abstract
The mechanisms of multicomponent transcription factor complex assembly are currently poorly defined. A paradigm for this type of complex is the ETS-domain transcription factor Elk-1 and the MADS-box transcription factor SRF which form a ternary complex with the c-fos serum response element (SRE). In this study we have analysed how a different ETS-domain transcription factor Fli-1 interacts with SRF to form ternary complexes with this element. Two regions of Fli-1 that are required for ternary complex formation have been identified. These SRF binding motifs are located on either side of the ETS DNA-binding domain. Hydrophobic amino acids within these motifs have been identified that play important roles in binding to SRF and ternary complex formation. By using Fli-1 derivatives with mutations in the N-terminal SRF binding motif, the significance of Fli-1–SRF interactions in recruitment of Fli-1 to the c-fos SRE in vivo has been demonstrated. Collectively our data provide a model of how Fli-1 interacts with SRF that differs significantly from the mechanism used by a different ETS-domain protein, Elk-1.
INTRODUCTION
ETS-domain proteins comprise a large family of eukaryotic transcription factors (1–3). These proteins are grouped due to the presence of the ETS DNA-binding domain but are further subclassified due to sequence conservation within this domain and the presence of other conserved domains. The activities of many of these proteins are subject to tight regulation and respond to signal transduction cascades such as the MAP kinase pathways. Moreover, in order to elicit cell-type and promoter-specific responses, ETS-domain proteins often function in multi-component transcription factor complexes. A paradigm for this type of complex is that formed by the ETS-domain protein Elk-1 and the MADS-box transcription factor SRF at the c-fos serum response element (SRE) (reviewed in 4,5). In this complex, Elk-1 forms both protein–DNA contacts via the ETS-domain and protein–protein contacts with SRF via a short conserved motif known as the B-box (6). The B-box is located ~50 amino acids C-terminal to the ETS-domain and is necessary for binding to SRF in the absence of DNA and ternary formation at the c-fos SRE. Hydrophobic amino acids in the B-box play critical roles in binding to SRF and are thought to form the face of an α-helix which acts as the SRF-interaction motif (7). The reciprocal binding surface on SRF has been mapped and forms a surface-exposed hydrophobic groove (8; see Fig. 1C).
Figure 1.

Fli-1 forms a ternary complex with wild-type SRF and mutant SRF (T196A) at the c-fos SRE. (A) Diagrammatic representation of full-length Fli-1 [Fli-1(1–451)] and the N-terminally truncated derivative Fli-1 (220–451). The ETS DNA-binding domain is shown as a grey box. (B) Gel retardation analysis of ternary complexes formed between the indicated Fli-1 derivatives, wild-type (lanes 1 and 2) and the T196A mutant form (lanes 3 and 4) of coreSRF and the c-fos SRE. Equal molar amounts of each Fli-1 construct were used in all the binding reactions. The location of the ternary (30) Fli-1–SRF–SRE and binary (20) SRF–SRE complexes are indicated by brackets and an open arrow respectively. (C) Structure of SRF (black) bound to the c-fos SRE (grey) (32). Residues comprising the Elk-1 binding surface are shown in white (8). The location of T196 within this surface is shown in grey. (D) Diagrammatic representation of the domain structure of Elk-1 and the truncated derivative Elk-1(1–168). The sequence of the B-box region used to derive peptides for competition assays as shown below these diagrams. (E) Gel retardation analysis of ternary complex formation by Fli-1(220–451) (lanes 1–6) and Elk-1(1–168) (lanes 7–12), wild-type coreSRF and the c-fos SRE in the presence of increasing amounts of competitor B-box peptide (120 pmol, lanes 1 and 8; 600 pmol, lanes 2 and 9) or a non-specific peptide (12 pmol, lanes 4 and 10; 600 pmol, lanes 5 and 11; 6 nmol, lanes 6 and 12). The locations of binary (20, open arrows) SRF–SRE and ternary (30, closed arrows) Elk-1/Fli-1–SRF–SRE complexes are indicated.
Fli-1 is a member of a different subfamily of ETS-domain transcription factors. It was originally cloned from the integration site of the Friend murine leukaemia virus (F-MuLV) in a murine erythroleukaemia cell line (9). This fusion leads to Fli-1 overexpression. Moreover, the majority of human chromosomal translocations that result in tumours related to Ewing’s sarcomas involve the fusion of the EWS gene to the fli-1 gene and result in the production of proteins comprised of the N-terminal region of EWS and a variable length of the C-terminal region (containing the ETS-domain) of Fli-1 (10). In humans, Fli-1 is expressed in the heart, lung, spleen and thymus and is overexpressed in many erythroleukaemias (9,11). During early embryogenesis, the mouse, xenopus and zebrafish fli-1 genes are expressed in the haemangioblast population, endothelial cells and possibly neural crest cells (12–15). In order to elicit promoter-specific responses, Fli-1 has been shown to interact with several transcription factors including Pax-5 (16), c-Jun (17) and the ETS-domain protein Tel (18). Recently, additional complexes have been detected between Fli-1 and a EWS–Fli-1 fusion protein with SRF on the c-fos and egr-1 SREs (19,20). In comparison to wild-type Fli-1, EWS–Fli-1 forms stronger complexes on the c-fos SRE, implicating the C-terminal end of Fli-1 in binding to SRF and suggesting a role for the N-terminus of Fli-1 in inhibiting complex formation (19).
In this study, we have investigated how Fli-1 interacts with SRF. Two SRF binding motifs (SBMs) have been mapped and are located on either side of the ETS DNA-binding domain. Subsequently, using mutational analysis the relevance of SRF binding to Fli-1 recruitment to SRF–SRE complexes in vitro and in vivo has been determined. Our results demonstrate that in comparison to the Elk-1–SRF paradigm, Fli-1 uses a novel mode of interaction with SRF, with two binding motifs, neither of which exhibit significant similarity to the SBM (B-box) of Elk-1.
MATERIALS AND METHODS
Plasmid constructs
The following plasmids were used for expressing GST fusion proteins in Escherichia coli. pAS58 (encoding coreSRF amino acids 132–222) (6) has been described previously. pAS818 (encoding GST-coreSRF with the point mutation T196A) was constructed by inserting a BamHI/XbaI cleaved PCR fragment (primer pair COR2/ADS569 on the template pAS430) into the same sites in pGEX-KG (21).
The following plasmids were used in mammalian cell transfections. pSRE-luc contains two copies of the c-fos SRE (nucleotides –357 to 275) upstream from a minimal tk promoter and the luciferase gene (22). pAS346 contains the E74-CAT reporter gene and has been described previously (7). pAS1005 and pAS1012 are CMV promoter-driven vectors encoding, wild type and the A224D/V225E mutant fusion proteins of full-length Fli-1 (amino acids 1–451) fused to the VP16 transcription activation domain, Fli-1[VP16] and Fli-1[VP16](A224D/V225E), respectively. pAS1005 and pAS1012 were constructed by inserting the HindIII/EcoRI fragments from pAS516 and pAS1009 into the same sites in pcDNA3. pAS513 was constructed by cloning a XhoI/XbaI cleaved PCR fragment (primer pair ADS705/ADS706, template pAS160; 15) into the same sites of pBS-SK+. pAS516 (encoding Fli-1[VP16] under the control of a T3 promoter) was constructed by ligating an NcoI/XhoI fragment from pAS513 into the same sites in pBUT2-VP16 (kindly provided by Adam Rodaway). pAS1009 (encoding Fli-1[VP16](A224D/V225E) under the control of a T3 promoter) was constructed by inserting a NcoI/XhoI cleaved PCR product (two-step PCR using the flanking primers FOR and REV, the mutagenic primer ADS659 and pAS513 as a template) into the same sites in pAS516.
The following plasmids were used for in vitro transcription/translation. All proteins are driven by T3 promoters. pAS430 [encoding coreSRF(T196A)] (8), pAS96 (encoding Elk-1–168; amino acids 1–168) (6), pAS302, pAS303, pAS304 and pAS305 [encoding Fli-1(220–372), Fli-1(276–451), Fli-1(276–372) and Fli-1(220–451) respectively] (15) have been described previously. pAS513 (encoding full-length Fli-1, amino acids 1–451) is described above. pAS811, pAS812, pAS813 and pAS814 [encoding Fli-1(229–451), Fli-1(242–451), Fli-1(256–451) and Fli-1(266–451) respectively], were constructed by inserting NcoI–BamHI cleaved PCR products (primer pairs FOR/ADS528, FOR/ADS529, FOR/ADS530 and FOR/ADS531 on the template pAS305) into the same sites in pAS37 (23). pAS1001, pAS1002, pAS1003, pAS1004 and pAS1010 [encoding Fli-1(220–451) with the site-directed mutations Y222D, A224D, V225E, W230R and A224D/V225E respectively] were constructed using fragments obtained from a two-step PCR protocol as described previously (6) with two flanking primers FOR and REVL, the template pAS305 and the mutagenic primers ADS560, ADS561, ADS562, ADS563 and ADS650 respectively. PCR derived fragments were cleaved with NcoI and BamHI and inserted into the same sites in pAS302. pAS1013, pAS1014, pAS1015, pAS1016, pAS1017 and pAS1018 [encoding Fli-1(276–451) with the site-directed mutations A392D, V397D, F399D, V400D, M407K and V409D respectively] were constructed using fragments obtained using the same two-step PCR protocol and flanking primers, the template pAS303 and the mutagenic primers ADS699, ADS700, ADS701, ADS702, ADS703 and ADS704 respectively. PCR derived fragments were cleaved with NcoI and BamHI and inserted into the same sites in pAS305. pAS1018 includes the additional PCR introduced mutation T410N.
Protein expression
The synthesis of proteins by in vitro transcription and translation was carried out using either sequential transcription and translation or the coupled TNT‘ reticulocyte lysate system (Promega) according to the manufacturer’s recommendations. Proteins were synthesised either directly from plasmid templates (see above) or from PCR products derived from these templates. The proteins Fli-1(276–391), Fli-1(276–411), Fli-1(276–431) and Fli-1(276–401) were made from templates synthesised by PCR using the primer pairs REV/ADS626, REV/ADS627, REV/ADS628, REV/ADS679 and pAS303 as a template. Newly synthesised 35S-labelled proteins were analysed by SDS–PAGE followed by visualisation and quantification by phosphorimage analysis (Fuji BAS-1500 phosphorimager and TINA 2.08e software). GST fusion proteins were purified as described previously (6). The purity and relative concentration of GST fusion proteins was determined following separation by SDS–PAGE.
In vitro protein–protein interaction assays and western analysis
Interactions between coreSRF(T196A) and in vitro translated Fli-1 derivatives were investigated using pulldown assays as previously described (6). Fli-1[VP16] fusion protein derivatives were detected by immunoblot analysis with an anti-VP16 mouse monoclonal antibody (kindly provided by P. O’Hare). Immune complexes were detected using horseradish peroxidase-conjugated secondary antibody followed by enhanced chemiluminescence (Amersham).
Gel retardation assays
Gel retardation assays were performed with 32P-labelled probes as described previously (24). The c-fos SRE and the E74 binding sites have been described previously (6). Bacterially expressed coreSRF and coreSRF(T196A) were used except in Figure 3B where in vitro translated coreSRF(T196A) was used. All Fli-1 derivatives were synthesised by in vitro translation. Generally, reactions were normalised for reticulocyte lysate content. In peptide competition experiments, different concentrations (final concentrations ~10–500 µM) of peptides were pre-incubated with the proteins for 15 min at room temperature prior to the addition of DNA. Protein–DNA complexes were analysed on non-denaturing 5% polyacrylamide gels cast in 0.5× Tris-borate–EDTA and visualised by autoradiography and phosphorimaging.
Figure 3.

Mapping of the N-terminal SRF binding motif. (A) Diagrammatic representation of N-terminally truncated Fli-1 proteins. Numbers represent the first and last amino acids in each construct. (B) Gel retardation analysis of ternary complex formation by the indicated Fli-1 derivatives, wild-type coreSRF (lanes 1–5), mutant coreSRF(T196A) (lanes 6–10) and the c-fos SRE. Binary complex formation by the same Fli-1 proteins with the E74 site in the absence of coreSRF is shown in lanes 11–15.
Cell culture transfection and reporter gene assays
NIH 3T3 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum (FBS) (Gibco BRL) and 25 mM glucose. Duplicate transfection experiments were carried out in six well plates using Superfect reagent according to the manufacturer’s recommendations (Qiagen). For reporter gene assays, SRE-driven reporters were cotransfected alongside vectors encoding wild type and mutant Fli-1[VP16] fusion proteins. DNA concentrations were normalised with pcDNA3 vector.
Transfected cells were incubated in Optimem‘ (Gibco BRL) for 24 h after transfection and lysed in 200 µl lysis buffer [100 mM K2PO4, pH 7.8, 0.2% Triton X-100, 0.5 mM dithiothreitol (DTT)]. Cell debris was removed by centrifugation at 14 000 g for 15 min at 4°C and the supernatant assayed for luciferase activity as described previously (25). Transfection efficiencies were standardised by normalising for the β-galactosidase activity from co-transfected pCH110 plasmid (0.5 µg) (Pharmacia KB Biotechnology Inc.). β-galactosidase activities were determined by the use of a chemiluminescent substrate in the Dual-light‘ reporter assay system (Tropix).
Figure generation
Figures were generated electronically from scanned images of autoradiographic images by using Picture Publisher (Micrografix) and Powerpoint 7.0 (Microsoft) software. Results are representative of the original autoradiographic images.
RESULTS
Ternary complex formation between Fli-1, SRF and the c-fos SRE
Both Fli-1 and the fusion protein EWS–Fli-1 have previously been shown to be capable of forming ternary complexes with SRF on SREs found in the egr-1 promoter. However, in contrast, only binding of EWS–Fli-1 to the c-fos SRE could be detected (20). In another study, binding of both Fli-1 and EWS–Fli-1 fusion proteins to the c-fos SRE could be detected (19). In order to confirm these results and probe for potential inhibitory domains in Fli-1, we tested the ability of full-length Fli-1 [Fli-1(1–451)] and an N-terminally truncated protein, Fli-1(220–451) (Fig. 1A), to form ternary complexes with SRF on the c-fos SRE. This N-terminal truncation endpoint was selected to correspond to the position of the Fli-1 moiety in the majority of EWS–Fli-1 fusion proteins. In these experiments, we used zebrafish Fli-1 as we reasoned that the critical interaction surfaces of Fli-1 should be evolutionarily conserved as the minimal DNA binding domains (and hence reciprocal binding surface) of zebrafish and human SRF (coreSRF) are 100% identical (26).
A ternary complex was observed between full-length Fli-1, SRF and the c-fos SRE (Fig. 1B, lane 1). Truncation of the N-terminal 220 amino acids of Fli-1 enhances its ability to form ternary complexes (Fig. 1B, lane 2). In contrast, neither of these proteins form complexes on the SRE in the absence of SRF (see Fig. 2; data not shown), implying that interactions with SRF are required for complex formation.
Figure 2.

Identification of two SRF binding domains on Fli-1. (A) Diagrammatic representation of truncated Fli-1 proteins. Numbers represent the first and last amino acids in each construct. (B) Gel retardation analysis of binary and ternary complex formation by the indicated Fli-1 derivatives in the absence (lanes 1, 3, 5 and 7) and presence (lanes 2, 4, 6 and 8) of coreSRF. The locations of the binary (20, open arrows) SRF–SRE and ternary (30, closed arrows) Fli-1–SRF–SRE complexes are indicated. (C) Gel retardation analysis of ternary complex formation by the indicated Fli-1 derivatives and coreSRF(T196A). The identities of the complexes are indicates as in (B). (D) GST pull-down analysis of the indicated 35S-labelled Fli-1 derivatives and either GST or GST coreSRF(T196A). Ten percent of the input proteins is shown in lanes 1, 4, 7 and 10.
We also tested the ability of these Fli-1 derivatives to form ternary complexes with an altered SRF protein which contains the point mutation T196A within the Elk-1 binding surface (8). In comparison to wild-type SRF, both Fli-1(1–451) and Fli-1(220–451) exhibit enhanced ternary complex forming ability with SRF(T196A) (Fig. 1B, lanes 3 and 4). This result further suggests that Fli-1 and SRF interact directly and that T196 defines part of the binding surface for both Elk-1 and Fli-1.
A peptide corresponding to the B-box region of Elk-1 (Fig. 1D) has previously been shown to disrupt the binding of Elk-1 to SRF–SRE complexes by acting as a competitor (7). In order to investigate whether the interaction surfaces of Fli-1 and Elk-1 on SRF overlap, we tested the ability of the B-box peptide to inhibit ternary complex formation by the high affinity SRF binding derivative of Fli-1, Fli-1(220–451). The B-box acts as a competitor for both Fli-1 and Elk-1 (compare Fig. 1E, lanes 2 and 3, and 8 and 9). In contrast, a non-specific peptide does not inhibit complex formation by either protein even at a 10-fold higher concentration (Fig. 1E, lanes 4–6 and 10–12). These results therefore indicate that Fli-1 and Elk-1 share similar binding surfaces on SRF. In support of this notion, in addition to T196A, several mutations on the Elk-1 binding surface of SRF have been shown to also affect ternary complex formation between Fli-1 and SRF (8; see Discussion). Collectively, these data show that Fli-1 forms ternary complexes with SRF and the c-fos SRE and that Fli-1 and Elk-1 bind to overlapping surfaces on SRF.
Mapping the SRF-binding domain(s) on Fli-1
A series of truncated Fli-1 proteins were created which contain the ETS DNA-binding domain but lack regions located either N- or C-terminally (Fig. 2A). Binding to the c-fos SRE could not be detected for any of these proteins in the absence of SRF (Fig. 2B). Moreover, binding by Fli-1(276–451) and Fli-1(276–372) could not be detected even in the presence of SRF (Fig. 2B, lanes 6 and 8). However, Fli-1(220–451) and to a lesser extent Fli-1(220–372) are able to form ternary complexes with SRF and the c-fos SRE (Fig. 2B, lanes 2 and 4). The region located N-terminal to the ETS-domain (amino acids 220–276) therefore appears to be sufficient to promote ternary complex formation although the region located C-terminal to the ETS-domain (amino acids 372–451) is required for maximal complex formation.
The ability of the same series of truncated Fli-1 protein to form ternary complexes at the c-fos SRE with the mutant protein SRF(T196A) was also tested (Fig. 2C). Interestingly, in addition to Fli-1(220–451), the N-terminally truncated protein Fli-1(276–451) is able to form a ternary complex with SRF(T196A) albeit at a lower level (Fig. 2C, lane 3). Weak complex formation with Fli-1(220–372) could also be observed (data not shown). This result indicates that upon mutation of SRF, the region C-terminal to the ETS-domain (amino acids 372–451) becomes necessary and sufficient for promoting ternary complex formation. The N-terminal region (amino acids 220–276) is also required for maximal complex formation.
By analogy with the Elk-1/SRF paradigm, the simplest explanation of how ternary complex formation between Fli-1 and SRF is mediated, is by direct protein–protein interactions. Interactions between truncated Fli-1 proteins and GST-coreSRF were analysed by GST pull-down assays. Of the truncated Fli-1 derivatives, only specific binding of Fli-1(220–451) to wild- type SRF could be detected (data not shown). We therefore used GST-coreSRF(T196A) due to its higher affinity for Fli-1. In agreement with the gel retardation analysis (Fig. 2C), both Fli-1(220–451) and Fli-1(276–451) bind to SRF(T196A) (Fig. 2D, lanes 2 and 5). In contrast, neither the isolated ETS-domain Fli-1(276–372) nor the C-terminally truncated protein Fli-1(220–372) bind to SRF in this assay. These results demonstrate that Fli-1 and SRF interact directly and confirm that the region located C-terminal to the ETS-domain is the major binding surface with the mutant SRF protein SRF(T196A). In the case of interaction with wild-type SRF, both the N- and C-terminal regions of Fli-1 are required for interaction with SRF in the absence of DNA.
Thus, it appears that Fli-1 contains two regions that interact with SRF. The region N-terminal to the ETS-domain is sufficient for interaction but requires the presence of the C-terminal region for maximal interaction. This C-terminal interaction motif is revealed by analysis of binding to the SRF(T196A) mutant where the reciprocal situation exists and the region N-terminal to the ETS-domain of Fli-1 is required for maximal binding.
Analysis of the ‘N-terminal’ SRF binding motif in Fli-1
One of the SBMs in Fli-1 is located between amino acids 220 and 276. In order to further define this interaction motif, a series of truncated Fli-1 proteins were produced in which the region located N-terminally to the ETS-domain was gradually deleted (Fig. 3A). Deletion to beyond amino acid 228 causes a reduction in binding of Fli-1 to SRF–SRE complexes (Fig. 3B, lanes 1–5), indicating that residues between positions 220 and 229 play critical roles in this process. However, all the deleted proteins bind efficiently to the E74 binding site in the absence of SRF, indicating that their DNA binding activity is not compromised (Fig. 3B, lanes 11–15). Furthermore, with the exception of Fli-1(266–451), all these proteins efficiently form complexes with SRF(T196A), indicating that their C-terminal region is still functional (Fig. 3B, lanes 6–10). The reduction in binding observed with Fli-1(266–451) indicates that amino acids 256–265 are required in conjunction with the C-terminal region for maximal binding to SRF(T196A) (see Discussion).
Site-directed mutagenesis was used in order to identify amino acids that are important in the binding process whilst retaining the integrity of the protein. By analogy with Elk-1 (7), it was anticipated that hydrophobic residues might play a major role in binding to SRF. Hydrophobic amino acids located before and after the deletion end point in Fli-1 (229–451) were therefore altered to charged residues to disrupt potential hydrophobic interaction surfaces (Fig. 4B). The amino acids selected exhibit a high degree of evolutionary conservation (Fig. 4A). The mutant proteins Fli-1(220–451) Y222D, A224D and V225E all exhibit reduced ternary complex forming ability (Fig. 4C, lanes 3–5). However, binding by the mutated Fli-1(220–451) W230R is not decreased (Fig. 4C, lane 6). All the mutant Fli-1 proteins bind to the E74 site efficiently, indicating that their DNA binding activity is not impaired (Fig. 4C, lanes 7–12). These data are therefore consistent with the deletion analysis and identify residues Y222, A224 and V225 within the region 220–228 as important determinants within the N-terminal SRF interaction motif.
Figure 4.

Identification of important residues within the N-terminal SRF interaction motif. (A) Alignment of the sequences surrounding the truncation endpoint in Fli-1(229–451) (indicated by a black arrow) from zebrafish, xenopus, mouse and human homologues. Residues conserved in three or more species are shaded grey, and fully conserved amino acids are indicated by asterisks. (B) Location and identity of the amino acids mutated in zebrafish Fli-1. (C) Gel retardation analysis of complex formation by the indicated truncated and mutant Fli-1 derivatives in the presence of coreSRF and the c-fos SRE (lanes 1–6) or in the absence of coreSRF on the E74 site (lanes 7–12). The locations of the ternary Fli-1–SRF–SRE (30, closed arrow), binary SRF–SRE (20, open arrow) and binary Fli-1–E74 (20, grey arrow) complexes are indicated.
Analysis of the ‘C-terminal’ SRF binding motif in Fli-1
A second SRF-binding motif in Fli-1 is located between amino acids 373 and 451. In order to further define this interaction motif, a series of truncated Fli-1 proteins were produced in which the region located C-terminally to the ETS-domain was gradually deleted in the context of the truncated Fli-1 derivative, Fli-1(276–451) (Fig. 5A). In this case, binding to the mutant protein SRF(T196A) was analysed. Deletion to beyond amino acid 411 results in a reduction of binding of Fli-1(276–451) to SRF–SRE complexes (Fig. 5B, lanes 1–5), indicating that residues between positions 392 and 411 play a critical role in this process. However, all the deleted proteins bind efficiently to the E74 site in absence of SRF, indicating that their DNA binding activity is not compromised (Fig. 5B, lanes 6–10). An additional deletion construct, Fli-1(276–401), was constructed to further define the SRF interaction motif. In comparison to Fli-1(276–451), this protein still retains the ability to bind to SRF–SRE complexes but with reduced affinity (Fig. 5C, lanes 1 and 3). This indicates that whilst the region encompassing amino acids 402–411 contains amino acids required for maximal interactions with SRF, residues located between amino acids 392 and 401 play important roles in mediating these interactions. Point mutations were introduced before and after amino acid 401 to identify residues that play an important role in binding to SRF. Again, conserved hydrophobic residues were targeted for mutation (Fig. 6A and B). Ternary complex formation by the mutant proteins Fli-1(276–451) A392D, V397D and F399D was not detectable (Fig. 6C, lanes 1–3). However, the mutant derivatives V400D, M407K and V409D still form complexes with comparable or greater efficiency than the wild-type protein (Fig. 6C, lanes 4–8). All the mutant proteins efficiently form binary complexes on the E74 site (Fig. 6C, lanes 9–16).
Figure 5.

Mapping of the C-terminal SBM. (A) Diagrammatic representation of C-terminally truncated Fli-1 proteins. Numbers represent the first and last amino acids in each construct. (B and C) Gel retardation analysis of ternary complex formation by the indicated Fli-1 derivatives, mutant coreSRF(T196A) [(B), lanes 1–5 and (C), lanes 1–3] and the c-fos SRE. Binary complex formation by the same Fli-1 proteins with the E74 site in the absence of coreSRF is shown in (B), lanes 6–10.
Figure 6.

Identification of important residues within the C-terminal SRF interaction motif. (A) Alignment of the sequences surrounding the truncation endpoints in Fli-1(276–391), Fli-1(276–401) and Fli-1(276–411) (indicated by black arrows) from zebrafish, xenopus, mouse and human homologues. Residues conserved in three or more species are shaded grey, and fully conserved amino acids are indicated by asterisks. (B) Location and identity of the amino acids mutated in zebrafish Fli-1. A summary of the results of the gel retardation assays is shown on the right. (C) Gel retardation analysis of complex formation by the indicated truncated and mutant Fli-1 derivatives in the presence of coreSRF(T196A) and the c-fos SRE (lanes 1–8) or in the absence of coreSRF on the E74 site (lanes 9–16). The locations of the ternary Fli-1–SRF–SRE (30, closed arrow), binary SRF–SRE (20, open arrow) and binary Fli-1–E74 (20, grey arrow) complexes are indicated.
Collectively, these data therefore indicate that residues A392, V397 and F399 within the region 392–401 are important determinants within the C-terminal SRF interaction motif although additional residues between amino acids 402 and 411 are required for maximal binding.
Interactions between Fli-1 and SRF are required for recruitment of Fli-1 into ternary complexes in vivo
To investigate the importance of the SBMs in vivo, mutations were first introduced into Fli-1 in the context of the full-length protein and the truncated version, Fli-1(220–451) (Fig. 7A). Consistent with the effects of the individual mutations, the introduction of the two mutations A224D/V225E into Fli-1 abrogates its ability to form ternary complexes with SRF and the c-fos SRE (Fig. 7B, lane 2).
Figure 7.

Mutations in the N-terminal SBM reduce Fli-1 recruitment to the c-fos SRE in vitro and in vivo. (A) Diagrammatic representation of wild-type and mutant, full-length and truncated Fli-1 constructs. (B) Gel retardation analysis of ternary complex formation by wild-type and A224D/V225E mutant Fli-1(220–451), wild-type coreSRF and the c-fos SRE. The locations of the ternary (30, closed arrow) Fli-1–SRF–SRE and binary (20, open arrow) SRF–SRE complexes are indicated. (C) Western analysis of the expression of wild-type Fli-1(1–451)[VP16] and mutant Fli-1(1–451)[VP16](A224D/V225E) in NIH 3T3 cells. Lysates are analysed that contain increasing amounts of each Fli-expression vector (0.25 µg, lanes 1 and 4; 1 µg, lanes 2 and 5; 5 µg, lanes 3 and 6) and expression was detected using an anti-VP16 antibody. (D) Activation of SRE-luciferase (left) and E74-CAT (right) reporters by wild-type and mutant Fli-1–VP16 fusion proteins. The SRE-luciferase reporter construct (50 ng) was transfected into NIH 3T3 cells either alone or with increasing amounts of wild-type and mutant A224D/V225E Fli-1(1–451)[VP16] (0.5 and 1 µg) and a β-galactosidase expression vector. For the E74-CAT transfections, 1 µg E74-CAT and 0.5 µg of wild-type or mutant Fli-1 were cotransfected.
The mutations A224D/V225E were also introduced into a Fli-1 fusion protein composed of Fli-1 amino acids 1–451 fused to the VP16 transactivation domain (Fig. 1D). This VP16 fusion protein represents a constitutively active version of Fli-1 that does not require activation by signalling pathways or co-regulatory proteins. An analogous Elk-1–VP16 fusion protein has been used to demonstrate Elk-1 recruitment to the c-fos SRE in vivo (7,27). The expression of wild-type Fli-1[VP16] and Fli-1[VP16](A224D/V225E) was monitored by western blotting. Both proteins were expressed to similar extents with Fli-1[VP16](A224D/V225E) being expressed to a slightly higher level (Fig. 1C).
The ability of the wild-type and mutant Fli-1 proteins to be recruited to the c-fos SRE in vivo was determined by transient transfection of the two Fli-1 derivatives with a c-fos SRE-luciferase reporter plasmid. Wild-type Fli-1[VP16] activates the c-fos SRE in a dose-dependent manner (Fig. 7D) in an analogous manner to Elk-1[VP16] (7; data not shown). However, no significant activation of the c-fos SRE could be observed by the Fli-1[VP16](A224D/V225E) mutant protein (Fig. 7D). In order to verify that the mutations into Fli-1 do not abrogate the ability of Fli-1[VP16](A224D/V225E) to activate transcription rather than its ability to interact with SRF, this protein was tested for its ability to activate an E74-CAT reporter. This reporter construct can be bound and activated by ETS-domain proteins in the absence of coregulatory partners (7). Both wild-type and mutant Fli-1 proteins can activate the E74-CAT reporter gene, albeit at a reduced level for the mutant protein (Fig. 7D, 8-fold and 4-fold respectively). At the same concentration of transfected DNA, no significant activation of the SRE is observed, demonstrating the specificity of the mutations in blocking activation in complexes with SRF. At higher concentrations of Fli-1 (A224D/V225E), transcriptional squelching was observed on both the SRE and E74 reporter constructs (Fig. 7D, data not shown), most likely reflecting the higher expression level of this mutant protein (Fig. 7C).
Together, these data indicate that Fli-1 can be recruited to the c-fos SRE in vivo and that disruption of the N-terminal SRF-interaction motif abrogates this recruitment. Thus, like Elk-1, SAP-1 and SAP-2, Fli-1 can be considered a ternary complex interaction partner for SRF at SREs.
DISCUSSION
It is becoming evident that multicomponent transcription complexes play a major role in regulating the expression of eukaryotic genes. In combination with molecular biological approaches, recent structural studies have elucidated the mechanisms of how several such complexes form (reviewed in 28). One such study demonstrated how the ankyrin repeats of GABP2 mediate interactions with the DNA-binding domain of the ETS-domain protein GABPα (29). Indeed, many ETS-domain proteins have been shown to interact with other transcription factors (reviewed in 1–3). In this study we have investigated how Fli-1 interacts with the MADS-box transcription factor SRF. Two SBMs have been identified, which are located on either side of the ETS DNA-binding domain of Fli-1 (Fig. 8A). Important amino acids within these motifs have been identified by using site-directed mutagenesis. By investigating Fli-1 proteins with mutations in SBM1, the importance of the interactions between Fli-1 and SRF in recruitment of Fli-1 to the c-fos SRE in vivo have been demonstrated.
Figure 8.
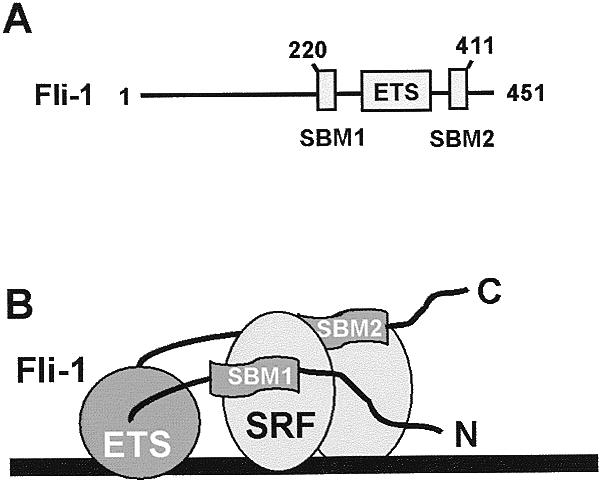
Model of the Fli–SRF complex. (A) Diagrammatic representation of Fli-1 showing the locations of the two SBMs relative to the ETS DNA-binding domain. The numbers of the amino acids mapped to the N- and C-terminal extents of these two motifs are indicated. (B) Model of the structure of the Fli-1–SRF complex. In this model, the two SBMs contact different SRF monomers although two contact points on the same monomer is also plausible (see Discussion). The ETS-domain and SBMs of Fli-1 are shown in dark grey whereas each SRF monomer is represented by light grey ellipses. DNA is represented by an extended black rectangle.
It has been previously shown that complexes can be formed between mammalian Fli-1 and SRF complexes at the c-fos (19) and egr-1 (20) SREs. In the latter study, Fli-1–SRF complexes could not be detected at the c-fos SRE, possibly due to the sensitivity of the assay system. However, deletion of the N-terminal region of Fli-1, either artificially or in EWS–Fli-1 fusion proteins, results in an enhancement of the ability of Fli-1 to form ternary Fli-1–SRF–SRE complexes (19). These observations suggest the presence of an inhibitory region at the N-terminus of Fli-1. Indeed, our results support this conclusion, as truncation of the N-terminus of the zebrafish Fli-1 homologue raises its affinity for both ternary complex formation (Fig. 1) and its ability to bind autonomously to the high affinity E74 site (15).
Here, we have mapped the important parts of the SBMs in Fli-1 to amino acids 220–228 (N-terminal to the ETS-domain) and amino acids 392–401 (C-terminal to the ETS-domain). Significantly, the N-terminal limit of SBM, corresponds almost exactly to the N-terminal end of the Fli-1 moiety contained in the most commonly found EWS–Fli-1 fusion proteins (10). Indeed, 94% of EWS–Fli-1 proteins contain SBM1. Furthermore, both SBM1 and SBM2 are highly conserved amongst vertebrate Fli-1 homologues (Figs 4A and 6A), further implying a functional importance for these motifs.
The mechanism of interaction of Fli-1 with SRF shows several key differences to the interaction of Elk-1 with SRF. Although hydrophobic residues appear to be important in both cases, neither SBM1 nor SBM2 show any significant homology with the SRF binding motif (B-box) in Elk-1 (compare Figs 1D, 4B and 6B). Moreover, the SBM1 maps to outside a region suggested to be related to the Elk-1 B-box (20). However, as no proline residues are found within SBM1 and SBM2, it is possible that as shown for Elk-1 (7), these motifs might be able to adopt an α-helical conformation for binding to SRF. The binding surfaces for Fli-1 and Elk-1 on SRF also appear to differ significantly. Although competition experiments clearly demonstrate an overlap in binding surfaces, mutagenic studies indicate that distinct residues play important roles in each case (8). For example, whilst the mutation V194E in SRF virtually abolishes ternary complex formation with Elk-1, the binding of Fli-1 is still readily detectable. A similar scenario is observed with the mutation T196A which enhances interactions with Fli-1 whilst leaving interactions with Elk-1 unaffected. Other mutations such as T196K result in reduced complex formation by both Fli-1 and Elk-1. Thus, whilst the binding surfaces overlap, further detailed mutagenesis is required to define the binding surface(s) on SRF to which Fli-1 binds.
The in vivo significance of Fli-1 interactions with SRF is currently unclear. However, in this study we provide the first evidence that Fli-1 can be recruited to the c-fos SRE in vivo and that interactions with SRF are critical in this process (Fig. 7). It should be emphasised that we do not envisage SRF and Fli-1 existing in non-DNA bound complexes in vivo and there is currently no evidence to support such a notion. It is likely that protein–DNA contacts by Fli-1 are essential for its recruitment into complexes with SRF in an analogous manner to the Elk-1–SRF complex. Indeed, higher affinity Fli-1–SRF complexes are likely to occur on other sites that contain more optimal ets binding motifs such as in the egr-1 promoter (20). Furthermore, due to the loss of the inhibitory N-terminal end of Fli-1, EWS–Fli-1 chimeras form higher affinity complexes at the c-fos and probably other SREs (Fig. 1; 19). Overexpression of Fli-1 as observed in erythroleukaemias, is also likely to drive the formation of ternary complexes with SRF. Finally, it is currently unclear whether complex formation by Fli-1 is regulated by signal transduction pathways in vivo as observed with the Elk-1–SRF complex (reviewed in 5) but such a mechanism could also lead to enhanced ternary complex formation.
Several models can be envisaged for how Fli-1 interacts with SRF at the c-fos SRE. One of these is depicted in Figure 8B, in which Fli-1 makes protein–DNA contacts with the ets motif in the SRE via its ETS DNA-binding domain and makes protein–protein contacts with SRF via SBM1 and SBM2. In this model, SBM1 and SBM2 bind different SRF monomers but it is equally likely that the two motifs bind to the same SRF monomer. Furthermore, additional complexities are suggested by the observation that the region immediately N-terminal to the ETS-domain (amino acids 256–266) is required for maximal binding by SBM2 (Fig. 3), suggesting that these regions co-operate in some manner. In summary, therefore, our data allow a model to be derived for how Fli-1 interacts with SRF. The mechanism of complex assembly shows several key differences to the Elk-1–SRF complex. As other transcription factors including NFκB (30) and SPIN/TFII-I (31) have also been shown to bind to SRF, it will also be interesting to determine how these diverse proteins interact with SRF. Transcription factor complexes containing SRF therefore serve as a useful model to further our understanding of how multicomponent transcription factor complexes are assembled.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Margaret Bell for excellent technical assistance and Lynne Smith for secretarial assistance. We would also like to thank Bob Liddell for DNA sequencing and Adam Rodaway, Roger Patient, Peter O’Hare and Richard Treisman for reagents. We are grateful to Alan Hair, Paul Shore and Shen-Hsi Yang for comments on the manuscript and members of our laboratory for helpful discussions. This work was supported by the Wellcome Trust and the UK Cancer Research Campaign [CRC]. A.D.S. is a Research Fellow of the Lister Institute of Preventative Medicine.
REFERENCES
- 1.Sharrocks A.D., Brown,A.L., Ling,Y. and Yates,P.R. (1997) Int. J. Biochem. Cell Biol., 29, 1371–1387. [DOI] [PubMed] [Google Scholar]
- 2.Graves B.J. and Petersen,J.M. (1998) In van de Woude,G. and Klein,G. (eds), Advances in Cancer Research. Academic Press, San Diego, CA.
- 3.Wasylyk B., Hagman,J. and Gutierrez-Hartmann,A. (1998) Trends Biochem. Sci., 23, 213–216. [DOI] [PubMed] [Google Scholar]
- 4.Treisman R. (1994) Curr. Opin. Genet. Dev., 4, 96–101. [DOI] [PubMed] [Google Scholar]
- 5.Yang S.-H., Yates,P.R., Mo,Y. and Sharrocks,A.D. (1998b) Gene Theor. Mol. Biol., 3, 355–371. [Google Scholar]
- 6.Shore P. and Sharrocks,A.D. (1994) Mol. Cell. Biol., 14, 3283–3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ling Y., Lakey,J., Roberts,E.C. and Sharrocks,A.D. (1997) EMBO J., 16, 2431–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ling Y., West,A.G., Roberts,E.C., Lakey,J.H. and Sharrocks,A.D. (1998) J. Biol. Chem., 273, 10506–10514. [DOI] [PubMed] [Google Scholar]
- 9.Ben-David Y., Giddens,E.B., Letwin,K. and Bernstein,A. (1991) Genes Dev., 5, 908–918. [DOI] [PubMed] [Google Scholar]
- 10.Zucman J., Melot,T., Desmaze,C., Ghysdael,J., Plougastel,B., Peter,M., Zucker,J.M., Triche,T.J., Sheer,D., Turc-Carel,C., Ambros,P., Combaret,V., Lenoir,G., Aorias,A., Thomas,G. and Delattre,O. (1993) EMBO J., 12, 4481–4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klemsz M.J., Maki,R.A., Papayannopoulou,T., Moore,J. and Hromas,R. (1993) J. Biol. Chem., 268, 5769–5773. [PubMed] [Google Scholar]
- 12.Meyer D., Stiegler,P., Hindelang,C., Mager,A.-M. and Remy,P. (1995) Int. J. Dev. Biol., 39, 909–919. [PubMed] [Google Scholar]
- 13.Melet F., Motro,B., Rossi,D.J., Zhang,L. and Bernstein,A. (1996) Mol. Cell. Biol., 16, 2708–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thompson M.A., Ransom,D.G., Pratt,S.J., MacLennan,H., Kieran,M.W., Detrich,H.W., Vail,B., Huber,T.L., Paw,B., Brownlie,A.J., Oates,A.C., Fritz,A., Gates,M.A., Amores,A., Bahary,N., Talbot,W.S., Her,H., Beier,D.R., Postlethwait,J.H. and Zon,L.I. (1998) Dev. Biol., 197, 248–269. [DOI] [PubMed] [Google Scholar]
- 15.Brown A.L., Rodaway,A., Schilling,T., Jowett,T., Ingham,P., Patient,R. and Sharrocks,A.D. (1999) Mech Dev., in press. [DOI] [PubMed] [Google Scholar]
- 16.Fitzsimmons D., Hodsdon,W., Wheat,W., Maira,S.-M., Wasylyk,B. and Hagman,J. (1996) Genes Dev., 10, 2198–2211. [DOI] [PubMed] [Google Scholar]
- 17.Bassuk A.G. and Leiden,J.M. (1995) Immunity, 3, 223–237. [DOI] [PubMed] [Google Scholar]
- 18.Kwiatkowski B.A., Bastian,L.S., Bauer,T.R.,Jr, Tsai,S., Zielinska-Kwiatkowska,A.G. and Hickstein,D.D. (1998) J. Biol. Chem., 273, 17525–17530. [DOI] [PubMed] [Google Scholar]
- 19.Magnaghi-Jaulin L., Masutani,H., Robin,P., Lipinski,M. and Harel-Bellan,A. (1996) Nucleic Acids Res., 24, 1052–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watson D.K., Robinson,L., Hodge,D.R., Kola,I., Papas,T.K. and Seth,A. (1997) Oncogene, 14, 213–221. [DOI] [PubMed] [Google Scholar]
- 21.Guan K. and Dixon,J.E. (1991) Anal. Biochem., 192, 262–267. [DOI] [PubMed] [Google Scholar]
- 22.Seth A., Gonzalez,F.A., Gupta,S., Raden,D.L. and Davis,R.J. (1992) J. Biol. Chem., 267, 24796–24804. [PubMed] [Google Scholar]
- 23.Sharrocks A.D., von Hesler,F. and Shaw,P.E. (1993a) Nucleic Acids Res., 21, 215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharrocks A.D., Gille,H. and Shaw,P.E. (1993b) Mol. Cell. Biol., 13, 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang S.-H., Whitmarsh,A.J., Davis,R.J. and Sharrocks,A.D. (1998) Mol. Cell. Biol., 18, 710–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vogel A.M. and Gerster,T. (1999) Mech. Dev., 81, 217–221. [DOI] [PubMed] [Google Scholar]
- 27.Price M.A., Rogers,A.E. and Treisman,R. (1995) EMBO J., 14, 2589–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolberger C. (1998) Curr. Opin. Genet. Dev., 8, 552–559. [DOI] [PubMed] [Google Scholar]
- 29.Batchelor A.H., Piper,D.E., de la Brousse,F.C., McKnight,S.L. and Wolberger,C. (1998) Science, 279, 1037–1041. [DOI] [PubMed] [Google Scholar]
- 30.Franzoso G., Carlson,L., Brown,K., Daucher,M.B., Bressler,P. and Siebenlist,U. (1996) EMBO J., 15, 3403–3412. [PMC free article] [PubMed] [Google Scholar]
- 31.Grueneberg D.A., Henry,R.W., Braver,A., Novina,C.D., Cheriyath,V., Roy,A.L. and Gilman,M. (1997) Genes Dev., 11, 2482–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pellegrini L., Tan,S. and Richmond,T.J. (1995) Nature, 376, 490–498. [DOI] [PubMed] [Google Scholar]