

Abstract

Regioselective phosphonation of pyridines is an interesting transformation in synthesis and medicinal chemistry. We report herein a metal-free approach enabling access to various 4-phosphonated pyridines. The method consists of simply activating the pyridine ring with a Lewis acid (BF3·OEt2) to facilitate the nucleophilic addition of a phosphine oxide anion. The formed sigma complex is subsequently oxidized with an organic oxidant (chloranil) to yield the desired adducts in good to excellent yields. We furthermore showed that access to C2-phosphoinated pyridines can be achieved in certain cases with strong Lewis basic phosphorus nucleophiles or with strong Lewis acidic pyridines. Both experimental and computational mechanistic investigations were undertaken and allowed us to understand the factors controlling the reactivity and selectivity of this reaction.
Keywords: C−H functionalization, DFT calculations, mechanisms, phosphonation, pyridines, reactivity
Introduction
Pyridines are key motifs in agrochemicals, functional materials, transition metal catalysis, and organocatalysis.1−4 Thus, the development of practical methods for the selective functionalization of these heterocycles remains an important challenge for both academia and pharmaceutical industry. Apart from the selective activation of C–H bonds of pyridine to forge C–C, C–N, and C–O bonds, the introduction of C–P bonds is of high importance as its enables access to phosphonates, phosphine oxides, and phosphines, which are highly important scaffolds in materials science, biochemistry, and catalysis.5 More importantly, the development of site-selective phosphonation has attracted much attention during the last decades, given the capability of those approaches to furnish organophosphorus compounds in straightforward and step-economical fashions.6,7
For instance, the addition of triphenylphosphine to a pyridinium ion, followed by deprotonation, has allowed the group of McNally to synthesize a large variety of phosphonium ions that were converted to highly useful C4-functionalized pyridines.8−10
Moreover, a plethora of interesting approaches, mainly using transition metal catalysis, allowing direct access to phosphosphonated pyridines from readily available starting materials have emerged. Recently, the field has gained much interest with the renaissance of the field of photoredox catalysis.11−16 For instance, the Hong group has elegantly achieved the C4-phosphonation of pyridines by combing N-ethoxypyridinium salts with secondary phosphine oxides in the presence of an oxidant (K2S2O8) and an organophotocatalyst under blue light irradiation (Scheme 1).17
Scheme 1. Different Approaches for the Site-selective Phosphonation of pyridines. TM: Transition Metal, PC: Photocatalyst.

In contrast to transition metal and photocatalytic methods outlined above, the use of polar chemistry to achieve selective C-4 phosphonation has not been extensively investigated.18 As part of our interest to develop mechanistically driven approaches for the formation of C–P,19−23 we hypothesized that the use of oxidative Chichibabin-type reaction, that is, nucleophilic addition of a phosphine oxide anion to an activated pyridine followed by oxidative aromatization, would provide a straightforward access to C-4 phosphonated pyridines through the mechanism outlined in Scheme 1.24
Results and Discussion
To test our hypothesis, we investigated the reaction of 3-cyanopyridine (1a) with diphenyl phosphine oxide (2a) in the presence of a Lewis acid, a base, and an oxidant. To ensure full activation of pyridine and to avoid the complexation of the nucleophile with the Lewis acid, 1.1 equiv of the latter was added to the pyridine in THF at −78 °C. To this mixture was added diphenylphosphine oxide anion, which was generated in situ by adding a base to diphenylphosphine oxide (2a) in THF. Finally, the oxidant was added to obtain the desired phosphonated pyridine (3a)
As shown in Table 1, we first evaluated the effect of the base on the reaction by taking BF3·OEt2 as the Lewis acid and chloranil as the oxidant. Unsurprisingly, no reaction took place when the relatively weak Brønsted bases such as NaHCO3 (entry 1) and NaOH (entry 2) were employed. However, 79% of 3a was isolated by column chromatography and characterized by X-ray diffraction.25 When tBuOK was used as a base, a good conversion was observed (entry 3). Unsurprisingly, the reaction did not proceed in the absence of a base (entry 4). Keeping tBuOK as the base of choice, we next examined the effect of the Lewis acid on the reaction. While no reaction occurred in the absence of BF3·OEt2 (entry 5), modest or low yields were obtained when BCl3 or 0.2 equiv of BF3·OEt2 was employed, respectively (entries 6 and 7). The nature of the oxidant turned out to be a key factor for the feasibility of the reaction as mild oxidants such as air, S8, O2, and I2 failed to drive the reaction (entries 8–11). Although 54% of the isolated yield of 3a was obtained when DDQ was used as the oxidant (entry 12), it remained less efficient as chloranil. Other solvents like acetonitrile and diethylether (entries 13 and 14) were tested and gave lower yields than that obtained with THF. Finally, increasing the temperature to −40 °C slightly diminished the yield of the reaction. Taken together, the best reaction conditions used to explore the scope were those depicted in entry 3 of Table 1.
Table 1. Optimization of the C4-Phosphonation of 3-Cyanopyridinea.

| entry | base | oxidant | solvent | 3a, yield [%]b |
|---|---|---|---|---|
| 1 | NaHCO3 | chloranil | THF | |
| 2 | NaOH | chloranil | THF | |
| 3 | tBuOK | chloranil | THF | 79 |
| 4 | chloranil | THF | ||
| 5 | tBuOKc | chloranil | THF | |
| 6 | tBuOKd | chloranil | THF | 52 |
| 7 | tBuOKe | chloranil | THF | 17 |
| 8 | tBuOK | air | THF | |
| 9 | tBuOK | S8 | THF | |
| 10 | tBuOK | O2 | THF | |
| 11 | tBuOK | I2 | THF | |
| 12 | tBuOK | DDQ | THF | 54 |
| 13 | tBuOK | chloranil | ACN | 64 |
| 14 | tBuOK | chloranil | Et2O | 62 |
| 15 | tBuOKf | chloranil | THF | 71 |
Reaction conditions: 3-cyanopyridine 1a (1 mmol, 1 equiv), BF3·OEt2 (1.1 mmol, 1.1 equiv), diphenylphosphine oxide 2a (1.2 mmol, 1.2 equiv), tBuOK (1.4 mmol, 1.4 equiv), chloranil (2 mmol), solvent (2 mL) at −78 °C, 10 min.
Isolated yield.
In the absence of BF3·OEt2.
BCl3 instead of BF3·OEt2.
0.2 equiv of BF3·OEt2.
T = −40 °C.
With the optimized reaction conditions in hand, we next explored the scope of the reaction (Figure 1). The parent pyridine gave only C-4 regioisomer 3b in 85% yield, and products resulting from C2- or C6-phosphonations could not be detected under the reaction conditions. The same regioselectivity was observed with different pyridines bearing either electron-withdrawing (3c–3g) or -donating (3h) groups at the C3-position. Importantly, halogens [chloro (3c), bromo (3d), fluoro (3e),26 and idodo (3f)] groups were all tolerated under our reaction conditions. The reaction proceeds smoothly with disubstituted pyridine, furnishing the C4-adduct (3i) in good yield (85%). Moreover, the reaction works well with 2-methylpyridine (3h, 82%), 6,7-dihydro-5H-cyclopenta[b]pyridine (3k, 83%), and 3-bromoquinoline (3l, 82%). We further tested other phosphine oxides with diphenyl phosphine oxide (2a). As shown in Figure 1, diaryl phosphine oxides (3m and 3n) resulted in good yields (79–86%). Finally, alkyl arylphosphine oxide and dialkyphosphine oxide were also compatible with the reaction conditions and gave the desired adducts 3o and 3p in good yields.
Figure 1.

Substrate scope for metal-free phosphonation.
Evidently, the C2-regioisomer could be obtained when the C4-position of the pyridine is occupied with cyano (3qa) or diphenylphosphine oxide groups (3qb). Interestingly, pyridine-bearing carbonyls at the C3 position led to the exclusive formation of C2-phosphonated pyridines 3qc and 3qd. However, unlike tert-butyl(phenyl)- and di-tert-butyl-phosphine oxide that gave only C4-adducts under the optimized reaction conditions (Figure 1, 3o and 3p), the C2-regioisomers were obtained when the reaction was carried out with ethyl(phenyl)phosphine oxide (3qe) and dicyclohexylphosphine oxide (3qf). The same C2-regioselectivity was observed with phosphinate (3qg) in good yield (73%). Importantly, highly diasteroselective C2-phosphonation (dr = 74%) could be achieved with a chiral phosphinate, leading to 3qh in fair yield (55%) (Figure 2). Crystallization of this mixture leads to the isolation of a pure diastereomer in 44% yield, which was characterized by X-ray diffraction.27
Figure 2.

C2-regioselectivity of the phosphonation of pyridines.
Having examined the scope of the phosphonation reaction, we turned our attention to the mechanism of this metal-free transformation. In accordance with previous work by Moore,28 the addition of BF3·OEt2 to pyridine 1c leads to a fast and quantitative formation of the expected Lewis base–Lewis base adduct (1c-BF3) as attested by 1H and 11B NMR spectroscopy experiments. When 1 equiv of tBuOK was added to diphenyl phosphine oxide (2a), a new signal relaxing at δP = 82.3 ppm corresponding to anion 2a–k was observed in deuterated THF (Scheme 2).
Scheme 2. Investigation of the Reaction Mechanism of the Phosphonation of Pyridine 1c by 31P NMR Spectroscopy.

The addition of this anion to the complex 1c-BF3 results in the immediate appearance of a new signal at 22.8 ppm as attested by 31P NMR. To elucidate the structure of the new species, 2D heteronuclear single quantum correlation (1H–31P HSQC) was performed in d8-THF at −50 °C. As shown in Figure 3, the phosphorus signal at 22.8 ppm correlates with all dihydropyridine protons (Figure 3). The 1H NMR spectrum shows coupling of these protons with the phosphorus nucleus. These spectroscopic results suggest the formation of sigma complex 4c.29
Figure 3.

1H–31P HSQC spectrum of intermediate 4c.
The spectroscopic information outlined above strongly support the mechanism depicted in Scheme 2, where the reaction starts with the formation of adduct 1-BF3. The regioselectivity (C4 vs C2) attack depends on the Lewis basicity of the phosphine oxide anion.30 Indeed, strong Lewis bases such as ethyl(phenyl)phosphine oxide- (1qe), di-cyclohexylphosphine oxide-anion (1qf), and ethylphenylphosphinate-anions react irreversibly at the C-2 position of 1-BF3. By contrast, weaker Lewis bases such as diarylphosphine oxide anions react reversibly at the C-2 position due to steric clash between substituent of the nucleophile and fluorine atoms of 1-BF3, which lead to the exclusive formation of the thermodynamic adducts (C-4) (Scheme 2). The high regioselectivity observed in the cases of 3qc and 3qd might be explained by the high Lewis acidity of the activated pyridine 1-BF3.
To further support the mechanistic proposal, we performed a computational investigation at the DLPNO-CCSD(T)/def2-TZVPPD/SMD(THF)//ωB97X-D/6-311+G(d,p) for selected 1-BF3 adducts. In line with the experimental findings discussed above, our calculations also predict a strong interaction energy between the free pyridine and BF3 (−79 < ΔG < −67 kJ mol–1). We then focused on the formation of the C–P bond as the key step of this transformation and considered a potential nucleophilic attack at C2, C4, and C6 of the activated pyridine (Table 2). In line with the high reactivity of both reaction partners, the computed barriers for this step are all relatively low and indicate rapid reactions even at lower temperatures. In all cases, phosphonation at C4 leads to the thermodynamically most stable sigma complexes 4, which are substantially more stable than the isomeric structures 5 and 6. In contrast, the kinetic preference is not as clear throughout this series. In line with the increasing electron deficiency, the activation free energies generally decrease within the series 1a → 1g → 1h. However, substantially smaller differences were calculated for the activation free energies of TSC2, TSC4, and TSC6 compared to the sigma complexes. Selected structures for these transition states are shown in Figure 4 for the reaction of the 3-fluorinated pyridine complex 1e-BF3. In all transition-state structures, the C–P bonds are very long (2.73–2.79 Å) indicating very early transition states. Even in the sigma complexes, these bonds are still slightly elongated (1.89–1.92 Å compared to 1.82–1.83 Å for the other CAr–P bonds). Interestingly, the most electron-deficient substrates studied computationally (1qc) now also features a substantial kinetic preference for an attack at either C2 or C6. The computational investigations predict a preferential kinetic attack at the C6 position, which is experimentally not observed (see Figure 2). Given the low calculated barriers and the bimolecular character of this reaction step, a substantial part of the activation energy stems from entropic contributions, which can be difficult to calculate accurately31,32 and could be responsible for this deviation. In general, the computational investigations further support the hypothesis of thermodynamic control of the phosphonation reaction.
Table 2. Computational Results for the Regioselective Phosphonationa.

| R | TSC2 | 5 | TSC4 | 4 | TSC6 | 6 |
|---|---|---|---|---|---|---|
| H (1b) | +59 | –4 | +54 | –31 | ||
| 3-F (1e) | +46 | –20 | +50 | –43 | +42 | –6 |
| 3-Ph (1g) | +45 | –18 | +40 | –36 | +49 | –15 |
| 3-COPh (1qc) | +28 | –55 | +37 | –83 | +23 | –59 |
DLPNO-CCSD(T)/def2-TZVPPD/SMD(THF)//ωB97X-D/6-311+G(d,p), kJ mol–1.
Figure 4.
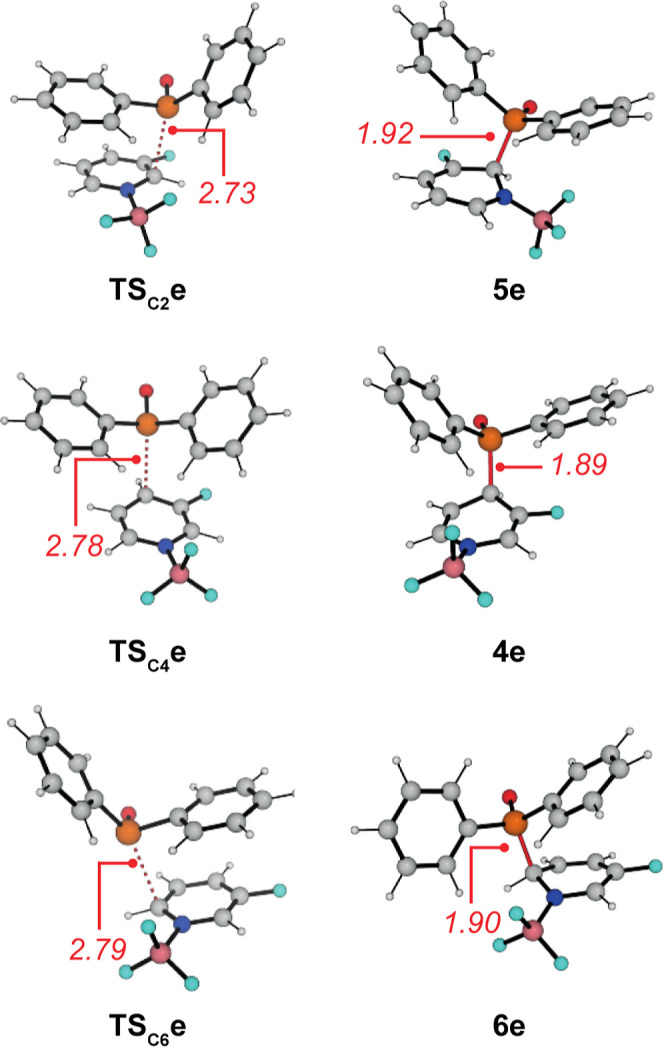
Calculated transition states and σ-complexed with selected bond length (in Å) for the phosphonation of 1e-BF3.
In conclusion, we have developed a metal-free BF3·OEt2-mediated phosphonation of various pyridines. The reaction is practically simple, highly yielding, and completely C4-regioselective. Mechanistic investigations have allowed the characterization of the sigma complex, formed from the nucleophilic addition of the phosphine oxide anion to activated pyridines (1-BF3), as a key intermediate in this transformation. Interestingly, the C2-regioselectivity observed with dialkyl and alkylaryl phopshine oxides was attributed to the high Lewis basicity of the corresponding anions. The use of this completely regioselective approach for the site-selective functionalization of pyridines with other nucleophiles is being studied in our laboratories and will be reported in due course.
Acknowledgments
The authors are grateful to the generous support from the “Ministère de la Recherche et des Nouvelles Technologies” (allocation to V.Q., T.H.V.N., and G.M.), Normandie Université, University of Toulouse, CNRS, “Région Normandie”, the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, BR5154/4-1), and the LABEX SynOrg (ANR-11-LABX-0029).
Data Availability Statement
The data underlying this study are available in the published article and its online Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsorginorgau.2c00055.
Reaction optimization studies, synthetic procedures, and characterization data for the phosphonation reaction, spectroscopic data for new compounds, and copies of NMR spectra (PDF)
Author Contributions
∥ V.Q. and T.H.V.N. contributed equally. V.Q., T.H.V.N., G.M., and S.C. performed the synthetic experiments; V.Q., S.C., and T.H.V.N performed NMR experiments; M.B. performed the DFT calculations, and S.L. designed and supervised the project. All authors have given approval to the final version of the manuscript. CRediT: Valentin Quint formal analysis (equal), investigation (equal), methodology (equal), writing-review & editing (equal); Gary Mathieu formal analysis (equal), investigation (equal).
The authors declare no competing financial interest.
Dedication
This article is dedicated to the memory of Prof. Mohamed Baker Rammah (University of Monastir) (1947–2022).
Supplementary Material
References
- Matolscy G.Pesticide Chemistry; Elseiver Scientific: Amsterdam, 1988. [Google Scholar]
- Leclerc N.; Sanaur S.; Galmiche L.; Mathevet F.; Attias A.-J.; Fave J.-L.; Roussel J.; Hapiot P.; Lemaître N.; Geffroy B. 6-(Arylvinylene)-3-bromopyridine Derivatives as Lego Building Blocks for Liquid Crystal, Nonlinear Optical, and Blue Light Emitting Chromophores. Chem. Mater. 2005, 17, 502. 10.1021/cm040358k. [DOI] [Google Scholar]
- Zafar M. N.; Atif A. H.; Nazar M. F.; Sumrra S. H.; Gul-E-Saba; Paracha R. Pyridine and related ligands in transition metal homogeneous catalysis. Russ. J. Coord. Chem. 2016, 42, 1. 10.1134/S1070328416010097. [DOI] [Google Scholar]
- Gujjarappa R.; Vodnala N.; Malakar C. C. Recent Advances in Pyridine-Based Organocatalysis and its Application towards Valuable Chemical Transformations. ChemistrySelect 2020, 5, 8745–8758. 10.1002/slct.202002765. [DOI] [Google Scholar]
- For a recent review, see:McNally R. D.; Dolewski M. C.; Hilton A. 4-Selective Pyridine Functionalization Reactions via Heterocyclic Phosphonium Salts. Synlett 2018, 29, 8–14. 10.1055/s-0036-1591850. [DOI] [Google Scholar]
- Feng C. G.; Ye M.; Xiao K. J.; Li S.; Yu J. Q. Pd(II)-Catalyzed Phosphorylation of Aryl C-H Bonds. J. Am. Chem. Soc. 2013, 135, 9322. 10.1021/ja404526x. [DOI] [PubMed] [Google Scholar]
- Xiang C. B.; Bian Y. J.; Mao X. R.; Huang Z. Z. Coupling Reactions of Heteroarenes with Phosphites under Silver Catalysis. J. Org. Chem. 2012, 77, 7706. 10.1021/jo301108g. [DOI] [PubMed] [Google Scholar]
- Hilton M. C.; Dolewski R. D.; McNally A. Selective Functionalization of Pyridines via Heterocyclic Phosphonium Salts. J. Am. Chem. Soc. 2016, 138, 13806. 10.1021/jacs.6b08662. [DOI] [PubMed] [Google Scholar]
- Zhang X.; McNally A. Phosphonium Salts as Pseudohalides: Regioselective Nickel-Catalyzed Cross-Coupling of Complex Pyridines and Diazines. Angew. Chem., Int. Ed. 2017, 56, 9833. 10.1002/anie.201704948. [DOI] [PubMed] [Google Scholar]
- Dolewski R. D.; Fricke P. J.; McNally A. Site-Selective Switching Strategies to Functionalize Polyazines. J. Am. Chem. Soc. 2018, 140, 8020. 10.1021/jacs.8b04530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanam J. M. R.; Stephenson C. R. J. Visible Light Photoredox Catalysis: Application in Organic Synthesis. Chem. Soc. Rev. 2011, 40, 102. 10.1039/B913880N. [DOI] [PubMed] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.-R.; Hu X.-Q.; Lu L.-Q.; Xiao W.-J. Exploration of Visible-Light Photocatalysis in Heterocycle Synthesis and Functionalization: Reaction Design and Beyond. Acc. Chem. Res. 2016, 49, 1911. 10.1021/acs.accounts.6b00254. [DOI] [PubMed] [Google Scholar]
- Romero N. A.; Nicewicz D. A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075. 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- Schultz D. M.; Yoon T. P. Solar Synthesis: Prospects in Visible Light Photocatalysis. Science 2014, 343, 1239176. 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y.; Yi H.; Lei A. Synthetic Applications of Photoredox Catalysis with Visible Light. Org. Biomol. Chem. 2013, 11, 2387. 10.1039/c3ob40137e. [DOI] [PubMed] [Google Scholar]
- Kim I.; Kang G.; Lee K.; Park B.; Kang D.; Jung H.; He Y.-T.; Baik M.-H.; Hong S. Site-Selective Functionalization of Pyridinium Derivatives via Visible-Light-Driven Photocatalysis with Quinolinone. J. Am. Chem. Soc. 2019, 141, 9239–9248. 10.1021/jacs.9b02013. [DOI] [PubMed] [Google Scholar]
- For a recent elegant work on Chichibabin phosphonylation of 2-(hetero)aryl pyridines, see:Yan X.; Sun R.; Zhang C.; Zhang Y.; Su Z.; Ge Y.; Chen H.; Fu H.; Li R. Chichibabin-Type Phosphonylationof 2-(Hetero)aryl Pyridines:SelectiveSynthesisof 4-PhosphinoylPyridinesvia an ActivatedN-BenzylpyridiniumSalt. Adv. Synth. Catal. 2022, 364, 2387–2394. 10.1002/adsc.202200289. [DOI] [Google Scholar]
- Quint V.; Morlet-Savary F.; Lohier J.-F.; Lalevée J.; Gaumont A.-C.; Lakhdar S. Metal-Free Visible Light-Photocatalyzed Synthesis of Benzo[b]phosphole Oxides: Synthetic and Mechanistic Investigations. J. Am. Chem. Soc. 2016, 138, 7436–7441. 10.1021/jacs.6b04069. [DOI] [PubMed] [Google Scholar]
- Quint V.; Chouchène N.; Askri M.; Lalevée J.; Gaumont A.-C.; Lakhdar S. Visible-Light-Mediated α-Phosphorylation of N-Aryl Tertiary Amines Through the Formation of Electron-Donor–Acceptor Complexes: Synthetic and Mechanistic Studies. Org. Chem. Front. 2019, 6, 41–44. 10.1039/C8QO00985F. [DOI] [Google Scholar]
- Lecroq W.; Bazille P.; Morlet-Savary F.; Breugst M.; Lalevée J.; Gaumont A.-C.; Lakhdar S. Visible-Light-Mediated Metal-Free Synthesis of Aryl Phosphonates: Synthetic and Mechanistic Investigations. Org. Lett. 2018, 20, 4164. 10.1021/acs.orglett.8b01379. [DOI] [PubMed] [Google Scholar]
- Noël-Duchesneau L.; Lagadic E.; Morlet-Savary F.; Lohier J.-F.; Chataigner I.; Breugst M.; Lalevée J.; Gaumont A.-C.; Lakhdar S. Metal-Free Synthesis of 6-Phosphorylated Phenanthridines: Synthetic and Mechanistic Insights. Org. Lett. 2016, 18, 5900–5903. 10.1021/acs.orglett.6b02983. [DOI] [PubMed] [Google Scholar]
- Pal S.; Gaumont A.-C.; Lakhdar S.; Gillaizeau I. Diphenyliodonium Ion/Et3N Promoted Csp2-H Radical Phosphorylation of Enamides. Org. Lett. 2019, 21, 5621–5625. 10.1021/acs.orglett.9b01963. [DOI] [PubMed] [Google Scholar]
- For an excellent example, see:Chen Q.; du Jourdin X. M.; Knochel P. Transition-metal-free BF3-mediated regioselective direct alkylation and arylation of functionalized pyridines using Grignard or organozinc reagents. J. Am. Chem. Soc. 2013, 135, 4958–4961. 10.1021/ja401146v. [DOI] [PubMed] [Google Scholar]
- CCDC—2212787. Contains the supplementary crystallographic data.
- CCDC—2212788. Contains the supplementary crystallographic data.
- CCDC—2212789. Contains the supplementary crystallographic data.
- Chénard E.; Sutrisno A.; Zhu L.; Assary R. S.; Kowalski J. A.; Barton J. L.; Bertke J. A.; Gray D. L.; Brushett F. R.; Curtiss L. A.; Moore J. S. Synthesis of Pyridine– and Pyrazine–BF3 Complexes and Their Characterization in Solution and Solid State. J. Phys. Chem. C 2016, 120, 8461–8471. 10.1021/acs.jpcc.6b00858. [DOI] [Google Scholar]
- For an excellent monograph, see: Terrier F.Modern Nucleophilic Aromatic Substitution; Wiley-VCH: Weinheim, 2013. [Google Scholar]
- Mayr H.; Ammer J.; Baidya M.; Maji B.; Nigst T. A.; Ofial A. R.; Singer T. Scales of Lewis Basicities toward C-Centered Lewis Acids (Carbocations). J. Am. Chem. Soc. 2015, 137, 2580–2599. 10.1021/ja511639b. [DOI] [PubMed] [Google Scholar]
- Ribeiro R. F.; Marenich A. V.; Cramer C. J.; Truhlar D. G. Use of Solution-Phase Vibrational Frequencies in Continuum Models for the Free Energy of Solvation. J. Phys. Chem. B 2011, 115, 14556–14562. 10.1021/jp205508z. [DOI] [PubMed] [Google Scholar]
- Grimme S. Supramolecular Binding Thermodynamics by Dispersion-Corrected Density Functional Theory. Chem.—Eur. J. 2012, 18, 9955–9964. 10.1002/chem.201200497. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its online Supporting Information.