

Abstract

Herein, we report a multistep one-pot reaction of substituted pyridines leading to N-protected tetrahydropyridines with outstanding enantioselectivity (up to 97% ee). An iridium(I)-catalyzed dearomative 1,2-hydrosilylation of pyridines enables the use of N-silyl enamines as a new type of nucleophile in a subsequent palladium-catalyzed asymmetric allylic alkylation. This telescoped process overcomes the intrinsic nucleophilic selectivity of pyridines to synthesize enantioenriched, C-3-substituted tetrahydropyridine products that have been otherwise challenging to access.
Aromatic compounds are stable feedstock chemicals available with numerous substitution patterns that find application in various areas, such as material science,1 pharmaceuticals,2 and many others. However, the number of methods for converting these readily available aromatic chemicals into value-added, enantioenriched, saturated molecules with increased complexity remains sparse.3 Transition-metal-catalyzed asymmetric allylic alkylation (AAA) reactions are established and reliable transformations to form tertiary and quaternary stereocenters via numerous combinations of nucleophiles and electrophiles.4,5 Because of the versatility of this transformation, a vibrant area of research has emerged to convert readily available aromatic compounds into enantioenriched, saturated substrates via dearomative, transition-metal-catalyzed asymmetric allylic alkylation reactions.6 Trost et al. reported the first enantioselective palladium-catalyzed dearomative allylic alkylation at the C-3 position of indoles in 2006 (Scheme 1A).7 Since then, the reactivity of numerous electron-rich heteroaromatics8a and electron-rich benzene derivates (phenols, anilines) have been explored.5 Recently in 2018, You et al. reported the first intramolecular, iridium-catalyzed dearomative allylation of simple benzenes, which further expanded this field (Scheme 1B).9 In 2014, You et al. published the intramolecular allylic N-alkylation of pyridines (Scheme 1C).10 However, the expansion of this field to electron-poor heterocycles, such as pyridines, remains elusive. In 2007, Hartwig et al. reported the allylic alkylation of terminal enamines under iridium catalysis, which leads to enantioenriched β-ketones after hydrolysis (Scheme 1D).11 The utilization of a similar enamine intermediate derived from the dearomative reduction of pyridines should allow for the asymmetric allylic alkylation of pyridines at the C-3 position, which would invert the inherent nucleophilic selectivity of the aromatic substrate. Among the most common motifs in pharmaceuticals are (partially) saturated N-heterocycles,12 while their synthesis remains challenging, especially in an enantioselective fashion. While the electrophilicity of the C-2 and C-4 positions of pyridines has been utilized with many nucleophiles, the functionalization of the C-3 position is less explored.13 Friedel–Crafts-like alkylations favor the 3-position, although they are mainly limited to pyridines bearing electron-donating substituents.
Scheme 1. Transition-Metal-Catalyzed Allylic Alkylations.

However, dearomative hydrosilylation reactions of pyridines and related heterocycles have been developed with several heterogeneous catalysts,14 transition metal catalysts (Ti, Ru, Ir, Zn),15 and main group metals (Ca)16 and organic catalysts (boranes).17 While the resulting N-silylated enamines are highly unstable and cannot be purified, their derivatization can lead to stable dihydropyridines. We hypothesized that N-silyl dihydropyridines, obtained by a dearomative 1,2-hydrosilylation, can act as enamine C-nucleophiles in an allylic alkylation reaction (Scheme 1E).
Here, we report a protocol that enables the dearomative C3-allylic alkylation of pyridines in high enantioselectivity. To the best of our knowledge, this is the first report on the utilization of pyridines as C-nucleophile precursors in enantioselective allylic alkylation reactions. For the initial reduction, we selected the iridium(I)-catalyzed hydrosilylation of aza-heteroaromatics that was reported by Chang et al. in 2016 (Scheme 2).15g
Scheme 2. Ir-Catalyzed Hydrosilylation of Pyridines by Chang et al.15g.

We tested our hypothesis by treating 3-fluoropyridine 1a with 1 mol % of [Ir(coe)2Cl]2 and 5 equiv of diethyl silane at 50 °C for 3 h. In accordance with Chang et al.’s results, we found almost quantitative conversion to the N-silyl dihydropyridine. This mixture was then added to a solution of Pd2(dba)3 (2.5 mol %), PPh3 (15 mol %), and cinnamyl methyl carbonate 2a (1.5 equiv) in CH2Cl2 (0.2 M) at 40 °C for 16 h. The desired dearomative allylation product 3a was observed in trace quantities (Table 1, entry 1). However, two side products were also detected. Namely, the rearomatized 3-alkylated pyridine was observed as a major product (4a, 11%) together with a bisalkylated side product (5a, <5%). Initial screening of monodentate phosphines revealed that neither more electron-rich or electron-deficient phosphines nor sterically more demanding monodentate phosphines could improve the outcome of this reaction. Having established a proof of concept for this reaction pathway, we turned our attention to developing an asymmetric variant. To control the stereochemistry at the C3-postion, several privileged chiral monodentate and bidentate phosphorus-containing ligands, such as PHOX, phosphoramidite, BINAP, DTBM-SegPhos, and DIOP were investigated [for details, see the Supporting Information (SI)]. Unfortunately, less than 5% of the desired product was observed in all reactions, but enantioinduction could be observed with up to 30% and 48% ee for L1 and Feringa’s phosphoramidite L2, respectively (Table 1, entries 2 and 3). Employing Trost-DACH ligand L3 (7 mol %) resulted in significantly increased conversion (38% of 3a) and excellent enantioselectivity (96% ee, entry 4). Modifications on the ligand did not result in an improved performance in the transformation. Pd(OAc)2 was found to be the optimal Pd source, while other precursors, such as Pd2dba3·CHCl3, gave similar results (Table 1, entries 4 and 5). CH2Cl2 was already the ideal solvent of the reaction, while lower conversion with slightly higher enantioselectivity was observed in benzene (Table 1, entry 6), THF, and 1,4-dioxane (for details, see the SI). Finally, an enhanced conversion for the desired product 3a was achieved though the addition of catalytic amounts of sodium fluoride (Table 1, entry 7). Further exploration of additional bases or hydride sources for the allylic alkylation step had no beneficial effect. Interestingly, with a reduced amount of hydrosilane, the alkylated pyridine 4b (Table 1, entry 8) was obtained as a major product (28% of 4a could be isolated; for details, see the SI). The ratio between the synergistic catalysts was crucial for the desired reactivity. Altering the amount of iridium dimer to 0.5 or 2.5 mol % resulted in a significantly decreased amount of 3a (Table 1, entries 9 and 10). Both catalysts are required for the reaction; otherwise, no product was observed in the control experiments without the Ir or Pd catalyst. In the reaction setting, the iridium-catalyzed hydrosilylation was performed under argon. After the indicated time, this reaction mixture was added to a prestirred mixture of palladium catalyst, ligand, additive, and carbonate in CH2Cl2 under argon and heated to 40 °C.
Table 1. Selected Optimization Reactionsa.

| # | deviation from standard reaction conditions | L | 3 (% ee) | 4 | 5 |
|---|---|---|---|---|---|
| 1 | Pd2(dba)3 2.5 mol % | PPh3 | 2 (n.d.) | 11 | <5 |
| 2 | Pd2(dba)3 2.5 mol % | L1 | <5 (30) | <5 | <5 |
| 3 | Pd2(dba)3 2.5 mol % | L2 | <5 (48) | <5 | |
| 4 | L3 | 38 (96) | 8 | 5 | |
| 5 | Pd2dba3·CHCl3 2.5 mol % | L3 | 38 (96) | 9 | 10 |
| 6 | Pd2dba3 2.5 mol % in PhH | L3 | 18 (98) | 11 | <5 |
| 7 | NaF 10 mol % | L3 | 49 (96) | 8 | 6 |
| 8b | Et2SiH2 2.5 equiv, NaF 10 mol % | L3 | 29 (n.d.) | 38 | 29 |
| 9b | [Ir] 2.5 mol %, NaF 10 mol % | L3 | 5 (n.d.) | <5 | <5 |
| 10b | [Ir] 0.5 mol %, NaF 10 mol % | L3 | 14 (n.d.) | 34 | 45 |

Reaction conditions: first step = 1a (0.2 mmol), Et2SiH2 (5 equiv), [Ir(coe)2Cl]2 (1 mol %), 50 °C, 3 h; second step = 2a (1.5 equiv), Pd(OAc)2 (5 mol %), L6 (7 mol %), CH2Cl2 (0.2 M), 40 °C, 16 h. LC/UV–vis yields were determined via calibration curve obtained using isolated products. L = ligand; n.d. = not determined.
3-Chloropyridine (1b) was used as substrate.
With the optimized conditions in hand, we investigated the substrate scope (Scheme 3). For simplified handling during purification, the product amines 3 were protected by N-acylation in an additional step in the same pot. Various electron-poor pyridines reacted smoothly to the desired products 6a–d and 6f–k in moderate yields between 21 and 54% but, in most cases, with excellent enantioselectivity of above 90% ee. Substituents were generally tolerated in the 3- and 4-position of the pyridine. Substituents on the 2- or 6-position of the pyridine did not yield any desired product likely because of the steric hindrance for the first hydrosilylation step. Motifs such as 4-aryl or 4-heteroaryl pyridines (6e, 6h, 6l, and 6m) also gave the desired products in moderate yields and excellent enantioselectivity. More electron-rich pyridines were challenging in this reaction mainly because of the significantly slower iridium-catalyzed hydrosilylation reaction, as previously observed.18
Scheme 3. Substrate Scope for the Enantioselective Allylic Alkylation Towards Chiral Products 6(19),
The iridium catalyst (1.5 mol %) was used with increased reaction times for the first step and by stepwise addition for more electron-rich pyridines; for details see the SI.
Standard reaction conditions: first step = 1 (0.5 mmol), Et2SiH2 (6.0 equiv), [Ir(coe)2Cl]2 (1 mol %), 50 °C, 1–6 h; second step = 2 (1.5 equiv) Pd(OAc)2 (5 mol %), L3 (7 mol %), CH2Cl2 [0.2 M], 40 °C, 16 h; third step = AcCl (3.0 equiv), pyridine (3.0 equiv), CH2Cl2, 25 °C, 16 h.
Residual pyridine did not inhibit the Pd-catalyzed alkylation. A control experiment even revealed some influence on the reaction selectivity, which is under further mechanistic investigation (see the SI for details).20 The electrophile scope; however, tolerates electron-donating as well as electron-withdrawing substituents on the arene, thereby giving the corresponding products in moderate yields and excellent enantioselectivity (6n–s). meta-Substitution on the arene also delivered the desired product (6t), while ortho-OCF3-substituted cinnamyl carbonate showed no conversion in this transformation (not shown). Fortunately, heteroaromatic carbonates could be applied in this reaction, as shown by products 6v and 6w. While simple alkyl-substituted allylic carbonates did not afford any product (see Scheme S7 for details), we found that a conjugated diene precursor delivered the diene product 6x in low yield but excellent enantioselectivity. The products 6 bearing a halogen handle also allowed further derivatization via cross-coupling chemistry, as demonstrated for 6b in Scheme 4. Typical Stille conditions with PhSnBu3 delivered the C–C cross-coupled product 8 in 82% yield, while a Ni-catalyzed Kumada reaction provided the N-deprotected C–C cross-coupled product 9 with similar yield (Scheme 4), which shows the synthetic utility of these motifs.
Scheme 4. Further Derivatization of 6b via Stille and Kumada Reactions.

From a synthetic perspective, the free NH products, such as 9 or 3a, are also of high interest. However, the direct purification by column chromatography after the second step, that is, the Pd-catalyzed allylic alkylation, proved to be challenging because of the polarity, as well as the complex reaction mixture. N-Boc protection after the allylic alkylation and a subsequent purification delivered an inseparable mixture of the N-Boc-protected desired product 3a, as well as the bisalkylated N-Boc side product 5a (Scheme 5, right). An acidic deprotection with TFA allowed the isolation of the pure NH product 3a in 22% yield over all four steps. Alternatively, the N-acetylated compounds 6b could be treated with PhMgBr to give the free NH product 3b in 88% yield (47% yield over four steps, Scheme 5, left).
Scheme 5. Isolation of the Free NH Product 3a,b.

On the basis of our results and understanding, the proposed reaction sequence (see Scheme 6) starts with the iridium-catalyzed 1,2-hydrosilylation of the pyridine substrate A, which leads to an N-silyl enamine B. The palladium-catalyzed allylic alkylation leads to a cyclic imine intermediate C (or as an N-silyl iminium ion). There are several plausible pathways from this key intermediate C. The excess of silane in the reaction mixture and the, therefore, overall reductive conditions in the presence of two transition metals can lead to a reduction of the imine, thereby leading to the enantioenriched products 3. Another pathway, which could explain the formation of the bisalkylated products 5, is the tautomerization of the imine C to the enamine D, which results in a nucleophile that participates in an additional alkylation event. Reduction of the product imine E leads to the bisalkylated derivatives 5. The observed rearomatized product 4 can potentially form at different stages during the proposed sequence, but would require an oxidant (e.g., air or a hydride transfer to π-allyl complexes).
Scheme 6. Proposed Reaction Sequence.

During the investigation of the reaction scope, we observed that the bisalkylated side products 5 occur in different ratios on the basis of the substrate. Since those motifs can be of synthetic interest, as well, the transformation was optimized toward these scaffolds using 3-chloropyridine 1b as the standard substrate.
The screening of alkylating reagents with leaving groups of variable basicity did not increase the overall yields of either 3b or 5b (for details, see the SI). It was found that catalytic amounts of benzoic acid (20 mol %) significantly shift the selectivity toward the bisalkylated product 5b (Scheme 7).21 The electronic properties of the alkylating reagent seem to have less of an influence on the outcome of the reaction (5c–f).
Scheme 7. Bisalkylated Products 5 and RCM.

The bisalkylated products 5 enable a potential ring-closing metathesis (RCM) to yield spiro compounds. Therefore, the N-benzyl product 10a was treated with typical RCM conditions under ruthenium catalysis. The spiro compound 11a was successfully formed in good yield of 73%, although as a 6:4 mixture of isomers that is formed by isomerization of the disubstituted olefin (see Scheme 7).
In conclusion, we have developed the first intermolecular asymmetric allylic alkylation (AAA) using electron-poor arenes, namely pyridines, as C-nucleophile precursors. A stepwise one-pot sequence allows rapid access to interesting molecular scaffolds in excellent enantioselectivities, although in moderate yields. The products are valuable building blocks for further exploration. The chlorine-substituted tetrahydropyridines are especially shown to be of particular use for the synthetic community as complex building blocks.
Acknowledgments
The authors thank Dr. Scott Virgil and Alex Cusumano for helpful discussions, David VanderVelde for NMR assistance, Dr. Michael Takase for XRD assistance, and Dr. Mona Shahgholi for mass spectrometry assistance. The authors thank the Beckman Institute for their support of the Caltech XRD facility, as well as the Dow Next Generation Instrument Grant.
Glossary
ABBREVIATIONS
- AAA
asymmetric allylic alkylation.
- RCM
ring-closing metathesis
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c02470.
Experimental procedures and spectroscopic data (1H NMR, 13C NMR, IR, and HRMS) (PDF)
S.G. and L.S. gratefully acknowledge the Alexander von Humboldt Foundation for a Feodor Lynen fellowship (2019–2020). The authors acknowledge the NIH-NIGMS (R35GM145239 and R01GM080269), as well as the Heritage Medical Research Investigator Program, for funding this research.
The authors declare no competing financial interest.
Supplementary Material
References
- For selected reviews, see:; a Aumaitre C.; Morin J.-F. Polycyclic Aromatic Hydrocarbons as Potential Building Blocks for Organic Solar Cells. Chem. Rec. 2019, 19 (6), 1142–1154. 10.1002/tcr.201900016. [DOI] [PubMed] [Google Scholar]; b Zhang L.; Cao Y.; Colella N. S.; Liang Y.; Brédas J.-L.; Houk K. N.; Briseno A. L. Unconventional, Chemically Stable, and Soluble Two-Dimensional Angular Polycyclic Aromatic Hydrocarbons: From Molecular Design to Device Applications. Acc. Chem. Res. 2015, 48 (3), 500–509. 10.1021/ar500278w. [DOI] [PubMed] [Google Scholar]
- For selected reviews, see:; a Ali Y.; Hamid S. A.; Rashid U. Biomedical Applications of Aromatic Azo Compounds. Mini-Rev. Med. 2018, 18 (18), 1548–1558. 10.2174/1389557518666180524113111. [DOI] [PubMed] [Google Scholar]; b Al-Mulla A. A Review: Biological Importance of Heterocyclic Compounds. Der Pharma Chem. 2017, 9 (13), 141–147. [Google Scholar]; c Saini M. S.; Kumar A.; Dwivedi J.; Singh R. A Review: Biological Significances of Heterocyclic Compounds. Int. J. Pharm. Sci. Res. 2013, 4 (3), 66–77. [Google Scholar]
- For selected reviews, see:; a Lovering F.; Bikker J.; Humblet C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52 (21), 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]; b Cox B.; Booker-Milburn K. I.; Elliott L. D.; Robertson-Ralph M.; Zdorichenko V. Escaping from Flatland:[2+ 2] Photocycloaddition; Conformationally Constrained Sp3-Rich Scaffolds for Lead Generation. ACS Medicinal Chem. Lett. 2019, 10 (11), 1512–1517. 10.1021/acsmedchemlett.9b00409. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lovering F. Escape from Flatland 2: Complexity and Promiscuity. MedChemComm. 2013, 4 (3), 515–519. 10.1039/c2md20347b. [DOI] [Google Scholar]
- For selected reviews, see:; a Trost B. M.; Van Vranken D. L. Asymmetric Transition Metal-Catalyzed Allylic Alkylations. Chem. Rev. 1996, 96, 395–422. 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]; b Trost B. M.; Crawley M. L. Asymmetric Transition-Metal-Catalyzed Allylic Alkylations: Applications in Total Synthesis. Chem. Rev. 2003, 103, 2921–2944. 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]; c Lu Z.; Ma S. Metal-Catalyzed Enantioselective Allylation in Asymmetric Synthesis. Angew. Chem., Int. Ed. 2008, 47, 258–297. 10.1002/anie.200605113. [DOI] [PubMed] [Google Scholar]; d Cheng Q.; Tu H.-F.; Zheng C.; Qu J.-P.; Helmchen G.; You S.-L. Iridium-Catalyzed Asymmetric Allylic Substitution Reactions. Chem. Rev. 2019, 119, 1855–1969. 10.1021/acs.chemrev.8b00506. [DOI] [PubMed] [Google Scholar]; e Liu Y.; Han S.-J.; Liu W.-B.; Stoltz B. M. Catalytic Enantioselective Construction of Quaternary Stereocenters: Assembly of Key Building Blocks for the Synthesis of Biologically Active Molecules. Acc. Chem. Res. 2015, 48, 740–751. 10.1021/ar5004658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected reviews and examples for Pd catalysis, see:; a Trost B. M.; Schultz J. E. Palladium-Catalyzed Asymmetric Allylic Alkylation Strategies for the Synthesis of Acyclic Tetrasubstituted Stereocenters. Synthesis 2019, 51, 1–30. 10.1055/s-0037-1610386. [DOI] [Google Scholar]; b Hong A. Y.; Stoltz B. M. The Construction of All-Carbon Quaternary Stereocenters by Use of Pd-Catalyzed Asymmetric Allylic Alkylation Reactions in Total Synthesis. Eur. J. Org. Chem. 2013, 2013, 2745–2759. 10.1002/ejoc.201201761. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Guerrero Rios I.; Rosas-Hernandez A.; Martin E. Recent Advances in the Application of Chiral Phosphine Ligands in Pd-Catalysed Asymmetric Allylic Alkylation. Molecules 2011, 16, 970–1010. 10.3390/molecules16010970. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Trost B. M.; Machacek M. R.; Aponick A. Predicting the Stereochemistry of Diphenylphosphino Benzoic Acid (DPPBA)-Based Palladium-Catalyzed Asymmetric Allylic Alkylation Reactions: A Working Model. Acc. Chem. Res. 2006, 39, 747–760. 10.1021/ar040063c. [DOI] [PubMed] [Google Scholar]; For selected examples, see:; e Trost B. M.; Dietsch T. J. New Synthetic Reactions. Asymmetric Induction in Allylic Alkylations. J. Am. Chem. Soc. 1973, 95, 8200–8201. 10.1021/ja00805a056. [DOI] [Google Scholar]; f Trost B. M.; Xu J.; Schmidt T. Palladium-Catalyzed Decarboxylative Asymmetric Allylic Alkylation of Enol Carbonates. J. Am. Chem. Soc. 2009, 131, 18343–18357. 10.1021/ja9053948. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Alexy E. J.; Zhang H.; Stoltz B. M. Catalytic Enantioselective Synthesis of Acyclic Quaternary Centers: Palladium-Catalyzed Decarboxylative Allylic Alkylation of Fully Substituted Acyclic Enol Carbonates. J. Am. Chem. Soc. 2018, 140, 10109–10112. 10.1021/jacs.8b05560. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Behenna D. C.; Stoltz B. M. The Enantioselective Tsuji Allylation. J. Am. Chem. Soc. 2004, 126, 15044–15045. 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]; i Mohr J. T.; Behenna D. C.; Harned A. M.; Stoltz B. M. Deracemization of Quaternary Stereocenters by Pd-Catalyzed Enantioconvergent Decarboxylative Allylation of Racemic β-Ketoesters. Angew. Chem., Int. Ed. 2005, 44, 6924–6927. 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]; j Keith J. A.; Behenna D. C.; Sherden N.; Mohr J. T.; Ma S.; Marinescu S. C.; Nielsen R. J.; Oxgaard J.; Stoltz B. M.; Goddard W. A. I. The Reaction Mechanism of the Enantioselective Tsuji Allylation: Inner-Sphere and Outer-Sphere Pathways, Internal Rearrangements, and Asymmetric C–C Bond Formation. J. Am. Chem. Soc. 2012, 134, 19050–19060. 10.1021/ja306860n. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Chen J.-P.; Ding C.-H.; Liu W.; Hou X.-L.; Dai L.-X. Palladium-Catalyzed Regio-, Diastereo-, and Enantioselective Allylic Alkylation of Acylsilanes with Monosubstituted Allyl Substrates. J. Am. Chem. Soc. 2010, 132, 15493–15495. 10.1021/ja106703y. [DOI] [PubMed] [Google Scholar]; l Behenna D. C.; Mohr J. T.; Sherden N. H.; Marinescu S. C.; Harned A. M.; Tani K.; Seto M.; Ma S.; Novák Z.; Krout M. R.; McFadden R. M.; Roizen J. L.; Enquist J. A. Jr.; White D. E.; Levine S. R.; Petrova K. V.; Iwashita A.; Virgil S. C.; Stoltz B. M. Enantioselective Decarboxylative Alkylation Reactions: Catalyst Development, Substrate Scope, and Mechanistic Studies. Chem.—Eur. J. 2011, 17, 14199–14223. 10.1002/chem.201003383. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Bai D.-C.; Yu F.-L.; Wang W.-Y.; Chen D.; Li H.; Liu Q.-R.; Ding C.-H.; Chen B.; Hou X.-L. Palladium/N-Heterocyclic Carbene Catalysed Regio and Diastereoselective Reaction of Ketones with Allyl Reagents via Inner-Sphere Mechanism. Nat. Commun. 2016, 7, 11806. 10.1038/ncomms11806. [DOI] [PMC free article] [PubMed] [Google Scholar]; n Behenna D. C.; Liu Y.; Yurino T.; Kim J.; White D. E.; Virgil S. C.; Stoltz B. M. Enantioselective Construction of Quaternary N-Heterocycles by Palladium-Catalysed Decarboxylative Allylic Alkylation of Lactams. Nat. Chem. 2012, 4, 130–133. 10.1038/nchem.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review, see:; a Zhuo C.-X.; Zheng C.; You S.-L. Transition-Metal-Catalyzed Asymmetric Allylic Dearomatization Reactions. Acc. Chem. Res. 2014, 47, 2558–2573. 10.1021/ar500167f. [DOI] [PubMed] [Google Scholar]; For selected examples, see:; b Trost B. M.; Bai W.-J.; Hohn C.; Bai Y.; Cregg J. J. Palladium-Catalyzed Asymmetric Allylic Alkylation of 3-Substituted 1H-Indoles and Tryptophan Derivatives with Vinylcyclopropanes. J. Am. Chem. Soc. 2018, 140, 6710–6717. 10.1021/jacs.8b03656. [DOI] [PubMed] [Google Scholar]; c Liu Y.; Du H. Pd-Catalyzed Asymmetric Allylic Alkylations of 3-Substituted Indoles Using Chiral P/Olefin Ligands. Org. Lett. 2013, 15, 740–743. 10.1021/ol3032736. [DOI] [PubMed] [Google Scholar]; d Kaiser T. M.; Yang J. Catalytic Enantioconvergent Decarboxylative Allylic Alkylation of Allyl Indolenin-3-Carboxylates. Eur. J. Org. Chem. 2013, 2013, 3983–3987. 10.1002/ejoc.201300515. [DOI] [Google Scholar]; e Gao R.-D.; Xu Q.-L.; Zhang B.; Gu Y.; Dai L.-X.; You S.-L. Palladium(0)-Catalyzed Intermolecular Allylic Dearomatization of Indoles by a Formal [4 + 2] Cycloaddition Reaction. Chem.—Eur. J. 2016, 22, 11601–11604. 10.1002/chem.201602691. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Quancard J. Palladium-Catalyzed Enantioselective C-3 Allylation of 3-Substituted-1H-Indoles Using Trialkylboranes. J. Am. Chem. Soc. 2006, 128, 6314–6315. 10.1021/ja0608139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review, see:; a Wu W.-T.; Zhang L.; You S.-L. Catalytic Asymmetric Dearomatization (CADA) Reactions of Phenol and Aniline Derivatives. Chem. Soc. Rev. 2016, 45, 1570–1580. 10.1039/C5CS00356C. [DOI] [PubMed] [Google Scholar]; Selected examples for phenols as nucleophile:; b Wu Q.-F.; Liu W.-B.; Zhuo C.-X.; Rong Z.-Q.; Ye K.-Y.; You S.-L. Iridium-Catalyzed Intramolecular Asymmetric Allylic Dearomatization of Phenols. Angew. Chem., Int. Ed. 2011, 50, 4455–4458. 10.1002/anie.201100206. [DOI] [PubMed] [Google Scholar]; c Yang Z.-P.; Wu Q.-F.; Shao W.; You S.-L. Iridium-Catalyzed Intramolecular Asymmetric Allylic Dearomatization Reaction of Pyridines, Pyrazines, Quinolines, and Isoquinolines. J. Am. Chem. Soc. 2015, 137, 15899–15906. 10.1021/jacs.5b10440. [DOI] [PubMed] [Google Scholar]; d Yang Z.-P.; Jiang R.; Zheng C.; You S.-L. Iridium-Catalyzed Intramolecular Asymmetric Allylic Alkylation of Hydroxyquinolines: Simultaneous Weakening of the Aromaticity of Two Consecutive Aromatic Rings. J. Am. Chem. Soc. 2018, 140, 3114–3119. 10.1021/jacs.8b00136. [DOI] [PubMed] [Google Scholar]; e Nemoto T.; Ishige Y.; Yoshida M.; Kohno Y.; Kanematsu M.; Hamada Y. Novel Method for Synthesizing Spiro[4.5]Cyclohexadienones through a Pd-Catalyzed Intramolecular Ipso-Friedel–Crafts Allylic Alkylation of Phenols. Org. Lett. 2010, 12, 5020–5023. 10.1021/ol102190s. [DOI] [PubMed] [Google Scholar]; f Xu Q.-L.; Dai L.-X.; You S.-L. Enantioselective Synthesis of Tetrahydroisoquinolines via Iridium-Catalyzed Intramolecular Friedel–Crafts-Type Allylic Alkylation of Phenols. Org. Lett. 2012, 14, 2579–2581. 10.1021/ol3008793. [DOI] [PubMed] [Google Scholar]; g Suzuki Y.; Nemoto T.; Kakugawa K.; Hamajima A.; Hamada Y. Asymmetric Synthesis of Chiral 9,10-Dihydrophenanthrenes Using Pd-Catalyzed Asymmetric Intramolecular Friedel–Crafts Allylic Alkylation of Phenols. Org. Lett. 2012, 14, 2350–2353. 10.1021/ol300770w. [DOI] [PubMed] [Google Scholar]; h Zhuo C.-X.; You S.-L. Palladium-Catalyzed Intermolecular Asymmetric Allylic Dearomatization Reaction of Naphthol Derivatives. Angew. Chem., Int. Ed. 2013, 52, 10056–10059. 10.1002/anie.201304591. [DOI] [PubMed] [Google Scholar]; i Zhuo C.-X.; You S.-L. Palladium-Catalyzed Intermolecular Allylic Dearomatization Reaction of α-Substituted β-Naphthol Derivatives: Scope and Mechanistic Investigation. Adv. Synth. Catal. 2014, 356, 2020–2028. 10.1002/adsc.201400154. [DOI] [Google Scholar]; j Zhao Z.-L.; Xu Q.-L.; Gu Q.; Wu X.-Y.; You S.-L. Enantioselective Synthesis of 4-Substituted Tetrahydroisoquinolines via Palladium-Catalyzed Intramolecular Friedel–Crafts Type Allylic Alkylation of Phenols. Org. Biomol. Chem. 2015, 13, 3086–3092. 10.1039/C4OB02574A. [DOI] [PubMed] [Google Scholar]
- Yang Z.-P.; Jiang R.; Wu Q.-F.; Huang L.; Zheng C.; You S.-L. Iridium-Catalyzed Intramolecular Asymmetric Allylic Dearomatization of Benzene Derivatives. Angew. Chem., Int. Ed. 2018, 57, 16190–16193. 10.1002/anie.201810900. [DOI] [PubMed] [Google Scholar]
- Yang Z.-P.; Wu Q.-F.; You S.-L. Direct Asymmetric Dearomatization of Pyridines and Pyrazines by Iridium-Catalyzed Allylic Amination Reactions. Angew. Chem., Int. Ed. 2014, 53, 6986–6989. 10.1002/anie.201404286. [DOI] [PubMed] [Google Scholar]
- Weix D. J.; Hartwig J. F. Regioselective and Enantioselective Iridium-Catalyzed Allylation of Enamines. J. Am. Chem. Soc. 2007, 129, 7720–7721. 10.1021/ja071455s. [DOI] [PubMed] [Google Scholar]
- a Vitaku E.; Smith D. T.; Njardarson J. T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]; b Heravi M. M.; Zadsirjan V. Prescribed Drugs Containing Nitrogen Heterocycles: An Overview. RSC Adv. 2020, 10, 44247–44311. 10.1039/D0RA09198G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bull J. A.; Mousseau J. J.; Pelletier G.; Charette A. B. Synthesis of Pyridine and Dihydropyridine Derivatives by Regio- and Stereoselective Addition to N-Activated Pyridines. Chem. Rev. 2012, 112, 2642–2713. 10.1021/cr200251d. [DOI] [PubMed] [Google Scholar]; b Bertuzzi G.; Bernardi L.; Fochi M. Nucleophilic Dearomatization of Activated Pyridines. Catalysts 2018, 8, 632. 10.3390/catal8120632. [DOI] [Google Scholar]; c Londregan A. T.; Jennings S.; Wei L. Mild Addition of Nucleophiles to Pyridine-N-Oxides. Org. Lett. 2011, 13, 1840–1843. 10.1021/ol200352g. [DOI] [PubMed] [Google Scholar]; d Wang D.; Wang Z.; Liu Z.; Huang M.; Hu J.; Yu P. Strategic C–C Bond-Forming Dearomatization of Pyridines and Quinolines. Org. Lett. 2019, 21, 4459–4463. 10.1021/acs.orglett.9b01247. [DOI] [PubMed] [Google Scholar]; e Comins D. L.; Abdullah A. H. Regioselective Addition of Grignard Reagents to 1-Acylpyridinium Salts. A Convenient Method for the Synthesis of 4-Alkyl(Aryl)Pyridines. J. Org. Chem. 1982, 47 (22), 4315–4319. 10.1021/jo00143a028. [DOI] [Google Scholar]
- For selected hydrosilylation reviews, see:; a Ríos P.; Rodríguez A.; Conejero S. Activation of Si–H and B–H Bonds by Lewis Acidic Transition Metals and p-Block Elements: Same, but Different. Chem. Sci. 2022, 13, 7392–7418. 10.1039/D2SC02324E. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lipke M. C.; Liberman-Martin A. L.; Tilley T. D. Electrophilic Activation of Silicon–Hydrogen Bonds in Catalytic Hydrosilations. Angew. Chem., Int. Ed. 2017, 56, 2260–2294. 10.1002/anie.201605198. [DOI] [PubMed] [Google Scholar]
- For examples with Ti, see:; a Hao L.; Harrod J. F.; Lebuis A.-M.; Mu Y.; Shu R.; Samuel E.; Woo H.-G. Homogeneous Catalytic Hydrosilylation of Pyridines. Angew. Chem., Int. Ed. 1998, 37, 3126–3129. . [DOI] [PubMed] [Google Scholar]; b Gutsulyak D. V.; van der Est A.; Nikonov G. I. Facile Catalytic Hydrosilylation of Pyridines. Angew. Chem., Int. Ed. 2011, 50, 1384–1387. 10.1002/anie.201006135. [DOI] [PubMed] [Google Scholar]; For examples with Ru, see:; c Lee S.-H.; Gutsulyak D. V.; Nikonov G. I. Chemo- and Regioselective Catalytic Reduction of N-Heterocycles by Silane. Organometallics 2013, 32, 4457–4464. 10.1021/om400269q. [DOI] [Google Scholar]; d Königs C. D. F.; Klare H. F. T.; Oestreich M. Catalytic 1,4-Selective Hydrosilylation of Pyridines and Benzannulated Congeners. Angew. Chem., Int. Ed. 2013, 52, 10076–10079. 10.1002/anie.201305028. [DOI] [PubMed] [Google Scholar]; e Bähr S.; Oestreich M. A Neutral RuII Hydride Complex for the Regio- and Chemoselective Reduction of N-Silylpyridinium Ions. Chem.—Eur. J. 2018, 24, 5613–5622. 10.1002/chem.201705899. [DOI] [PubMed] [Google Scholar]; f Behera D.; Thiyagarajan S.; Anjalikrishna P. K.; Suresh C. H.; Gunanathan C. Ruthenium(II)-Catalyzed Regioselective 1,2-Hydrosilylation of N-Heteroarenes and Tetrel Bonding Mechanism. ACS Catal. 2021, 11, 5885–5893. 10.1021/acscatal.1c01148. [DOI] [Google Scholar]; For examples with Ir, see:; g Jeong J.; Park S.; Chang S. Iridium-Catalyzed Selective 1,2-Hydrosilylation of N-Heterocycles. Chem. Sci. 2016, 7, 5362–5370. 10.1039/C6SC01037G. [DOI] [PMC free article] [PubMed] [Google Scholar]; For examples with Zn, see:; h Lortie J. L.; Dudding T.; Gabidullin B. M.; Nikonov G. I. Zinc-Catalyzed Hydrosilylation and Hydroboration of N-Heterocycles. ACS Catal. 2017, 7, 8454–8459. 10.1021/acscatal.7b02811. [DOI] [Google Scholar]; i Wang X.; Zhang Y.; Yuan D.; Yao Y. Regioselective Hydroboration and Hydrosilylation of N-Heteroarenes Catalyzed by a Zinc Alkyl Complex. Org. Lett. 2020, 22, 5695–5700. 10.1021/acs.orglett.0c02082. [DOI] [PubMed] [Google Scholar]; j Prybil J. W.; Wallace R.; Warren A.; Klingman J.; Vaillant R.; Hall M. B.; Yang X.; Brennessel W. W.; Chin R. M. Silylation of Pyridine, Picolines, and Quinoline with a Zinc Catalyst. ACS Omega 2020, 5, 1528–1539. 10.1021/acsomega.9b03317. [DOI] [PMC free article] [PubMed] [Google Scholar]; For the theoretical prediction of a Ni(I) catalyst for the hydrosilylation of pyridine, see:; k Singh V.; Sakaki S.; Deshmukh M. M. Theoretical Prediction of Ni(I)-Catalyst for Hydrosilylation of Pyridine and Quinoline. J. Comput. Chem. 2019, 40, 2119–2130. 10.1002/jcc.25864. [DOI] [PubMed] [Google Scholar]; l Ma X.; Mane M. V.; Cavallo L.; Nolan S. P. Ruthenium-catalyzed regioselective 1,2-hydrosilylation of N-heteroarenes. Eur. J. Org. Chem. 2023, 26, 1466. 10.1002/ejoc.202201466. [DOI] [Google Scholar]
- Intemann J.; Bauer H.; Pahl J.; Maron L.; Harder S. Calcium Hydride Catalyzed Highly 1,2-Selective Pyridine Hydrosilylation. Chem.—Eur. J. 2015, 21, 11452–11461. 10.1002/chem.201501072. [DOI] [PubMed] [Google Scholar]
- For selective examples, see; a Gandhamsetty N.; Park S.; Chang S. Selective Silylative Reduction of Pyridines Leading to Structurally Diverse Azacyclic Compounds with the Formation of Sp3 C–Si Bonds. J. Am. Chem. Soc. 2015, 137, 15176–15184. 10.1021/jacs.5b09209. [DOI] [PubMed] [Google Scholar]; b Petrushko W. D.; Nikonov G. I. Mono(Hydrosilylation) of N-Heterocycles Catalyzed by B(C6F5)3 and Silylium Ion. Organometallics 2020, 39, 4717–4722. 10.1021/acs.organomet.0c00697. [DOI] [Google Scholar]
- For more electron-rich pyridines (1i, 1j, 1l, and 1m), an increase in the Ir catalyst loading to 2 mol % and Pd catalyst loading to 10 mol % leads to an increase in the yield of the corresponding amine product 6, but at lower enantioselectivity. See the SI for details.
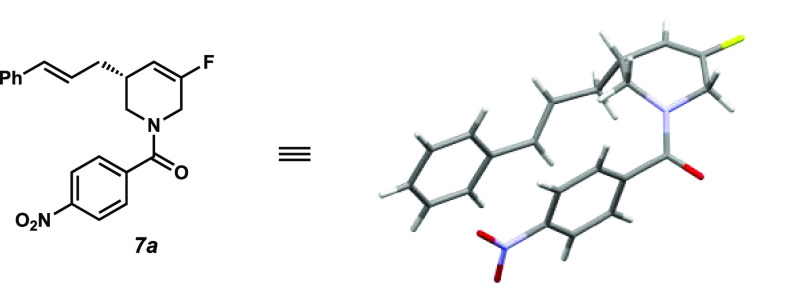
- The substrate scope
was undertaken to unambiguously confirm the structure of the newly
formed products (i.e., 6) and to identify the absolute
stereochemistry of these products. By analogy, the absolute configuration
was adopted for the remaining scope entries. For the detailed procedure
and conditions, see the Supporting Information and CCDC 2234165.
- In control experiments, doping the reaction with an additional equivalent of 3-halopyridine during the Pd-catalyzed allylic alkylation step resulted in an increase in yield and selectivity of the desired monoalkylation product for both pyridines investigated (see Scheme S6 for details).
- We believe that benzoic acid assists in the tautomerization of the imine species formed after the initial Pd-catalyzed asymmetric allylic alkylation to a nucleophilic enamine species (e.g., Scheme 6C→D).

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.