


Abstract
The hepatocyte nuclear factor (HNF)4α, a member of the nuclear receptor superfamily, regulates genes that play a critical role in embryogenesis and metabolism. Recent studies have shown that mutations in the human HNF4α gene cause a rare form of type 2 diabetes, maturity onset diabetes of the young (MODY1). To investigate the properties of these naturally occurring HNF4α mutations we analysed five MODY1 mutations (R154X, R127W, V255M, Q268X and E276Q) and one other mutation (D69A), which we found in HepG2 hepatoma cells. Activation of reporter genes in transfection assays and DNA binding studies showed that the MODY1-associated mutations result in a variable reduction in function, whereas the D69A mutation showed an increased activity on some promoters. None of the MODY mutants acted in a dominant negative manner, thus excluding inactivation of the wild-type factor as a critical event in MODY development. A MODY3-associated mutation in the HNF1α gene, a well-known target gene of HNF4α, results in a dramatic loss of the HNF4 binding site in the promoter, indicating that mutations in the HNF4α gene might cause MODY through impaired HNF1α gene function. Based on these data we propose a two-hit model for MODY development.
INTRODUCTION
In vertebrates the tissue-specific transcription factor hepatocyte nuclear factor (HNF)4α is expressed in liver, kidney, gut and endocrine pancreas (1,2). HNF4α as a member of the nuclear receptor superfamily is designated NR2A1 (3) and belongs to the 2A subfamily together with HNF4β (4) and HNF4γ (5) as its closest related genes in Xenopus and mammals, respectively. HNF4α was initially described as an orphan receptor lacking an identified ligand, but recently fatty acyl-CoA thioesters have been proposed as ligands (6). The DNA-binding domain of HNF4α consists of two zinc finger motifs that specifically interact with a DNA element found in many promoters and enhancers of target genes. These target genes encode serum proteins as well as enzymes involved in lipid, amino acid and glucose metabolism, but also the tissue-specific transcription factor HNF1α (1). Based on the HNF4 binding site in the HNF1α promoter, a regulatory hierarchy has been proposed with HNF4α playing a superior role (7). However, as the HNF4α promoter contains an HNF1 binding site, a reverse regulatory cascade may also be possible (8). The gene regulatory potential of HNF4α has been clearly documented in transfection experiments where the forced expression of HNF4α activates several endogenous genes, but the extent of activation is determined by the type of recipient cell used (7,9–11).
HNF4 also plays an important role in the early phases of embryogenesis since it is present as a maternal component in the egg of the amphibian species Xenopus (12) as well as in Drosophila (13). In the mouse, embryonic expression of HNF4α can already be observed in the primary endoderm at day 4.5 (14). Furthermore, in undifferentiated murine embryonic stem cells, the HNF4α gene is expressed by apparently using an alternative promoter (15). The essential early function of HNF4α in mammals is proven by the fact that disruption of the murine HNF4α gene leads to embryonic lethality due to a dysfunction of the visceral endoderm (16,17).
Lack of HNF4α expression is a typical feature of human renal cell carcinoma (18), but so far no tumour-associated mutations have been found (our unpublished data). In contrast, mutations in the human HNF4α gene cause maturity onset diabetes in the young (MODY1), a rare form of non-insulin-dependent diabetes mellitus (19). This disease is inherited in an autosomal dominant pattern, affects people usually under the age of 25 years and is characterised by defective secretion of insulin by the β-cells of the pancreas (reviewed in 20). The mutants described so far include two nonsense mutations, R154X (21) and Q268X (19), and two frameshift mutations, F75delT (22) and K99fsdelAA (23), which all result in truncated HNF4α proteins. In addition, five missense mutations have been identified, G115S, R127W (24,25), V255M (26), E276Q (27) and V393I (28), that affect various domains of the HNF4α protein. As HNF4α is not only expressed in the β-cells (1), mutated HNF4α proteins are expected to have pleiotropic phenotypes. However, no additional severe clinical phenotypes have been reported in mutant carriers, except secondary complications associated with diabetes itself. Recently, a MODY1-associated decrease in triglyceride concentration was observed in diabetic patients that is considered to be an effect of impaired expression of hepatic apoCIII and apoB (23). MODY3, with identical clinical features to MODY1 patients, is associated with mutations in the gene encoding the tissue-specific transcription factor HNF1α (reviewed in 20). This fact supports the hypothesis that HNF4α and HNF1α cooperate in a network of transcription factors (2) and is strengthened by the finding of a MODY3 mutation in the HNF4 binding site of the HNF1α promoter (29).
In the present work we have analysed the functional activity of several MODY-associated HNF4α mutations as well as a novel mutation found in the human hepatoma cell line HepG2.
MATERIALS AND METHODS
Plasmid constructions
Expression vectors. To construct the pOP-mycHNF4α2 expression vector the myc tag containing a hexameric repeat of the Myc epitope was amplified from the pCS2+MT vector (30) with the 5′ primer (5′-ATAAGAATGCGGCCGCGCTATGG-AGCAAAAGCTC-3′) and 3′ primer (5′-ATAAGAATGGCGGCCGCTCTAGAGGGGTACCAGGCCTTG-3′) introducing two NotI (underlined) sites and a KpnI site (italic), respectively. The NotI fragment of the PCR product was used to replace the cat gene of the pOP13CAT expression vector (Stratagene). The human HNF4α2 cDNA was amplified from the RcHNF4α2 expression vector (5) with a 5′ primer generating a KpnI site (italic) (5′-GGGGTACCATGCGACTCTCCAA-AACC-3′) and the SP6 primer (5′-CTATTTAGGTGACACTATAG-3′) and cloned as a KpnI–NotI fragment in-frame downstream of the myc tag. PCR was performed with Taq DNA polymerase (Gibco BRL) under standard conditions.
HNF4α mutants were generated by site-directed mutagenesis (Quick Change™ site-directed mutagenesis kit; Stratagene) on the pOP-mycHNF4α2 construct using the following primer pairs: D69A, 5′-GAGCTGTGCCGGCTGCAAG-3′, 5′-CTT-GCAGCCGGCACAGCTC-3′; R154X, 5′-GTCCTGTCCTG-ACAGATCACC-3′, 5′-GGTGATCTGTCAGGACAGGAC-3′; V255M, 5′-GATGAGCCGGATGTCCATACG-3′, 5′-CGTA-TGGACATCCGGCTCATC-3′; E276Q, 5′-GATGACAATC-AGTATGCCTACC-3′, 5′-GGTAGGCATACTGATTGTCA-TC-3′; Q268X, 5′-CGATCTGCAGCTCCTAGAAGGG-3′, 5′-CCCTTCTAGGAGCTGCAGATCG-3′; R127W, 5′-CGG-GACTGGATCAGCACTCG-3′, 5′-CGAGTGCTGATCCAG-TCCCG-3′. The authenticity of all DNA fragments amplified by PCR was verified by sequencing.
Reporter gene constructs. The H4-tk-luc (31) and HNF1α(–594/–207)lucI (32) reporter gene constructs were described pre-viously. To establish the rat apo-AI(–1000/+14)lucI reporter gene the apo-AI promoter (33,34) was cloned from genomic DNA of C2 cells by PCR, with the primer (5′-CGCGGATCCGCGTTAGCGTTGCCAACACAGGGT-3′ and 5′-CGTAA-GCTTCTCCCCCGGAGCTCTCCAACA-3′) introducing BamHI and HindIII restriction sites (underlined), respectively, that were used to insert the promoter fragment (nucleotides –1000 to +14) into the pXP1 luciferase vector (35). The human HNF1α promoter was amplified from a 6 kb genomic fragment subcloned into Bluescript SK+ that was previously isolated from a genomic λ phage library. The primers used for this PCR were a T3 primer (5′-CATTAACCCTCACTAAAGGGA-3′) and a primer introducing a 5′ HindIII restriction site at +138 nt from the transcription start site (36). A SacI–HindIII fragment (nucleotides –325/+138) was cloned into a luciferase reporter gene vector based on pGL-Basic II (a gift from Ludger Klein-Hitpass) and named hHNF1(–325/+138)lucII. The hHNF1(–325/+138)mut reporter was made by site-directed mutagenesis as described above using the primers 5′-GCTGAAGTCCCAAGTTCAGTCCC-3′ and 5′-GGGACTG-AACTTGGGACTTCAGC-3′. The exchanged nucleotides are underlined and represent the MODY3 mutation found in the HNF1α promoter (29). The authenticity of all DNA fragments amplified by PCR was verified by sequencing.
Cell culture, transfection and immunofluorescence
293 E1A-transformed embryonic human kidney cells and HeLa cells were cultured at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) with penicillin (100 U/ml), streptomycin (100 U/ml) and 10% heat-inactivated foetal calf serum (Biochrome). For transient transfection of 293 cells the DNA calcium phosphate co-precipitation method was used (37). Twenty micrograms of expression vector were transfected in a 10 cm cell culture dish with 0.1 mM chloroquine to obtain optimal transfection efficiency.
HeLa cells were transfected with Lipofectamine (Gibco BRL) using 2 µg reporter gene, 0.3 µg expression vector and 6 µl Lipofectamine in a 3.3 cm cell culture dish. In all transfection experiments the final DNA concentration was equalised by the addition of pOP13 vector.
Immunofluorescence was performed essentially as previously described (38) using HeLa or 293 cells transfected in a 3.3 cm dish. An aliquot of 250 µl of hybridoma supernatant of the monoclonal myc-specific antibody (9E10) diluted 1:4 with culture medium was used as the primary antibody. The Cy3-conjugated secondary antibody (code no. 415-166-100; Jackson ImmunoResearch) was visualised by fluorescence microscopy.
Extract preparation and gel shifts
High salt nuclear extracts of cultured cells and gel shift assays were performed as described (4). The reaction mixture for gel shifts contained 2 µg of total protein, 150 ng of salmon sperm DNA and 104 c.p.m. of 32P-labelled H4 oligonucleotide (37). For competition experiments oligonucleotides containing the wild-type and the mutated HNF4 binding site of the human HNF1α promoter were used (5′-CGGCTGAAGTCCAAAGTTCAGTCCCTTCGT-3′, 5′-CTAGACGAAGGGACTGAAC-TTTGGACTTCAGCCGAGCT-3′). In the mutated oligonucleotide the underlined base was changed from an A and T to a C and G residue, respectively. Competition was performed with 0.5 µl rat liver nuclear extracts per reaction (39) using labelled wild-type oligonucleotide (104 c.p.m.) and increasing amounts of unlabelled wild-type or mutant oligonucleotide. The total amount of unlabelled DNA was kept constant at 150 ng by adding salmon sperm DNA.
Western blotting
Western blotting was performed using the anti-myc tag antibody (9E10) as the primary antibody and a horseradish peroxidase antibody as the secondary antibody, which was detected using the ECL system (Amersham). Pellets were incubated with DNase I prior to SDS–PAGE.
RESULTS
Mutation in the HNF4α gene of the human hepatoma cell line HepG2
In a search for mutations in the HNF4α gene in various tumour cell lines of human origin we observed altered migration of the PCR-amplified fragment of exon 2 upon denaturing gel electrophoresis. Direct sequencing of this PCR product revealed an A as well as a C residue at a position 206 bp downstream of the translation start site in the coding strand of the HepG2 DNA, whereas only the nucleotide A was found in the SK-RC-8 cell line that was used as a control. Thus codon 69 encodes the usual aspartic acid (GAC = D) as well as the amino acid alanine (GCC = A). The heterozygosity at codon 69 was initially found in a HepG2 cell line maintained in our laboratory for several years, but was also present in a HepG2 sample obtained from CLS (passage 30; Cell Line Services, Heidelberg, Germany) as well as the HepG2 variant C3A obtained from the ATTC (CRL-10741; American Tissue Type Collection, Manassas, USA). Thus, we conclude that the heterozygous D69A mutation is a feature specific for the human hepatoma cell line HepG2.
The mutation D69A changes an amino acid in the P-box (CDGCKG) of the first zinc finger of the DNA-binding domain (Fig. 1). This element is identical in all members of the HNF4 gene family and has been considered as a hallmark for this nuclear receptor subfamily (1). To investigate whether the mutated allele is transcribed, we amplified cDNA from HepG2 RNA using a primer pair specific for exons 2 and 4 of the HNF4α gene. Sequencing of the PCR product revealed an A as well as a C at nucleotide position 206, implying transcription of both alleles.
Figure 1.
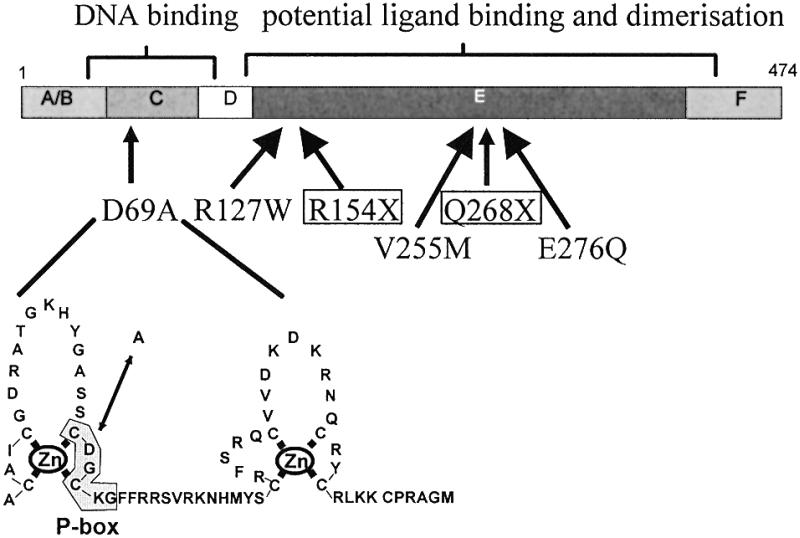
Naturally occurring mutations within the human HNF4α gene. The domain structure of the HNF4α protein is given (1). The D69A mutation found in HepG2 cells affects an amino acid within the P-box of the first zinc finger of the DNA-binding domain. The diabetes-associated mutations whose functional properties are analysed in this paper are given.
The D69A mutation has not been detected in any MODY-associated family (Fig. 1; 22–24,28).
A MODY3-associated mutation destroys the HNF4 binding site in the HNF1α promoter
The concept that HNF4α and HNF1α act together in the same pathways is strengthened by the fact that HNF1α represents the MODY3 gene. In MODY3 patients mutations have not only been found in the open reading frame of the HNF1α gene, but also at nucleotide –58 of the promoter region (29). This region is highly conserved between Xenopus and mammals and corresponds to a HNF4 binding site (37). To analyse the effect of this mutation we prepared a HNF1α promoter–luciferase reporter gene containing the sequence from –325 to +138 of the human HNF1α gene as well as a corresponding construct containing the MODY3 mutation (Fig. 2A). In transfection experiments using HeLa cells that lack endogeneous HNF4 proteins, the wild-type HNF1α promoter-driven luciferase activity was stimulated up to 35-fold with increasing amounts of an expression vector encoding HNF4α. In contrast, the reporter containing the MODY3 mutation was stimulated by, at most, 4-fold with addition of the HNF4α expression vector (Fig. 2B). This demonstrates that the single nucleotide change at –58 leads to a dramatic loss in the activation potential of the HNF1α promoter by HNF4α. To analyse whether this effect mainly represented the loss of binding of HNF4α to this site we performed bandshift assays using the corresponding labelled oligonucleotide and rat liver nuclear extracts. Figure 2C demonstrates a DNA–protein complex up-shifted with an antibody specific for HNF4α. This complex was competed out efficiently by adding increasing amounts of the unlabelled oligonucleotide, whereas the addition of an unlabelled competitor representing the mutated HNF4 binding site had no effect. From the input of the oligonucleotides as competitors, we estimate that the MODY3 mutation in the HNF4 binding site reduces the binding affinity of HNF4α by at least a factor of 30. Based on this finding the HNF1α promoter is a valuable reporter to analyse the function of HNF4α derivatives within a natural promoter context.
Figure 2.
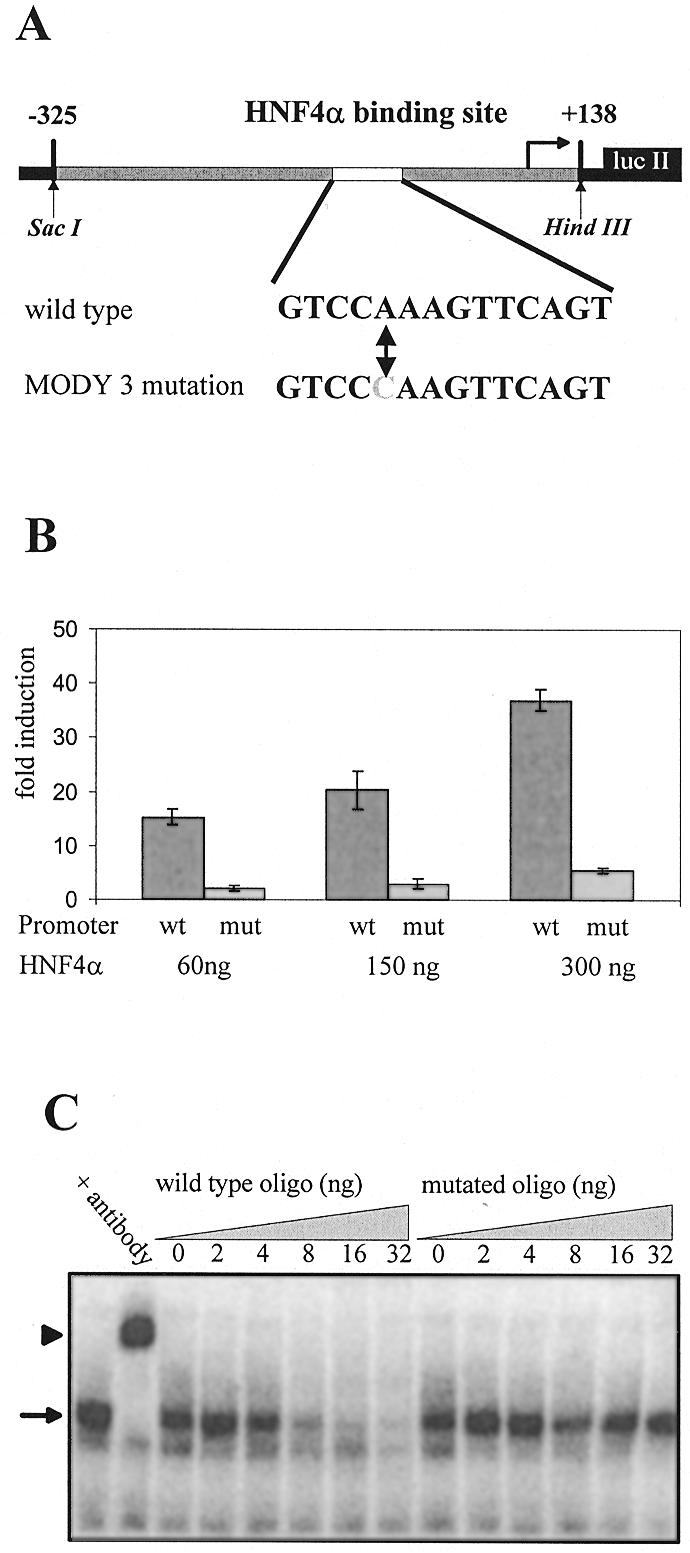
The MODY3 mutation of the HNF4 binding site in the HNF1α promoter results in a loss of function. (A) Schematic representation of the hHNF1(–325/+138)lucII construct containing the human HNF1α promoter is given. The HNF4 binding site with the MODY3 mutation at –58 of the human HNF1α promoter (29) is marked. (B) Comparison of transactivation of the wild-type (wt) and mutant (mut) HNF1α promoter in transient transfection assays by increasing amounts of expression vector encoding wild-type HNF4α in HeLa cells. The total amount of transfected DNA was adjusted by adding pOP13 vector DNA. Fold induction refers to the activity seen without HNF4α expression vector. The error bars indicate the standard deviation of six determinations. (C) Gel shift experiments were done with 0.5 µl of rat liver extract using a labelled HNF4 binding site as probe. The retarded DNA–protein complex and the supershift obtained by an HNF4α-specific antibody are marked with an arrow and an arrowhead, respectively. The addition of increasing amounts of unlabelled wild-type or mutated oligonucleotide representing the HNF4 binding site in (A) is given.
The transactivation potentials of the HNF4α mutants
To assess the functional consequences of the mutations in the HNF4α gene we introduced the six mutations shown in Figure 1 into an expression vector encoding a myc-tagged human HNF4α protein. Two of these mutants, R154X and Q268X, are nonsense mutants resulting in truncated proteins, whereas the others are missense mutants. Initially we transfected saturating amounts of the expression vector together with the luciferase reporter gene containing either four HNF4 binding sites in front of the thymidine kinase promoter or the human HNF1α promoter extending from –325 to +138. The data given in Figure 3 show that the missense mutant R127W retains ~50% of wild-type activity whereas the three missense mutants, V255M, E276Q and D69A, are as active as the wild-type. In contrast, both nonsense mutants, R154X and Q268X, have completely lost their transactivation potential. To look for more subtle changes in the activity of the mutated HNF4α proteins we performed saturation curves with the missense mutants using increasing concentrations of the corresponding expression vectors and compared them to the wild-type. Figure 4A shows that the R127W mutant was less active at all concentrations tested, whereas the V255M and E276Q mutants gave decreased transactivation values only at low expression vector input. However, the performance of the D69A mutant was identical to the wild-type protein. The experiments shown in Figure 4A were performed with the luciferase reporter containing the human HNF1α promoter, but identical results were obtained using the reporter containing four HNF4 binding sites linked to the thymidine kinase promoter. As the mutant D69A did not show any difference in its transactivation potential, we tested two further luciferase reporter constructs known to be activated by HNF4. Using the Xenopus HNF1α promoter (37) as well as the rat apolipoprotein AI promoter (33,34) the D69A mutant was 2- to 3-fold more active than the wild-type (Fig. 4B).
Figure 3.

Transactivation potential of wild-type and mutated HNF4α in transient transfection experiments at saturation. The reporter constructs hHNF1(–325/+138)lucII (A) and H4-tk-luc (B) were co-transfected into HeLa cells with saturating amounts of expression vector (300 ng) encoding wild-type or the mutated HNF4α. Fold induction refers to the activity without any HNF4 derivative. The error bars indicate standard deviation of six determinations.
Figure 4.

Saturation curves of transactivation potential of wild-type and mutated HNF4α in transfection assays. (A) Increasing amounts of the expression vectors encoding the HNF4α mutants as given were co-transfected with the reporter construct hHNF1(–325/+138)lucII. The dotted and the solid lines represent the activity of the mutant and the wild-type, respectively. (B) The expression vector encoding the HNF4α mutant D69A was co-transfected with either the Xenopus HNF1(–594/207) promoter construct or the rat apoAI (–1000/+14)lucI reporter. Fold induction refers to the activity without any HNF4 derivative and the error bars indicate standard deviation of six determinations.
The mutated HNF4α proteins do not act as dominant negative factors
As HNF4α acts as a homodimer, we wondered whether the mutated factors have a dominant negative effect on the wild-type protein. Using the HNF1α promoter-driven luciferase reporter we transfected the expression vector encoding wild-type and/or the R127W mutant into HeLa cells. As Figure 5A illustrates, the transactivation potential of a mixture containing 30 ng wild-type and 30 ng mutant gave transactivation as expected from the potentials of both components alone. Even when the amount of mutant compared to the wild-type was increased, transactivation was always as expected from the individual components (data not shown). Identical data were obtained for all the other missense mutants. Using the nonsense mutant R154X, which has no transactivation potential (see Fig. 3), the addition of 30 ng mutant to 30 ng of the wild-type did not alter transactivation of the wild-type factor, as shown in Figure 5B. Even a 3-fold excess of mutant to wild-type factor failed to change the activity mediated by wild-type HNF4α (data not shown). Similarly, the Q268X mutant also did not interfere with the transactivation potential of wild-type HNF4α in co-transfection experiments (data not shown).
Figure 5.

Transactivation potential of co-transfected wild-type and mutant HNF4α on the human HNF1 promoter. Co-transfection of wild-type HNF4α and either R127W (A) or R154X (B) was done with the amount of expression vector given. In each experiment the amount of reporter gene construct and total DNA transfected was constant. Fold induction refers to the activity with no HNF4α derivative and the error bars indicate standard deviation of six determinations.
The nonsense mutants R154X and Q268X are deficient in DNA binding
The nonsense mutations R154X and Q268X encode truncated HNF4α proteins that retain the entire DNA-binding domain (see Fig. 1). To investigate the DNA binding properties of the mutants we prepared extracts from 293 cells transfected with the corresponding expression vector. In western blots using an antibody specific for the myc tag we could detect the R154X mutant protein in the nuclear salt extract and the insoluble pellet, as for all the other mutants. However, the Q268X mutant protein could only be seen in the insoluble pellet and not in the nuclear salt extract (Fig. 6A). This insolubility of the Q268X protein has also been reported elsewhere using the rat protein as backbone (40). Therefore, in band shift assays no binding to an oligonucleotide containing an HNF4 binding site was detected with nuclear salt extracts for the Q268X mutant. In contrast, the R154X mutant showed a retarded band, although exclusively in the lane with the antibody, indicating a weak DNA binding that is stabilised upon antibody binding. Efficient binding was obtained by all extracts transfected with either the wild-type transcription factor or the missense mutants of HNF4α (Fig. 6B).
Figure 6.
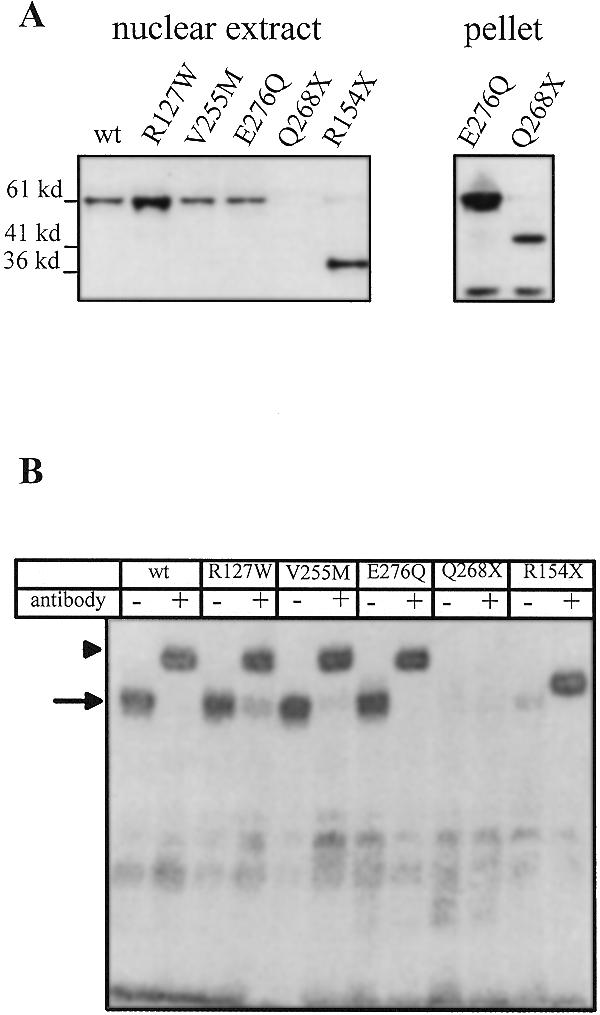
Western blot and gel shift assay with nuclear extracts from transfected 293 cells. 293 human embryonic kidney cells were transfected with expression vectors encoding wild-type or mutant HNF4α and nuclear salt extracts were prepared after 24 h for western blots (A) or gel retardation assays (B). The pellet fractions of the salt extracts were solubilised by DNase I digestion and used for western blots. Only the pellet fractions of E276Q and Q268X are shown. To visualise the transfected HNF4α variants the myc-specific antibody 9E10 was used in the western blots, whereas the monoclonal antibody H4/55, raised against amino acids 1–114 of rat HNF4α (18) was used in the gel retardation assays. The band of <36 kDa detected in the western blot of the pellet fractions is non-specific as it is also seen in untransfected cells.
Cellular localisation of the HNF4α mutants
To monitor whether the mutated HNF4α proteins show a distinct distribution within the cell, we determined the cellular localisation by immunofluorescence in transfected HeLa and 293 cells using the antibody specific for the myc tag. Wild-type HNF4α was found in 95–98% in the nuclear compartment and the same distribution was found for all the missense mutants (data not shown). On the other hand the myc tag antibody rarely detected any signal derived from the transfected nonsense mutants R154X and Q268X in HeLa cells (data not shown). As both truncated proteins can be detected in western blots derived from transfected cells, we assume that the myc tag is not accessible for immunohistochemistry by the antibody in both these constructs.
DISCUSSION
By screening the HNF4α gene in human carcinoma cell lines we identified the D69A mutation of the HepG2 hepatoma cell line. As this mutation affects an allele that is expressed in the cell line and increases the transactivation potential of the HNF4α protein on some promoters, this finding is most relevant, as HepG2 cells are one of the most frequently used human models for hepatic cell function. The HepG2 cell line was established in 1980 (41). Our efforts to identify whether the mutation was already in the germline or occurred either during tumour development or during establishment of the cell line were unsuccessful due to a lack of appropriate material. However, this mutation has not been observed in any MODY patient to date and we did not identify any other mutation in the HNF4α gene in other human hepatoma cells analysed (unpublished data in collaboration with D. Simon and B.B. Knowles). The D69A mutation alters an amino acid in the P-box of the first zinc finger. As this residue is conserved in all HNF4 proteins from insects to mammals (1), we would assume that it plays a crucial role. Our transfection experiments showed that it has an increased activity on some promoters. However, the effect is quite small and one may wonder why this residue has been conserved throughout evolution.
We also investigated the functional consequence of a mutation in the HNF4 binding site of the promoter of the HNF1α gene that was identified in a MODY3 family (29). By gel retardation assays we could show that the mutated HNF4 binding site has an at least 30-fold lower affinity for HNF4α compared to the wild-type binding site. Consistent with this dramatically reduced binding, in transfection experiments we observed that the mutated promoter can hardly be stimulated by HNF4α. This finding establishes that the MODY3 mutation generating a mutated HNF4 binding site largely destroys the ability for transactivation by HNF4α and thus represents a loss of function mutation of the HNF1α gene. It gives further evidence that HNF4α and HNF1α cooperate in β-cells of the pancreas and thus both genes constitute a sensitive target for the development of diabetes. Furthermore, the result indicates that the HNF1α promoter is a relevant reporter to assay the function of HNF4α mutants in transfection experiments. The importance of the HNF4 binding site in the HNF1α promoter has been shown in transgenic Xenopus, as a mutated HNF4 binding site of the Xenopus HNF1α promoter precludes expression of the HNF1α promoter transgenes in the pronephros (42).
Our functional analysis of mutated HNF4α proteins that occur in MODY patients reveals a broad range of effects on the performance of this transcription factor. The two nonsense mutants R154X and Q268X lack any transactivation potential and do not interfere with the activity of the wild-type factor. This conclusion had previously been made in a report investigating Q268X function (40,43). As Q268X has the potential to heterodimerise with the wild-type HNF4α protein in vitro, it has been proposed that the inability of the mutant to affect the activity of the wild-type in vivo may be due to an altered subcellular localisation of the mutant compared to the wild-type. The truncated proteins derived from Q268X and R154X both retain the DNA-binding domain, but only R154X binds DNA in a gel retardation assay (Fig. 6B). This finding is in agreement with a systematic deletion analysis of the various domains of HNF4α (44): in the absence of the C-terminal sequence (amino acids 340–465) the sequences located carboxyl to residue 174 inhibit DNA binding of these truncated HNF4α variants. According to this result the DNA binding activity observed for the R154X mutant is expected.
In contrast to the nonsense mutants, the three missense mutants also associated with diabetes retain substantial transactivation potential (Fig. 4A). In fact, at saturation the mutants V255M and E276Q gave identical transactivation compared to the wild-type and only differed by a decreased activity at low expression vector input. Under the same experimental conditions the mutant R127W gave a 2- to 3-fold lower transactivation at all concentrations tested. After completion of our work a functional analysis of the same three missense mutants has been published (45). The lack of any functional defect in the R127W and V255M mutants in this report may have been overlooked by the use of a single concentration of expression vector in the transfection assay. More significantly, these authors found no transactivation for the E276Q mutant. This discrepancy may be explained by the fact that they introduced the mutation into the HNF4α protein of the rat, since the human protein carrying the same mutation was found to be active by another group (46), in agreement with our finding. In fact, these authors (46) have established that a major effect of the E276Q mutant involves an impaired synergy of the mutated factor with the chicken ovalbumin upstream transcription factor II (COUP TFII). A fourth MODY missense mutant of HNF4α, V393I, has been shown to have a 2-fold reduced transactivation activity (28). Thus all missense mutants associated with diabetes have a reduced transactivation potential, whereas the missense mutant we found in HepG2 cells has an increased activity on at least some promoters (Fig. 4B).
Taken together these functional data on the various HNF4α mutants associated with diabetes show a very broad effect on the activity of the transcription factor. The only consistent feature that can be observed is a loss of function. Mixing a transcription factor with reduced transactivation potential with the wild-type factor yields an intermediate activity if assayed at saturation but an additive value at sub-saturating conditions (Fig. 5A). Such a behaviour is expected for a transcription factor acting as a dimer. Surprisingly, none of the naturally occurring mutants analysed exhibited a clear dominant negative effect (Fig. 5B) that would involve inactivation of the wild-type factor, although such a mechanism seems quite probable for a transcription factor acting as a dimer. Since different artificial mutants of HNF4α have been generated in several laboratories that act as dominant negative factors on the wild-type factor (43,47–49), we speculate that such mutations are not compatible with human development, as they would inactivate essential functions of the wild-type factor. This assumption is strengthened by the fact that knockout mice die at early embryonic stages prior to gastrulation (16). However, it is quite puzzling that a loss of function mutation of one allele in the presence of a wild-type allele leads to the development of a disease. Assuming a gene dosage effect one would expect clinical differences between the nonsense mutations that destroy the gene, e.g. R154X and Q268X, and the missense mutations that hardly affect the function of HNF4α. Such differences are not apparent, but could be hidden by the individual differences between the various patients. As MODY1 patients are not born with diabetes and initially have no measurable abnormal function in the β-cells of the pancreas, it seem unlikely that the mutated HNF4α is deficient in a specific function such as interaction with COUP-TF (46). It is more probable that additional events occur with time. Therefore, we speculate that the function of the wild-type allele is occasionally lost in β-cells, involving either a somatic mutation or some epigenetic event. We imagine that this loss of function of the wild-type allele leads to some selective advantage, thus allowing overgrowth of the original β-cell population. Such a scenario is reminiscent of the development of tumours in inherited cancer syndromes where one tumour suppressor allele is mutated in the germline and inactivation of the wild-type allele is a major step in tumour formation (50,51). Such a two-hit model also seems to occur in non-malignant diseases, most notably in inherited human autosomal dominant polycystic kidney disease, which is characterised by loss of or a mutation in the normal allele (52,53). Clearly, such a speculation on the mechanism of MODY development has to be verified by more direct approaches. It would be crucial to show that expression of the wild-type HNF4α gene in β-cells of MODY patient is gradually lost. However, such an analysis cannot be readily performed in the human system. Furthermore, one should identify HNF4α-dependent processes that maintain the balance between proliferation, differentiation and apoptosis within the pancreatic β-cell population.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Hassan Nakhei for preparing the rat apo-AI(–1000/+14)lucI reporter, Fabian Esser for liver nuclear extracts and Bernhard Horsthemke for the human genomic library to isolate the HNF1α promoter. This work was supported by the Deutsche Forschungsgemeinschaft (Ry 5/4-1) and the British Diabetic Association.
REFERENCES
- 1.Sladek F.M. (1994) Liver Specific Gene Expression. R.G. Landes Co., Austin, TX.
- 2.Cereghini S. (1996) FASEB J., 10, 267–282. [PubMed] [Google Scholar]
- 3.Nuclear Receptors Nomenclature Committee (1999) Cell, 97, 161–163. [DOI] [PubMed] [Google Scholar]
- 4.Holewa B., Zapp,D., Drewes,T., Senkel,S. and Ryffel,G.U. (1997) Mol. Cell. Biol., 17, 687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drewes T., Senkel,S., Holewa,B. and Ryffel,G.U. (1996) Mol. Cell. Biol., 16, 925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hertz R., Magenheim,J., Berman,I. and Bar-Tana,J. (1998) Nature, 392, 512–516. [DOI] [PubMed] [Google Scholar]
- 7.Kuo C.J., Conley,P.B., Chen,L., Sladek,F.M., Darnell,J.E.,Jr and Crabtree,G.R. (1992) Nature, 355, 457–461. [DOI] [PubMed] [Google Scholar]
- 8.Zhong W., Mirkovitch,J. and Darnell,J.E.,Jr (1994) Mol. Cell. Biol., 14, 7276–7284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bulla G.A. (1997) Nucleic Acids Res., 25, 2501–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bailly A., Spaeth,G., Bender,V. and Weiss,M.C. (1998) J. Cell Sci., 111, 2411–2421. [DOI] [PubMed] [Google Scholar]
- 11.Spaeth G.F. and Weiss,M. (1998) J. Cell Biol., 140, 935–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holewa B., Pogge v. Strandmann,E., Zapp,D., Lorenz,P. and Ryffel,G.U. (1996) Mech. Dev., 54, 45–57. [DOI] [PubMed] [Google Scholar]
- 13.Zhong W., Sladek,F.M. and Darnell,J.E.,Jr (1993) EMBO J., 12, 537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duncan S.A., Manova,K., Chen,W.S., Hoodless,P., Weinstein,D.C., Bachvarova,R.F. and Darnell,J.E.,Jr (1994) Proc. Natl Acad. Sci. USA, 91, 7598–7602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakhei H., Lingott,A., Lemm,I. and Ryffel,G.U. (1998) Nucleic Acids Res., 26, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen W.S., Manova,K., Weinstein,D.C., Duncan,S.A., Plump,A.S., Prezioso,V.R., Bachvarova,R.F. and Darnell,J.E.,Jr (1994) Genes Dev., 8, 2466–2477. [DOI] [PubMed] [Google Scholar]
- 17.Duncan S.A., Nagy,A. and Chan,W. (1997) Development, 124, 279–287. [DOI] [PubMed] [Google Scholar]
- 18.Sel S., Ebert,T., Ryffel,G.U. and Drewes,T. (1996) Cancer Lett., 101, 205–210. [DOI] [PubMed] [Google Scholar]
- 19.Yamagata K., Furuta,H., Oda,N., Kaisaki,P.J., Menzel,S., Cox,N.J., Fajans,S.S., Signorini,S., Stoffel,M. and Bell,G.I. (1996) Nature, 384, 458–460. [DOI] [PubMed] [Google Scholar]
- 20.Hattersley A.T. (1998) Diabet. Med., 15, 15–24. [DOI] [PubMed] [Google Scholar]
- 21.Lindner T., Gragnoli,C., Furuta,H., Cockburn,B.N., Petzold,C., Rietzsch,H., Weiss,U., Schulze,J. and Bell,G.I. (1997) J. Clin. Invest., 100, 1400–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moller A.M., Dalgaard,L.T., Ambye,L., Hansen,L., Schmitz,O., Hansen,T. and Pedersen,O. (1999) J. Clin. Endocrinol. Metab., 84, 367–369. [DOI] [PubMed] [Google Scholar]
- 23.Lehto M., Bitzen,P.O., Isomaa,B., Wipemo,C., Wessman,Y., Forsblom,C., Tuomi,T., Taskinen,M.R. and Groop,L. (1999) Diabetes, 48, 423–425. [DOI] [PubMed] [Google Scholar]
- 24.Malecki M.T., Yang,Y., Antonellis,A., Curtis,S., Warram,J.H. and Krolewski,A.S. (1999) Diabet. Med., 16, 193–200. [DOI] [PubMed] [Google Scholar]
- 25.Furuta H., Iwasaki,N., Oda,N., Hinokio,Y., Horikawa,Y., Yamagata,K., Yano,N., Sugahiro,J., Ogata,M., Ohgawara,H. et al. (1997) Diabetes, 46, 1652–1657. [DOI] [PubMed] [Google Scholar]
- 26.Moller A.M., Urhammer,S.A., Dalgaard,L.T., Reneland,R., Berglund,L., Hansen,T., Clausen,J.O., Lithell,H. and Pedersen,O. (1997) Diabetologia, 40, 980–983. [DOI] [PubMed] [Google Scholar]
- 27.Bulman M.P., Dronsfield,M.J., Frayling,T., Appleton,M., Bain,S.C., Ellard,S. and Hattersley,A.T. (1997) Diabetologia, 40, 859–862. [DOI] [PubMed] [Google Scholar]
- 28.Hani E.H., Suaud,L., Boutin,P., Chevre,J.C., Durand,E., Philippi,A., Demenais,F., Vionnet,N., Furuta,H., Velho,G. et al. (1998) J. Clin. Invest., 101, 521–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gragnoli C., Lindner,T., Cockburn,B.N., Kaisaki,P.J., Gragnoli,F., Marozzi,G. and Bell,G.I. (1997) Diabetes, 46, 1648–1651. [DOI] [PubMed] [Google Scholar]
- 30.Rupp R.A., Snider,L. and Weintraub,H. (1994) Genes Dev., 8, 1311–1323. [DOI] [PubMed] [Google Scholar]
- 31.Drewes T., Clairmont,A., Klein-Hitpass,L. and Ryffel,G.U. (1994) Eur. J. Biochem., 225, 441–448. [DOI] [PubMed] [Google Scholar]
- 32.Weber H., Holewa,B., Jones,E.A. and Ryffel,G.U. (1996) Development, 122, 1975–1984. [DOI] [PubMed] [Google Scholar]
- 33.Dai P.H., Lan,S.S., Ding,X.H. and Chao,Y.S. (1990) Eur. J. Biochem., 190, 305–310. [DOI] [PubMed] [Google Scholar]
- 34.Chan J., Nakabayashi,H. and Wong,N.C. (1993) Nucleic Acids Res., 21, 1205–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nordeen S.K. (1988) Biotechniques, 6, 454–458. [PubMed] [Google Scholar]
- 36.Kaisaki P.J., Menzel,S., Lindner,T., Oda,N., Rjasanowski,I., Sahm,J., Meincke,G., Schulze,J., Schmechel,H., Petzold,C. et al. (1997) Diabetes, 46, 528–535. [DOI] [PubMed] [Google Scholar]
- 37.Zapp D., Bartkowski,S., Holewa,B., Zoidl,C., Klein-Hitpass,L. and Ryffel,G.U. (1993) Mol. Cell. Biol., 13, 6416–6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pogge v. Strandmann E., Zoidl,C., Nakhei,H., Holewa,B., Pogge,V., Strandmann,R., Lorenz,P., Klein-Hitpass,L. and Ryffel,G.U. (1995) Protein Eng., 8, 733–735. [DOI] [PubMed] [Google Scholar]
- 39.Döbbeling U., Ross,K., Klein-Hitpass,L., Morley,C., Wagner,U. and Ryffel,G.U. (1988) EMBO J., 7, 2495–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sladek F.M., Dallas-Yang,Q. and Nepomuceno,L. (1998) Diabetes, 47, 985–990. [DOI] [PubMed] [Google Scholar]
- 41.Knowles B.B., Howe,C.C. and Aden,D.P. (1980) Science, 209, 497–499. [DOI] [PubMed] [Google Scholar]
- 42.Ryffel G.U. and Lingott,A. (2000) Mech. Dev., 90, 65–75. [DOI] [PubMed] [Google Scholar]
- 43.Stoffel M. and Duncan,S.A. (1997) Proc. Natl Acad. Sci. USA, 94, 13209–13214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hadzopoulou-Cladaras M., Kistanova,E., Evagelopoulou,C., Zeng,S.Y., Cladaras,C. and Ladias,J.A.A. (1997) J. Biol. Chem., 272, 539–550. [DOI] [PubMed] [Google Scholar]
- 45.Navas M.A., Munoz-Elias,E.J., Kim,J., Shih,D. and Stoffel,M. (1999) Diabetes, 48, 1459–1465. [DOI] [PubMed] [Google Scholar]
- 46.Suaud L., Hemimou,Y., Formstecher,P. and Laine,B. (1999) Diabetes, 48, 1162–1167. [DOI] [PubMed] [Google Scholar]
- 47.Taylor D.G., Haubenwallner,S. and Leff,T. (1996) Nucleic Acids Res., 24, 2930–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fraser J.D., Keller,D., Martinez,V., Santisomere,D., Straney,R. and Briggs,M.R. (1997) J. Biol. Chem., 272, 13892–13898. [DOI] [PubMed] [Google Scholar]
- 49.Taraviras S., Schütz,G. and Kelsey,G. (1997) Eur. J. Biochem., 244, 883–889. [DOI] [PubMed] [Google Scholar]
- 50.Eng C. and Ponder,B.A. (1993) FASEB J., 7, 910–919. [DOI] [PubMed] [Google Scholar]
- 51.Fearon E.R. (1997) Science, 278, 1043–1050. [DOI] [PubMed] [Google Scholar]
- 52.Qian F., Watnick,T.J., Onuchic,L.F. and Germino,G.G. (1996) Cell, 87, 979–987. [DOI] [PubMed] [Google Scholar]
- 53.Qian F. and Germino,G.G. (1997) Am. J. Hum. Genet., 61, 1000–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]