

ABSTRACT
The implementation of process analytical technologies is positioned to play a critical role in advancing biopharmaceutical manufacturing by simultaneously resolving clinical, regulatory, and cost challenges. Raman spectroscopy is emerging as a key technology enabling in-line product quality monitoring, but laborious calibration and computational modeling efforts limit the widespread application of this promising technology. In this study, we demonstrate new capabilities for measuring product aggregation and fragmentation in real-time during a bioprocess intended for clinical manufacturing by applying hardware automation and machine learning data analysis methods. We reduced the effort needed to calibrate and validate multiple critical quality attribute models by integrating existing workflows into one robotic system. The increased data throughput resulting from this system allowed us to train calibration models that demonstrate accurate product quality measurements every 38 s. In-process analytics enable advanced process understanding in the short-term and will lead ultimately to controlled bioprocesses that can both safeguard and take necessary actions that guarantee consistent product quality.
KEYWORDS: automation, clinical manufacturing, high throughput process development, liquid handling robotics, machine learning, process analytical technology, Raman spectroscopy
Introduction
The biopharmaceutical industry has a proven record of effectively identifying and meeting challenges in clinical manufacturing.1 The evolution of pharmaceutical manufacturing in response to market demands has been progressive, from the emergence of monoclonal antibodies (mAbs)2 to the transition to advanced modalities,3 advancements in low-cost clinical sequencing,4 and new challenges introduced by the COVID-19 pandemic.5 Recent advancements in biopharmaceutical manufacturing include continuous manufacturing6 with advanced process analytical technologies (PAT) for product quality control,7 as well as improvements to existing platform strategies, high throughput screening,5 and process intensification.8 In addition to financial constraints, regulatory agencies have updated commitments to increased processing understanding and controls through updated guidelines, in the form of ICH Q12 guidelines on established conditions and Q13 guidelines on continuous manufacturing.9 Because of these ongoing pressures, innovations in PAT have become attractive solutions that will potentially meet wider clinical, regulatory, and ultimately business needs.7
As the scope of development possibilities widens to include ever expanding types of antibody therapeutics that deviate from traditional platform concepts10 and manufacturing processes,11 the Design Space concept that relies on product and process knowledge12 is being phased out in favor of PAT implementations that seek to control process engineering outputs to allow for a wider range of input process parameters and more flexible product characteristics. Although PAT will become a necessary component of continuous manufacturing systems of the future,13 with a heavy concentration of recent work on the affinity purification loading step14–16 due to its high immediate business value,7 there is considerable interest in developing PAT technologies for a wider range of applications, including off-line sample testing17–19 and preparative ion-exchange chromatography.20,21 Thus, to meet the diverse challenges facing the industry, PAT is positioned to make a large impact.
Raman spectrometry is becoming a widely used analytical tool for in-line monitoring of protein concentration and product quality attributes, such as aggregation.17,18 Compared with alternative methods for monitoring high molecular weight species, such as nuclear magnetic resonance (NMR),22 dynamic light scattering (DLS),23 or variable path-length UV spectroscopy,21 Raman spectroscopy provides more independent analytical features, typically 3101 variables,16 different vibrational modes, and less interference from water.24 Most studies using Raman spectrometry14,15,17–19,23,25–28 rely on hardware that collect spectra on longer timescales not currently feasible for use in downstream preparative chromatography applications. Recently, optical improvements in quantum efficiency through virtual slit technology29 have allowed Raman signals to be collected on the order of seconds, enabling investigation into downstream protein purification applications where shorter timescale changes are criticalfor example, monitoring breakthrough concentrations during affinity product capture.16 Leveraging this recent technology, we aimed to investigate whether Raman spectrometry is able to measure simultaneously, in real time, multiple-quality attributes, including product molecular size variants, charge variants, glycosylation, and oxidation,19 during downstream unit operations, such as affinity purification,14 hydrophobic interaction chromatography (HIC),30 ion-exchange chromatography (IEX),20,21 and ultrafiltration/diafiltration (UFDF).31 We prioritized affinity capture as the unit operation of interest due to its operational criticality,7 and size variants as the quality attribute of interest due to its general criticality in bioprocessing.32 We sought to accomplish our aim by designing a Tecan automation system that generated large-scale calibration datasets that could then be used to build models capable of deconvoluting Raman spectra collected in real time during affinity capture into in-line quality attribute measurements that were comparable to off-line analytical results.
Results
Calibration sample mixing and Raman spectra preprocessing
We studied an industrial process for the purification of an investigative pharmaceutical IgG1 mAb drug substance during affinity chromatography. We were interested in monitoring product molecular weight variants during the elution phase since this part of the process provides a crucial opportunity for minimizing potentially critical product impurities. We performed affinity chromatography on harvested cell culture fluid (HCCF) from two clones derived from Chinese hamster ovary (CHO) cells and collected 25 fractions for Raman calibration (Figure 1a). We started with an existing approach for calibrating computational models33,34 and modified it to greatly increase the number of effective calibration data points without increasing off-line analytical testing burden by using a mixing series (Figure 1b). By mixing different proportions of each fraction n with the adjacent fraction n + 1, we generated six additional calibration samples from two original fractions. This strategy of mixing 25 fractions generated a total of 169 calibration points and was not fundamentally different from a conventional dilution series, except in this case the mixture is between two samples of known analytical product quality attributes instead of between a stock solution and a dilution buffer. We created a custom integrated Raman-Tecan system to automate the calibration process following this mixing strategy, generating 169 sets of Raman spectra each matched with corresponding sets of off-line analytical product quality attributes in under 48 h (Figure S1). We calculated 169 expected product attributes by weighing each of 25 empirical analytical results by the mixing ratios and could thus visualize the averaged Raman spectra for different values of product aggregate and fragment proportions (Figure 1c). Important features of the Raman spectra were qualitatively noticeable, including the sapphire peak at 418 cm−1, the major glycine peak at 898 cm−1, and minor glycine peaks at 1332, 1412, and 1444 cm−1. The aggregates content was in the range of 1.6 ± 0.1% to 7.1 ± 0.3% in the first training run, and 1.3 ± 0.2% to 5.9 ± 0.4% in the second run, used for testing. For the fragments content, the training dataset ranged from 2.8 ± 0.2% to 5.1 ± 0.1% and the testing dataset ranged from 1.9 ± 0.05% to 5.4 ± 0.7%. A standard deviation of 2.1 ± 1.5 g/L was observed in the elution fraction concentrations due to evaporation. The robotic mixing accuracy was validated off-line by mixing a dilution series up to 2 g/L for 80 samples (R2 = 0.9996, error = 0.009 g/L).
Figure 1.

Raman spectroscopy calibration dataset constructed from one Affinity Chromatography run.
Notes: (a) Chromatogram of the elution phase of an affinity chromatography unit operation during downstream bioprocessing of a pharmaceutical monoclonal antibody drug product, showing the off-line concentration (shaded region indicates the 95% CI), and in-line pH and conductivity measurements. One fraction represented 12 mL elution volume or 35 s. (b) Fractions collected during elution were mixed to generate calibration samples. Each elution fraction and its adjacent fraction were mixed to generate eight equally spaced calibration points. (c) Preprocessed Raman spectra for each of the two sets of 171 calibration points are shown. Lines are colored by high molecular weight variants fraction (top) or by low molecular weight variants (bottom) as measured off-line by analytical UPLC-SEC.
Our Raman-Tecan integrated system required a suitable data preprocessing pipeline, thus we evaluated several existing algorithms for eliminating spectral distortions present at smaller scales with lower flow rates.35,36 We started by applying a high-pass digital Butterworth filter followed by sapphire peak (418 cm−1) maximum normalization, and tested alternative combinations of commonly used baseline removal, filtering, and smoothing methods (Table 1). We calculated the average variation in the pre-processed spectra collected under flow rates ranging from 0.1 to 7 mL/min (Figure S2) and aimed to find preprocessing steps that consistently produced stable spectra despite different flow rates. The average error in the normalized Raman spectra due to differences in flow rates was lowered after implementing the optimized preprocessing pipeline from 0.019 to 0.001, representing a 19-fold reduction of noise. We repeated this analysis with a second buffer system and obtained similar preprocessing results. We found empirically that a value of 2 for the Butterworth filter (see Supplementary Materials) was optimally suited to filter out spectral interferences from variations in flow rate (Table 1).
Table 1.
Effect of preprocessing methods on reducing variances from variable flow rates.
| Buffer A |
Buffer B |
|||
|---|---|---|---|---|
| Method | Mean Error | Std Error | Mean Error | Std Error |
| Raw | 0.0059 | 0.0093 | 0.0049 | 0.0075 |
| Polynomial | 0.0099 | 0.0091 | 0.0084 | 0.0075 |
| Differential | 0.0193 | 0.0168 | 0.0190 | 0.0164 |
| Butterworth-0.5 | 0.0247 | 0.0357 | 0.0210 | 0.0299 |
| Butterworth-2 | 0.0012 | 0.0011 | 0.0011 | 0.0010 |
| Butterworth-4 | 0.0013 | 0.0011 | 0.0013 | 0.0011 |
Preprocessing methods were distinguished based on their robustness to changes in flow rates. Errors were measured as the standard deviation for each of 3101 Raman wavenumbers across 213 spectra collected under different flow rates. The error calculation was performed for two different buffers. The Butterworth-2 method consistently produced the lowest mean errors.
We performed several controls and regularization steps to ensure the robustness of calibration results. Affinity chromatography elution fractions provided training examples of product monomers and variants, but did not contain samples with no product or variant conditions. To prevent the prediction of protein samples under blank buffer conditions, we performed a blank affinity chromatography run without HCCF load, keeping all other variables constant. The in-line Raman spectra collected during the elution phase was then processed using the same methodology and appended to the calibration dataset. After harmonizing the training dataset with a preprocessing pipeline (see Supplementary Materials), we applied noise augmentation to reduce effects from model over-fitting and trained a panel of regression models including a convolution neural network (CNN),16 support vector regressor (SVR)17 with a nonlinear radial basis function kernel, principal component analysis regressor (PCR),25,27 and partial least squares (PLS) regressor14,19,37 (Table 2). We used a value of 2 for the number of latent variables used by the PCR and PLS models based on values used in previous studies.21 Qualitatively, the k-Nearest Neighbor (KNN) and CNN regressors were best able to predict product quality attributes that were comparable to off-line analytical results (Figure S3).
Table 2.
Regression model training fit accuracy metrics.
| Model | R2 | R | MSE | MAE | MAPE |
|---|---|---|---|---|---|
| Aggregates (HMW%) | |||||
| CNN | 0.91 | 0.95 | 0.19 | 0.29 | 10% |
| SVR | 0.22 | 0.47 | 1.64 | 0.99 | 23% |
| PCR | 0.03 | 0.17 | 2.03 | 1.19 | 27% |
| PLS | 0.08 | 0.28 | 1.92 | 1.15 | 26% |
| Fragments (LMW%) | |||||
| CNN | 0.92 | 0.96 | 0.08 | 0.20 | 8% |
| SVR | 0.08 | 0.28 | 0.83 | 0.69 | 25% |
| PCR | 0.00 | 0.01 | 0.81 | 0.76 | 25% |
| PLS | 0.02 | 0.14 | 0.80 | 0.76 | 25% |
Training metrics are illustrated for measuring product aggregates and fragments for different models where MSE: mean squared error, MAE: mean absolute error, MAPE: mean absolute percent error (detailed scatter plots are illustrated in Figure S3). Each model was trained on 169 off-line calibration points plus control blank chromatography run. The trivial case of the training performance of the KNN model is not shown.
Prediction of in-line product quality results from Raman spectra
After testing a panel of models on the training dataset, we asked whether it would be possible to predict in-line product quality during the elution phase of a second affinity chromatography run with HCCF from a different clone of the cultivated cell line. The KNN model performed better than all tested models for predicting both the in-line aggregates content and in-line fragments content (Figure 2a). When compared to the performance of the commonly used PLS model, the KNN model reduced the mean absolute error (MAE) three-fold from 1.8% high molecular weight (HMW) content to 0.5% HMW, and from 0.7% low molecular weight (LMW) content to 0.4% LMW (Table 3). Qualitatively, predictions made by the KNN model showed good linearity (Figure 2b). The distribution of product quality attributes measurements was normal and did not show signs of statistical bias (Figure 2c). The distributions of product quality values from the first calibration run were not significantly different to that of the second run for both aggregates measurements (p = 0.07, n = 171, two-sided Kolmogorov–Smirnov test) and for fragment measurements (p = 0.12, n = 171, two-sided KS test), indicating that the two calibration datasets were statistically comparable. To test whether these results were unique to our chosen preprocessing method, we applied an alternative preprocessing pipeline composed of a first-order differential followed by Savitzky-Golay filtering (Table 1, Figure S2) and re-calculated the prediction accuracies while keeping all other variables constant (Table S1). Under the alternative preprocessing method, the KNN prediction MAE increased from 0.53 to 0.83 (+57%) for the %HMW content and increased from 0.35 to 0.74 (+111%) for the %LMW content, while the PLS prediction MAE decreased from 1.78 to 1.01 (−43%) for the %HMW content and decreased from 0.74 to 0.61 (−18%) for the %LMW content. Thus, we demonstrate that our choice of preprocessing was critical in ensuring higher prediction accuracies that were not achievable using other methods found in previous studies.
Figure 2.

In-line prediction of product aggregate and fragment content during affinity chromatography.
Notes: (a) Prediction performance of five models trained on one chromatography run tested against measurements from a separate chromatography run. Accuracy was measured by the MAE: mean absolute error, using models including the KNN: k-nearest neighbors regressor (green), CNN: convolutional neural network, SVR: support vector regressor, PCR: principal component regressor (gray), and PLS: partial least squares regressor (yellow). Error bars represent 95% CI. (b) Scatter plots of in-line prediction of high (left) and low (right) molecular weight variants using the KNN regressor, error bars represent 95% CI. (c) Histograms showing probability distributions of size variants in the overall dataset.
Table 3.
Predictive in-line monitoring accuracy for five regression models.
| Model | Q2 | Q | MSE | MAE | MAPE |
|---|---|---|---|---|---|
| Aggregates (HMW%) | |||||
| KNN | 0.86 | 0.93 | 0.37 | 0.53 | 20% |
| CNN | 0.81 | 0.90 | 0.75 | 0.76 | 27% |
| SVR | 0.81 | 0.90 | 1.36 | 1.00 | 24% |
| PCR | 0.09 | 0.31 | 3.39 | 1.47 | 31% |
| PLS | 0.29 | 0.53 | 4.65 | 1.78 | 39% |
| Fragments (LMW%) | |||||
| KNN | 0.65 | 0.80 | 0.33 | 0.35 | 13% |
| CNN | 0.68 | 0.82 | 0.53 | 0.58 | 34% |
| SVR | 0.66 | 0.81 | 0.35 | 0.44 | 15% |
| PCR | 0.12 | 0.34 | 0.78 | 0.75 | 24% |
| PLS | 0.49 | 0.70 | 0.74 | 0.74 | 24% |
Following the results shown in Figure 2a, different metrics of model predictive performance are shown, including Q: the predictive correlation coefficient, MSE: mean squared error, MAE: mean absolute error, MAPE: mean absolute percent error.
The performance of the in-line product quality prediction model ultimately depended on the successful integration of preprocessing methods, automated large-scale data acquisition, and regression modeling selection. We found that model selection was especially critical for in-line measurement accuracy, where the KNN model yielded more accurate product aggregation and fragmentation proportions than the conventional PLS model (Figure 3a). The critical transition phase between the first half of the elution, which was composed mostly of product, was accurately predicted by the KNN model. Prediction accuracy was consistent across both product aggregates and fragments proportions. Combining the monitored variables together allowed visualization of the in-line chromatogram for the KNN model (Figure 3b), and for all other models (Figure S4). Deviations in model prediction arose from multiple predictions under multiple instances of statistical sampling that was part of the data augmentation strategy for reducing model overfitting (see Methods). In-line Raman product quality measurements were collected every 37.5 s, providing sufficiently rapid feedback feasible for real-time process control. Alternatively, product quality monitoring metrics were considered in terms of absolute product aggregate and fragment concentrations measured in grams per liter instead of percentages. We repeated our analyses with this alternative absolute metric and achieved comparable predictive results (Figure S5, Table S2).
Figure 3.
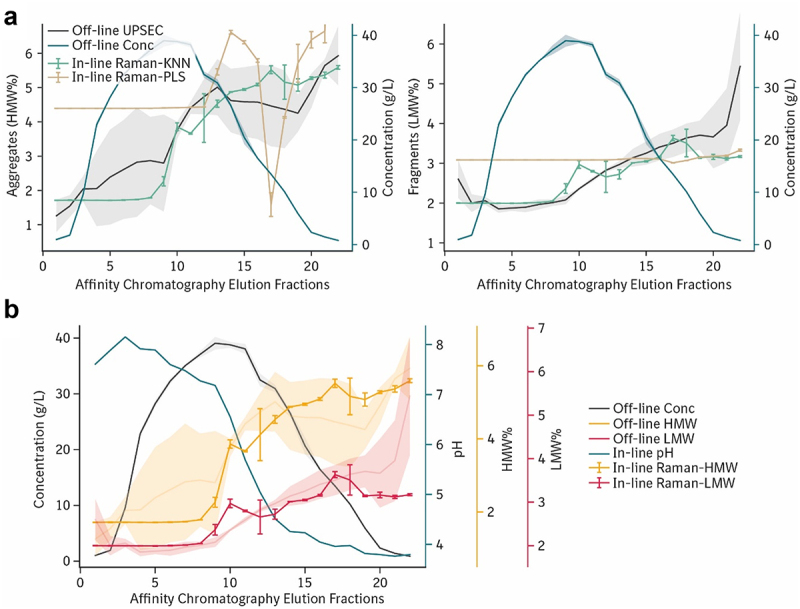
In-line measurement of product aggregates and fragments were comparable to off-line UPLC-SEC analytics.
Notes: (a) Chromatograms of the second affinity chromatography elution phase including off-line concentration and aggregates (left) or fragments (right) plotted with error regions representing 95% CIs. Predictions of the off-line size variants are plotted as green lines for the k-Nearest Neighbor regressor, the best-performing model overall, and as gold lines for the Partial Least Squares regressor, the conventional calibration model. (b) Following the format in panel A, the chromatogram for KNN model prediction is illustrated together with pH and concentration.
A larger dataset enabled by hardware Raman-Tecan integration (Figure S1) was important to achieve model predictive accuracy. To test the effect of dataset size on product quality prediction accuracy, we randomly removed fractions of the dataset and reevaluated the prediction Q2 accuracy. For predicting aggregates with the KNN regressor, the prediction accuracy decreased to Q2 = 0.5 at 20% dataset size, and for fragments, the prediction accuracy decreased to Q2 = 0.1 at 35% dataset size (Figure 4a). Thus, we determined that larger dataset sizes were necessary for maintaining higher model accuracies. Experimental data are naturally subject to technical and methodological variations, thus we aimed to determine whether our methods were disturbed by input noise. To test the robustness of the accuracy prediction results, we added random noise in the form of gaussian windows (σ = 5 cm−1, amplitude = 20%) across the wavenumber domain and computed the Q2 accuracy for the simulated perturbation dataset. The KNN regression accuracy remained relatively unchanged in the lower wavenumber regions and was minimally sensitive to noise perturbation in general (Figure 4b). Some wavenumbers were especially robustfor example, 321, 414, 774 cm−1, while others, such as 1055, 1658, 1725, 2940 cm−1, were moderately robust. Taken as a whole, we demonstrate that the KNN model is capable of monitoring product aggregation and fragmentation in-line. This capability depends on larger dataset sizes and was robust to random noise perturbations.
Figure 4.

Performance improvements from larger calibration dataset size and robustness to noise perturbations.
Notes: (a) The in-line aggregates (left) and fragments (right) prediction accuracy Q-squared is plotted against simulated dataset size. Predictive performance scaled with the size of the calibration dataset in general and was more pronounced for the KNN model. (b) Random noise was added to preprocessed Raman spectra every 2 cm−1 and the corresponding KNN accuracy is shown. Dotted lines indicate the starting prediction accuracy before noise addition, shaded regions indicate 95% CI.
Discussion
Using a process for the purification of mAbs from HCCF, we demonstrated the capability of using an in-line Raman spectrometer, preprocessing pipelines, automated off-line calibration system, and KNN regression models to provide real-time analytics for protein quality every 38 s. A mixing procedure was implemented which leveraged the higher data throughput of automation (Figure 1) and increased the efficiency of off-line analysis by a factor of 7 (Figure S1). A new preprocessing pipeline leveraging a high-pass filter for baseline removal followed by sapphire peak maximum normalization (Table 1) resulted in better reduction of confounding flow rate effects (Figure S2). Model fits were then obtained for a panel of commonly used regression models for translating the preprocessed Raman spectra into product quality measurements (Table 2, Figure S3). Using a second in-line affinity chromatography run as the testing dataset, prediction values were calculated for the panel of regression models, with the KNN model having the lowest MAE (Figure 2, Table 3). These results were dependent on the larger dataset size enabled by the Tecan-Raman integration, and we demonstrated by statistical modeling that our accuracy results significantly decreased after re-analysis with simulated smaller datasets (Figure 4a). We additionally demonstrate that prediction accuracy was robust to random noise perturbations to the Raman spectra (Figure 4b). Taken together, these results suggest the immediate possibility of implementing process controls during affinity purification of therapeutic mAbs.
Advancements in optics have made it possible for Raman spectrometers to increase signal throughput by re-focusing excess light which would otherwise have been lost due to dispersion,29 resulting in much more analytical data output useful for bioprocess monitoring. The HyperFlux Pro Plus spectrometer (Tornado Spectral Systems, Mississauga, Ontario, Canada) used in this study leverages this recent optical improvement and formed the basis for significant advancements in Raman bioprocess monitoring in this and recent studies.16,38 Although accurate models may still be achievable with longer Raman acquisition timesfor example, the up to 22.5 min read time19 required by conventional Raman spectrometers, faster acquisition times will play a central role in enabling widespread industrial implementation of previously demonstrated feasibilities.14,15,25–27 Alternatively, in applications, such as clinical and commercial manufacturing where the monitoring accuracy required for implementation has not yet been achieved, the application of optical improvements will improve signal throughput and lead to higher model accuracies while keeping all other variables constant. Thus, a time window longer than the currently reported read time of 37.5 s may better suit applications operating at larger scales or under good manufacturing practices (GMP). Partly due to the relatively longer read time of conventional Raman spectrometers, previous efforts have focused on off-line analysis with limited extension into in-line monitoring during bioprocessing, especially in downstream applications. While previous efforts have demonstrated the ability to measure product quality attributes for pharmaceutical mAbs off-line,19 protein concentration in-line,14–16 and average molecular weight in-line by multi-angle light scattering,30 this study extends the measurement of product aggregates and fragments to real-time product quality monitoring during an industrial process without adjustments to the off-line calibration model.
A key success factor for in-line product quality measurements by Raman spectrometry is the choice of preprocessing methods such as baseline correction.35,36 More sophisticated baseline correction methods have been used in previous studies and may be a further source of preprocessing optimization.39 The prominent sapphire peak was observed consistently at a wavenumber of 418 cm−1, consistent with similar observations in existing studies employing the same Raman hardware.16,40 Many glycine peaks were visible in the calibration dataset (Figure 1c), matching wavenumbers of glycine in solution (898 cm−1).41 Although the objective for optimizing the Butterworth filter cutoff frequency was to reduce noise due to flow rate differences (Table 1), the preservation of the glycine band integrity likely played a role in the final accuracy of the models (Tables 2 and 3), given that a slight, but sharp decrease in accuracy can be observed in the noise perturbation results around 898 cm−1 for both aggregates and fragments prediction (Figure 4b). Due to the lower accuracy of the SVR model (Figure S4), which derived its features from the glycine peak, the Raman signature of glycine was not sufficient to explain all the prediction accuracy, despite glycine concentration acting as a proxy for the elution time, since it is the unique ingredient in the elution buffer (see Methods). Apart from the clear sapphire and glycine peaks, several other unidentified peaks were also important for prediction accuracy; thus, work on elucidating the exact mechanisms of Raman distortions caused by protein aggregation and fragmentation and which mechanisms are the causal drivers of the corresponding distortions remain to be carried out. Accuracy of the calibration model (Tables 2 and 3) depends on variations of analytical label values present in the training dataset. These variations were dependent on undesirable changes to sample attributes during calibration sample preparation (Figures 2b and 3). Improvements to sample stability and automation hardware robustness are expected to improve measurement prediction accuracy. Accuracy requirements may be different depending on implementation strategies.
Raman spectroscopy is one of the several PAT technologies that have generated substantial industrial interest and implementation is being evaluated through all units of bioprocessing operation.7,13 One challenge of general interest which we have addressed through the exploitation of hardware automation is the so-called “Low-N problem”.42 Combined with the analytical efficiency leveraged by mixing (Figure 1b) and assuming a 10-fold increase in analytical leverage due to the noninvasive in-line nature of Raman spectra collection, we expect the combined analytical leverage to be around 70-fold per unit of off-line ultra-performance liquid size-exclusion chromatography (UPSEC) analysis. Although the total calibration dataset size in this study (n = 415) was comparable to that of a similar study, which measured product quality by Raman (n = 384),19 our off-line analytical effort was meaningfully reduced through automation (n = 50). Our scale can be further decreased by a factor of 4 if a commercially available 45 µL flow-cell is used instead of our 200 µL cell. Given that analytical testing of elution fractions may overlap with existing process development activities, there is potentially zero additional analytical testing required to implement in-line quality monitoring as described in this study. In addition to operational improvements, the mixing of elution fractions generated more analytically representative Raman spectra of product aggregates and fragments (Figure 2c).
Product impurities used in this study were sampled directly from a real process in contrast to existing approaches that use designs of experiments19 in combination with forced degradation study samples43 generated through heat incubation.17 Thus, mixed real-process samples used for off-line calibration were more biochemically similar to analytes found in-line, which ultimately facilitated the successful measurement of product quality attributes during affinity chromatography. The advantage of this increase in representativeness can be demonstrated by the robustness to dataset size reduction in the prediction of product aggregates (Figure 4a). A consequence of using elution fractions for calibration is that quantitative modeling is limited by the impurity range of the process capability, in other words the calibrated maximum of the dynamic range can be no higher than what is observed during elution sampling. Bioprocess development mainly aims to improve process parameters such that impurities are minimized, thus it may be assumed that calibration models built at earlier stages of development would simultaneously maximally enrich process development by supplying a wealth of in-line data and be sufficient to cover later stage applications where lower impurity concentrations are generally expected. The constant need for re-calibration as dynamic range specifications evolve to fit changing development priorities is thus avoidable when process performance is constantly improving. The number of fractionation samples used for calibration is a critical parameter determining model representativeness and subsequent risk of inaccurate measurements. The real-time continuous elution profile of impurities is effectively reduced to linear representations between fractionation sampling points, thus while a smaller number of samples may reduce analytical burden, it could lead to a misrepresentation of the true elution behavior of product variants and lead to a misrepresented calibration training dataset. Variable sampling strategies, such as more frequent sampling over periods of greater product variant concentration gradients, would improve representativeness for smaller numbers of fractionation. Furthermore, the elution peak should be included as a sampling point to ensure that product variant maximum concentrations are captured by the calibration training dataset.
It would be feasible to generalize the main results from this study to monitor additional product variants apart from product aggregates and fragments given previously documented successes in measuring product attributes such as oxidation by Raman.19 Although we focused mainly on reporting quality measurements in terms of conventional aggregate and fragment percentages, we also report the main findings in terms of absolute concentrations in units of grams per liter (Figure S5). We emphasize this ability to measure analyte concentrations individually to highlight that our methodology is scalable to include several quality attribute output variables, limited only by off-line testing capacity. We aim in the future to expand calibration models presented here to include factors such as product electrostatic charge variants, N-glycan variants, and total host cell protein (HCP) concentrations. To accommodate the expansion of the number of product variants to monitor, additional orthogonal analytical techniques may be considered,20,22,30,37,44,45 as well as data fusion to complement individual strengths and weaknesses between approaches. The intensive process of training and interpreting convolutional neural networks16 may be complemented by less sophisticated modeling methods not investigated in this studyfor example, by adopting a Gaussian process regression approach26 paired with a suitable kernel function. Another potentially fruitful approach could be to combine recent developments in mechanistic modeling,33,34 with Raman modeling15 using Kalman filtering or alternative Bayesian approaches.40
In conclusion, we demonstrate a labor-efficient method of building and calibrating a Raman spectrometry regression model for measuring potentially critical product quality attributes directly in-line during downstream bioprocessing every 38 s and without generating additional off-line analytical workload. This technology has the potential to dramatically increase process understanding and encourage widespread adoption of noninvasive in-process control testing, and to ultimately stimulate our conception of advanced manufacturing for biopharmaceuticals in the future.
Materials and methods
Protein A affinity capture fractionation experiments: Two affinity chromatography unit operations were conducted to generate calibration and validation datasets. These runs were used for both in-line and off-line analytics. A third run was carried out as the buffer blank and followed the same procedures as the previous two, except for the exemption of a load phase. Protein A affinity chromatography resin Mab Select PrismA was packed for operation on an Akta Avant 150 system (Cytiva, Uppsala, Sweden) in a 2.6 cm diameter column to a height of 19.4 cm (103 mL column volume). Equilibration was performed at 25 mM Tris, pH 7.5, 1.98 mS/cm, at a volumetric flow rate of 20.6 mL/min for five column volumes (CV). For the first calibration run, 1.274 L harvest material at 4.85 g/L (6.2 g) was loaded, while for the second validation run 1.483 L harvest material at 4.17 g/L (6.2 g) was loaded, for a column load density of 60 mg/mL. Following the loading phase, the column was washed with 3 CV of equilibration buffer, followed by 3 CV of 50 mM Tris, pH 7.5 wash buffer, followed by 3 CV of equilibration buffer. Following the washing phase, 3 CV of pH 3.5, 1.25 mS/cm buffer was used for elution. Fractionation was performed every 12 mL until the end of elution during the 26th fraction and a constant flow rate was maintained throughout fractionation as is typical of industrial processes. Following the elution phase, the column was regenerated and stored following conventional procedures.
In-line process analytical measurements: Raman acquisition settings were critical for maximizing signal-to-noise ratio for a given dynamic range of the experimental system.16 A HyperFlux Pro Plus Raman spectrometer (Tornado Spectral Systems, Mississauga, Ontario, Canada) was equipped with a standard 785 nm laser and was operated at the maximum power 495 mW. A flow cell with a dead volume of 200 µL (Marqmetrix, Seattle, Washington, USA) was equipped with the spectrometer and was connected in-line to the Akta Avant 150 system directly downstream of the built-in UV flow cell. The measurement of product-related impurities, the minority of species present in the cell harvest material, was prioritized over measuring total concentration. We performed multiple auto-exposure scans on a panel of both preparative chromatography buffers and process product samples, and we determined that 500 ms exposure with an averaging of 15 exposures per scan was optimal for maximizing the signal-to-noise ratio of protein aggregates and fragments. However, detector saturation was occasionally observed during peak elution, and thus we did not focus on concentration prediction. Although product size variant percentages may be concentration dependent, we trained our model to focus on indirect secondary effects of increased protein aggregation, such as changes to water flow which are detectable using flow water proton NMR.22 We emphasize that a process characterization study demonstrating the minimization of process parameter risks, such as the potential for errors in in-line monitoring due to small variations in process flows, cannot be omitted from any implementation strategies resulting from the methods described in this study.
Off-line process analytical measurements: To generate analytical data for off-line calibration, product quality was assessed in each of 25 fractions from each of the two chromatography runs. Each fraction was analyzed for concentration by Lunatic plate reader (Unchained Labs, Pleasanton, California, USA), and for molecular weight variants by UPSEC with an Acquity BEH SEC 200 system (Waters Corporation, Milford, MA, USA). These analyses were repeated approximately 48 h after the initial measurement due to long dwell times at room temperature, then the mean and standard deviations were estimated and used for training and validation. Long dwell times were due to a lack of optimization of the integrated Tecan-Raman automation system and should be addressed in future prototypes.
Regression model training: Regression models were built and validated using Python packages sklearn and tensorflow. The KNN regressor was constructed with the distance metric as weights. The hyperparameter k was not tuned directly; however, since the noise augmentation coefficient controlled the dataset size (see Supplementary Materials), which indirectly controls the effect of k, the resulting effective k value was different than the tuned value. The optimal k value was 42 and sampling coefficient value was 10, resulting in an effective k value of 4. Hyperparameter optimization was performed by random search and accuracy metrics were weighed equally. The SVR was built using the Raman intensity in the region of the strongest glycine peak (887–907 cm−1, inclusive) as features. Because the elution is driven by the introduction of the glycine buffer, we intended to use the SVR as a control model that exclusively uses the glycine peak for prediction. A radial basis function was used, with C = 0.6. A latent variable of 2 was used to maximize combined training and validation performance for both the PLS and PCR models. The convolutional neural network (CNN) regressor architecture was adapted from a previous study,16 with three outputs for concentration, aggregates, and fragments, learning rate of 0.002, batch size of 1, learning decay rate of 3 × 10−6, momentum of 0.8, and a training duration of 600 epochs. Model hyperparameters were optimized by assessing optimal training performance over 13,613 random initializations.
Supplementary Material
Funding Statement
This work was funded by Boehringer Ingelheim Pharma GmbH & Co. KG.
Abbreviations
- CHO
Chinese hamster ovary
- CNN
Convolution neural network
- CV
Column volumes
- DLS
Dynamic light scattering
- GMP
Good manufacturing practices
- HCCF
Harvested cell culture fluid
- HCP
Host cell protein
- HIC
Hydrophobic interaction chromatography
- HMW
High molecular weight
- IEX
Ion-exchange chromatography
- KNN
k-Nearest Neighbor
- LMW
Low molecular weight
- mAbs
Monoclonal antibodies
- MAE
Mean absolute error
- NMR
Nuclear magnetic resonance
- PAT
Process analytical technologies
- PCR
Principal component regressor
- PLS
Partial least squares
- SVR
Support vector regressor
- UFDF
Ultrafiltration/diafiltration
- UPSEC
Ultra-performance liquid size exclusion chromatography
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request and require legal agreements prior to sharing.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2023.2220149
References
- 1.CMC_Biotech_Working_Group . A-Mab: a case study in bioprocess development. 2009;
- 2.Scannell JW, Blanckley A, Boldon H, Warrington B.. Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discov. 2012;11(3):191–11. doi: 10.1038/nrd3681. [DOI] [PubMed] [Google Scholar]
- 3.Ringel MS, Scannell JW, Baedeker M, Schulze U. Breaking Eroom’s Law. Nat Rev Drug Discov. 2020;19:833–34. doi: 10.1038/d41573-020-00059-3. [DOI] [PubMed] [Google Scholar]
- 4.Ochoa D, Karim M, Ghoussaini M, Hulcoop DG, McDonagh EM, Dunham I. Human genetics evidence supports two-thirds of the 2021 FDA-approved drugs. Nat Rev Drug Discov. 2022;21(8):551–551. doi: 10.1038/d41573-022-00120-3. [DOI] [PubMed] [Google Scholar]
- 5.Kelley B. Developing therapeutic monoclonal antibodies at pandemic pace. Nat Biotechnol. 2020;38:540–45. doi: 10.1038/s41587-020-0512-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farid SS, Baron M, Stamatis C, Nie W, Coffman J. Benchmarking biopharmaceutical process development and manufacturing cost contributions to R&D. MAbs. 2020;12(1):1754999. doi: 10.1080/19420862.2020.1754999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gillespie C, Wasalathanthri DP, Ritz DB, Zhou G, Davis KA, Wucherpfennig T, Hazelwood N. Systematic assessment of process analytical technologies for biologics. Biotechnol Bioeng. 2022;119(2):423–34. doi: 10.1002/bit.27990. [DOI] [PubMed] [Google Scholar]
- 8.Crowell LE, Rodriguez SA, Love KR, Cramer SM, Love JC. Rapid optimization of processes for the integrated purification of biopharmaceuticals. Biotechnol Bioeng. 2021;118:3435–46. doi: 10.1002/bit.27767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.ICH . Continuous manufacturing of drug substances and drug products Q13. The International Council for Harmonisation (ICH). 2021. [Google Scholar]
- 10.Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the “high-hanging fruit. Nat Rev Drug Discov. 2018;17(3):197–223. doi: 10.1038/nrd.2017.227. [DOI] [PubMed] [Google Scholar]
- 11.Erickson J, Baker J, Barrett S, Brady C, Brower M, Carbonell R, Charlebois T, Coffman J, Connell‐Crowley L, Coolbaugh M, et al. End‐to‐end collaboration to transform biopharmaceutical development and manufacturing. Biotechnol Bioeng. 2021;118(9):3302–12. doi: 10.1002/bit.27688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eon‐Duval A, Broly H, Gleixner R. Quality attributes of recombinant therapeutic proteins: an assessment of impact on safety and efficacy as part of a quality by design development approach. Biotechnol Progr. 2012;28(3):608–22. doi: 10.1002/btpr.1548. [DOI] [PubMed] [Google Scholar]
- 13.Pedro MNS, Klijn ME, Eppink MH, Ottens M. September. Process analytical technique (PAT) miniaturization for monoclonal antibody aggregate detection in continuous downstream processing. J Chem Technology Biotechnology. 97(9):2022. [Google Scholar]
- 14.Feidl F, Garbellini S, Vogg S, Sokolov M, Souquet J, Broly H, Butté A, Morbidelli M. A new flow cell and chemometric protocol for implementing in‐line Raman spectroscopy in chromatography. Biotechnol Progr. 2019;35(5):e2847. doi: 10.1002/btpr.2847. [DOI] [PubMed] [Google Scholar]
- 15.Feidl F, Garbellini S, Luna MF, Vogg S, Souquet J, Broly H, Morbidelli M, Butté A. Combining mechanistic modeling and raman spectroscopy for monitoring antibody chromatographic purification. Process. 2019;7(10):683. doi: 10.3390/pr7100683. [DOI] [Google Scholar]
- 16.Rolinger L, Rüdt M, Hubbuch J. Comparison of UV‐ and Raman‐based monitoring of the protein a load phase and evaluation of data fusion by PLS models and CNNs. Biotechnol Bioeng. 2021;118(11):4255–68. doi: 10.1002/bit.27894. [DOI] [PubMed] [Google Scholar]
- 17.Zhang C, Springall JS, Wang X, Barman I. Rapid, quantitative determination of aggregation and particle formation for antibody drug conjugate therapeutics with label-free Raman spectroscopy. Anal Chim Acta. 2019;1081:138–45. doi: 10.1016/j.aca.2019.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldrick S, Umprecht A, Tang A, Zakrzewski R, Cheeks M, Turner R, Charles A, Les K, Hulley M, Spencer C, et al. High-throughput raman spectroscopy combined with innovate data analysis workflow to enhance biopharmaceutical process development. Process. 2020;8(9):1179. doi: 10.3390/pr8091179. [DOI] [Google Scholar]
- 19.Wei B, Woon N, Dai L, Fish R, Tai M, Handagama W, Yin A, Sun J, Maier A, McDaniel D, et al. Multi-attribute Raman spectroscopy (MARS) for monitoring product quality attributes in formulated monoclonal antibody therapeutics. MAbs. 2021;14(1):2007564. doi: 10.1080/19420862.2021.2007564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akhgar CK, Ebner J, Spadiut O, Schwaighofer A, Lendl B. QCL–IR spectroscopy for in-line monitoring of proteins from preparative ion-exchange chromatography. Anal Chem. 2022;94(14):5583–90. doi: 10.1021/acs.analchem.1c05191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brestrich N, Rüdt M, Büchler D, Hubbuch J. Selective protein quantification for preparative chromatography using variable pathlength UV/Vis spectroscopy and partial least squares regression. Chem Eng Sci. 2018;176:157–64. doi: 10.1016/j.ces.2017.10.030. [DOI] [Google Scholar]
- 22.Taraban MB, Briggs KT, Merkel P, Yu YB. Flow water proton NMR: in-line process analytical technology for continuous biomanufacturing. Anal Chem. 2019;91(21):13538–46. doi: 10.1021/acs.analchem.9b02622. [DOI] [PubMed] [Google Scholar]
- 23.Zhou C, Qi W, Lewis EN, Carpenter JF. Concomitant Raman spectroscopy and dynamic light scattering for characterization of therapeutic proteins at high concentrations. Anal Biochem. 2015;472:7–20. doi: 10.1016/j.ab.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 24.Smith E, Dent G. 25 February. 2019. Modern Raman Spectroscopy: A Practical Approach. John Wiley & Sons Ltd. 9781119440550. [Google Scholar]
- 25.McAvan BS, Bowsher LA, Powell T, O’Hara JF, Spitali M, Goodacre R, Doig AJ. Raman spectroscopy to monitor post-translational modifications and degradation in monoclonal antibody therapeutics. Anal Chem. 2020;92(15):10381–89. doi: 10.1021/acs.analchem.0c00627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tulsyan A, Wang T, Schorner G, Khodabandehlou H, Coufal M, Undey C. Automatic real‐time calibration, assessment, and maintenance of generic Raman models for online monitoring of cell culture processes. Biotechnol Bioeng. 2020;117(2):406–16. doi: 10.1002/bit.27205. [DOI] [PubMed] [Google Scholar]
- 27.Yilmaz D, Mehdizadeh H, Navarro D, Shehzad A, O’Connor M, McCormick P. Application of Raman spectroscopy in monoclonal antibody producing continuous systems for downstream process intensification. Biotechnol Progr. 2020;36(3):e2947. doi: 10.1002/btpr.2947. [DOI] [PubMed] [Google Scholar]
- 28.McDaniel DL, Kadaub E, Wei B, Maier AJ, Magill GE. 12 January. Use of genetic algorithms to determine a model to identity sample properties based on Raman spectra. US20230009725. 2023.
- 29.Meade JT, Hajian AR, Behr BB, Cenko AT. 26 February. Optical slicer for improving the spectral resolution of a dispersive spectrograph. US8384896B2. 2013.
- 30.Patel BA, Gospodarek A, Larkin M, Kenrick SA, Haverick MA, Tugcu N, Brower MA, Richardson DD. Multi-angle light scattering as a process analytical technology measuring real-time molecular weight for downstream process control. MAbs. 2018;10:945–50. doi: 10.1080/19420862.2018.1505178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rolinger L, Rüdt M, Diehm J, Chow-Hubbertz J, Heitmann M, Schleper S, Hubbuch J. Multi-attribute PAT for UF/DF of proteins—monitoring concentration, particle sizes, and buffer exchange. Anal Bioanal Chem. 2020;412:2123–36. doi: 10.1007/s00216-019-02318-8. [DOI] [PubMed] [Google Scholar]
- 32.Rolinger L, Rüdt M, Hubbuch J. A critical review of recent trends, and a future perspective of optical spectroscopy as PAT in biopharmaceutical downstream processing. Anal Bioanal Chem. 2020;412:2047–64. doi: 10.1007/s00216-020-02407-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saleh D, Wang G, Müller B, Rischawy F, Kluters S, Studts J, Hubbuch J. Straightforward method for calibration of mechanistic cation exchange chromatography models for industrial applications. Biotechnol Progr. 2020;36(4):e2984. doi: 10.1002/btpr.2984. [DOI] [PubMed] [Google Scholar]
- 34.Rischawy F, Saleh D, Hahn T, Oelmeier S, Spitz J, Kluters S. Good modeling practice for industrial chromatography: mechanistic modeling of ion exchange chromatography of a bispecific antibody. Comput Chem Eng. 2019;130:106532. doi: 10.1016/j.compchemeng.2019.106532. [DOI] [Google Scholar]
- 35.Ryabchykov O, Guo S, Bocklitz T. Analyzing Raman spectroscopic data. Phys Sci Rev. 2018;4:20170043. doi: 10.1515/psr-2017-0043. [DOI] [Google Scholar]
- 36.Guo S, Popp J, Bocklitz T. Chemometric analysis in Raman spectroscopy from experimental design to machine learning–based modeling. Nat Protoc. 2021;16:5426–59. doi: 10.1038/s41596-021-00620-3. [DOI] [PubMed] [Google Scholar]
- 37.Ramakrishna A, Prathap V, Maranholkar V, Rathore AS. Multi-wavelength UV-based PAT tool for measuring protein concentration. J Pharmaceut Biomed. 2022;207:114394. doi: 10.1016/j.jpba.2021.114394. [DOI] [PubMed] [Google Scholar]
- 38.Graf A, Lemke J, Schulze M, Soeldner R, Rebner K, Hoehse M, Matuszczyk J. A novel approach for non-invasive continuous in-line control of perfusion cell cultivations by raman spectroscopy. Front Bioeng Biotech. 2022;10:719614. doi: 10.3389/fbioe.2022.719614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Z-M, Chen S, Liang Y-Z. Baseline correction using adaptive iteratively reweighted penalized least squares. Analyst (Lond). 2010;135:1138–46. doi: 10.1039/b922045c. [DOI] [PubMed] [Google Scholar]
- 40.Rolinger L, Hubbuch J, Rüdt M. Monitoring of ultra- and diafiltration processes by Kalman-filtered Raman measurements. Anal Bioanal Chem. 2023;415:841–54. doi: 10.1007/s00216-022-04477-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu G, Zhu X, Fan Q, Wan X. Raman spectra of amino acids and their aqueous solutions. Spectrochimica Acta Part Mol Biomol Spectrosc. 2011;78(3):1187–95. doi: 10.1016/j.saa.2010.12.079. [DOI] [PubMed] [Google Scholar]
- 42.Tulsyan A, Garvin C, Undey C. Industrial batch process monitoring with limited data. J Process Contr. 2019;77:114–33. doi: 10.1016/j.jprocont.2019.03.002. [DOI] [Google Scholar]
- 43.Nowak C, Cheung JK, Dellatore SM, Katiyar A, Bhat R, Sun J, Ponniah G, Neill A, Mason B, Beck A, et al. Forced degradation of recombinant monoclonal antibodies: a practical guide. MAbs. 2017;9(8):1217–30. doi: 10.1080/19420862.2017.1368602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.AL de F e S, Elcoroaristizabal S, Ryder AG. Multi-attribute quality screening of immunoglobulin G using polarized excitation emission matrix spectroscopy. Anal Chim Acta. 2020;1101:99–110. doi: 10.1016/j.aca.2019.12.020. [DOI] [PubMed] [Google Scholar]
- 45.Akhgar CK, Ebner J, Alcaraz MR, Kopp J, Goicoechea H, Spadiut O, Schwaighofer A, Lendl B. Application of quantum cascade laser-infrared spectroscopy and chemometrics for in-line discrimination of coeluting proteins from preparative size exclusion chromatography. Anal Chem. 2022;94(32):11192–200. doi: 10.1021/acs.analchem.2c01542. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request and require legal agreements prior to sharing.