


Abstract
The stability of collagen α1(I) mRNA is regulated by its 5′ stem–loop, which binds a cytoplasmic protein in a cap-dependent manner, and its 3′-untranslated region (UTR), which binds αCP. When cultured in a three-dimensional gel composed of type I collagen, mouse fibroblasts had decreased collagen α1(I) mRNA steady-state levels, which resulted from a decreased mRNA half-life. In cells cultured in gel, hybrid mouse–human collagen α1(I) mRNA with a wild-type 5′ stem–loop decayed faster than the same mRNA with a mutated stem–loop. When the 5′ stem–loop was placed in a heterologous mRNA, the mRNA accumulated to a lower level in cells grown in gel than in cells grown on plastic. This suggests that the 5′ stem–loop down-regulates collagen α1(I) mRNA. Protein binding to the 5′ stem–loop was reduced in cells grown in gel, which was associated with destabilization of the collagen α1(I) mRNA. In addition to the binding of a cytoplasmic protein, there was also a nuclear binding activity directed to the collagen α1(I) 5′ stem–loop. The nuclear binding was increased in cells grown in gel, suggesting that it may negatively regulate expression of collagen α1(I) mRNA. Binding of αCP, a protein involved in stabilization of collagen α1(I) mRNA, was unchanged by the culture conditions.
INTRODUCTION
Type I collagen is the most abundant protein in mammals and is composed of two α1(I) chains and one α2(I) chain (1). Separate genes encode these polypeptides, the expression of which is coordinately regulated (2,3). The α1(I) gene is regulated at the transcriptional and post-transcriptional level in various cell types (4–10). Transcription of the α1(I) gene is driven by proximal promoter sequences, which bind several ubiquitous transcription factors (11–13). A region of 2300 or 440 nt of the promoter has been found to be sufficient for transcription of a reporter gene in transgenic mice in several studies (14–17). However, to achieve expression similar to the endogenous gene in collagen-producing tissue, the transgene had to be present in multiple copies (10–12 copies) in these animals (14). At the same time the mice had overexpression of the transgene in tissues that normally produce low levels of collagen, like liver, spleen, thymus and lungs. Clearly, the proximal promoter of the α1(I) gene cannot fully account for the regulated expression in transgenic animals. Distal DNase I hypersensitive sites were described in the α1(I) gene locus as potential binding sites for enhancer-like or locus controlling region factors, but the functional significance of these sites is not known (18).
Expression of the α1(I) gene is also regulated at the post-transcriptional level (6–10,19,20). We measured the rate of transcription of this gene in quiescent hepatic stellate cells (hscs), which produce a small amount of α1(I) mRNA, and in activated hscs, where this mRNA is expressed at a 60- to 70-fold higher level. The transcription rate was increased only 3-fold (8). However, the half-life of the α1(I) mRNA was increased ~16-fold in activated hscs compared to quiescent hscs, suggesting a predominantly post-transcriptional regulation of the α1(I) gene in this model. αCP, a protein involved in stabilization of α-globin mRNA (21,22) and tyrosine hydroxylase mRNA (23,24), binds to the collagen α1(I) mRNA 3′-untranslated region (UTR) in activated hscs and may be involved in its post-transcriptional regulation (8).
The 5′-UTRs of α1(I) mRNA, α2(I) mRNA and α1(III) mRNA contain an evolutionarily conserved stem–loop structure (9,25). These three mRNAs are coordinately up-regulated in fibrotic processes of various organs (3,26–29). We analyzed a regulatory role of the 5′ stem–loop in the expression of reporter genes in quiescent and activated hscs (9). We found that the stem–loop prevented expression of the reporter genes in quiescent hscs but allowed expression in activated hscs. Reporter genes with a mutated stem–loop were constitutively expressed in both cell types. This inhibitory effect of the stem–loop was in part mediated by a decreased half-life of the corresponding mRNAs (9). In activated hscs, as well as in mouse fibroblasts, there are protein factors that bind the stem–loop in a cap-dependent manner. Because this binding is absent in quiescent hscs, we postulated that it is required for stabilization of collagen α1(I) mRNA (9). These studies have shown the importance of two regions of collagen α1(I) mRNA, the αCP binding site in the 3′-UTR and the stem–loop in the 5′-UTR, as well as their cognate protein factors, in post-transcriptional regulation of collagen α1(I) mRNA.
Culture conditions of fibroblasts often have a pronounced effect on gene expression and cell morphology (19,20,30–34). Cells grown within or on a matrix composed of extracellular proteins extracted from Engelbreth–Swarm–Holms tumor (matrigel) or pure collagen type I gels are in a more physiological environment than cells grown on plastic. Several studies have shown that primary human fibroblasts down-regulate collagen α1(I) mRNA steady-state levels when cultured within a collagen gel matrix (19,20,32,33). This down-regulation was partly due to an increased turnover of α1(I) mRNA and was accompanied by a more elongated appearance of the cells (19,20). Similarly, activated hscs reverse to a more quiescent phenotype when cultured in matrigel (35).
In this paper we used mouse fibroblast cell lines and a matrix gel composed of collagen type I to study the role of previously identified cis-acting elements in the regulation of collagen α1(I) mRNA. We found that collagen α1(I) mRNA is destabilized in cells grown within a matrix and that its increased turnover requires the presence of the 5′ stem–loop. Previously identified protein factors that bind the stem–loop are reduced in cells grown in the gel. However, a novel stem–loop binding activity was found in nuclear extracts and was increased in cells grown in the gel. We propose a complex regulation of collagen α1(I) mRNA by two differentially expressed RNA binding activities.
MATERIALS AND METHODS
Cell culture
NIH 3T3 and Swiss 3T3 cells were grown in DMEM supplemented with 10% calf serum. For culture in the collagen matrix, 106 cells were suspended in 12 ml of a 1 mg/ml solution of collagen type I (Collaborative Biomedical Products) in DMEM/10% calf serum and seeded in a 100 mm plate, as described (33). The collagen gel formed before the cells had settled and within 2–3 min. For measuring mRNA half-life, actinomycin D (Sigma) was added to the medium or on the gel at 15 µg/ml. Cells were harvested from plates by scraping and from the gel by digestion of the matrix with 50 µl of 2% collagenase B (Boehringer), for 30 min at 37°C. Transient transfections were performed by the calcium phosphate method using 30 µg of DNA per 150 mm plate. Twenty-four hours after addition of the precipitate the medium was changed and after an additional 24 h the cells were pooled and seeded into a gel matrix in several plates. Adenovirus-mediated gene transfer was carried out at an MOI of 1000 and the cells were processed as above.
Plasmid constructs
Construction of the adenoviruses has been described (9). Hybrid mouse–human collagen genes were made as follows. A full-size human collagen α1(I) cDNA was obtained from ATCC (clone no. 61322). From that clone, a 2.5 kb EcoRI–BamHI fragment was subcloned into the HindIII and BamHI sites of vector pGL3 (Promega), after blunting the EcoRI and HindIII sites. Then, a 2.3 kb BamHI–BamHI fragment of ATCC 61322 was cloned into the BamHI site of the above pGL3 clone. A 330 nt BglII–XbaI fragment of the promoter and the first exon of the mouse collagen α1(I) gene, either with the wild-type sequence or with a mutation in the stem–loop, was cloned into the BglII–XbaI sites of the pGL3 clone, generating the chimeric mouse–human collagen α1(I) genes mWT/hCOLL and mMUT/hCOLL, respectively. Both of these genes contain only the first polyadenylation signal of the human collagen α1(I) gene. Riboprobes used to map expression of luciferase reporter genes and the endogenous collagen α1(I) gene have been described (9). The template for the riboprobe for mouse GAPDH was obtained from Ambion and in experiments with hybrid mouse–human genes was used after linearization with StyI. The riboprobe used to analyse the hybrid mouse–human collagen mRNA was made by subcloning a 142 nt XbaI–KpnI fragment of ATCC 61322 into the KpnI–XbaI sites of vector Bluscript SK. After linearization with SacI and transcription with T7 RNA polymerase the template gives a riboprobe of 173 nt which protects 150 nt of human collagen α1(I) mRNA. Short RNA with the sequence of the mouse collagen α1(I) 5′ stem–loop or RNA with this stem–loop inverted have been described (9). These RNAs were made with or without the 7mG cap structure as described (9).
Preparation of cell extracts and electrophoretic mobility shift assay
Cytoplasmic cellular extracts were prepared as before (9). Nuclear extracts were made according to Digman (36). Mobility shift assays were performed by incubating 40 000 c.p.m. of labeled RNA probe with 60–70 µg of extract in 25 µl of 25 mM KCl, 10 mM Tris–HCl, pH 7.6, 10 mM MgCl2, 200 ng/µl tRNA, 10% glycerol. Competitor RNA was added as indicated. For mobility shift experiments with the αCP probe (8), 10 µg of extract was used and different antibodies added as indicated. For experiments with nuclear extracts, 60 µg of extract was used together with 4 µg of HeLa total RNA to suppress non-specific binding. After a 20 min incubation on ice, samples were resolved on a 6% native acrylamide gel.
RNase protection assay
This assay was done as described (8,9) using 25 µg of total RNA. The riboprobe for the test gene and that for GAPDH, as an internal control, were always hybridized together. Intensities of experimental bands were quantitated by phosphorimaging and normalized to their GAPDH control bands. In experiments with hybrid mouse–human genes, the digestion mixture contained RNase ONE (1 U/ml), in addition to RNase A and RNase T1.
RESULTS
Primary human fibroblasts decrease their steady-state levels of collagen α1(I) mRNA when cultured in a type I collagen matrix (19,20,32,33). To see if the same result is obtained with mouse fibroblast lines, we cultured Swiss 3T3 and NIH 3T3 cells on plastic or within a gel composed of pure collagen type I. Three days after plating, the cells were analyzed for collagen α1(I) mRNA by RNase protection assays and expression was compared to the internal standard GAPDH mRNA (Fig. 1). In Swiss 3T3 cells grown in gel, collagen α1(I) mRNA was decreased to 20% of the level seen in cells grown on plastic (lanes 1 and 2). For NIH 3T3 cells this decrease was ~50% (lanes 3 and 4). Thus, mouse fibroblast lines down-regulate collagen α1(I) mRNA in a three-dimensional matrix similarly to primary fibroblasts. We have consistently observed a greater effect with Swiss 3T3 cells than with NIH 3T3 cells. Cultured in gel, both cells had an elongated shape with long cytoplasmic protrusions, as described for primary fibroblasts (not shown) (33). Contraction of the collagen gel was minimal after 3 days in culture. Based on this result, we decided to use this model to study the mechanism that decreases collagen α1(I) mRNA in more detail.
Figure 1.
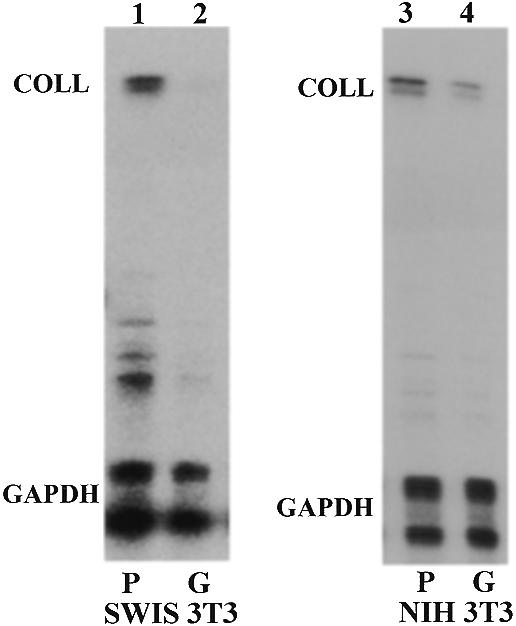
Steady-state level of collagen α1(I) mRNA in mouse fibroblasts grown on plastic or in gel. Swiss 3T3 (lanes 1 and 2) or NIH 3T3 fibroblasts (lanes 3 and 4) were cultured for 3 days on plastic (P, lanes 1 and 3) or in a collagen type I gel (G, lanes 2 and 4). Total RNA was probed with riboprobes specific for mouse collagen α1(I) mRNA and GAPDH mRNA, as an internal control. Migration of the protected bands is indicated.
Decreased steady-state levels of mRNA can be a result of its decreased synthesis or increased degradation or both. Since there is some evidence that primary cells in matrix destabilize collagen α1(I) mRNA (10,19,20), we measured the half-life of endogenous α1(I) mRNA in Swiss 3T3 fibroblasts grown on plastic and in gel. Figure 2 shows the results of two independent experiments. In cells grown on plastic there was no decrease in collagen α1(I) mRNA level up to 12 h after transcription block. This is consistent with a long half-life of this mRNA in Swiss 3T3 fibroblasts, as described previously (10). However, in cells grown in gel, collagen α1(I) mRNA decayed faster, with a half-life of ~12 h. Therefore, increased turnover of collagen α1(I) mRNA is, at least in part, responsible for its lower steady-state level in the mouse cells grown within a collagen matrix.
Figure 2.

Half-life of collagen α1(I) mRNA in Swiss 3T3 fibroblasts cultured on plastic (plate, closed circles) or in gel (gel, open circles). Swiss 3T3 cells were cultured for 3 days on plastic or in gel, actinomycin D was then added and after the indicated times total RNA was extracted and analyzed as in Figure 1. Expression of collagen α1(I) mRNA was normalized to GAPDH mRNA and the results of two independent experiments are shown. Expression at time 0 was set as 100%.
We have previously described two cis-acting elements that are involved in post-transcriptional regulation of collagen α1(I) mRNA in hscs, the αCP binding site in its 3′-UTR (8) and the stem–loop structure in its 5′-UTR (9). To demonstrate a role for the stem–loop in the down-regulation of collagen α1(I) mRNA in cells grown in gel, we infected Swiss 3T3 fibroblasts with adenoviruses expressing reporter genes and subsequently cultured these cells on plastic or in gel for 3 days. The reporter genes used in this experiment, WT/LUC and MUT/LUC, are driven by the SV40 promoter and contain the luciferase open reading frame and 3′ flanking sequence (9) (Fig. 3A). The WT/LUC gene has the wild-type collagen α1(I) stem–loop structure in its 5′-region, while the MUT/LUC gene has a mutation of the stem–loop. Figure 3B shows a representative RNase protection analysis of the expression of these genes. This experiment was repeated three times with similar results. The WT/LUC reporter gene was expressed at a much lower level than the MUT/LUC gene (compare lanes 1 and 2 to lanes 3 and 4), as described previously (9). The WT/LUC gene was expressed at a similar level in cells grown in gel (lane 2) as in cells grown on plastic (lane 1). The MUT/LUC gene was expressed at 2- to 4-fold higher levels in cells grown in gel (lane 4) than in cells grown on plastic (lane 3). This increase probably reflects an increased activity of the viral promoter, since when we infected the cells with virus expressing β-galactosidase under control of the CMV promoter, the β-galactosidase activity was found to be higher in cells grown in gel than in cells grown on plastic (not shown). Clearly, the presence of the 5′ stem–loop can abrogate the increase in expression in gel, and we conclude that the collagen stem–loop structure can down-regulate a heterologous mRNA in cells grown in the matrix.
Figure 3.
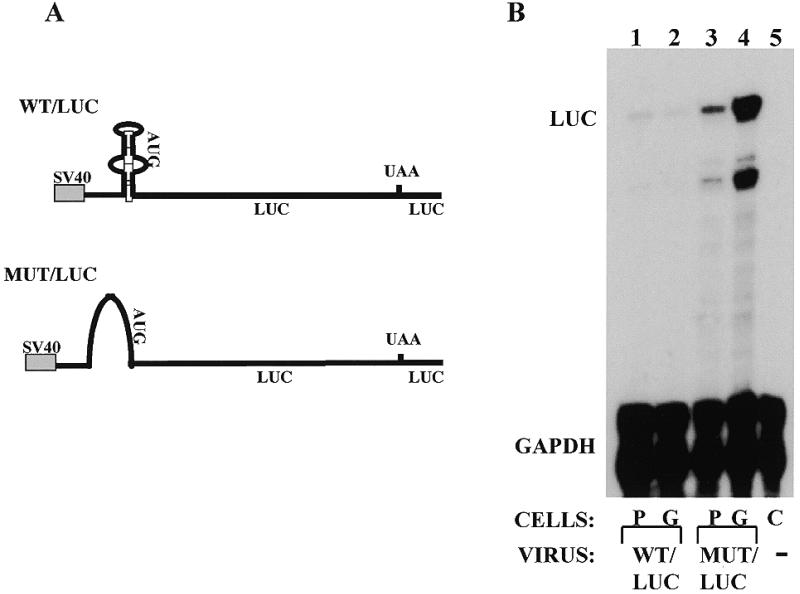
Expression of reporter mRNAs in Swiss 3T3 fibroblasts grown on plastic or in gel. (A) Constructs delivered by adenovirus-mediated gene transfer. Both genes have been described before (9) and are driven by the SV40 promoter. The WT/LUC gene has 79 nt of the mouse collagen α1(I) mRNA 5′-UTR including the stem–loop fused in-frame with the luciferase gene. The MUT/LUC gene is identical exept that it has a substitution of 23 nt in the stem–loop. The 3′-UTR is supplied by the luciferase vector. (B) RNase protection assay showing expression of the reporter genes in cells grown on plastic (P) or in gel (G). The WT/LUC (lanes 1and 2) and MUT/LUC genes (lanes 3 and 4) were expressed in Swiss 3T3 fibroblasts by adenoviral infection, the cells were divided and plated on plastic (lanes 1 and 3) or in gel (lanes 2 and 4) and expression analyzed after 3 days by RNase protection assay. Riboprobes specific for the test gene (LUC) and internal standard gene (GAPDH) were simultaneously hybridized and the protected bands are indicated. Lane 5 is non-infected control cells.
To provide insight into the mechanism by which the stem–loop decreases steady-state levels of mRNA and to show that it has the same effect in the authentic collagen mRNA, we constructed two hybrid mouse–human collagen genes (Fig. 4A). These genes are driven by 220 nt of the mouse collagen α1(I) promoter, followed by 120 nt of mouse 5′-UTR sequence with either the wild-type or a mutated stem–loop, ligated to the full-size human collagen cDNA and its 3′ flanking region (genes mWT/hCOLL and mMUT/hCOLL). These genes synthesize mRNAs with the collagen α1(I) sequence, with or without a stem–loop structure, and their expression can be distinguished from the endogenous mouse α1(I) gene by a sensitive RNase protection assay using a human-specific riboprobe. Thus, the role of the stem–loop can be analyzed in its natural RNA context. These genes were transiently transfected into NIH 3T3 cells, since the transfection efficiency into Swiss 3T3 cells was too low to allow such an analysis. After transfection, the cells were seeded into the collagen gel for 3 days and the half-life of the mRNAs was estimated (Fig. 4B). Comparison of lanes 1 and 4 shows the specificity of the RNase protection assay using a riboprobe specific for the human sequences of the transgenes. Bands representing mWT/hCOLL mRNA are present in transfected cells (COLL, lanes 1–3). There was no cross-reactivity with the endogenous mouse α1(I) mRNA in non-transfected cells (lane 4). A specific band of a slightly higher electrophoretic mobility was seen with mMUT/hCOLL mRNA using the same probe (lanes 5–7), presumably resulting from slightly extended hybridization of the probe to the mutated sequence. Again, the probe was specific for the transgene (compare lanes 5–7 to lane 8). The mWT/hCOLL mRNA was less stable, with a half-life of ~6 h (lanes 1–3), than the mMUT/hCOLL mRNA, which showed no decay after 6 h (lanes 5–7). This clearly demonstrates that the stem–loop is required for accelerated decay of collagen α1(I) mRNA in gel. In this experiment there was no correction for transfection efficiency of the two constructs, so the steady-state level of the mRNAs at time 0 may not accurately represent their accumulation. A different riboprobe for GAPDH was used in this experiment than in the experiments shown in Figures 1 and 3, which produces a shorter protected band, so that the positions of the bands appear slightly different. GAPDH mRNA is a stable RNA and showed no decay within 6 h, as described previously (8).
Figure 4.
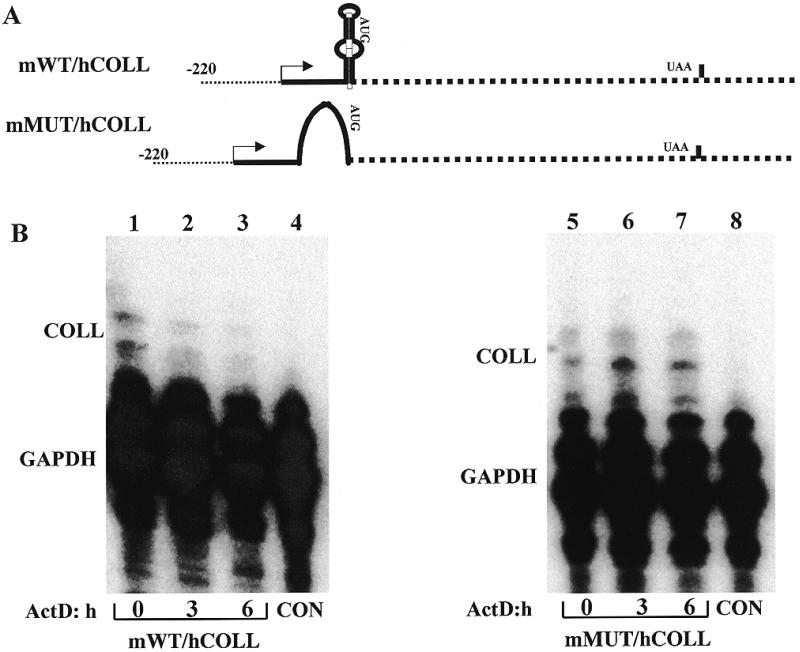
The 5′ stem–loop destabilizes collagen α1(I) mRNA. (A) Hybrid mouse–human collagen α1(I) genes. The genes are driven by the mouse collagen α1(I) promoter and contain the mouse 5′-UTR with either the wild-type or mutated stem–loop, ligated to the human collagen α1(I) cDNA and 3′-UTR (genes mWT/hCOLL and mMUT/hCOLL, respectively). The promoter is shown as a dotted line, the mouse sequence as a full line and the human sequence as a dashed line. The transcription start site is indicated by an arrow. The genes contain only the first polyadenylation signal of the human gene. (B) Stability of chimeric mouse–human collagen α1(I) mRNAs with and without the 5′ stem–loop. The genes in (A) were transiently transfected into NIH 3T3 fibroblasts, followed by culturing the cells in gel for 3 days. Actinomycin D was added and after the indicated times total RNA was extracted. RNase protection assays using a riboprobe specific for human α1(I) sequence (COLL) and internal control gene (GAPDH) were performed. Lanes 1–3, stability of mWT/hCOLL mRNA; lanes 5–7, stability of mMUT/hCOLL mRNA. Lanes 4 and 8 are control non-transfected cells.
The collagen α1(I) stem–loop binds cytosolic proteins in a cap-dependent manner in activated hscs and fibroblasts (9). This binding abrogates the negative effect of the stem–loop and allows for mRNA accumulation. Therefore, we compared the stem–loop binding activity in extracts of Swiss 3T3 fibroblasts cultured on plastic and in gel by incubating the extracts with 7mG capped 5′ stem–loop RNA (5′SL) or capped RNA with an inverted stem–loop (INV), which served as a control. The RNA–protein complexes were resolved on a native acrylamide gel (Fig. 5A). The specific stem–loop binding activity was detected in extracts of cells grown on plastic (lane 2), as before (9). A non-specific complex was also formed in this extract. The 5′ stem–loop specific binding activity was greatly reduced in extracts of cells grown in gel (lane 3). As expected, the INV RNA bound only cap binding proteins in both extracts (lanes 5 and 6), as well as the non-specific complex from the plate extract. Formation of this non-specific complex was associated with a reduction in the fast migrating cap binding complex CBP (lane 5), so it may represent a higher order cap binding complex (37). Taking this into account, the cap binding proteins seem to be equally abundant in the two extracts, and can serve as a control for loading.
Figure 5.
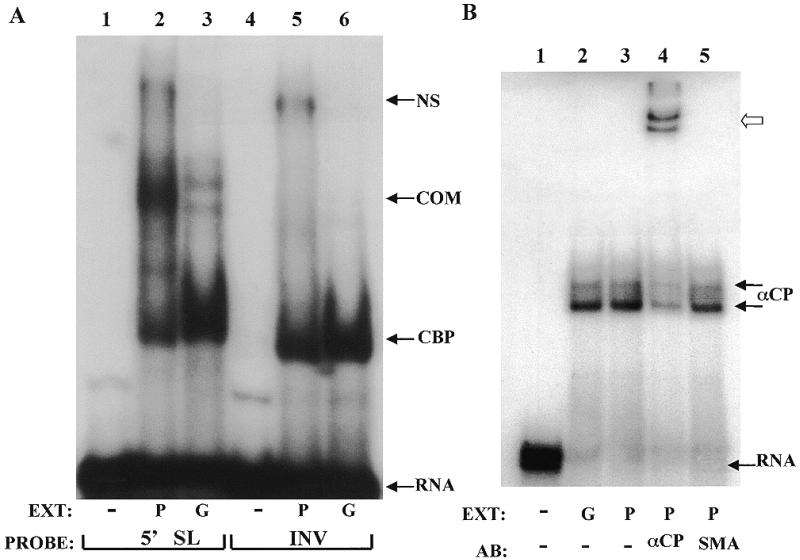
Protein binding to the collagen α1(I) mRNA in cytoplasmic extracts from cells grown on plastic or in gel. (A) Binding to the 5′ stem–loop. Swiss 3T3 fibroblasts were cultured on plastic (P) or in gel (G) for 3 days and cytoplasmic extracts were prepared. Aliquots of 60 µg of the extracts were incubated with radiolabeled 7mG capped 5′SL RNA (lanes 2 and 3) or 7mG capped INV RNA (lanes 5 and 6) and resolved on native acrylamide gels. Lanes 1 and 4 are probes alone. Migration of the free RNA (RNA), cap binding complex (CBP) and specific complex (COM) are indicated. NS indicates a non-specific complex. (B) αCP binding to the mouse collagen α1(I) 3′-UTR. Aliquots of 10 µg of the same extracts as in (A) were incubated with a 33 nt probe containing the αCP binding site (8) and complexes were resolved on a native acrylamide gel. Lane 1, no extract; lane 2, extract from cells cultured in gel (G); lanes 3–5, extract from cells cultured on plastic (P). In lane 4 an antibody to αCP was added and in lane 5 a control antibody (SMA) was added. Migration of the RNA–αCP complexes (double arrow, αCP) and supershifted complexes (open arrow) as well as free probe (RNA) are indicated.
In our previous work we found that αCP, a protein involved in stabilization of α-globin (21,22) and tyrosine hydroxylase mRNAs (24), binds to the 3′-UTR of collagen α1(I) mRNA and proposed its role in stabilization of this mRNA (8). Therefore, we performed a gel mobility shift experiment with the same extracts, but used the αCP binding sequence as a probe (Fig. 5B). There was no difference in binding activity of αCP in cells grown on plastic or in gel (lanes 2 and 3). The two complexes seen in this experiment probably represent alternatively spliced isoforms or different modifications of αCP. Addition of polyclonal antibody specific to both isoforms of αCP (22) (a kind gift of S. Liebhaber) resulted in a supershift of the complexes (lane 4, open arrow), while addition of an antibody to smooth muscle actin (SMA) had no effect (lane 5). We conclude from this experiment that culture of mouse fibroblasts in gel decreases binding activity to the collagen 5′ stem–loop. This is associated with a decreased stability of this mRNA and a reduced steady-state level. No change in the binding activity of αCP was observed.
After synthesis in the nucleus, some mRNAs associate with sequence-specific RNA binding proteins, which have important regulatory roles (38,39). We wanted to determine if a nuclear protein binds the collagen 5′ stem–loop in a sequence-specific manner. We prepared nuclear extract from Swiss 3T3 fibroblasts grown on plastic and incubated the extract with 5′SL RNA as a probe. This 5′SL RNA probe did not contain 7mG cap as in the experiments with cytoplasmic extracts. As shown in Figure 6A, lane 2, an RNA–protein complex was formed. This complex was efficiently competed out with an excess of the same unlabeled RNA (lanes 3 and 4). It was not significantly effected by an excess of INV RNA, which served as a non-specific competitor (lanes 5 and 6). This result suggests that there is a nuclear protein which recognizes the collagen α1(I) mRNA 5′ stem–loop, and that binding of this protein does not require a 7mG cap on the RNA. This feature makes it clearly distinct from the cytoplasmic stem–loop binding protein described in Figure 5 and Stefanovic et al. (9). Next, we made nuclear extracts from cells grown in gel and on plastic and compared their binding activities. The nuclear extract made from Swiss 3T3 cells grown in gel contained more of the stem–loop binding activity than the extract from cells grown on plastic (Fig. 6B, compare lanes 2 and 3). Therefore, culturing cells in the collagen matrix regulates the nuclear 5′ stem–loop binding activity differently than the cytoplasmic binding activity.
Figure 6.
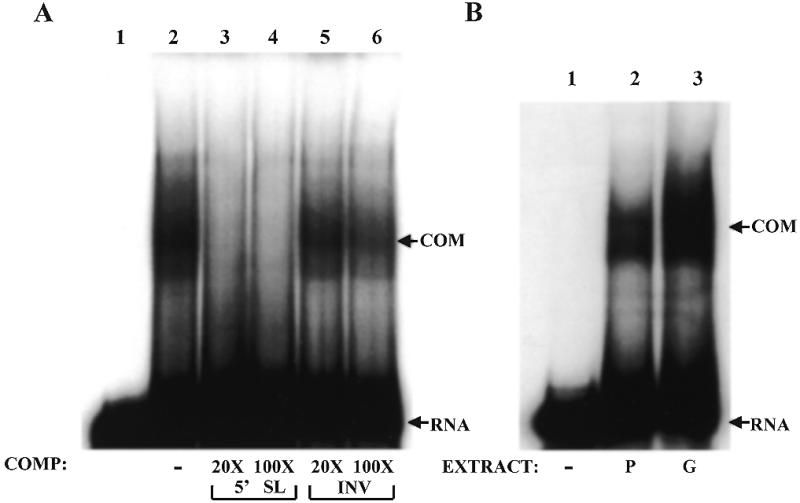
Nuclear 5′ stem–loop binding activity. (A) Sequence-specific binding to the 5′SL RNA in nuclear extracts of Swiss 3T3 fibroblasts. Aliquots of 60 µg of nuclear extract were incubated with radiolabeled 5′SL RNA (no cap) (lanes 2–6) and no competitor RNA was added (lane 2) or the indicated molar excess of specific competitor RNA (5′SL, lanes 3 and 4) and non-specific competitor RNA (INV, lanes 5 and 6) was added. Complexes were resolved on native acrylamide gels. Lane 1 is probe alone. Migration of free RNA (RNA) and RNA–protein complexes (COM) is indicated. (B) Nuclear binding activity is increased in cells grown in gel. Nuclear extracts were prepared from Swiss 3T3 cells grown on plastic (P) or in gel (G) for 3 days and RNA binding assays were performed as in (A) using 5′SL RNA as probe. Lane 1 is probe alone. Migration of the specific complex is indicated (COM).
DISCUSSION
Expression of collagen α1(I) gene is a complex process involving both transcriptional and post-transcriptional mechanisms (6–10,19,20). We have previously reported the importance of post-transcriptional regulation of this gene in cells which differentiate from the quiescent phenotype (quiescent hscs) to the activated phenotype (activated hscs), as well as the importance of the 5′ stem–loop structure in this process (8,9). In this paper we demonstrate a role of the 5′ stem–loop in the opposite process, in cells which revert to a more quiescent phenotype after being cultured within a collagen type I matrix. A change in cell morphology upon culture within a three-dimensional matrix has been reported for primary human fibroblasts (33). This was associated with a decreased steady-state level of collagen α1(I) mRNA and its increased turnover (9,19,20). Here we show that rodent fibroblast cell lines exhibit a similar property; a morphological change to a more elongated form and a 50–80% decrease in collagen α1(I) mRNA accumulation (Fig. 1). The decreased mRNA level was due to selective destabilization of this mRNA (Fig. 2). Collagen α1(I) mRNA is a stable mRNA in rodent fibroblasts with a reported half-life ranging from 8 to >18 h, depending on cell type, culture conditions and method of measurement (6,10,40,41). We reported a half-life of this mRNA in activated hscs of ~24 h (8). In Swiss 3T3 fibroblasts grown on plastic there was no decay 12 h after addition of actinomycin D, while in cells grown in gel the half-life was ~12 h (Fig. 2). Prolonged exposure to actinomycin D proved to be toxic, so we could not quantitate the mRNA destabilization in these cells. Experiments with DRB also did not result in any measurable decay of α1(I) mRNA in cells grown on plastic. Nevertheless, it is clear that in rodent fibroblasts the turnover of collagen α1(I) mRNA is significantly modulated by the culture conditions employed. For primary human fibroblasts the half-life of collagen α1(I) mRNA was decreased from 4.4 to 2.1 h in cells grown on plastic and in gel, respectively (19).
Two cis-acting elements in collagen α1(I) mRNA regulate its metabolism; the 5′ stem–loop (9) and the αCP binding site in the 3′-UTR (8). The 5′ stem–loop is a negative regulatory element, which prevents expression of reporter genes in quiescent hscs. In activated hscs a cytosolic protein factor(s) of unknown identity binds to the stem–loop and requires a 7mG cap for binding. This binding activity parallels mRNA expression (9). In Swiss 3T3 fibroblasts grown in gel the cytoplasmic 5′ stem–loop binding activity was greatly reduced (Fig. 5). Concomitantly, the expression of endogenous collagen α1(I) mRNA (Fig. 1) was down-regulated. These results strongly suggest that the 5′ stem–loop binding activity is required for high level expression in this system and is in agreement with the results using hscs. A reporter mRNA with a mutated stem–loop synthesized under the control of the SV40 promoter was expressed at a 2- to 4-fold higher level in cells grown in gel than on plastic (Fig. 3, lanes 3 and 4). Since experiments with a β-galactosidase reporter gene utilizing a CMV promoter also gave increased expression in cells grown in gel (not shown), it is likely that the viral promoters are stimulated in cells grown in matrix. Such stimulation was, however, completely abrogated by placing the 5′ stem–loop in the reporter mRNA (Fig. 3, lanes 1 and 2).
To provide further evidence for the role of the 5′ stem–loop, we constructed hybrid mouse–human collagen α1(I) genes (Fig. 4). These genes allow synthesis of the full-size collagen α1(I) mRNA and analysis of the effects of the stem–loop mutation in its natural context. However, these genes contain only the first polyadenylation signal of the collagen α1(I) gene. Since collagen α1(I) mRNA exists as two species, 4.7 and 5.7 kb mRNA (42), the results obtained may not pertain to regulation of the 5.7 kb collagen α1(I) mRNA. We analyzed the stability of this hybrid mouse–human collagen α1(I) mRNA in NIH 3T3 cells grown in gel. When the 5′ stem–loop was mutated, the hybrid mouse–human collagen α1(I) mRNA was more stable than when this stem–loop was intact, suggesting that in its natural context the 5′ stem–loop is required for decay of collagen α1(I) mRNA (Fig. 4). The half-life of the hybrid mouse–human collagen α1(I) mRNA was ~6 h, which is shorter than that observed for endogenous collagen α1(I) mRNA in Swiss 3T3 cells (12 h; Fig. 2). As mentioned above, this may be due to different decay rates of the 4.7 and 5.7 kb collagen α1(I) mRNAs (6), which are both measured as endogenous collagen α1(I) mRNA, while only the 4.7 kb mRNA is represented by the hybrid mRNA. The difference may also result from the different cell type used, NIH 3T3 versus Swiss 3T3 fibroblasts, or be due to the transient transfection procedures.
The mechanism by which the stem–loop accelerates the decay of collagen α1(I) mRNA is not clear. Cytoplasmic factors which bind this structure seem to reverse its destabilizing effect. Here we have also described a nuclear protein that binds the stem–loop in a sequence-specific manner (Fig. 6). This activity seems to be different from the cytoplasmic activity. It can be detected only in nuclear extracts, it does not require the presence of a 7mG cap for binding in vitro and it has a different electrophoretic mobility in native gels. However, some subunits participating in this binding activity may be common in cytoplasmic and nuclear complexes. The nuclear binding inversely correlates with accumulation of collagen α1(I) mRNA (Fig. 6B), suggesting that it is a negative modulator of collagen α1(I) mRNA expression. It is possible that after synthesis, the collagen 5′ stem–loop binds this nuclear factor, which may negatively affect transcriptional elongation (43,44), splicing or nuclear export. Upon export into the cytoplasm, the 5′ stem–loop is recognized by the cytoplasmic binding protein(s). If this cytoplasmic protein(s) is absent, as in quiescent hscs (9), or reduced, as in fibroblasts grown in gel, α1(I) mRNA may be targeted for degradation. If it is present, the α1(I) mRNA may be stabilized and directed for translation, as in activated hscs (9) and fibroblasts grown on plastic. Collagen type I is a heterotrimer composed of two α1(I) chains and one α2(I) chain (1). Therefore, its assembly in endoplasmic reticulum would be more efficient, due to a higher local concentration of the chains, if both mRNAs are translated on a subset of ribosomes closely positioned on the rough endoplasmic reticulum. Cytoplasmic stem–loop binding activity may target collagen α1(I) mRNA to such a subset.
Binding of αCP to the 3′-UTR of collagen α1(I) mRNA correlates with stabilization of this mRNA in hscs (8). In this work there was no difference in αCP binding activity in cells grown on plastic or in gel (Fig. 5B). Perhaps, the difference in the culture conditions described here is not a sufficiently strong stimulus to change binding of this factor, as is differentiation of hscs (8). However, a role of αCP in stimulating translation of some viral mRNAs has been documented (45,46) and preliminary results from our laboratory using the yeast two-hybrid system suggest that it can interact with poly(A) binding protein, a known stimulator of translation (47,48). Therefore, it is possible that αCP further enhances translation of collagen α1(I) mRNA, and this is currently under investigation.
Based on the results presented in this paper it is likely that collagen α1(I) mRNA is regulated by a complex interaction with sequence-specific RNA binding proteins in both the nucleus and the cytoplasm. Modulation of these interactions is associated with changes in mRNA stability and accumulation.
REFERENCES
- 1.Kivirikko K.I. (1998) Matrix Biol., 16, 355–356. [DOI] [PubMed] [Google Scholar]
- 2.Slack J.L., Liska,D.J. and Bornstein,P. (1993) Am. J. Med. Genet., 45, 140–151. [DOI] [PubMed] [Google Scholar]
- 3.Vuorio E. and de Crombrugghe,B. (1990) Annu. Rev. Biochem., 59, 837–872. [DOI] [PubMed] [Google Scholar]
- 4.Focht R.J. and Adams,S.L. (1984) Mol. Cell. Biol., 4, 1843–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Panduro A., Shalaby,F., Biempica,L. and Shafritz,D.A. (1988) Hepatology, 8, 259–266. [DOI] [PubMed] [Google Scholar]
- 6.Pentinnen R.P., Kobayashi,S. and Bornstein,P. (1988) Proc. Natl Acad. Sci. USA, 85, 1105–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raghow R., Postlethwaite,A.E., Keski-Oja,J., Moses,H.L. and Kang,A.H. (1987) J. Clin. Invest., 79, 1285–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stefanovic B., Hellerbrand,C., Holcik,M., Briendl,M., Liebhaber,S.A. and Brenner,D.A. (1997) Mol. Cell. Biol., 17, 5201–5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stefanovic B., Hellerbrand,C. and Brenner,D.A. (1999) Mol. Cell. Biol., 19, 4334–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhawan J., Lichtler,A., Rowe,D. and Farmer,S.R. (1991) J. Biol. Chem., 266, 8470–8475. [PubMed] [Google Scholar]
- 11.Nehls M.C., Rippe,R.A., Veloz,L. and Brenner,D.A. (1991) Mol. Cell. Biol., 11, 4065–4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nehls M.C., Grapilon,M.L. and Brenner,D.A. (1992) Mol. Cell. Biol., 11, 443–452. [Google Scholar]
- 13.Rippe R.A., Almounajed,G. and Brenner,D.A. (1995) Gastroenterology, 22, 241–251. [Google Scholar]
- 14.Slack J.L., Liska,D.J. and Bornstein,P. (1991) Mol. Cell. Biol., 11, 2066–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Houglum K., Buck,M., Alcorn,J., Contreras,S., Bornstein,P. and Chojkier,M. (1995) J. Clin. Invest., 96, 2269–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sokolov B.P., Ala-Kokko,L., Dhulipala,R., Arita,M., Khillan,J.S. and Prockop,D.J. (1995) J. Biol. Chem., 270, 9622–9629. [DOI] [PubMed] [Google Scholar]
- 17.Rossert J., Eberspaecher,H. and de Crombrugghe,B. (1995) J. Cell Biol., 129, 1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salimi-Tari P., Cheung,M., Safar,C., Tracy,J., Tran,I., Harbers,K. and Breindl,M. (1997) Genes Dev., 198, 61–72. [DOI] [PubMed] [Google Scholar]
- 19.Eckes B., Mauch,C., Huppe,G. and Krieg,T. (1996) FEBS Lett., 318, 129–133. [DOI] [PubMed] [Google Scholar]
- 20.Eckes B., Mauch,C., Huppe,G. and Krieg,T. (1996) Biochem. J., 315, 549–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X., Kiledjian,M., Weiss,I.M. and Liebhaber,S.A. (1995) Mol. Cell. Biol., 15, 1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kiledjian M., Wang,X. and Liebhaber,S.A. (1995) EMBO J., 14, 4357–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holcik M. and Liebhaber,S.A. (1997) Proc. Natl Acad. Sci. USA, 94, 2410–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paulding W. and Czyzyk-Kyzeska,M. (1999) J. Biol. Chem., 274, 2532–38. [DOI] [PubMed] [Google Scholar]
- 25.Yamada Y., Mudryj,M. and de Crombrugghe,B. (1983) J. Biol. Chem., 258, 14914–14919. [PubMed] [Google Scholar]
- 26.Nakatsukasa H., Nagy,P., Evarts,R.P., Chu-Chieh,H., Marsden,E. and Thorgeirsson,S.S. (1990) J. Clin. Invest., 85, 1833–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masuda H., Fukumoto,M., Hirayoshi,K. and Nagata,K. (1994) J. Clin. Invest., 94, 2481–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Milani S., Herbst,H., Schuppan,D., Kim,K.Y., Riecken,E.O. and Stein,H. (1990) Gastroenterology, 98, 175–184. [DOI] [PubMed] [Google Scholar]
- 29.Schupan D. (1990) Semin. Liver Dis., 10, 1–10. [DOI] [PubMed] [Google Scholar]
- 30.Maher J.J., Zia,S. and Tzagarakis,C. (1994) Alcohol Clin. Exp. Res., 18, 403–409. [DOI] [PubMed] [Google Scholar]
- 31.Contard P., Jacobs,L.I., Perlish,J. and Fleischmajer,R. (1993) Cell Tissue Res., 273, 571–575. [DOI] [PubMed] [Google Scholar]
- 32.Sato M., Ishikawa,O. and Miyachi,Y. (1998) Br. J. Dermatol., 138, 938–943. [DOI] [PubMed] [Google Scholar]
- 33.Mauch C., Hatamochi,A., Scharffetter,K. and Krieg,T. (1988) Exp. Cell Res., 178, 493–503. [DOI] [PubMed] [Google Scholar]
- 34.Ishikawa O., Kondo,A., Okada,K., Miyachi,Y. and Furumura,M. (1997) Br. J. Dermatol., 136, 6–11. [PubMed] [Google Scholar]
- 35.Friedman S.L., Roll,F.J., Boyles,J., Arenson,D.M. and Bissell,D.M. (1989) Proc. Natl Acad. Sci. USA, 264, 10756–10762. [PubMed] [Google Scholar]
- 36.Dignam J.D., Lebovitz,R.M. and Roeder,R.G. (1983) Nucleic Acids Res., 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haghighat A. and Sonenberg,N. (1997) J. Biol. Chem., 272, 21677–21680. [DOI] [PubMed] [Google Scholar]
- 38.Wang Z., Whitfield,M., Ingledue,T.I., Dominski,Z. and Marzluff,W.F. (1996) Genes Dev., 10, 3028–3040. [DOI] [PubMed] [Google Scholar]
- 39.Kao S., Calman,A., Luciw,P.A. and Peterlin,B. (1987) Nature, 330, 489–493. [DOI] [PubMed] [Google Scholar]
- 40.Hamalainen L., Oikarinen,J. and Kivirikko,K.I. (1985) J. Biol. Chem., 260, 720–725. [PubMed] [Google Scholar]
- 41.Slack J.L., Parker,M.I., Robinson,V.R. and Bornstein,P. (1992) Mol. Cell. Biol., 12, 4714–4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mooslehner K. and Harbers,K. (1988) Nucleic Acids Res., 16, 773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krumm A., Meulia,T. and Groudine,M. (1993) Bioessays, 15, 659–665. [DOI] [PubMed] [Google Scholar]
- 44.Krumm A., Hickey,L.B. and Groudine,M. (1995) Genes Dev., 9, 559–572. [DOI] [PubMed] [Google Scholar]
- 45.Niepmann M., Petersen,A., Meyer,K. and Beck,E. (1997) J. Virol., 71, 8330–8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gamarnik A.V. and Andino,R. (1997) RNA, 3, 882–892. [PMC free article] [PubMed] [Google Scholar]
- 47.Gallis B. (1991) Genes Dev., 5, 2108–2116. [DOI] [PubMed] [Google Scholar]
- 48.Coller J., Gray,N.K. and Wickens,M. (1998) Genes Dev., 12, 3226–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]