


Abstract
Mutations in the chromosome 8p WRN gene cause Werner syndrome (WRN), a human autosomal recessive disease that mimics premature aging and is associated with genetic instability and an increased risk of cancer. All of the WRN mutations identified in WRN patients are predicted to truncate the WRN protein with loss of a C-terminal nuclear localization signal. However, many of these truncated proteins would retain WRN helicase and/or nuclease functional domains. We have used a combination of immune blot and immune precipitation assays to quantify WRN protein and its associated 3′→5′ helicase activity in genetically characterized WRN patient cell lines. None of the cell lines from patients harboring four different WRN mutations contained detectable WRN protein or immune-precipitable WRN helicase activity. Cell lines from WRN heterozygous individuals contained reduced amounts of both WRN protein and helicase activity. Quantitative immune blot analyses indicate that both lymphoblastoid cell lines and fibroblasts contain ~6 × 104 WRN molecules/cell. Our results indicate that most WRN mutations result in functionally equivalent null alleles, that WRN heterozygote effects may result from haploinsufficiency and that successful modeling of WRN pathogenesis in the mouse or in other model systems will require the use of WRN mutations that eliminate WRN protein expression.
INTRODUCTION
Werner syndrome (WRN; MIM #277700) is an uncommon autosomal recessive disease that results from mutational inactivation of the human RecQ family helicase encoded by the chromosome 8p WRN gene (1,2). The Werner phenotype resembles premature aging: after puberty, patients rapidly develop premature graying and loss of hair, scleroderma-like skin changes, osteoporosis, atherosclerosis, bilateral cataract formation, diabetes mellitus and hypogonadism (3,4). Spontaneous genetic instability has been identified in Werner patients, and may be an important determinant of the increased cancer risk in patients (reviewed in 2,5). This increased cancer risk is largely restricted to five tumor types: soft tissue sarcomas, osteosarcoma, thyroid carcinoma, acral lentigenous melanoma and meningioma (3,6). Most Werner patients die prematurely of cancer or cardiovascular disease, with an average age at death of 47 years (4).
The WRN gene, located at 8p11-12, encodes a human RecQ family helicase that has both 3′ → 5′ helicase and exonuclease activities (7–10). WRN is one of five human RecQ helicases, and one of three that has been associated with a heritable human disease. BLM mutations that reduce or eliminate BLM RecQ helicase activity have been identified in Bloom syndrome patients (11), and a subset of patients with Rothmund–Thomson syndrome have truncating mutations in the human RECQL4 gene (12).
All of the mutations thus far identified in WRN patients are predicted to truncate the WRN protein with a loss of up to 1256 amino acid residues, including the C-terminal nuclear localization signal (NLS), from the 1432 residue native protein (reviewed in 2). We have used a combination of immune blot and immune precipitation assays to detect and quantify WRN protein and WRN helicase activity in cell lines from genetically characterized Werner patients, heterozygotes and controls. Cell lines from patients harboring four different WRN mutations contained no detectable WRN protein or WRN helicase activity; heterozygous individuals had reduced levels of both WRN protein and helicase activity. These results have implications for Werner syndrome pathogenesis, the mechanistic basis for WRN heterozygote effects and the modeling of WRN pathogenesis in murine and other model organisms.
MATERIALS AND METHODS
Cell culture
The Epstein–Barr virus (EBV) transformed B-lymphoblastoid cell lines (LCLs) from Werner pedigrees and the primary and SV40-transformed WRN and control fibroblasts used in this study are detailed in Table 1 and have been previously described (13,14). The mouse hybridoma cell line 9E10 that produces a monoclonal antibody directed against the myc epitope tag has been previously described (15). Suspension cells were grown in RPMI 1640 medium, and attached cells in Dulbecco’s modified minimal essential medium (DMEM) containing 4.5 g/l glucose. Both media were supplemented with 10% fetal calf serum (Hyclone), 100 U/ml penicillin G sulfate and 100 µg/ml streptomycin sulfate. All cultures were grown in a humidified, 5% CO2 incubator at 37°C.
Table 1. Cells and cellular WRN content as a function of WRN genotype.
| WRN | Predicted | WRN content | ||
|---|---|---|---|---|
| Cell line/straina | genotype | mutationb | WRN proteinc | (×10–15 g/cell)d |
| LGS90610 |
–/– |
c.1336C>T |
R369X |
not detected |
| LGS90625 |
+/– |
c.1336C>T |
R369X |
5.5 ± 1.6 |
| LGS90635 |
+/– |
c.1336C>T |
R369X |
8.0 ± 0.9 |
|
SYR10011 |
–/– |
4 bp deletion |
1246X |
not detected |
|
SYR10009 |
+/– |
4 bp deletion |
1246X |
4.1 ± 3.0 |
|
SYR10010 |
+/+ |
none |
1432 |
13.9 ± 3.8 |
| TUR90010 |
–/– |
c.3724C>T |
Q1165X |
not detected |
| TUR90060 |
+/– |
c.3724C>T |
Q1165X |
5.4 ± 0.6 |
| TUR90050 |
+/+ |
none |
1432 |
9.2 ± 3.5 |
| ZM90630 |
–/– |
IVS25-1G>C |
1060X |
not detected |
| ZM90634 |
+/– |
IVS25-1G>C |
1060X |
4.8 ± 1.0 |
| ZM90633 |
+/+ |
none |
1432 |
22.8 ± 10.3 |
| AG11395 |
–/– |
c.1336C>T |
R369X |
not detected |
| GM639 |
+/+ |
none |
1432 |
16.0 ± 1.4 |
| HDF88-1 | +/+ | none | 1432 | 16.0 ± 7.1 |
aCell lines/strains: AG11395 (also known as WS780) and GM639 are SV40-transformed fibroblast cell lines; HDF88-1 is a primary human diploid fibroblast cell strain. All other cells are Epstein–Barr virus-transformed B-lymphoblastoid cell lines.
bWRN mutation: mutations in cell lines are given following suggested nomenclature of the Nomenclature Working Group (41), using the WRN cDNA as a numbering reference. Mutations are described in detail at http://www.pathology.washington.edu/werner/ , the WRN Locus-Specific Mutational Database. A print version of the database is also available (2).
cPredicted WRN protein: the number indicates remaining amino acid residues in predicted WRN proteins encoded by mutant alleles, and letters indicate the coding change(s) produced by the indicated mutation in single letter amino acid code with ‘X’ signifying a termination codon. The native WRN protein contains 1432 amino acid residues.
dWRN content: the value shown is the mean and standard deviation of at least three independent determinations per cell line. ‘not detected’ indicates that in comparable assays WRN could not be detected at a sensitivity level of ≥0.1 × 10–15 g/cell.
Plasmids and transfection
A cDNA encoding full-length WRN was inserted in-frame with the N-terminal myc epitope tag of plasmid pCS2+MT (16) to create plasmid pMM229. This plasmid was used as a substrate for oligonucleotide-directed mutagenesis (Transformer system, Clontech) to generate plasmid pMM247 containing a c.3724C>T mutation—and a resulting new Q1165X termination codon—in the WRN open reading frame. Plasmid pMM248, encoding a myc epitope-tagged K577M WRN protein, was generated by replacing an internal fragment of pMM229 with a comparable WRN cDNA fragment containing a c.1961A>T substitution (7). Plasmid DNAs were transfected into the SV40-transformed WRN fibroblast cell line AG11395 (WS780) by using SuperFect (Qiagen).
Antisera and antibody generation
Rabbit polyclonal antisera SAM1 and SAM2 were raised against recombinant tagged WRN protein. The protein expression vector consisted of pRSETB (Invitrogen), into which a WRN cDNA fragment encoding protein residues 722–1432 was inserted in-frame with an N-terminal hexahistidine tag. Recombinant protein expressed in Escherichia coli BL21(DE3) pLysS was purified by Ni2+ affinity chromatography on a BioCad Sprint MC20 column (PerSeptive BioSystems) as previously described (17). The peak fraction, eluting at ~300 mM imidazole, was mixed at a 1:1 ratio with Freund’s complete adjuvant and used to immunize two female NZ White rabbits. A third rabbit polyclonal antiserum, 9130J, was generated against full-length recombinant WRN protein expressed from a baculoviral vector in insect cells as described (10). IgG fractions were prepared from all three antisera by standard methods (18). Mouse hybridoma 9E10 supernatant containing an IgG1 that recognizes the myc epitope tag was adjusted to pH 8 with 1 M Tris–HCl and used without further purification.
Immune blot analyses
Cell pellets, resuspended in SDS sample buffer at 1–2 × 107 cells/ml, were lysed by heating for 3 min at 100°C. Lysate proteins were separated by electrophoresis through 6% polyacrylamide–SDS gels, and then transferred onto PVDF membrane by electroblotting (90 min at 100 V in Tris–glycine buffer containing 20% methanol; 19). Non-specific binding sites on membranes were blocked with TBS-T buffer (25 mM Tris–HCl, pH 7.5, 500 mM NaCl, 0.1% Tween-20) containing 10% non-fat dry milk (NFDM) prior to the addition of SAM1 or SAM2 IgG (1:7500) or 9130J IgG (1:1000) diluted in TBS-T containing 1% NFDM. Bound antibody was detected using a goat anti-rabbit horseradish peroxidase-conjugated antiserum (1:1000 dilution) and enhanced Luminol reagent (NEN Life Sciences). The chemiluminescence signal was recorded on BioMAX MR film (Kodak). WRN protein copy number determinations were performed in triplicate, using extracts from 0.5–1.5 × 105 cells and purified recombinant full-length WRN protein of known concentration as a standard (10). Immune blot films were scanned and converted to TIF files using Photoshop 4.01 (Adobe) prior to quantification using NIH Image (Wayne Rasband, NIH). Experiments in which recombinant WRN protein standards fit a linear equation were used to quantify WRN in cell extracts.
Immune precipitation and helicase activity assays
Frozen cells (~108; wet vol 0.1 ml) were resuspended in 4 vol of ice-cold extraction buffer (20 mM Tris–HCl, pH 8.0, 0.5 M NaCl, 1 mM EDTA, 0.5 mM DTT, 0.5% NP-40, 25% glycerol, 0.2 mM PMSF and 10 µg/ml each of aprotinin, pepstatin and leupeptin), then incubated on ice for 20 min prior to disruption in a teflon-glass homogenizer. The homogenate was centrifuged at 20 000 g for 15 min at 4°C. This procedure reproducibly solubilized 50% of the WRN protein contained in cells. An aliquot of the supernatant (1 mg total protein) was pre-cleared by incubation with a 10% (v/v) suspension of Pansorbin (Staphylococcus aureus cells; CalBiochem) in IP buffer (20 mM Tris–HCl, pH 8.0, 150 mM NaCl, 25% glycerol and 0.5% NP-40, 0.05% sodium deoxycholate and 0.005% SDS) for 30–60 min on ice. Cells were pelleted at 20 000 g for 5 min. The resulting pre-cleared supernatant was incubated with an excess of 9130J WRN antiserum or with pre-immune serum from the same animal for 60 min at 4°C. WRN:IgG complexes were precipitated by the addition of a 10% (v/v) suspension of S.aureus cells and collected by centrifugation as described above. The pellet was washed three times with 0.5 ml of IP buffer and resuspended in 15 µl of 25 mM Tris–HCl buffer, pH 8.0, 0.5 mM EDTA, 1 mM DTT, 0.05% NP-40 and 25% glycerol. The suspension was assayed immediately for helicase activity, detected by the displacement of a 32P-5′-labeled 20mer oligonucleotide from a 20mer/46mer partial DNA duplex as previously described (7). Helicase assays in which activity was compared among patient, heterozygote and control immune precipitates were performed in the linear range of the unwinding assay (<10% of substrate consumed).
RESULTS
WRN polyclonal antisera
The ability of three newly generated rabbit polyclonal antisera to detect WRN was determined by immune blot analyses of cell extracts prepared by transfecting WRN cell line AG11395 with a WRN cDNA expression vector that encoded myc epitope-tagged full-length WRN or a Q1165X truncated WRN protein. The Q1165X protein was generated from a WRN cDNA that contained the identical mutation found in the TUR Werner pedigree (1,2; see Table 1). The IgG fraction of rabbit polyclonal 9130J antiserum, raised against full-length WRN protein, was able to detect both native and Q1165X WRN protein with comparable sensitivity (Fig. 1A, top panel). IgG prepared from the SAM1 and SAM2 antisera, raised against the C-terminal half of WRN, readily detected full-length WRN and, with lower sensitivity, Q1165X WRN (Fig. 1A, bottom panel and additional results not shown). 9E10 monoclonal antibody directed against the myc epitope tag of the recombinant full-length and Q1165X WRN proteins was used to determine the relative abundance of each protein, and thus allow an estimate of the sensitivity of detection of each protein by WRN antisera (Fig. 1B; compare band intensities with 1A). SAM1 IgG was the most sensitive of the three WRN IgG fractions: it was able to detect as little as 40 pg (~2.5 × 108 molecules) of purified recombinant WRN.
Figure 1.
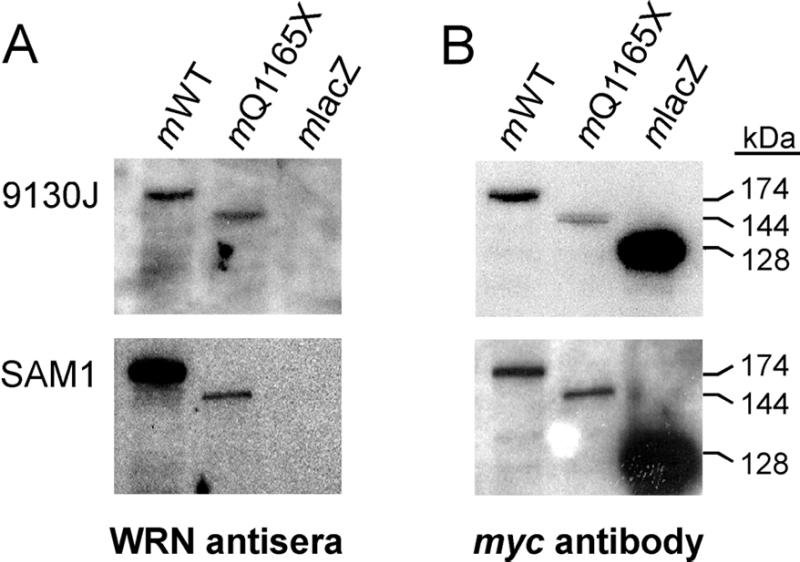
WRN antisera detect full-length and truncated WRN proteins. (A) IgG fractions of two rabbit polyclonal antisera, 9130J (top) raised against full-length WRN, and SAM1 (bottom) raised against the C-terminal half of WRN, were used in immune blot analyses to detect three myc epitope-tagged proteins. Each lane contains equivalent amounts of total cell extract from AG11395 WRN fibroblasts expressing full-length WRN (mWT; 174 kDa), truncated Q1165X WRN (mQ1165X; 144 kDa) or control bacterial β-galactosidase (mlacZ; 128 kDa). The myc epitope comprises 11.7 kDa of each tagged protein. WRN proteins are detected by each antiWRN IgG fraction, though neither detected the β-galactosidase control. (B) Immune blots shown in (A) were dried to inactivate the peroxidase-conjugated secondary antibody, then rehydrated and incubated with 9E10 monoclonal antibody against the myc epitope tag of each protein. This allows the relative abundance of each protein to be determined, and the comparative sensitivity of detection of the antiWRN IgG fractions to be estimated (see Results).
Immune blot analyses
We used the most sensitive of the WRN antisera, SAM1 IgG, in immune blot analyses to search for WRN protein in cell line extracts from Werner patients carrying four different WRN mutations. The lengths of the predicted WRN proteins encoded by these mutant alleles ranged from 368 to 1245 amino acids (Table 1). Three of the four predicted proteins should be readily detected by the SAM1 IgG fraction, as they share epitopes with the C-terminal half of WRN that was used to raise the SAM1 antiserum. However, the SAM1 IgG did not detect the predicted WRN proteins in extracts from any of the Werner patient cell lines we studied (Fig. 2A, lanes 2–5). Immune blot analyses using 9130J IgG raised against full-length WRN also failed to detect the predicted R369X WRN protein in patient cells from the LGS Werner pedigree (Table 1; additional data not shown).
Figure 2.
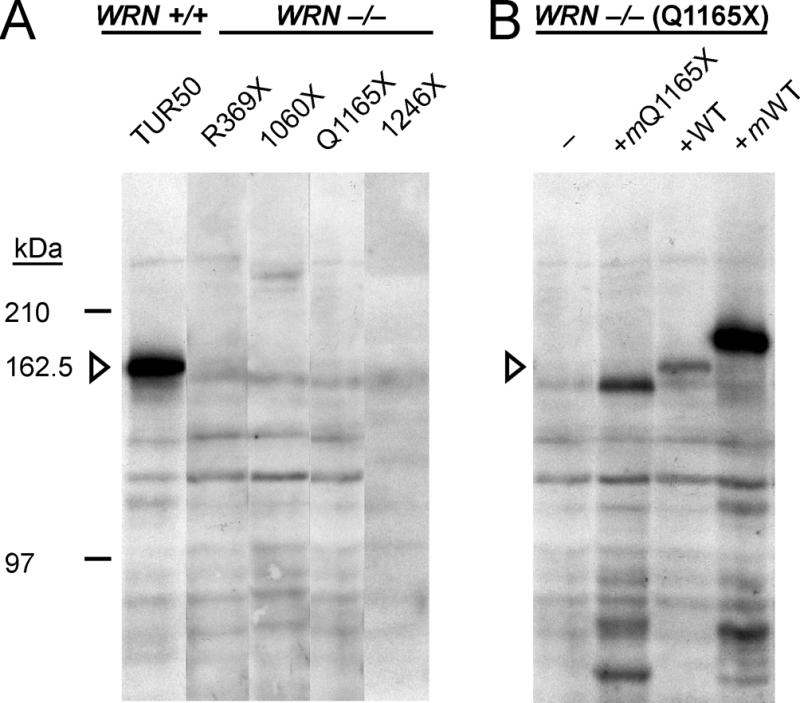
WRN protein is not detected in WRN patient cell lines. (A) Whole cell extracts prepared from 106 control (WRN +/+; TUR90050) cells (lane 1) or from 2 × 106 Werner patient (WRN –/–) cells (lanes 2–5) containing four different WRN mutations (Table 1) were used for immune blot analyses. WRN protein was detected only in the WRN +/+ control (lane 1). Additional cross-reacting bands of lower or higher molecular weight do not appear to be WRN-derived: they are present whether WRN protein is detected or absent, and do not differ between cell lines carrying different WRN mutations. (B) Full-length and truncated WRN proteins have comparable stability in WRN patient cells or cell extracts. Extract from 2 × 106 WRN patient (WRN –/– ;TUR90010) cells lacking detectable Q1165X WRN protein (lane 1) was mixed with extracts from: 105 transfected AG11395 WRN fibroblasts expressing myc epitope-tagged recombinant Q1165X protein (lane 2, +mQ1165X); 105 WRN +/+ cells from a within-pedigree control (TUR90050, lane 3; +WT); or extract from 105 transfected AG11395 WRN fibroblasts expressing myc epitope-tagged recombinant full-length WRN protein (lane 4, +mWT) prior to immune blot analysis. The position of full-length native WRN is indicated by the open arrowhead in both panels. The SAM1 antiWRN IgG fraction was used for both analyses.
We were able to detect full-length WRN protein in reduced amount in cell lines derived from WRN heterozygotes in all four pedigrees. A detection sensitivity of ≥2% was established by determining the number of control (WRN +/+) cells that, when added to 2 × 106 Werner patient cells, allowed the consistent detection of WRN in immune blot analyses. This was 4 × 104 cells, or 2% of the cell number, included in immune blot analyses (Fig. 2B, lanes 1 and 3; additional results not shown). A comparable sensitivity of detection for truncated WRN was established using extracts from AG11395 WRN fibroblasts that had been transiently transfected with plasmids encoding myc epitope-tagged Q1165X or full-length WRN protein (Fig. 2B, lanes 2 and 4; additional results not shown). A majority of the recombinant protein expressed in patient cells consisted of the predicted full-length native or truncated WRN protein (Figs 2B and 4A). Low molecular weight, cross-reacting bands detected in extracts from transfected cells may represent degradation products of full-length or Q1165X WRN protein (Fig. 2B, lowest bands in lanes 2 and 4).
Figure 4.
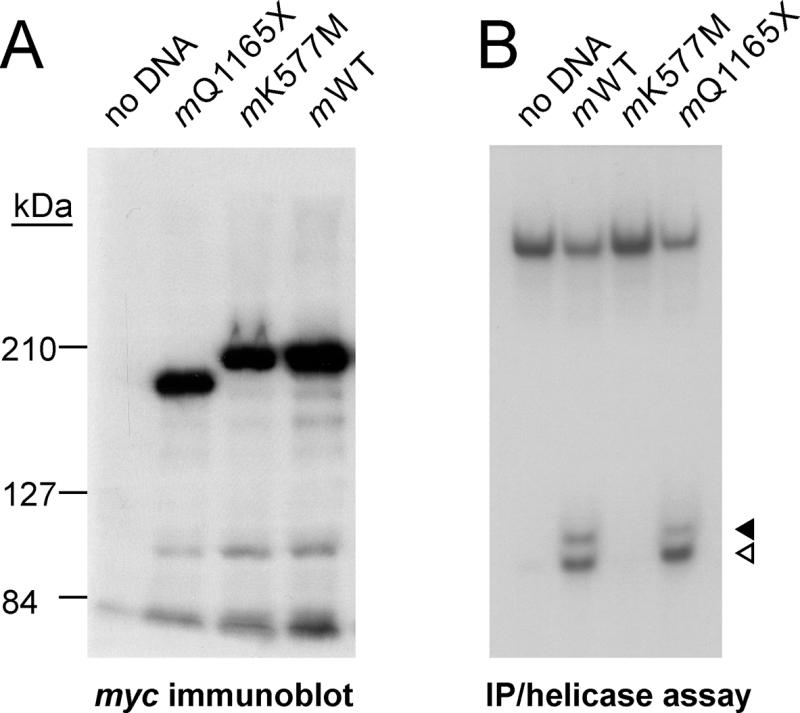
Truncated WRN proteins can be identified and distinguished by immune blot and immune precipitation/helicase assays. (A) myc epitope-tagged Q1165X WRN (mQ1165X, lane 2), K577M WRN (mK577M, lane 3) and full-length native WRN (mWT, lane 4) proteins were expressed in the WRN cell line AG11395 by transient transfection, and then immune precipitated from AG11395 cell extracts with 9130J antiWRN antiserum as described in the text. Precipitated proteins were detected in an immune blot analysis by mouse 9E10 hybridoma supernatant containing an IgG that recognizes the myc epitope tag. Lane 1 (no DNA) extract was prepared from mock-transfected AG11395 cells. Comparable results were obtained by immune blot analysis of cell extracts (additional results not shown). (B) Helicase activity assays of immune precipitates of transfected cell extracts containing the WRN proteins shown in (A). The assay was performed as described in Materials and Methods and the legend to Figure 3. Helicase activity was detected in immune precipitates expressing full-length WRN (mWT, lane 2) or Q1165X truncated WRN (mQ1165X, lane 4) protein, but not in immune precipitates from mock-transfected cells (no DNA, lane 1) or cells expressing K577M WRN protein (mK577M, lane 3). The displaced 20mer and degradation products generated by WRN-associated exonuclease activity are indicated by open and closed arrowheads, respectively.
Immune precipitation analyses
We performed helicase activity assays on immune precipitates to detect and quantify WRN helicase activity in cell extracts. In three of the four Werner pedigrees analyzed, mutant WRN alleles are predicted to encode truncated proteins that retain both the WRN helicase and N-terminal nuclease functional domains (ZM, TUR and SYR pedigrees; Table 1). However, we could not detect helicase activity in immune precipitates from any of the four Werner pedigrees studied (Fig. 3). Pre-immune serum did not precipitate detectable activity from WRN-positive extracts, and extracts from heterozygotes having one intact WRN allele had consistently reduced levels of helicase activity when compared with within-pedigree controls (Fig. 3; additional data not shown).
Figure 3.
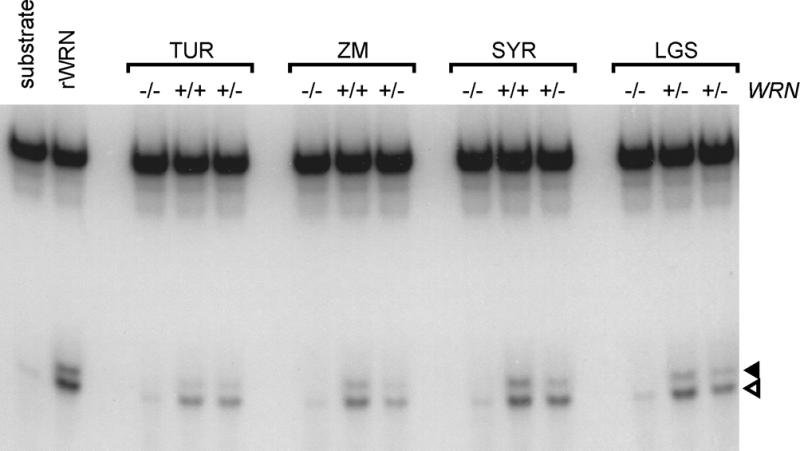
Absence of WRN helicase activity in WRN patient cell lines. WRN protein in cell extracts from WRN patients or within-pedigree controls was precipitated with 9130J WRN antiserum. Immune precipitates were assayed for helicase activity by monitoring the displacement of a 5′-32P end-labeled 20mer from a partial DNA duplex (7). The 20mer displaced by helicase activity was separated on a 12% non-denaturing polyacrylamide gel and visualized by autoradiography. Open and closed arrowheads indicate, respectively, the displaced 20mer and degradation products generated by WRN exonuclease activity. Exonucleolytic degradation products of 19 or fewer bases run slower than the 20mer and are not resolved (10). rWRN, recombinant full-length WRN expressed in insect cells; TUR, ZM, SYR and LGS, Werner pedigrees carrying different WRN mutations; and + and – symbols refer to WRN alleles in cell lines used from each pedigree (Table 1). LGS pedigree heterozygotes were LGS90625 (LGS lane 2) and LGS90635 (LGS lane 3; Table 1).
Insect cell extracts over-expressing WRN (7) and AG11395 WRN cell extracts containing overexpressed myc epitope-tagged full-length, K577M missense or Q1165X truncated WRN proteins were used to demonstrate the ability of immune precipitation to detect the helicase activity of different length WRN proteins that retain an active helicase domain. The expression and subsequent immune precipitation of epitope-tagged WRN proteins from WRN cell line AG11395 was verified by immune blot analyses (for example Fig. 4A). These analyses indicated that each of the tagged proteins could be synthesized in a WRN cell line that lacked detectable WRN protein, and that a majority (>90%) of each synthesized protein was of predicted length. We were able to detect helicase activity in immune precipitates from extracts containing full-length or Q1165X WRN (Fig. 4B, lanes 2 and 4). In contrast, K577M WRN protein could be immune-precipitated, though lacked detectable helicase activity (Fig. 4B, compare lanes 2 and 3). The K577M WRN protein has a methionine substituted for the catalytically important lysine at residue 577 in the WRN helicase consensus domain I/Walker A box. This substitution has been previously shown to inactivate WRN helicase activity (7,10).
Cellular WRN content
We used quantitative immune blotting versus a purified recombinant WRN protein standard to determine the number of WRN copies in different patient and control cell extracts (Fig. 5). Control LCLs from different Werner pedigrees contained 9.2–22.8 × 10–15 g of WRN protein/cell (Table 1). This corresponds to 3.4–8.4 × 104 WRN molecules/cell. The variation in WRN content/cell was <3-fold between unrelated individuals, and heterozygous cell lines contained reduced amounts of WRN protein as compared with within-pedigree controls (Table 1). There was a 50% reduction in protein level in cell lines containing stop codons within a WRN exon (LGS and TUR; Table 1), and a reduction of >50% in cell lines containing mutations that altered splice sites (ZM and SYR; Table 1). The WRN content of SV40-transformed control fibroblast cell line GM639 and the primary human diploid fibroblast strain HDF88-1, ~16 × 10–15 g/cell, was comparable to that observed in control LCLs (Table 1).
Figure 5.
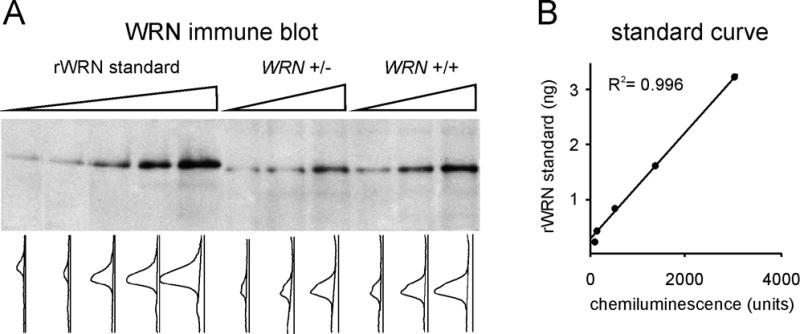
WRN protein copy number determination by quantitative immune blot analysis. (A) Recombinant WRN standard (lanes 1–5; range 0.2–3.2 ng) and whole cell extract prepared from 0.5, 1.0 and 1.5 × 105 cells from a WRN heterozygote (WRN +/– TUR90060; lanes 6–8) and a within-pedigree control (WRN +/+ TUR90050; lanes 9–11) were used for immune blot analysis with the SAM1 WRN IgG. Gels were scanned to convert the chemiluminescence signal in each lane into an intensity curve. (B) The area under each lane intensity curve was integrated, and these values were used to generate a standard curve relating chemiluminescence intensity (in arbitrary units) to known amounts of recombinant WRN standard. Linear least-squares regression was used to fit the regression line and determine the regression coefficient, R2.
DISCUSSION
We have analyzed WRN helicase expression in genetically characterized WRN patient, heterozygote and control cell lines by immune blot and immune precipitation-helicase activity assays. The cell lines we used contained four different, mutant WRN alleles that encode predicted WRN proteins of 368 to 1245 amino acid residues (Table 1). None of these predicted proteins could be detected in immune blot or immune precipitation-helicase activity assays. A recent study corroborates the results of our immune blot analyses of cell lines from the TUR and SYR pedigrees encoding, respectively, Q1165X and 1246X WRN proteins (20). Further, it supports the suggestion that most disease-associated WRN mutations are functionally equivalent null alleles: only an uncommon c.4146–4147insA single base insertion, the most C-terminal WRN mutation identified in a patient, led to the synthesis of readily detectable truncated WRN protein.
The low levels of WRN protein in Werner patient cells could be explained by a failure to synthesize WRN protein, by degradation of newly synthesized, truncated WRN protein, or by a combination of these mechanisms. Two lines of evidence argue that the first of these mechanisms is the most likely. First, both epitope-tagged full-length and truncated WRN proteins can be synthesized in WRN patient cells, and these recombinant proteins as well as native WRN appear to be comparably stable in WRN patient cells or cell extracts. Matsumoto et al. have obtained comparable results by overexpressing GFP-tagged native and mutant WRN proteins in HeLa cells (21). Second, all of the WRN mutations found in WRN patients create premature stop codons within the WRN open reading frame. These new internal stop codons could target mutant WRN mRNA for nonsense-mediated degradation (22,23) and thus explain the low levels of mutant WRN mRNA observed in WRN patient cell lines (24 and J.Oshima, unpublished results).
The absence of detectable WRN protein from patient cells has implications for the pathogenesis of WRN, the origins of clinical and pathologic heterogeneity among WRN patients, and for successful modeling of WRN pathogenesis in the mouse or in other model systems. The undetectable levels of WRN protein in patient cells indicate that the Werner syndrome develops when both the WRN helicase and nuclease activities are severely reduced. The common biochemical phenotype of different WRN mutations also indicates that clinical and pathologic differences between WRN patients or patient groups (for example 25) are best explained by genetic background or environmental exposure differences, rather than the expression of different, mutant WRN alleles.
The absence of detectable WRN protein from patient cells also indicates that the accurate modeling of WRN pathogenesis will require the use of mutations that eliminate or sharply reduce WRN protein expression. A recently published mouse model of Werner syndrome (26) may be problematic in this regard: the targeted alleles containing an in-frame deletion of the murine WRN helicase consensus domain produce a readily detectable, truncated protein that retains the WRN nuclease domain and C-terminal NLS (26). Further characterization of this mouse model, and the generation of additional mice in which WRN helicase or nuclease function is selectively inactivated by missense mutations, should allow us to determine whether the loss of WRN helicase or nuclease function alone generate clinical or cellular phenotypes that differ from those observed in WRN.
Several heterozygote effects have been observed in cells and cell lines from WRN patients. These include the intermediate sensitivity of WRN heterozygous lymphoblastoid cell lines to the DNA damaging agents 4NQO (27) and camptothecin (28,29), and the presence of genetic instability in vivo in the erythroid (red blood cell) lineage of WRN heterozygotes (M.Moser et al., unpublished results). The presence of reduced amounts of native WRN and the absence of truncated mutant WRN protein in WRN heterozygote cell lines (Fig. 2 and additional data not shown) indicates that the simplest model for these WRN heterozygote effects—haploinsufficiency—is likely to be correct.
As part of the immune blot analyses of WRN expression in cell lines from WRN pedigrees, we also determined the number of WRN molecules per cell. This was done by performing quantitative immune blot analyses using patient or control cell extracts and purified recombinant WRN protein as a standard. The number of WRN molecules/cell, ~60 000 in control (WRN +/+) lymphoblastoid and fibroblast lineage cells, is comparable to several other well-characterized nuclear proteins (e.g. c-Myc, 30; topoisomerases IIα and IIβ; 31). In contrast, two proteins that may interact with WRN, topoisomerase I and RPA, are ~10-fold more abundant (31 and M.S.Wold, personal communication).
The basis for between-individual variation in WRN content is unclear. EBV-transformed B-lymphoblastoid cell lines were used for these comparisons. EBV transformation upregulates WRN expression, but is not associated with appreciable differences in expression across the cell cycle (32,33). Fibroblast lineage cells contain equivalent amounts of WRN, but in contrast to lymphoblastoid cell lines SV40 transformation does not appear to upregulate WRN expression (Table 1 and additional unpublished results). There are suggestions that WRN polymorphisms may affect allele-specific WRN expression and perhaps function, although these ideas have not as yet been rigorously examined (34,35 and J.Oshima, unpublished results).
Recent results have begun to suggest functions for WRN in human somatic cells. A role for WRN in DNA repair is suggested by the observation that Werner cells are selectively sensitive to the DNA damaging agents 4-nitroquinoline 1-oxide and camptothecin (27–29,36). A second clue to in vivo function comes from identification of the Xenopus WRN homolog, FFA-1, as a protein required for replication focus formation in Xenopus egg extracts (37). This observation is particularly intriguing, as Werner cells and cell lines display DNA replication and S-phase abnormalities (reviewed in 5). A third clue to function comes from the recent finding that WRN and BLM can unwind G′2 triplet repeat and G4 tetraplex DNAs, respectively (38,39). These observations in concert—of selective sensitivity to DNA damaging agents, S-phase or replication abnormalities and the ability to unwind unusual DNA topologies—suggest that WRN may process unusual structures that arise during DNA replication, recombination or DNA repair at unusual sites (e.g. triplet repeat or telomeric DNAs; 40). Further biochemical and functional analyses should indicate how a reduction or loss of the WRN helicase and exonuclease activities promotes elevated levels of somatic mutation and the development of both neoplastic and non-neoplastic disease in specific human cell lineages.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Tom Kunkel and the Animal Facilities of the NIEHS Laboratory of Molecular Genetics for help in generating the SAM antisera, the International Registry of Werner Syndrome at the University of Washington and Sue Fredell for generating or providing Werner patient and control cell lines, and Drs Lawrence Loeb, James Shen, Ann Blank and two anonymous reviewers for helpful comments on the manuscript. This work was supported by grants from the NCI and NIA.
REFERENCES
- 1.Yu C.-E., Oshima,J., Fu,Y.-H., Wijsman,E.M., Hisama,F., Ouais,S., Nakura,J., Miki,T., Martin,G.M., Mulligan,J. and Schellenberg,G.D. (1996) Science, 272, 258–262. [DOI] [PubMed] [Google Scholar]
- 2.Moser M.J., Oshima,J. and Monnat,R.J.,Jr (1999) Hum. Mutat., 13, 271–279. [DOI] [PubMed] [Google Scholar]
- 3.Epstein C.J., Martin,G.M., Schultz,A.L. and Motulsky,A.G. (1966) Medicine, 45, 177–221. [DOI] [PubMed] [Google Scholar]
- 4.Goto M. (1997) Mech. Ageing Dev., 98, 239–254. [DOI] [PubMed] [Google Scholar]
- 5.Monnat R.J. Jr, (1992) Exp. Gerontol., 27, 447–453. [DOI] [PubMed] [Google Scholar]
- 6.Goto M., Miller,R.W., Ishikawa,Y. and Sugano,H. (1996) Cancer Epidemiol. Biomarkers Prev., 5, 239–246. [PubMed] [Google Scholar]
- 7.Gray M.D., Shen,J.-C., Kamath-Loeb,A.S., Blank,A., Sopher,B.L., Martin,G.M., Oshima,J. and Loeb,L.A. (1997) Nature Genet., 17, 100–103. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki N., Shimamoto,A., Imamura,O., Kuromitsu,J., Kitao,S., Goto,M. and Furuichi,Y. (1997) Nucleic Acids Res., 25, 2973–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang S., Li,B., Gray,M.D., Oshima,J., Mian,I.S. and Campisi,J. (1998) Nature Genet., 20, 114–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen J.-C., Gray,M.D., Oshima,J., Kamath-Loeb,A.S., Fry,M. and Loeb,L.A. (1998) J. Biol. Chem., 273, 34139–34144. [DOI] [PubMed] [Google Scholar]
- 11.Ellis N.A., Groden,J., Ye,T.-Z., Straughen,J., Lennon,D.J., Ciocci,S., Proytcheva,M. and German,J. (1995) Cell, 83, 655–666. [DOI] [PubMed] [Google Scholar]
- 12.Kitao S., Shimamoto,A., Goto,M., Miller,R.W., Smithson,W.A., Lindor,N.M. and Furuichi,Y. (1999) Nature Genet., 22, 82–84. [DOI] [PubMed] [Google Scholar]
- 13.Oshima J., Yu,C.-E., Piussan,C., Klein,G., Jabkowski,J., Balci,S., Miki,T., Nakura,J., Ogihara,T., Ells,J., Smith,M., Melaragno,M.I., Fraccaro,M., Scappaticci,S., Matthews,J., Ouais,S., Jarzebowicz,A., Schellenberg,G.D. and Martin,G.M. (1996) Hum. Mol. Genet., 5, 1909–1913. [DOI] [PubMed] [Google Scholar]
- 14.Fukuchi K., Martin,G.M. and Monnat,R.J.,Jr (1989) Proc. Natl Acad. Sci. USA, 86, 5893–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evan G.I., Lewis,G.K., Ramsay,G. and Bishop,J.M. (1985) Mol. Cell. Biol., 5, 3610–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rupp R.A.W., Snider,L. and Weintraub,H. (1994) Genes Dev., 8, 1311–1323. [DOI] [PubMed] [Google Scholar]
- 17.Bennett S.E., Umar,A., Kodama,S., Barrett,J.C., Monnat,R.J.,Jr and Kunkel,T.A. (1999) In Bohr,V.A., Clark,B.F.C. and Stevnsner,T. (eds), Molecular Biology of Aging. Alfred Benzon Symposium 44, Munksgaard, Copenhagen, pp. 214–224.
- 18.Harlow E. and Lane,D. (1988) Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 19.Coligan J.E., Dunn,B.M., Ploegh,H.L., Speicher,D.W. and Wingfield,P.T. (1995) Current Protocols in Protein Science. John Wiley & Sons, New York, NY.
- 20.Goto M., Yamabe,Y., Shiratori,M., Okada,M., Kawabe,T., Matsumoto,T., Sugimoto,M. and Furuichi,Y. (1999) Hum. Genet., 105, in press. [DOI] [PubMed] [Google Scholar]
- 21.Matsumoto T., Shimamoto,A., Goto,M. and Furuichi,Y. (1997) Nature Genet., 16, 335–336. [DOI] [PubMed] [Google Scholar]
- 22.Hentze M.W. and Kulozik,A.E. (1999) Cell, 96, 307–310. [DOI] [PubMed] [Google Scholar]
- 23.Culbertson M.R. (1999) Trends Genet., 15, 74–80. [DOI] [PubMed] [Google Scholar]
- 24.Yamabe Y., Sugimoto,M., Satoh,M., Suzuki,N., Sugawara,K., Goto,M. and Furuichi,Y. (1997) Biochem. Biophys. Res. Commun., 236, 151–154. [DOI] [PubMed] [Google Scholar]
- 25.Ishikawa Y., Sugano,H., Matsumoto,T., Furuichi,Y., Miller,R.W. and Gossen,M. (1999) Cancer, 85, 1345–1352. [PubMed] [Google Scholar]
- 26.Lebel M. and Leder,P. (1998) Proc. Natl Acad. Sci. USA, 95, 13097–13102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogburn C.E., Oshima,J., Poot,M., Chen,R., Hunt,K.E., Gollahon,K.A., Rabinovitch,P.S. and Martin,G.M. (1997) Hum. Genet., 101, 121–125. [DOI] [PubMed] [Google Scholar]
- 28.Okada M., Goto,M., Furuichi,Y. and Sugimoto,M. (1998) Biol. Pharm. Bull., 21, 235–239. [DOI] [PubMed] [Google Scholar]
- 29.Poot M., Gollahon,K.A. and Rabinovitch,P.S. (1999) Hum. Genet., 104, 10–14. [DOI] [PubMed] [Google Scholar]
- 30.Rudolph C., Adam,G. and Simm,A. (1999) Anal. Biochem., 269, 66–71. [DOI] [PubMed] [Google Scholar]
- 31.Meyer K.N., Kjeldsen,E., Straub,T., Knudsen,B.R., Hickson,I.D., Kikuchi,A., Kreipe,H. and Boege,F. (1997) J. Cell Biol., 136, 775–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiratori M., Sakamoto,S., Suzuki,N., Tokutake,Y., Kawabe,Y., Enomoto,T., Sugimoto,M., Goto,M., Matsumoto,T. and Furuichi,Y. (1999) J. Cell Biol., 144, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitao S., Ohsugi,I., Ichikawa,K., Goto,M., Furuichi,Y. and Shimamoto,A. (1998) Genomics, 54, 443–452. [DOI] [PubMed] [Google Scholar]
- 34.Ye L., Miki,T., Nakura,J., Oshima,J., Kamino,K., Rakugi,H., Ikegami,H., Higaki,J., Edland,S.D., Martin,G.M. and Ogihara,T. (1997) Am. J. Hum. Genet., 68, 494–498. [DOI] [PubMed] [Google Scholar]
- 35.Castro E., Ogburn,C.E., Hunt,K.E., Tilvis,R., Louhija,J., Penttinen,R., Erkkola,R., Panduro,A., Riestra,R., Piussan,C., Deeb,S.S., Wang,L., Edland,S.D., Martin,G.M. and Oshima,J. (1999) Am. J. Med. Genet., 82, 399–403. [PubMed] [Google Scholar]
- 36.Gebhart E., Bauer,R., Raub,U., Schinzel,M., Ruprecht,K.W. and Jonas,J.B. (1988) Hum. Genet., 80, 135–139. [DOI] [PubMed] [Google Scholar]
- 37.Yan H., Chen,C.-Y., Kobayashi,R. and Newport,J. (1998) Nature Genet., 19, 375–378. [DOI] [PubMed] [Google Scholar]
- 38.Fry M. and Loeb,L.A. (1999) J. Biol. Chem., 274, 12797–12802. [DOI] [PubMed] [Google Scholar]
- 39.Sun H., Karow,J.K., Hickson,I.D. and Maizels,N. (1998) J. Biol. Chem., 273, 27587–27592. [DOI] [PubMed] [Google Scholar]
- 40.Chakraverty R.K. and Hickson,I.D. (1999) Bioessays, 21, 286–294. [DOI] [PubMed] [Google Scholar]
- 41.Antonarakis S.E. and Nomenclature Working Group (1998) Hum. Mutat., 11, 1–3. [DOI] [PubMed] [Google Scholar]