


Abstract
Sorting isozymes are encoded by single genes, but the encoded proteins are distributed to multiple subcellular compartments. We surveyed the predicted protein sequences of several nucleic acid interacting sorting isozymes from the eukaryotic taxonomic domain and compared them with their homologs in the archaeal and eubacterial domains. Here, we summarize the data showing that the eukaryotic sorting isozymes often possess sequences not present in the archaeal and eubacterial counterparts and that the additional sequences can act to target the eukaryotic proteins to their appropriate subcellular locations. Therefore, we have named these protein domains ADEPTs (Additional Domains for Eukaryotic Protein Targeting). Identification of additional domains by phylogenetic comparisons should be generally useful for locating candidate sequences important for subcellular distribution of eukaryotic proteins.
INTRODUCTION
Eukaryotes are typified by the possession of organelles, generating numerous subcellular locations separated from one another by one or more membranes. Generally the different subcellular compartments carry out unique biochemical reactions. However, sometimes the same catalytic activity is found in more than one subcellular compartment. There are three different mechanisms used by eukaryotic cells to deliver the same enzymatic activity to more than one subcellular location. First, the same catalytic activity may be encoded by dissimilar genes. For example, cognate mitochondrial and cytosolic aminoacyl-tRNA synthetases can be quite distinct (1,2). Second, a catalytic activity may be encoded by multiple similar genes, each coding an isozyme with unique subcellular distribution. The yeast genes, ADH1, ADH2 and ADH3, provide an example of this type of mechanism (3). Finally, a single gene may encode two or more isozymes with different subcellular distributions. These proteins are called ‘sorting isozymes’ and are involved in many important metabolic processes (for a review see 4,5).
Sorting isozymes must contain information necessary for protein distribution to different compartments without compromising catalytic activity. Cellular mechanisms that achieve this are varied. In some cases, alternative transcriptional initiation generates mRNAs that encode the catalytic portion with or without signals for specific compartments. In other cases, the same end is achieved by alternative translational initiation or alternative splicing. Finally post-translational modifications can also alter the targeting information without altering catalytic activity (for a review see 4,5). In this report we focus on the cis-acting signals responsible for sorting isozyme distribution.
Genome sequencing efforts have generated information for several archaeal (six are complete and a few others are nearing completion; TIGR, http://www.tigr.org/tdb/mdb/mdb.html ), many eubacterial (19 are complete and many others are well underway), many, many viral and several eukaryotic nuclear as well as over 100 mitochondrial and 11 chloroplast organellar genomes [see Entrez Genomes at NCBI, http://www.ncbi.nlm. nih.gov/Entrez/Genome/org.html ). Indeed, the sequences of two eukaryotic nuclear genomes are virtually complete (6,7). If one assumes that sequences important to catalytic function will be conserved, then comparisons of eukaryotic sorting isozymes to their counterpart proteins in non-eukaryotic organisms might reveal the regions of the proteins serving the sorting function.
To test this assumption we conducted phylogenetic comparisons of five proteins. We chose genes that had been functionally characterized by cell biology and molecular biology experiments for their nuclear and mitochondrial targeting signals and some for cytoplasmic retention/nuclear export signals. We used three criteria to choose these proteins. (i) At least one eukaryotic member of the family has been shown directly to be a sorting isozyme and there is detailed information regarding the cis-acting sequences involved in subcellular distribution (Fig. 1). (ii) The catalytic functions are found in phylogenetically distinct organisms. (iii) The proteins interact with nucleic acids.
Figure 1.
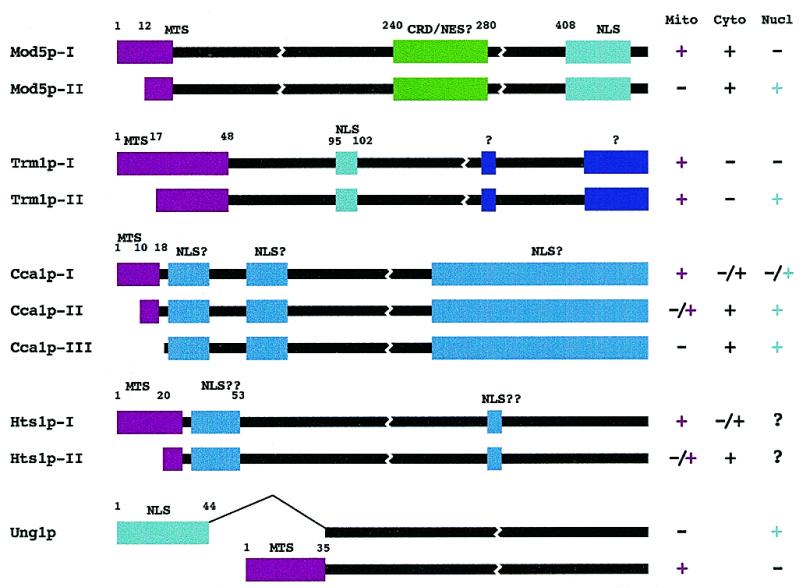
Location of information for subcellular distribution of sorting isozymes. Known and presumed targeting signals are represented as colored boxes. Magenta boxes represent known mitochondrial targeting information. Teal boxes and blue boxes represent known and presumed (NLS?) nuclear targeting information, respectively. Purple boxes may target Trm1p to a subnuclear location and the green boxes in Mod5p may be responsible for the predominantly cytosolic distribution of this protein. CRD, cytoplasmic retention domain; NES, nuclear export signal. The black lines represent the conserved regions of each protein and are not to scale. The subcellular distributions of the various forms of each protein are also indicated. For Hts1p, –/+ refers to locations detected upon protein over-expression.
Five sorting isozymes that fit our criteria are: (i) Mod5p catalyzing the modification of A37 to i6A37 on tRNA; (ii) Trm1p catalyzing the modification of G34 to m22G34 on tRNA; (iii) Hts1p the histidyl-tRNA synthetase; (iv) Cca1p catalyzing the addition of C, C and A to the 3′ ends of tRNAs; and (v) Ung1p a uracil-DNA glycosylase involved in DNA repair. Searches of databases demonstrate that eukaryotic counterparts of these proteins have domains in the same places, that archaeal/eubacterial counterparts do not. These comparisons, coupled with previous functional characterization of the protein domains, in at least one case led us to conclude that the additional information can serve to direct the eukaryotic proteins to the appropriate subcellular destination. We have named the eukaryotic additions ADEPTs (Additional Domains in Eukaryotes for Protein Targeting). We speculate that identification of ‘additional domains’ by phylogenetic comparisons and multiple sequence alignment will provide predictive information to locate unknown sequences important for the cellular distribution of eukaryotic proteins. Such analyses might also provide information for characterizing novel protein targeting motifs.
METHODS AND EXPLANATION OF ALIGNMENTS
Protein sequences were compared employing several databases (GenBank, EMBL, DDBJ, PDB, SWISS-PROT, PIR, PRF, dbEST, dbSTS, GSS and HTGS) using the BLAST (8) server at NCBI (http://www.ncbi.nlm.nih.gov/BLAST/ ). Similar proteins were identified, retrieved and used to search for additional matches. The retrieved sequences were aligned using either Clustal W or X (9,10). The aligned sequences were adjusted manually and shaded based on the BLOSUM 62 scoring matrix (11) with some weighting based on physical properties of the amino acids (12).
Table 1 lists the organisms and accession numbers of the peptides used in the alignments. An expanded version of this table (Table S1) is available as Supplementary Material at NAR Online. When the prokaryotic peptides used in the alignments originate from an incomplete genomic sequence and do not have an official accession number, the table is linked to the relevant genome sequencing center. For each of the individual alignments, not all organisms contain a peptide entry.
Table 1. Accession numbers.


º Indicates additional entries are available in Table S1.
The data are presented in two ways. Figures S1–S5 available as Supplementary Material at NAR Online, show the actual amino acid sequence alignment information. A score of ≥1 from the BLOSUM 62 matrix is designated as similar while a score of 0 is considered a weak similarity. Amino acids are grouped and colored as follows: aromatic amino acids phenylalanine, tyrosine and tryptophan (FYW) are magenta; hydrophobic amino acids isoleucine, valine, leucine and methionine (IVLM) are cyan; charged/polar amino acids aspartic acid, glutamic acid, glutamine, lysine, arginine, asparagine and histidine (DEQKRNH) are red; small amino acids glycine, alanine, cysteine, serine and threonine (GACST) are green; and proline (P) is blue. Three or more of a given amino acid yields upper case and color is turned on when at least five of a given amino acid or three of a given amino acid plus at least three amino acids from the same group with a score ≥1 are present. For the consensus lines 17–49% identity results in a lower case letter, 50–74% identity results in an upper case letter and 75–100% identity results in an upper case underlined letter.
Figures 2–6 show schematic diagrams of the protein alignments based on the sequence alignments described above. Blocks of similar color represent blocks of sequence similarity and are not a representation of any structural information. Different colored boxes represent uninterrupted regions of similarity (at least 35%) between the proteins from different organisms. Black lines represent eukaryotic sequences not generally similar to each other. Gray lines represent prokaryotic sequences not generally similar to each other or the eukaryotic sequences. Not all the sequences depicted are complete and some of the eukaryotic peptides judged to be too incomplete are not shown in the schematic diagrams. Eight eukaryotes were selected to represent the domain Eukarya: Homo sapiens, Mus musculus, Caenorhabditis elegans, Plasmodium falciparum, Schizosaccharomyces pombe, Saccharomyces cerevisiae and Candida albicans. Plants are usually represented as a composite diagram due to the lack of complete sequence information. An I to the right of the schematics designates incomplete information and a C designates complete cDNA or genomic DNA sequence information. The lengths of the polypeptide chains are indicated and where a composite schematic is shown the lengths of the individual polypeptide chains are separated by slashes. The eubacterial and archaeal schematics are derived from consensus sequences and the number of peptides used to generate the consensus is also indicated. Where information is available concerning the site of intron–exon junctions, the locations of introns are marked with an x.
Figure 2.

Schematic diagram of Mod5p alignment. Not all of the eukaryotic homologs are shown in this schematic. A sequence alignment of all identified Mod5p homologs and the eubacterial MiaA proteins can be found in Figure S1. The eubacterial MiaA peptides (46) are represented as a consensus sequence. No similar proteins were identified in the archaeal domain. Regions of uninterrupted sequence similarity (at least 35%) are shown as crosshatched colored boxes. See Methods for additional explanations.
Figure 6.

Schematic diagram of Ung1p alignment. A sequence alignment of all identified Ung1p homologs can be found in Figure S5. Ung1p was not identified in the archaeal domain. Thirty-three homologs were identified in the eubacterial domain and a consensus schematic is presented for these homologs. The schematic for plant in this figure is a composite of poplar and tomato. Regions of uninterrupted sequence similarity are shown as crosshatched colored boxes. Alternatively spliced exons are also indicated. See Methods for additional explanations.
RESULTS AND DISCUSSION
Mod5p homologs and conservation of regions for subcellular distribution
We previously reported an alignment of Mod5p/MiaA from 33 eubacteria and three eukaryotes (13). Our continued search for Mod5p homologs has now uncovered Mod5p/MiaA in 45 eubacteria (see Table 1). Two eubacterial organisms do not contain a miaA gene (Mycoplasma genitalium and Mycoplasma pneumoniae) while one, Porphyromonas gingivalis, contains two miaA genes. Seventeen eukaryotic homologs were identified in fifteen organisms (H.sapiens, M.musculus, Drosophila melanogaster, C.elegans; P.falciparum, Cryptosporidium parvum, Leishmania major, Trypanosoma brucei, Arabidopsis thaliana, Oryza sativa, S.pombe, S.cerevisiae, C.albicans, Kluyveromyces lactis and Neurospora crassa). Saccharomyces cerevisiae and C.elegans have only one gene encoding this protein. Only eight of the eukaryotic Mod5ps are shown in Figure 2 and the 46 eubacterial MiaA homologs are represented in Figure 2 as a consensus schematic. The entry for plants in Figure 2 represents a composite of three A.thaliana homologs and one homolog from rice. No homologs were identified in archaea, consistent with the fact that i6A has not been found on tRNAs isolated from organisms in the archaeal domain (14,15).
By alternative translational starts the S.cerevisiae MOD5 gene encodes two proteins, Mod5p-I and Mod5p-II (16), which are differentially partitioned between the cytoplasm, mitochondria and nucleus (17). Mod5p-I is located in the mitochondrial and cytosolic compartments whereas Mod5p-II is in the cytosol and the nucleus. Amino acids 1–20 comprise a mitochondrial targeting sequence (MTS) necessary for distribution of Mod5p-I to the mitochondria (17).
MTSs are usually located at the N-terminus, contain basic and hydrophobic amino acids and are predicted to form amphiphilic α-helices; however, there is no linear consensus sequence for mitochondrial targeting information (18,19). To assess whether other eukaryotes may utilize the same strategy as that for S.cerevisiae, we investigated the N-terminal regions of the other eukaryotic Mod5 proteins. Five of the eukaryotic homologs (S.cerevisiae, C.elegans, C.albicans, P.falciparum and one of the homologs from A.thaliana) contain multiple ATGs at the beginning of the coding region (Fig. 2), while for most of the other eukaryotes there is insufficient information available to predict whether or not multiple translation initiations give rise to different isozymes. The amphiphilic nature of these N-terminal peptides was investigated by plotting them on a helical wheel projection (not shown). In addition to S.cerevisiae, the C.elegans and C.albicans N-terminal regions resemble other MTSs (18,19). Thus, we predict that the C.elegans and C.albicans Mod5 proteins will also be sorted between the cytoplasm and mitochondria. The N-terminal regions of the P.falciparum homolog and the A.thaliana homolog with an N-terminal extension (Fig. S1, Athaippt) do not resemble other MTSs. In general, the eubacterial proteins do not have this N-terminal extension bolstering the idea that this extra domain found in the eukaryotic proteins is used for targeting.
Arabidopsis thaliana has at least three genes predicted to encode Mod5 proteins; therefore, different genes may well provide the same catalytic activity to different compartments for this organism. While additional information concerning A.thaliana and other eukaryotic organisms will be required to determine how mitochondrial/chloroplast/cytoplasmic/nuclear sorting may be achieved, it appears that for the Mod5p family sometimes one gene codes a catalytic activity found in multiple compartments whereas in other cases, two or more genes may code the isozymes.
Nearly all of the eukaryotic Mod5 proteins possess ~50 amino acids at the C-terminus that are not present in the eubacterial MiaA proteins (Fig. 2). The S.cerevisiae Mod5p nuclear localization sequence (NLS) maps within this ‘additional domain’ (amino acids 408–428; 13). In all of the other eukaryotes where sufficient sequence information is available (Fig. 2; S.pombe, C.albicans, C.elegans, rice and one of the A.thaliana homologs), the C-terminal region is similar leading to the prediction that they all contain a NLS and that a portion of the Mod5p pool in these organisms will also be located in the nucleus. Only one of the three A.thaliana homologs contains this NLS region while the others lack it (Fig. S1, not shown in Fig. 2), again suggesting that multiple genes encode differently located Mod5p in A.thaliana.
Besides the N-terminal and C-terminal additional domains, the eukaryotic Mod5 proteins also contain internal domains not found in the eubacterial homologs (Fig. 2). These internal additions overlap the region between amino acids 240 and 280 that were previously mapped to function in maintenance of the yeast Mod5p cytosolic pool (13). As all the eukaryotic sequences contain a similar region, we predict each of the eukaryotic counterparts also has a cytosolic pool of this protein.
A portion of the S.cerevisiae Mod5p-II resides in the nucleolus (13). The information used for nucleolar location has not been mapped. If, like the NLS and MTS, the nucleolus targeting/retention information resides in motifs absent from the eubacteria counterparts, then candidate locations for nucleolar targeting are between amino acids 303 and 345 and/or 373 and 408.
Trm1p homologs and conservation of regions for subcellular distribution
TRM1 genes are found in eukaryotes and archaea, but are generally not present in eubacteria (Fig. 3). In addition to the Trm1p homologs that have already been reported (20,21; six from the archaeal domain, Aquifex aeolicus, S.cerevisiae, S.pombe, C.elegans and human) our searches revealed three additional archaeal homologs and incomplete sequences for mouse, rat, zebrafish, D.melanogaster, P.falciparum, C.parvum, T.brucei, A.thaliana, rice, Brassica, Zea mays and C.albicans. There is only a single eubacterial organism, A.aeolicus, that contains a trm1 gene and this is likely a result of horizontal transfer (22–24). In agreement with our alignments, previous studies of tRNA modification have failed to uncover m22G in eubacterial tRNAs (14,15,25).
Figure 3.

Schematic diagram of Trm1p alignment. A sequence alignment of all identified Trm1p homologs can be found in Figure S2. Nine archaeal Trm1 peptides were identified and are represented as a consensus sequence. One trm1 homolog was identified in the eubacterial domain. The schematic for plant in this figure is a composite of A.thaliana, O.sativa and Brassica. Regions of uninterrupted sequence similarity are shown as crosshatched colored boxes. See Methods for additional explanations.
Eukaryotic and archaeal Trm1 proteins have considerable sequence similarity. However, like Mod5p, the eukaryotic proteins contain extra sequence information at the N- and C-termini and internally. The S.cerevisiae TRM1 gene contains ATG codons at positions 1 and 17. Human Trm1p contains two ATGs within the first 37 codons while mouse Trm1p contains three ATGs within the first 32 codons. Of the eukaryotic genes that have been sequenced at the N-terminus, only two, from C.elegans and D.melanogaster do not have multiple ATGs within the first 50 codons.
Some mitochondrial tRNAs of S.cerevisiae are modified by Trm1p and amino acids 1–48 of the S.cerevisiae Trm1p are sufficient to target this protein to mitochondria whereas amino acids 1–16 are not sufficient (26). There are several reports of m22G in mitochondrial and chloroplast tRNAs (27), but unfortunately the TRM1 genes have not been sequenced for the organisms demonstrated to contain m22G modified mitochondrial or chloroplast tRNAs. The N-terminus of the human Trm1p contains no acidic amino acids (Fig. S2) and when projected upon a helical wheel, it is predicted to have an amphiphilic structure, characteristic of MTSs (19). Thus, the human gene could encode a Trm1p that sorts to the mitochondria. The rodent homologs are very similar to the human in this region and the C.albicans Trm1p N-terminus contains what appears to be a very good MTS. As the C.elegans genome contains only a single TRM1 gene, it is likely that this gene provides the mitochondrial pool of tRNA (guanine-26,N2-N2) methyltransferase, if this modification occurs in C.elegans mitochondria.
Saccharomyces cerevisiae Trm1p is also targeted to the nucleus and an efficient NLS resides between amino acids 95 and 102 (28). All the other eukaryotic Trm1 proteins contain extra sequence information in this same region (Fig. 3, black region between 103 and 156 of human Trm1p). The C.elegans (21) and D.melanogaster proteins contain basic amino acids resembling the simple basic type of NLS in this region (see the review in 29), perhaps indicating a functional role in nucleus location. The corresponding extra sequences in human, mouse and S.pombe are not nearly as basic as the S.cerevisiae Trm1p sequence and neither a simple nor bipartite basic NLS motif can be identified in this region. However, it has recently become apparent that there are multiple nuclear import receptors in eukaryotic cells that have substrate specificities not yet delineated (see the review in 30). If the ADEPT regions of human, mouse and S.pombe Trm1p are used to sort this protein to the nucleus, as is the case in S.cerevisiae, then phylogenetic comparisons and sequence alignments may be a useful means to delineate non-conventional NLS motifs.
The eukaryotic genes also predict a large C-terminal region and a smaller region (between amino acids 346 and 367 in S.cerevisiae) not found in the archaeal proteins (Fig. 3). A zinc finger is present in the eukaryotic proteins (amino acids 348–387 human Trm1p) that is present in only half of the prokaryotic proteins. When present in prokaryotic proteins, the finger loop is much smaller than that found in eukaryotic proteins. The nuclear pool of Trm1p in S.cerevisiae is located at the inner surface of the nuclear membrane (28,31). If location at this subnuclear site is achieved via an ADEPT, then we predict that the targeting information will map to either the large C-terminal or the smaller upstream eukaryotic additional sequences (Fig. 1, purple boxes and Fig. 3).
Others (32) have reported results both consistent and inconsistent with our hypothesis. Deletion of the first 44 amino acids of S.cerevisiae Trm1p does not influence enzymatic activity, which is in accord with previous work demonstrating that this region contains targeting information (26) as well as our prediction that this region of the other eukaryotic proteins will supply targeting information. However, a deletion of just five amino acids at the C-terminus of S.cerevisiae Trm1p causes a significant reduction in activity (32). This result is inconsistent with our model in that all of the prokaryotic trm1 proteins lack this region and thus it is not expected to influence enzymatic activity. It is conceivable that an alteration in this region of the eukaryotic proteins may effect the higher order structure of the protein and interfere with activity.
Hts1p homologs and conservation of regions for subcellular distribution
HTS1 encodes histidine-tRNA synthetase, which is known as HisS in prokaryotes. Forty-five eubacterial and eight archaeal homologs were identified and 30 eukaryotic homologs were found. This enzyme is very similar in all three taxonomic domains (Fig. 4). Signature sequences can be identified that distinguish the eubacterial and archaeal proteins, and in some regions the archaeal signature is more similar to that of eukaryotes than to that of eubacteria.
Figure 4.

Schematic diagram of Hts1p alignment. A sequence alignment of all identified Hts1p homologs can be found in Figure S3. Forty-five eubacterial and eight archaeal HisS peptides were identified and are represented as consensus sequences. Additionally, a nuclear encoded organellar form of Hts1p from A.thaliana and a chloroplast genome encoded Hts1p from P.purpurea are shown in this diagram. The schematic for plant in this figure is a composite of A.thaliana, O.sativa, wheat and corn. Regions of uninterrupted sequence similarity are shown as crosshatched colored boxes. See Methods for additional explanations.
Six of the eukaryotic homologs contain multiple ATGs in their 5′ regions. However, the majority of the eukaryotic sequences are incomplete in this region and therefore we are unable to predict whether they encode proteins that differ at the N-terminus. In humans there are two genes arranged head-to-head that code for histidine-tRNA synthetases (Fig. 4, HumHARS and HumH03). The proteins encoded by these two genes are very similar (90%), except at the N-terminus where the similarity is only 38%. The N-terminus of HumHARS (residues 1–17) is acidic whereas that of HumH03 is not. Therefore, these two genes could provide the non-mitochondrial and mitochondrial forms of histidine-tRNA synthetase; however, this has yet to be determined.
Like Mod5p and Trm1p, where sufficient sequence information is available, the eukaryotic synthetases contain extra N-terminal information not present in the eubacterial or archaeal proteins. This region is precisely where the mitochondrial targeting sequence has been mapped for S.cerevisiae (33). In the red algae Porphyra purpurea, a gene for histidine-tRNA synthetase is present in the chloroplast genome. It is very similar to the eubacterial genes and does not code an extra N-terminal region. A nuclear HTS1 gene from A.thaliana that codes the organellar (mitochondrial and chloroplast) synthetase has been reported (34). It is more similar to archaeal genes, however it does code extra N-terminal amino acids.
Both Xenopus oocytes (35) and S.cerevisiae (36) aminoacylate tRNAs inside the nucleus as well as in the cytosol. Therefore, there must be nuclear pools of aminoacyl-tRNA synthetases. If Hts1p indeed possesses information that directs it to the nuclear interior, the targeting information could be located in the N-terminal region (Fig. S3, amino acids 20–53 of HumHARS). The additional sequences at this location in eukaryotic proteins contain basic residues resembling conventional NLS motifs (37). Fine mapping of the MTS in this region has not been completed and it is not yet clear where the MTS ends and where a putative NLS could begin. The MTS and NLS signals could also overlap. The majority of the eukaryotic sequences in this N-terminal region contain a higher charge density than does the S.cerevisiae sequence. Alternatively, the information could reside in the additional information located between amino acids 343 and 366 (S.cerevisiae numbering). The fungal counterparts are basic in this region while proteins from other eukaryotes are not.
Eukaryotic aminoacyl-tRNA synthetases tend to be larger than their prokaryotic counterparts and these extensions tend to be at the N- or C-terminus (38–41). The prevailing hypothesis is that these extensions are in part responsible for promoting the assembly of tRNA synthetase complexes found in eukaryotes (42). We and others (37) suggest that some portion of the extra information found in eukaryotic tRNA synthetases may be responsible for subcellular targeting.
Cca1p homologs and conservation of regions for subcellular distribution
Organisms in all three domains contain ATP (CTP): tRNA nucleotidyltransferase activity. However, the archaeal Cca proteins differ extensively from the eubacterial and eukaryotic Cca proteins (43). Nevertheless, all possess ‘nucleotidyltransferase’ motifs. Of the proteins we studied Cca1p is the least well conserved between eubacteria and eukaryotes. Large regions of sequence similarity, as found for the other proteins in our analysis, are lacking in this family. Sixteen eukaryotic homologs were identified in the following organisms: S.cerevisiae, S.pombe, C.albicans, human, mouse, rat, C.elegans, D.melanogaster, A.thaliana, Lupine, rice, Glycine max, L.major, Brugia malayi and P.falciparum. Eight archaeal homologs and 65 eubacterial homologs were identified. The latter have been grouped into three classes (Cca, Pap and PcnB) based on the sequence alignments as well as previous nomenclature. A consensus schematic is shown for each of these three classes of eubacterial proteins in Figure 5.
Figure 5.

Schematic diagram of Cca1p alignment. A sequence alignment of all identified Cca1p homologs can be found in Figure S4. Eight archaeal Cca peptides were identified and are represented as a consensus schematic. Sixty-five homologs were identified in the eubacterial domain. The eubacterial homologs fall into three classes and a consensus schematic is presented for each class: Cca-14, Pap-32 and PcnB-16. The schematic for plant in this figure is a composite of A.thaliana, O.sativa, lupine and G.max. Regions of uninterrupted sequence similarity are shown as crosshatched colored boxes. See Methods for additional explanations.
In S.cerevisiae the CCA1 gene encodes three proteins (Cca1p-I, Cca1p-II and Cca1p-III) that result from differential translation starts at three in-frame AUGs (44). Eight of the eukaryotic genes have multiple ATGs in this N-terminal region (Fig. S4), suggesting that multiple forms of Cca1p could also be produced by these genes.
Cca1p-I from S.cerevisiae is located primarily in mitochondria whereas Cca1p-II and Cca1p-III are located both in the cytosol and the nucleus (45). Like Mod5p, Trm1p and Hts1p the N-terminus of S.cerevisiae Cca1p contains mitochondrial targeting information. For each of the other eukaryotes where there is sufficient information, the eukaryotic Cca1p counterparts have an N-terminal extension that is absent or different in the eubacterial and archaeal proteins. This region most likely directs the non-plant Cca1p to mitochondria. Plant Cca1p should also be directed to the chloroplast. As chloroplast targeting information also is usually located at the N-terminus and resembles mitochondrial targeting information (46; for a review see 47), it is difficult to predict the function of the plant N-terminal Cca1p extensions. Also, since no plant genome has been completely sequenced there could be different genes for mitochondrial and chloroplast CCA activities.
The location of other targeting information for Cca1p is unknown, but there are other regions that contain additions not found in eubacteria (94–103; 109–114 S.cerevisiae numbering). There are also extensive regions of the proteins that are dissimilar between eukaryotes and prokaryotes (Fig. 5) that could contain nuclear targeting information.
Ung1p homologs and conservation of regions for subcellular distribution
Uracil-DNA glycosylase (UNG or UDG) is a DNA repair enzyme. The ung gene is found in 33 eubacteria, but is not present in archaea. Thus, either another gene product supplies this function or this function is not required. Interestingly, of the 19 complete eubacterial genomes, the ung gene is absent from six (Rickettsia prowazekii, Clostridium acetobutylicum, Treponema pallidum, A.aeolicus, Thermotoga maritima and Synechocystis), again suggesting that this function may not be required. Also of note is that within the genus Clostridium one organism, Clostridium difficile, contains a ung gene while C.acetobutylicum does not. UNG genes are also present in some viruses and consensus sequences for the Ung protein from 23 Herpes simplex viruses and five pox viruses are shown in Figure S5.
The human homolog of this enzyme is the most thoroughly studied. BLAST searches revealed Ung homologs in 11 other eukaryotes. The mouse homolog is very similar to the human (90% similarity) and both sort this enzyme between the nucleus and mitochondria via a mechanism that depends on alternative splicing (48,49; Fig. 6). This mechanism may also be used in C.elegans as there is an extra ‘exon’ upstream of the UNG gene which could be used to supply additional targeting information. However, this putative exon does not resemble known MTS or NLS motifs. Disregarding this putative exon the C.elegans ORF contains four in-frame ATGs. Downstream of AUG2 there is a sequence resembling a MTS, but we were unable to identify a classical simple or bipartite-like NLS in the N-terminal region. In S.cerevisiae there are four methionines within the first 50 amino acids and alternative transcription or translation start sites could provide the sorting mechanism for this enzyme; however, the available data (50; P.Burgers, personal communication) indicate that Ung1p is solely nuclear and unlikely to sort to mitochondria in yeast.
Since Ung1p should function within the nucleus of eukaryotes, there should be information to target this enzyme to the nucleus. Most of the eukaryotic and viral Ung proteins contain extra N-terminal sequence information not found in the bacterial counterparts. The human and mouse nuclear targeting information resides within this region and S.cerevisiae and P.falciparum appear to contain conventional bipartite NLSs within this region.
CONCLUSIONS
We surveyed five families of proteins containing at least one confirmed sorting isozyme. Four of these protein families have members that are highly conserved across taxonomic domains and the eukaryotic proteins contain additional sequences not found in the eubacterial or archaeal counterparts. Although the fifth protein, Cca1p, fits the pattern established by the other proteins in a limited sense, large portions of this protein are dissimilar when compared across taxonomic domains.
Additional information can be located at the N- or C-termini or it can be located internally. The location of additional sequence information is conserved, but the sequences are not necessarily similar. It has been proposed that intron locations correspond to positions separating independent functional domains of proteins (51,52). Although our data set is limited, our analysis does not appear to support this view. In general, ADEPTs do not correspond to genomic spliced regions.
We summarize the evidence that the additional sequences can encode information to sort the isozymes to appropriate subcellular locations (Fig. 1). The data lead us to propose the ADEPT hypothesis that similarly located extra information in other eukaryotic homologs will serve the same roles in protein subcellular distribution. We present this type of analysis as a predictive tool. Our results suggest that phylogenetic comparison/multiple sequence alignment will be a useful tool for predicting the cell biological information content of protein sequences. Future mechanistic tests of the sequences identified here will be necessary to determine how accurate these predictions are. However, data to date are quite consistent with the ADEPT concept.
SUPPLEMENTARY MATERIAL
See Supplementary Material available at NAR Online. Update to the published Supplementary Material will be available at http://www.collmed.psu.edu/labs/ahopper/DRS/ADEPTs/sortpaper.htm
Acknowledgments
ACKNOWLEDGEMENTS
We would like to express our gratitude to all members of the various genome sequencing projects (TIGR, Sanger Centre, OU-ACGT, Washington University GSC, BSNR, ChGP, Diversa, GTC, PGP, ASTRA, KDRI, NITE, Genome Therapeutics, Stanford University, UC Berkeley, University of Heidelberg, Uppsala University, University of Minnesota CBC, University of Wisconsin and the Utah Genome Center) for making information available and to the various funding agencies involved with these projects. Web links to the respective sequencing center are provided for each entry in Table S1. We would also like to thank all the members of the Hopper laboratory for their thoughtful discussions regarding this work. This work was supported by grants from the National Science Foundation to A.K.H. and N.C.M. and from the National Institutes of Health to A.K.H.
REFERENCES
- 1.Schneller J.M., Schneider,C. and Stahl,A.J. (1978) Biochem. Biophys. Res. Commun., 85, 1392–1399. [DOI] [PubMed] [Google Scholar]
- 2.Tzagoloff A. and Shtanko,A. (1995) Eur. J. Biochem., 230, 582–586. [DOI] [PubMed] [Google Scholar]
- 3.Pilgrim D. and Young,E.T. (1990) Mol. Cell. Biol., 7, 294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin N.C. and Hopper,A.K. (1994) Biochimie, 76, 1161–1167. [DOI] [PubMed] [Google Scholar]
- 5.Danpure C.J. (1995) Trends Cell Biol., 5, 231–237. [DOI] [PubMed] [Google Scholar]
- 6.Goffeau A., Aert,R., Agostini-Carbone,M.L., Ahmed,A., Aigle,M., Algerghina,L., Albermann,K., Albers,M., Aldea,M., Alexandraki,D. et al. (1997) Nature, 387 (suppl.), 5–105. [Google Scholar]
- 7.The C.elegans Sequencing Consortium(1998) Science, 282, 2012–2018.9851916 [Google Scholar]
- 8.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Higgins D.G., Thompson,J.D. and Gibson,T.J. (1996) Methods Enzymol., 266, 383–402. [DOI] [PubMed] [Google Scholar]
- 10.Thompson J.D., Gibson,T.J., Plewniak,F., Jeanmougin,F. and Higgins,D.G. (1997) Nucleic Acids Res., 25, 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henikoff S. and Henikoff,J.G. (1992) Proc. Natl Acad. Sci. USA, 89, 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Livingstone C.D. and Barton,G.J. (1996) Methods Enzymol., 266, 497–512. [DOI] [PubMed] [Google Scholar]
- 13.Tolerico L.H., Benko,A.L., Aris,J.P., Stanford,D.R., Martin,N.C. and Hopper,A.K. (1999) Genetics, 151, 57–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maden B.E.H. (1998) In Grosjean,H. and Benne,R. (eds), Modification and Editing of RNA. ASM Press, Washington, DC, pp. 421–440.
- 15.Winkler M.E. (1998) In Grosjean,H. and Benne,R. (eds), Modification and Editing of RNA. ASM Press, Washington, DC, pp. 441–468.
- 16.Gillman E.C., Slusher,L.B., Martin,N.C. and Hopper,A.K. (1991) Mol. Cell. Biol., 11, 2382–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boguta M., Hunter,L.A., Shen,W.-C., Gillman,E.C., Martin,N.C. and Hopper,A.K. (1994) Mol. Cell. Biol., 14, 2298–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Attardi G. and Schatz,G. (1988) Annu. Rev. Cell. Biol., 4, 289–333. [DOI] [PubMed] [Google Scholar]
- 19.von Heijne G. (1986) EMBO J., 5, 1335–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Constantinesco F., Benachenhou,N., Motorin,Y. and Grosjean,H. (1998) Nucleic Acids Res., 26, 3753–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu J., Zhou,G.-Q. and Straby,K.B. (1999) Gene, 226, 73–81. [DOI] [PubMed] [Google Scholar]
- 22.Syvanen M. (1994) Annu. Rev. Genet., 28, 237–261. [DOI] [PubMed] [Google Scholar]
- 23.Gray M.W., Burger,G. and Lang,B.F. (1999) Science, 283, 1476–1481. [DOI] [PubMed] [Google Scholar]
- 24.Doolittle W.F. (1999) Science, 284, 2124–2128. [DOI] [PubMed] [Google Scholar]
- 25.Grosjean H., Sprinzl,M. and Steinberg,S. (1995) Biochimie, 77, 139–141. [DOI] [PubMed] [Google Scholar]
- 26.Ellis S.R., Hopper,A.K. and Martin,N.C. (1989) Mol. Cell. Biol., 9, 1611–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sprinzl M., Horn,C., Brown,M., Ioudovitch,A. and Steinberg,S. (1998) Nucleic Acids Res., 26, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rose A.M., Joyce,P.B., Hopper,A.K. and Martin,N.C. (1992) Mol. Cell. Biol., 12, 5652–5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dingwall C. and Laskey,R.A. (1991) Trends Biochem. Sci., 16, 478–481. [DOI] [PubMed] [Google Scholar]
- 30.Ohno M., Fornerod,M. and Mattaj,I.W. (1998) Cell, 92, 327–336. [DOI] [PubMed] [Google Scholar]
- 31.Rose A.M., Belford,H.G., Shen,W.C., Greer,C.L., Hopper,A.K. and Martin,N.C. (1995) Biochimie, 77, 45–53. [DOI] [PubMed] [Google Scholar]
- 32.Liu J., Liu,J. and Straby,K.B. (1998) Nucleic Acids Res., 26, 5102–5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiu M.I., Mason,T.L. and Fink,G.R. (1992) Genetics, 132, 987–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akashi K., Grandjean,O. and Small,I. (1998) FEBS Lett., 431, 39–44. [DOI] [PubMed] [Google Scholar]
- 35.Lund E. and Dahlberg,J.E. (1998) Science, 282, 2082–2085. [DOI] [PubMed] [Google Scholar]
- 36.Sarkar S., Azad,A.K. and Hopper,A.K. (1999) Proc. Natl Acad. Sci. USA, 96, 14366–14371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schimmel P. and Wang,C.-C. (1999) Trends Biochem. Sci., 24, 127–128. [DOI] [PubMed] [Google Scholar]
- 38.Mirande M. and Waller,J.P. (1988) J. Biol. Chem., 263, 18443–18451. [PubMed] [Google Scholar]
- 39.Mirande M. (1991) Prog. Nucleic Acid Res. Mol. Biol., 40, 95–142. [DOI] [PubMed] [Google Scholar]
- 40.Kisselev L.L. and Wolfson,A.D. (1994) Prog. Nucleic Acid Res. Mol. Biol., 48, 83–142. [DOI] [PubMed] [Google Scholar]
- 41.Yang D.C.H. (1996) Curr. Top. Cell. Regul., 34, 101–135. [DOI] [PubMed] [Google Scholar]
- 42.Francklyn C., Musier-Forsyth,K. and Martinis,S.A. (1997) RNA, 3, 954–960. [PMC free article] [PubMed] [Google Scholar]
- 43.Yue D., Maizels,N. and Weiner,A.M. (1996) RNA, 2, 895–908. [PMC free article] [PubMed] [Google Scholar]
- 44.Wolfe C.L., Lou,Y.C., Hopper,A.K. and Martin,N.C. (1994) J. Biol. Chem., 269, 13361–13366. [PubMed] [Google Scholar]
- 45.Wolfe C.L., Hopper,A.K. and Martin,N.C. (1996) J. Biol. Chem., 271, 4679–4686. [DOI] [PubMed] [Google Scholar]
- 46.von Heijne G., Steppuhn,J. and Herrmann,R.G. (1989) Eur. J. Biochem., 180, 535–545. [DOI] [PubMed] [Google Scholar]
- 47.Cline K. and Henry,R. (1996) Ann. Rev. Cell. Dev. Biol., 12, 1–26. [DOI] [PubMed] [Google Scholar]
- 48.Otterlei M., Haug,T., Nagelhus,T.A., Slupphaug,G., Lindmo,T. and Krokan,H.E. (1998) Nucleic Acids Res., 26, 4611–4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muller-Weeks S., Mastran,B. and Caradonna,S. (1998) J. Biol. Chem., 273, 21909–21917. [DOI] [PubMed] [Google Scholar]
- 50.Percival K.J., Klein,M.B. and Burgers,P.M. (1989) J. Biol. Chem., 264, 2593–2598. [PubMed] [Google Scholar]
- 51.Go M. (1981) Nature, 291, 90–93.7231530 [Google Scholar]
- 52.de Souza S.J., Long,M., Schoenbach,L. and Gilbert,W. (1996) Proc. Natl Acad. Sci. USA, 93, 14632–14636. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.