


Abstract
The mutagenic and carcinogenic effects of simple alkylating agents are mainly due to O6-alkylation of guanine in DNA. This lesion results in transition mutations. In both prokaryotic and eukaryotic cells, repair is effected by direct reversal of the damage by a suicide protein, O6-alkylguanine-DNA alkyltransferase. The alkyltransferase removes the alkyl group to one of its own cysteine residues. However, this mechanism for preserving genomic integrity limits the effectiveness of certain alkylating anticancer agents. A high level of the alkyltransferase in many tumour cells renders them resistant to such drugs. Here we report the X-ray structure of the human alkyltransferase solved using the technique of multiple wavelength anomalous dispersion. This structure explains the markedly different specificities towards various O6-alkyl lesions and inhibitors when compared with the Escherichia coli protein (for which the structure has already been determined). It is also used to interpret the behaviour of certain mutant alkyltransferases to enhance biochemical understanding of the protein. Further examination of the various models proposed for DNA binding is also permitted. This structure may be useful for the design and refinement of drugs as chemoenhancers of alkylating agent chemotherapy.
INTRODUCTION
The DNA repair protein O6-alkylguanine-DNA alkyltransferase (AT) plays a pivotal role in the cellular defence against alkylating agents. Although a relatively minor product in relation to other sites of DNA alkylation, alkyl adducts at the O6-position of guanine represent one of the most harmful lesions (reviewed in 1). If not repaired, such lesions lead to GC→AT transition mutations and are associated with mutagenic, toxic and carcinogenic effects.
AT proteins act to remove alkyl groups from the O6-position of guanine through stoichiometric, irreversible, single-step transfer of adducts to an active site cysteine residue. Found in both eukaryotic and prokaryotic organisms, the alkyltransferase has been identified in at least 28 different species according to a PSI-BLAST search (2) in August 1999. All sequences exhibit the active site consensus sequence V(I)PCHRV(I). The best characterised AT proteins are those from Escherichia coli and human. The 21 kDa human protein (hAT) is homologous to the C-terminal domain of the Ada protein from E.coli (Ada-C), for which the crystal structure has been determined (3). More recently, the crystal structure for the 19.5 kDa hyperthermostable AT from Pyrococcus kodakaraensis (PkAT) has also been solved (4).
Despite the critical role that ATs play in maintaining DNA integrity, its presence is now well established as a leading cause of resistance of tumour cells to certain chemotherapeutic alkylating agents (5–7). Where lower levels of hAT activity are associated with a greater risk of tumour development, such tumours are correspondingly more sensitive to alkylating agent chemotherapy: indeed, low tumour hAT levels are correlated with better responses to treatment and greater survival (8,9).
Modulation of AT activity has been mooted as a possible chemoenhancing strategy since the early 1980s, resulting in the synthesis of many series of compounds as potential inhibitors (10–17). One such compound, O6-benzylguanine (BzG), has been studied extensively. When given in non-toxic doses, BzG has been shown to enhance the sensitivity of human tumour cells (in vitro and in animal xenograft models) to several alkylating agents, including certain chloroethylnitrosoureas and methylating compounds (7). Although BzG is now in phase I clinical trials, the suboptimal nature of its pharmaceutical and pharmacokinetic properties suggest that other inhibitors of the protein will be required for the true potential of AT modulation to be realised. In conjunction with the development of BzG analogues, there is also considerable interest in designing BzG-resistant mutants for gene therapy protection of the bone marrow during treatment (reviewed in 7).
While the crystal structure of Ada-C has been instructive with regard to the design of resistant mutants, significant substrate specificity differences between the human and Ada protein would indicate that there is structural diversity between their active sites. Such differences, typified by the resistance of Ada to BzG, suggest that structural information for the human protein is needed to enable rational design of inhibitors.
To this end, we present the X-ray crystal structure of hAT truncated at the C-terminus (hAT-G177stop, Fig. 1A). The 28 C-terminal residues of the full-length hAT (207 residues) have been shown to be dispensable for activity (18–20). More recently, this truncated form of hAT has been further characterised and is fully functional, repairing O6-methylguanine-containing oligonucleotides at a similar rate to the wild-type protein (21). Thus, with the three-dimensional structure of hAT-G177stop, it may now be possible to design inhibitors for the human protein. Furthermore, through analysis of some of the extensive mutational studies, this structure promotes understanding of substrate differences between mammalian and bacterial species and of the role of some invariant residues within the alkyltransferase family.
Figure 1.

(A) Sequence alignment of hAT (G177stop) with the C-terminal domain of Ada constructed according to topological equivalence. Fully conserved residues in the AT family are shown boxed (purple). Secondary structural elements and their labels as referred to in the text are shown above and below the sequences for hAT and Ada, respectively. These elements are colour coded as follows: α-helices are shown in blue except for H3 and H4 forming the helix–turn–helix element (green), β-sheets in red and a single turn of helix is shown in cyan. The Ada-C sequence is numbered by renaming T176 in the full-length Ada as residue 1. The numbers running above the hAT sequence refer to residue numbering for hAT only. (B) Stereo drawing of the human O6-alkylguanine-DNA alkyltransferase showing the principal secondary structural elements colour coded as in (A). In addition, the four residue turn of the active site residues C145–R148 are shown in yellow and labelled -CHR-. The N- and C-terminal residues are marked, as are the sequence numbers 36 and 42, indicating the undefined loop residues. (C) A stereo representation of the α-carbon coordinates of hAT (magenta) superimposed on those of the Ada-C (gold) crystal structure. This figure illustrates differences in topology between the N-terminal lobes of the two proteins and highlights the similarity of their C-terminal lobes. The residue numbering used to orient the illustration refers to the hAT protein. (B) and (C) were made using Molscript (54) and Raster3D (55).
MATERIALS AND METHODS
Production of native and selenomethionyl proteins
The details of expression and initial purification of mutant and wild-type protein have been described previously (22,23). Briefly, a clone of a truncated form of hAT (hAT-G177stop) was expressed in JM109 cells using the pQE30 vector that adds a His tag sequence, MRGS(H)6GS, to the N-terminus of the protein. Following initial purification, the protein was further purified using anion exchange FPLC and, if necessary, concentrated to 8 mg/ml using a Centricon concentrator (Amicon Co.). The seleno-l-methionine isoform of hAT-G177stop was produced in the same strain using a method adapted from Budisa et al. (24). Cells from a 30 ml overnight culture in LB were added to 1 litre of new minimal medium (24) containing the necessary trace elements, 100 µg/ml Se-Met and 100 µg/ml ampicillin. Suppression of methionine biosynthesis was achieved by adding lysine, phenylalanine and threonine at 100 µg/ml and isoleucine, leucine and valine at 50 µg/ml. Proline (50 µg/ml) was required for growth. After 12–15 h (OD600 = 0.8) the cultures were induced with 0.5 mM isopropyl-d-thiogalactopyranoside and the cells harvested after a further 12 h. Purification was performed as described above. The results from electrospray mass spectrometry were consistent with high levels of substitution at the five methionine sites. The proteins were kept in a Tris buffer (20 mM Tris, 200 mM NaCl, 10 mM DTT) at 4°C before use or stored at –80°C.
Crystallisation
The crystallisation conditions used for Ada-C (25) were tried unsuccessfully for hAT. Crystals of hAT, His tag remaining, were grown through hanging drop vapour diffusion of 4 µl of an 8 mg/ml protein solution (20 mM Tris, 200 mM NaCl, 10 mM DTT) with an equal volume of solution A [1.75 M (NH4)2SO4, 135 mM Tris, pH 7.5] against a well containing 600 µl of solution A at 19°C. These conditions readily produced high quality, well-diffracting crystals of native hAT. The Se-Met isoform was crystallised in the same way, although a lower yield and growth rate was observed. The crystals from both protein forms belong to space group P3121, with one molecule in the asymmetric unit and a solvent content of 55% (26). Unit cell dimensions for the Se-Met and native crystals used were a = b = 71.54 Å, c = 73.91 Å and a = b = 71.88 Å, c = 72.58 Å, respectively. Both forms show significant crystal non-isomorphism, with c varying between 72.5 and 75.0 Å.
Data collection and processing
A three-wavelength MAD dataset was collected on a single Se-Met hAT crystal on beam line BM14 at the ESRF (Grenoble, France). The crystal was transported in cryobuffer (40% maltose in mother liquor) pre-mounted and frozen in liquid nitrogen. X-ray fluorescence was used to select the three optimal wavelengths around the K absorption edge of selenium. With the crystal mounted to optimise collection of Friedel mates, data to 2.3 Å were collected at 100 K using a MARCCD CCD detector. At each wavelength, 120° of data were recorded using a 0.5° oscillation per image. A further dataset to 1.9 Å was collected for a native crystal on beam line 9.6 at the CLRC Synchrotron (Daresbury, UK). Diffraction intensities from the datasets were processed using the DENZO and SCALEPACK (27) and CCP4 (28) programs (see Table 1).
Table 1. Data collection statistics.
| Data collection | Edge | Peak | Remote | Native |
|---|---|---|---|---|
| Wavelength (Å) | 0.97898 | 0.97900 | 0.88550 | 0.870 |
| Se: f′, f′′ | –10.2, 3.06 | –6.20, 6.00 | –3.02, 4.40 | n/a |
| dmin (Å) | 2.3 | 2.3 | 2.3 | 1.9 |
| Total reflections | 211785 | 221638 | 205361 | 213536 |
| Unique reflections | 10092 | 10107 | 10055 | 17402 |
| Redundancy | 21.0 | 21.9 | 20.4 | 12.2 |
| Completenessa | 98.8 (98.7) | 98.7 (98.7) | 99.1 (99.3) | 99.5 (99.9) |
| I > 3σ (I) (%)a | 86.6 (61.0) | 87.0 (62.7) | 87.2 (61.9) | 93.3 (80.9) |
| Rmerge (%)ab | 4.5 (19.0) | 4.1 (17.1) | 4.4 (17.3) | 5.0 (19.3) |
N/a, not applicable.
aValues in parentheses correspond to the highest resolution shell (2.35–2.30 Å for MAD data; 1.97–1.90 Å for native data).
bRmerge ΣΣ|I – Ij |/ΣIj.
Structure determination and refinement
Data from the three-wavelength experiment were processed to 2.3 Å using SOLVE (29). The mean figure of merit for phasing from SOLVE was 0.72 to 2.3 Å and a readily interpretable electron density map was calculated for the P3121 enantiomer. The program found the three ordered selenium atoms expected in the asymmetric unit, permitting unambiguous orientation of the molecule. The two remaining selenium atoms, substituted into methionine residues either side of the N-terminal His tag, were assumed to be disordered. Map calculation and model building were carried out using the facilities within the XtalView package (30). Structural refinements were made using CNS v.0.5 (31) and progress was monitored throughout the experiment with a randomly generated 5% of reflections used to calculate Rfree. Successive rounds of rebuilding and simulated annealing enabled almost complete structural assignment from electron density maps with σA-weighted Fourier coefficients (32). CNS was used to extend the Rfree set to 1.9 Å. To allow for non-isomorphism between the native and Se-Met crystals, a single round of rigid body fitting and simulated annealing preceded initial calculation of 1.9 Å maps. Water molecules were added gradually during further rounds of side chain adjustment coupled with positional and B factor refinement. The refinement statistics for the final model are shown in Table 2. The model consists of 1264 protein atoms and 173 water molecules with a crystallographic R of 19.8% and an Rfree of 21.9% for reflections between 23.0 and 1.9 Å. The residues of the His tag tail, the first four N-terminal residues and loop residues G37–A41 were not included in the model as the electron density was not interpretable. Stereochemistry was analysed using PROCHECK (33); all non-glycine residues with the exception of I141 were in the favoured/allowed regions of the Ramachandran plot. I141 is present on a tight turn near to the active site C145; this residue is conserved and found with similar disallowed geometry in the crystal structures of both Ada-C and PkAT.
Table 2. Phasing and refinement statistics.
| Figure of merit (SOLVE) 2.3 Å | 0.72 |
| Rwork (Rfree)a | 19.8 (21.9) |
| r.m.s.d.: bonds (Å)/angles (°)b | 0.006/1.37 |
| Number of protein atoms/water molecules | 1264/173 |
| Average B factor (Å2) | 24.4 |
| Per cent residues: most favoured/additionally allowedc | 93.2/99.2 |
a Rfree = Rwork calculated on 5% of data excluded from refinement.
br.m.s.d., root mean square deviation.
cAccording to Ramachandran plot.
Determination of a binding pocket
The Multiple Copy Simulation Search (MCSS) (34) package within Quanta (35) was used to investigate favourable binding sites for guanine analogues in the active site region of hAT. A sphere with a radius of 12 Å centred on the active site cysteine was flooded with 500 copies of a guanine molecule. Guanine was used as a probe to mimic the approximate size and the common chemical attributes of the O6-substituted analogues. The energetically feasible positions and local minima of copies within this region were examined further.
RESULTS AND DISCUSSION
Tertiary structure
The hAT protein (Fig. 1B) is made up of distinct N-terminal (residues 4–85) and C-terminal (92–176) lobes. The N-terminal lobe is formed from a β-sheet of three classical β-strands, B1–B3, which packs against a single helix, H1. The β-sheet is connected to H1 by a loop between L33 of B3 and E57 of H1. Within this region, a stretch of residues E45–G55 runs anti-parallel with B1 along the solvent-exposed edge of the β-sheet. This region is surprisingly devoid of polar residues and could be said to form a ‘hydrophobic handle’. Such an element may serve to anchor hAT to other proteins. The electron density for residues E45–G55 is well defined and reveals a conformation stabilised by some β-sheet-like, and side chain hydrophobic, interactions with B1: the conformation may be promoted by the tight intermolecular packing in this region of the crystal lattice. Conversely, for the loop connecting B3 and residues E45–G55, the electron density was absent for residues G37–A41 and weak for residues L34–K36 and E42–V44, suggesting conformational flexibility at this apex (Fig. 1B). Connecting the N- and C-terminal lobes is a loop region including one turn of regular helix. The C-terminal lobe consists of four α-helices (H2–H5), of which H3 and H4 form the proposed DNA-binding, helix–turn–helix element. Between H4 and H5 is a stretch of coil and loop that includes the active site -C145HR- motif in a four residue turn (36).
Comparison with other ATs
Despite unsuccessful molecular replacement using either the crystal structure of the homologous Ada-C (3) or a human homology model based on the Ada-C structure (37), the overall topology of hAT closely resembles that of the bacterial protein (Fig. 1C). The major exception to this lies in the N-terminal lobe topology, where structural diversity in this region is reflected in the lower sequence identity between the two proteins (Fig. 1A, 14% over residues hAT 1–85 and Ada 8–83). In Ada-C, two helices sandwich the β-sheet, of which the second, H2, is topologically equivalent to H1 in hAT: Ada H2 and hAT H1 also rest at slightly different angles relative to their β-sheets. The first α-helix in Ada-C (H1) is replaced by the loop region that joins B3 and H1 in hAT. The topology in this region is also a site of structural variation between PkAT and Ada-C, where H1 in Ada is six residues shorter than the equivalent helix in PkAT. As predicted in the homology modelling of hAT, the β-sheet, together with the β-bulge (38) in B3, is conserved between hAT and Ada. In previous comparative analyses of hAT and Ada-C sequences it was predicted that a six residue insert in hAT would be located in the inter-domain loop (18,37). It is now evident that this ‘insert’ is partly accommodated as a single turn of α-helix in the centre of the loop (Fig. 1B and C).
In contrast, the two proteins have superimposable topology in the C-terminal lobe containing the active site and DNA-binding motifs. This is reflected in a much higher sequence identity (Fig. 1A, 42% over residues hAT 92–174 and Ada-C 94–175). The close structural similarity between the two proteins is confirmed by a root mean square difference of 1.2 Å2 as calculated over the α-carbon positions of 82 residues in topologically conserved regions, i.e. for hAT (Ada-C) B1-3 (B1-3), H2 (H3), H5 (H6) and residues Y114–V148 (Y115–V149). Over the 55 topologically equivalent residues in the C-terminal lobe only, this decreases to 0.9 Å2. Thus, structural and functional differences in this catalytic domain are likely to be determined by the nature of the amino acid side chains.
Homology modelling of hAT based on the coordinates of the Ada-C structure (37) was most successful in predicting the structure of the highly homologous C-terminal half of the protein. Within 8 Å of the active site cysteine residue, the homology model was found to be directly superimposable on the crystal structure of hAT. With the exception of the β-sheet, prediction of the N-terminal lobe of hAT was less accurate. It had been expected during the modelling process that the well-defined topology of the N-terminal lobe of Ada-C would be maintained between species.
With regard to similarity to other mammalian ATs, it is evident from sequence identity (60–70% for the rat, mouse and hamster proteins) that these proteins should share an almost identical fold with hAT. The only significant sequence difference is the presence of a four residue insert (PEGV) in the other mammalian proteins. This insert, between residues G55 and P56 of hAT, is located N-terminal to the start of H1 in hAT and is likely to extend the loop preceding the helix in these other proteins.
Identification of a binding pocket
MCSS (34,35) in the proximity of the active site cysteine has identified a potential binding cleft in hAT that is large enough to accept a guanine substrate and which could be used in the design of analogues as inhibitors. Examination of 287 energetically feasible copies from the MCSS trial with a guanine probe reveals a single site of interaction relevant to alkyltransfer. A tight cluster of 22 copies is located in a potential active site cavity existing between H4 and ‘wing’ residues 150–157 (Fig. 2A). Upon examination, it was found that the three lowest energy molecules in this group were in appropriate orientations to support alkyltransfer to the active site cysteine and embrace protein–substrate interactions. Furthermore, on examining possible DNA binding modes, a flipped-out base is predicted to enter and occupy this same cavity (discussed below). The remainder of the probe molecules (265 copies) are found on the external surface of the protein, inaccessible to the active site cysteine.
Figure 2.

(A) Stereo view of the active site region of hAT to show the position of BzG in the potential binding cleft as defined by a Multiple Copy Simulation Search. (The orientation of this view can be cross-referenced to Fig. 4.) The side chain positions of residues Y114, P138, P140, C145, V148 (main chain carbonyl only), N157, Y158, S159 and K165 with respect to the BzG molecule (magenta) are presented. The α-carbon coordinates of the remaining residues are shown in a coil representation. Side chains and secondary structure are shown in blue apart from Y114 on the HTH element (green). Atom colours are: nitrogen, blue; oxygen, red; sulphur, yellow. The side chain of C145 is in an appropriate position to carry out alkyltransfer by nucleophilic attack on the electron-deficient benzylic carbon. The imino ring of P140 presents a favourable hydrophobic packing surface for the benzyl group. Moreover, some of the experimental observations relating to the preferred attributes of guanine-based inhibitors (10–17) can be accommodated in this binding pocket. These include the preference for: (i) adequate space for p-phenylBzG and a wide variety of N-9 substituents; (ii) the absence of substituents at the 7-position; (iii) the presence of a 2-amino group. Substituents at the O6- and 9-positions of the molecule are directed towards the protein surface such that there is little constraint on the size of group that can be accommodated at these positions. S159 may act as a hydrogen bond donor to N-7 of BzG after a degree of side chain movement on binding. The amide side chain of N157 is also a candidate for this protein–inhibitor interaction. The 2-amino group of BzG is positioned to be able to hydrogen bond to the main chain carbonyl groups of C145 and V148. K165, a fully conserved residue essential for BzG sensitivity (results not shown), may also interact with the 2-amino group. The ability of K165 mutants to repair methylated DNA supports this, as the 2-amino group is of lesser importance for binding MeG in oligonucleotides (39,40). Moreover, it can be seen that the side chain of K165 plays an important role in maintaining the ring position of Y158 (involved here in an aromatic stacking interaction with the phenyl ring of BzG). Mutation of K165 to the majority of amino acids, particularly those with branched side chains, would serve to push the phenyl ring of Y158 into the proposed Bz group-binding pocket, explaining BzG resistance. For the K165A and K165G resistant mutants the absence of an extended side chain underpinning the orientation of Y158 may weaken the favourable packing interaction of the phenyl rings. (B) Stereo presentation of the equivalent view of Ada-C to illustrate factors contributing to BzG resistance in Ada [colour key as for (A)]. The BzG molecule has been imported in the corresponding position as determined for hAT (i.e. relative to the superimposable α-carbon backbones). The side chains of Y115, C146, K166 and V149 (main chain carbonyl only) conserved between hAT and Ada are indicated. The tryptophan side chain of W161, stabilised by aromatic interactions with Y159 and R160, is shown to impinge on the benzyl group-binding pocket proposed for hAT. The side chains of K139 and A141 corresponding to residues P138 and P140 in hAT are also shown. Figure 2 was made using Molscript (54) and Raster3D (55).
It has been suggested that the interactions in the binding cleft for hAT confer greater substrate specificity compared with Ada (39). In hAT, the N-1, N-3, N-7 and O6-positions of O6-methylguanine (MeG) in oligonucleotides were identified as important for reaction, whereas for Ada only the N-1 and O6-positions appear to be necessary (39,40). In the binding cleft proposed here (Fig. 2A), the side chain of S159 in hAT is in the vicinity and may serve as a hydrogen bond donor to N-7. Its counterpart in Ada-C, R160, is orientated away from the cleft and is unlikely to function in this way (Fig. 2B). Y114 may interact with the N-3 position of the guanine base: the contribution of this residue to the alkyltransfer reaction may depend on other local effects, including the role of this residue in DNA binding. Moreover, the side chain of residue N157 in hAT, a glycine in Ada, might also furnish interactions with either the N-3 or N-7 positions following side chain reorientation after inhibitor binding. Structure–activity studies of guanine analogues (10–17) also support the analysis of the proposed cleft (see legend to Fig. 2A).
However it should be noted that, first, while the MCSS search may identify the binding cleft, precise delineation of an active site binding pocket for free base analogues or analogue-containing oligonucleotides is not possible in the absence of structural information on AT–inhibitor complexes. Secondly, the precise binding interactions for the free base compounds may well differ in the presence of DNA and from those for the oligonucleotide analogues. Finally, the irreversible and rapid nature of alkyltransfer and absence of kinetic data for the protein and its mutants continues to hinder distinction between the binding and alkyltransfer components of the repair process.
Active site and substrate specificity
There are significant substrate specificity differences between the Ada and human ATs despite their structural similarity. Understanding the determinants could lead to the design of both more effective inhibitors and the BzG-resistant mutants required for bone marrow protection. Of note is the finding that, while hAT is more sensitive to BzG (IC50 = 0.2 µM) than MeG (IC50 = 350 µM), Ada is resistant to BzG (10,23,41). BzG sensitivity is shared with the other mammalian proteins and the smaller, constitutive, bacterial proteins, as typified by E.coli Ogt (42).
It had been proposed that the sequence change from G160 in hAT to W161 in Ada might pose a steric barrier to BzG in the bacterial protein (37). However, a W161A/G mutation in Ada only imparted BzG sensitivity when combined with an A141P mutation (23,43). Indeed, studies with chimeric proteins (hAT-C146-Ada and Ada-C146-hAT) confirm that residues on both sides of the active site cysteine are necessary for resistance (43). To further complicate the issue, the G160W mutant of hAT displays increased BzG sensitivity (44).
By comparison of the Ada-C and hAT crystal structures, such findings can now be explained in terms of steric and electrostatic components. (i) It appears that, in Ada-C, W161 in combination with R160 does form a steric barrier to BzG and contributes to resistance. The mutation of G160W in hAT enhances BzG sensitivity for different reasons. (ii) Sensitivity to BzG is also governed by a combination of local electrostatic effects in the active site.
With regard to the steric determinants of BzG sensitivity, the accessibility of the active site cysteine from the protein surface appears to be greater in hAT than in Ada. Contributing to the internalisation of the reactive cysteine in Ada is the tryptophan W161 side chain held in place by stacking interactions with Y159 and R160 (44) (see Fig. 2B). In addition, if the predicted BzG-binding cavity for hAT is considered with respect to Ada-C, the side chain of W161 occupies part of the benzyl group-binding region (Fig. 2B). That such a steric block exists is also suggested by studies with the BzG analogue 2,4-diamino-6-benzyloxy-5-nitrosospyrimidine (BzPy), in which the W161A mutant was shown to impart sensitivity (ED50 = 150 µM) to the otherwise BzPy-resistant, wild-type Ada (43). Being more potent than BzG, this compound may be a better agent for probing the AT active site.
The paradoxical enhancement in BzG sensitivity displayed by the hAT G160W mutant is also explained by the crystal structure: the tryptophan side chain is likely to be accommodated outside the BzG-binding pocket involved in stacking interactions with K32 and/or R135. In such a position, the W160 side chain could also present favourable aromatic stacking interactions in the binding of guanine-based analogues, as first suggested by Rafferty et al. using the hAT homology model (44). The key role this side chain plays in substrate specificity is further demonstrated by the finding that at least 14 of the 19 possible amino acid substitutions at G160 render hAT resistant to BzG (45). Although probably a combination of effects, it is possible that, unlike tryptophan, the smaller hydrophobic and polar residues enter the BzG-binding cavity and cause a degree of steric hindrance and resistance that is dependent on size. Charged residues, of which basic residues confer considerably more resistance than acidic residues, are also likely to enter the hydrophobic binding cavity and perturb the binding surface. The lesser resistance shown by negatively charged residues may be either a reflection of their shorter side chains or of their ability to form ion pairs with K32 or R135 outside the BzG-binding cavity.
In the absence of an overt conformational difference between the two proteins in the active site, it is clear that substrate specificity must also be determined by local side chain and electrostatic changes. Of note, mutational analysis has shown that P138 and P140 in the human protein confer BzG sensitivity, where in Ada the equivalent residues are lysine (K139) and alanine (A141), respectively. From studying the proposed binding pocket, it is evident that the side chain ring of the proline at position 140 could furnish a hydrophobic surface against which the benzyl group of BzG would pack (see Fig. 2A). The methyl group of A141 in Ada would contribute less to this interaction. That charge perturbations in the active site can alter BzG sensitivity is emphasised by the finding that, while the hAT mutants P140R, G160K/R and Y158H repair methylated DNA, they are resistant to inactivation by BzG (22,45,46).
Prior to solution of the hAT crystal structure, it was presumed that the proline residues were associated with an alteration in the local structure promoting a larger active site. However, confounding this assumption is the finding that the main chain conformation in this region is highly conserved in the two crystal structures (hAT 138–144; Ada-C 139–145). Indeed, in both proteins these residues form part of a loop providing the C-terminal half of the hydrophobic core and furnish inter-domain interactions with residues of the N-terminal lobe. The two proline residues are used in hAT to maintain the conformation effected by side chain interactions among local residues in Ada-C. In particular, it appears that where the hydrophobic bulk and main chain constraint of the proline rings is absent in Ada-C, two tryptophan residues are placed to compensate within the hydrophobic core; W101 for P138 and W161 for P140 (Fig. 3A and B). It had been predicted from sequence alignment that this apparently fully conserved tryptophan residue, W101 (W100 in hAT), should occupy the same relative position within the AT proteins. However, topological comparison of hAT and Ada-C reveals that this residue in hAT is shifted one residue C-terminal to that in Ada and, being present on an α-helix, is orientated into a different part of the hydrophobic core. Indeed, after further structural comparison of Ada-C, hAT and PkAT, and in the light of newly emerging sequences, it can be stated that W101 is not a conserved residue in the AT proteins. In PkAT, the equivalent tryptophan is placed two residues C-terminal to that of Ada-C: PkAT has two prolines in the equivalent positions to P138 and P140 in hAT.
Figure 3.

The α-carbon representation (far left in both figures) shows the location of the hydrophobic cores within (A) hAT and (B) Ada with the boxed regions indicating the area enlarged in the respective stereo views. The two proline residues in hAT and two tryptophan residues in Ada, important for maintaining core integrity, are depicted as a Van der Waal’s surface. The stereo views (enlarged area) for (A) hAT and (B) Ada show the principal interacting residues in the hydrophobic core. The protein backbone is shown in a coil representation (green) with the hydrophobic residues of the core shown in orange. Figure 3 was made using Molscript (54) and Raster3D (55).
Thus in hAT it would appear that P140 furnishes a sufficiently hydrophobic surface for acceptance of BzG without extending too far into the active site cavity to compromise available space and, together with P138, stabilises a fold likely to be conserved throughout the AT family.
DNA binding
As yet there is no structural information regarding the DNA binding mode of the AT family of proteins, although it is known that hAT forms a specific complex with MeG-containing oligonucleotides not competed for by the G-containing analogue (21). Following solution of the Ada-C crystal structure, various DNA binding models have been proposed for the AT proteins. These include the involvement of a helix–turn–helix element (hAT, H3/H4; Ada, H4/H5) (3), involvement of the C-terminal helix (3) and a helix–loop–wing motif (47). The pivotal consideration is how the relatively buried active site cysteine can interact with an alkyl lesion in DNA. Consistent with this is the proposition by Spratt and Campbell in 1994 (40), supported by the structural findings of Moore et al. (3), that the AT proteins may induce flipping of the modified base in the manner of HhaI cytosine methyltransferase (48). In support of this is the evidence that hAT binds to single-stranded DNA with greater affinity than double-stranded DNA (49).
The evidence for involvement of the HTH motif in DNA binding is very strong, not only because this region contains several of the fully conserved amino acids, of which R128 and Y114 have been shown to be essential for DNA binding (50). In addition, topological comparison with HTH motifs in other proteins reveals remarkable similarities. Most HTH motifs comprise three helices A, B and C, where the recognition helix (C) and B form the HTH element; A lies orthogonal to C with its N-terminus directed towards the DNA backbone. Various HTH motifs have been classified according to topology (51). Topologically, the hAT and Ada crystal structures show the greatest similarity to CAP (catabolite activator protein; 52). The root mean square difference between the HTH motifs in hAT and CAP, as calculated over 21 α-carbons of helices A–C (hAT, H2, H3 and H4), is only 0.85 Å2. This value is 1.6 Å2 in a comparison with the class represented by histone H5 (53).
In their model of DNA binding for hAT, Vora et al. (47) adopted the model proposed for MuA (51) on the basis that the two proteins shared structural and sequence similarities over helices B and C. This model uses a helix–loop–wing arrangement, whereby binding is effected by the recognition helix (C) of hAT (H4) and wing residues 151–157, and methyltransfer by flipping of the modified base. However, MuA differs in topology from the classical HTH proteins whereby helix A is actually transposed to the C-terminus of the motif and lies at a different angle to helices B and C, such that its N-terminus does not interact with DNA. Thus we propose that, in addition to using the wing residues, the DNA binding mode for the ATs will resemble that of CAP in employing all three helices of the HTH motif (Fig. 4).
Figure 4.
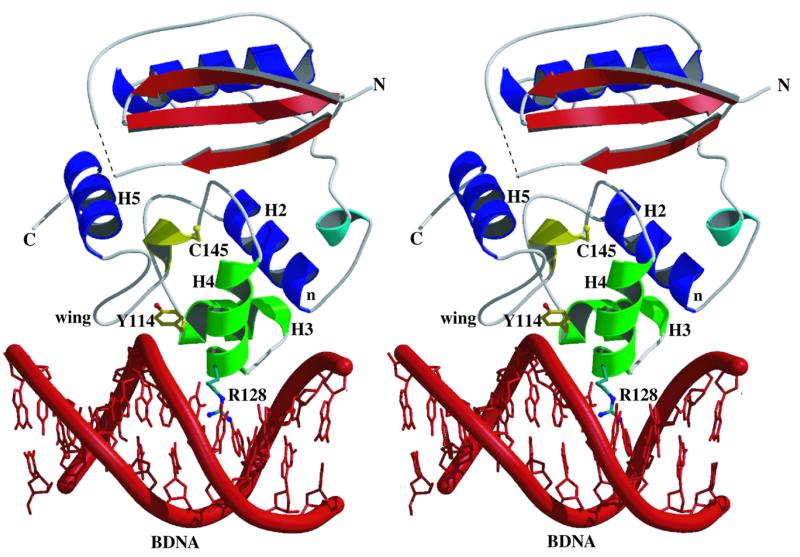
Stereo view of the proposed DNA binding model for hAT. It is probable that alkyl lesions are removed from DNA in a base-flipping manner, but the mechanism of this has yet to be determined. The model here is based on overlaying the helices of the HTH domain of hAT (H2, H3 and H4) onto the counterpart helices in CAP (51), followed by docking into a piece of standard B-DNA (red, with the phosphate backbone emphasised as a thick coil representation). Colour coded as described in Figure 1B, the N-termini of H2, H3 and the recognition helix H4 (denoted by the letter n and the residues Y114 and R128, respectively) can be seen to be oriented towards the phosphodiester backbone. The wing residues (151–157) are also shown to contact DNA (as proposed in 47). The side chains of the two fully conserved residues involved in DNA binding, Y114 (yellow) and R128 (green), are shown in ball and stick fashion. Two other fully conserved residues, A118 and V130 (side chains not shown), are involved in a hydrophobic interface between H3 and H4. The dotted line in the N-terminal lobe is used to represent residues 37–41 lacking interpretable electron density. Figure 4 was made using Molscript (54) and Raster3D (55).
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank Dr Sreenivas Kanugula (Department of Cellular and Molecular Physiology, Pennsylvania State University) for the design of the hAT-G177stop clone and Dr Kathryn Lilley (Protein and Nucleic Acid Chemistry Laboratory, University of Leicester) for protein mass spectrometry. We also wish to thank Dr Andrew Thompson and Dr Gordon Leonard for help with data collection on BM14 (ESRF, Grenoble, France) and EMBL for a travel grant. We are also grateful for beam time on station 9.6 at the CLRC SRS, Daresbury, UK. This work was supported by the UK Cancer Research Campaign.
PDB accession no. 1QNT
REFERENCES
- 1.Saffhill R., Margison,G.P. and O’Connor,P.J. (1985) Biochim. Biophys. Acta, 823, 111–145. [DOI] [PubMed] [Google Scholar]
- 2.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore M.H., Gulbis,J.M., Dodson,E.J., Demple,B. and Moody,P.C.E. (1994) EMBO J., 13, 1495–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hashimoto H., Inoue,T., Nishioka,M., Fujiwara,S., Tagaki,M., Imanaka,T. and Kai,Y. (1999) J. Mol. Biol., 292, 707–716. [DOI] [PubMed] [Google Scholar]
- 5.Pegg A.E., Dolan,M.E. and Moschel,R.C. (1995) Prog. Nucleic Acid Res. Mol. Biol., 51, 167–233. [DOI] [PubMed] [Google Scholar]
- 6.Chaney S.G. and Sancar,A. (1996) J. Natl Cancer Inst., 88, 1346–1360. [DOI] [PubMed] [Google Scholar]
- 7.Dolan M.E. and Pegg,A.E. (1997) Clin. Cancer Res., 3, 837–847. [PubMed] [Google Scholar]
- 8.Belanich M., Pastor,M., Randall,T., Guerra,D., Kitbitel,J., Alas,L., Li,B., Citron,M., Wasserman,P., White,A. et al. (1996) Cancer Res., 56, 783–788. [PubMed] [Google Scholar]
- 9.Hotta T., Saito,T., Fujita,H., Mikami,T., Kuriso,K., Kiya,K., Vozumi,T., Isowa,G., Ishizaki,K. and Ikenaga,M. (1994) J. Neurooncol., 21, 135–140. [DOI] [PubMed] [Google Scholar]
- 10.Moschel R.C., McDougall,M.G., Dolan,M.E., Stine,L. and Pegg,A.E. (1992) J. Med. Chem., 35, 4486–4491. [DOI] [PubMed] [Google Scholar]
- 11.Chae M.-Y., McDougall,M.G., Dolan,M.E., Swenn,K., Pegg,A.E. and Moschel,R.C. (1994) J. Med. Chem., 37, 342–347. [DOI] [PubMed] [Google Scholar]
- 12.Chae M.-Y., Swenn,K., Kanugula,S., Dolan,M.E., Pegg,A.E. and Moschel,R.C. (1995) J. Med. Chem., 38, 359–365. [DOI] [PubMed] [Google Scholar]
- 13.Arris C.E., Bleasdale,C., Calvert,A.H., Curtin,N.J., Dalby,C., Golding,B.T., Griffin,R.J., Lunn,J.M., Major,G.N. and Newell,D.R. (1994) Anticancer Drug Des., 9, 401–408. [PubMed] [Google Scholar]
- 14.Cussac C., Rapp,M., Mounetou,E., Madelmont,J.C., Maurizis,J.C., Godeneche,D., Dupuy,J.M., Sauzieres,J., Baudry,J.P. and Veyre,A. (1994) J. Pharmacol. Exp. Ther., 271, 1353–1358. [PubMed] [Google Scholar]
- 15.Mineura K., Fukuchi,M., Kowada,M., Terashima,I. and Kohda,K. (1995) Int. J. Cancer, 63, 148–151. [DOI] [PubMed] [Google Scholar]
- 16.Mounetou E., Debiton,E., Buchdahl,C., Gardette,D., Gramain,J.C., Maurizis,J.C., Veyre,A. and Madelmont,J.C. (1997) J. Med. Chem., 40, 2902–2909. [DOI] [PubMed] [Google Scholar]
- 17.McElhinney R.S., Donnelly,D.J., McCormick,J.E., Kelly,J., Watson,A.J., Rafferty,J.A., Elder,R.H., Middleton,M.R., Willington,M.A., McMurray,T.B.H. and Margison,G.P. (1998) J. Med. Chem., 41, 5265–5271. [DOI] [PubMed] [Google Scholar]
- 18.Elder R.H., Tumelty,J., Douglas,K.T., Margison,G.P. and Rafferty,J.A. (1992) Biochem. J., 285, 707–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morgan S.E., Kelley,M.R. and Pieper,R.O. (1993) J. Biol. Chem., 255, 19802–19809. [PubMed] [Google Scholar]
- 20.Crone T.M., Goodztova,K., Edara,S. and Pegg,A.E. (1994) Cancer Res., 54, 6221–6227. [PubMed] [Google Scholar]
- 21.Hazra T.K., Roy,R., Biswas,T., Grabowski,D.T., Pegg,A.E. and Mitra,S. (1997) Biochemistry, 36, 5769–5776. [DOI] [PubMed] [Google Scholar]
- 22.Edara S., Kanugula,S., Goodtzova,K. and Pegg,A.E. (1996) Cancer Res., 56, 5571–5575. [PubMed] [Google Scholar]
- 23.Goodtzova K., Kanugula,S., Edara,S., Pauly,G.T., Moschel,R.C. and Pegg,A.E. (1997) J. Biol. Chem., 13, 8332–8339. [DOI] [PubMed] [Google Scholar]
- 24.Budisa N., Steipe,B., Demange,P., Eckerskorn,C., Kellerman,J. and Huber,R. (1995) Eur. J. Biochem., 230, 788–796. [DOI] [PubMed] [Google Scholar]
- 25.Moody P. and Demple,B. (1990) J. Mol. Biol., 200, 751–752. [DOI] [PubMed] [Google Scholar]
- 26.Matthews B.W. (1968) J. Mol. Biol., 33, 491–497. [DOI] [PubMed] [Google Scholar]
- 27.Otwinowski Z. and Minor,W. (1997) Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 28.Collaborative Computational Project Number 4 (1994) Acta Crystallogr., D50, 760–763. [Google Scholar]
- 29.Terwilliger T.C. and Berendzen,J. (1999) Acta Crystallogr., D55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McRee D.E. (1992) J. Mol. Graphics, 10, 44–46. [Google Scholar]
- 31.Brünger A.T., Adams,P.D., Clore,G.M., Delano,W.L., Gros,P., Grosse-Kunstleve,R.W., Jiang,J.-S., Kuszewski,J., Nilges,M., Pannu,N.S. et al. (1998) Acta Crystallogr., D54, 905–931. [DOI] [PubMed] [Google Scholar]
- 32.Read R.J. (1986) Acta Crystallogr., A42, 140–149. [Google Scholar]
- 33.Laskowski R.A., MacArthur,M.W., Moss,D.S. and Thornton,J.M. (1993) J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- 34.Miranker A. and Karplus,M. (1991) Proteins, 11, 29–34. [DOI] [PubMed] [Google Scholar]
- 35. Molecular Simulations Inc. (1997) QUANTA97. The QUANTA Program. Molecular Simulations Inc., San Diego, CA.
- 36.Kabsch W. and Sander,C. (1983) Biopolymers, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- 37.Wibley J.E.A., McKie,J.H., Embrey,K., Marks,D.S., Douglas,K.T., Moore,M.H. and Moody,P.C.E. (1995) Anticancer Drug Des., 10, 75–95. [PubMed] [Google Scholar]
- 38.Richardson J.S., Getzoff,E.D. and Richardson,D.C. (1978) Proc. Natl Acad. Sci. USA, 75, 2574–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spratt T.E., Wu,J.D., Levy,D.E., Kanugula,S. and Pegg,A.E. (1999) Biochemistry, 38, 6801–6806. [DOI] [PubMed] [Google Scholar]
- 40.Spratt T.E. and Campbell,C.R. (1994) Biochemistry, 33, 11364–11371. [DOI] [PubMed] [Google Scholar]
- 41.Dolan M.E., Pegg,A.E., Dumenco,L.L., Moschel,R.C. and Gerson,S.L. (1991) Carcinogenesis, 12, 2305–2309. [DOI] [PubMed] [Google Scholar]
- 42.Potter P.M., Wilkinson,M.C., Fitton,J., Carr,F.J., Brennand,J., Cooper,D.P. and Margison,G.P. (1987) Nucleic Acids Res., 15, 9177–9193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crone T.M., Kanugula,S. and Pegg,A.E. (1995) Carcinogenesis, 16, 1687–1692. [DOI] [PubMed] [Google Scholar]
- 44.Rafferty J.A., Wibley,J.E.A., Speers,P., Hickson,I., Margison,G.P., Moody,P.C.E. and Douglas,K.T. (1997) Biochim. Biophys. Acta, 1343, 90–102. [DOI] [PubMed] [Google Scholar]
- 45.Welliver M.X., Leitao,J., Kanugula,S. and Pegg,A.E. (1999) Cancer Res., 59, 1514–1519. [PubMed] [Google Scholar]
- 46.Christians F.C., Dawson,B.J., Coates,M.M. and Loeb,L.A. (1997) Cancer Res., 57, 2007–2012. [PubMed] [Google Scholar]
- 47.Vora R.A., Pegg,A.E. and Ealick,S.E. (1998) Proteins, 32, 3–6. [PubMed] [Google Scholar]
- 48.Klimasauskas S., Kumar,S., Roberts,R.J. and Cheng,X. (1994) Cell, 76, 357–369. [DOI] [PubMed] [Google Scholar]
- 49.Fried M.G., Kanugula,S., Bromberg,J.L. and Pegg,A.E. (1996) Biochemistry, 35, 15295–15301. [DOI] [PubMed] [Google Scholar]
- 50.Kanugula S., Goodtzova,K., Edara,S. and Pegg,A.E. (1995) Biochemistry, 34, 7113–7119. [DOI] [PubMed] [Google Scholar]
- 51.Clubb R.T., Omichinski,J.G., Savilahti,H., Mizuuchi,K., Gronenborn,A.M. and Clore,M. (1994) Structure, 2, 1041–1048. [DOI] [PubMed] [Google Scholar]
- 52.Schultz S.C., Shields,G.C. and Steitz,T.A. (1991) Science, 253, 1001–1007. [DOI] [PubMed] [Google Scholar]
- 53.Ramakrishnan V., Finch,J.T., Graziano,V., Lee,P.L. and Sweet,R.M. (1993) Nature, 362, 219–223. [DOI] [PubMed] [Google Scholar]
- 54.Kraulis P.J. (1991) J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- 55.Merrit,E.A. and Murphy M.E. (1994) Acta Crystallogr., D50, 869–873. [Google Scholar]