


Abstract
DNA probes with conjugated minor groove binder (MGB) groups form extremely stable duplexes with single-stranded DNA targets, allowing shorter probes to be used for hybridization based assays. In this paper, sequence specificity of 3′-MGB probes was explored. In comparison with unmodified DNA, MGB probes had higher melting temperature (Tm) and increased specificity, especially when a mismatch was in the MGB region of the duplex. To exploit these properties, fluorogenic MGB probes were prepared and investigated in the 5′-nuclease PCR assay (real-time PCR assay, TaqMan assay). A 12mer MGB probe had the same Tm (65°C) as a no-MGB 27mer probe. The fluorogenic MGB probes were more specific for single base mismatches and fluorescence quenching was more efficient, giving increased sensitivity. A/T rich duplexes were stabilized more than G/C rich duplexes, thereby leveling probe Tm and simplifying design. In summary, MGB probes were more sequence specific than standard DNA probes, especially for single base mismatches at elevated hybridization temperatures.
INTRODUCTION
We have been developing the chemistry and applications of minor groove binder-oligodeoxynucleotide conjugates (MGB-ODNs) which form hyper-stabilized duplexes with complementary DNA (1–6). NMR studies showed that a conjugated MGB, dihydrocyclopyrroloindole tripeptide (DPI3), folds into the minor groove formed by the terminal 5–6 bp (7). The crescent shaped DPI3 (8) is isohelical with the deep and narrow minor groove of B-form DNA where it is stabilized mainly by van der Waals forces. Increases in melting temperature (Tm) of as much as 49°C were observed for A/T-rich octanucleotides (6). Both 3′- and 5′-modified MGB-ODNs were shown to hybridize with greater sequence specificity than natural (no-MGB) ODNs and the 5′-MGB probes arrested primer extension (3). The term probe is used to indicate an ODN which has been modified for a specific hybridization assay.
In this paper we explored the sequence specificity of 3′-MGB-ODNs and their performance as fluorogenic probes for PCR. 3′-MGB-ODNs are easier to prepare than 5′-conjugates since MGB-modified solid supports and automated DNA synthesis can be used. These methods are discussed. A DNA duplex formed with MGB-ODN1 and a 23mer ODN target was studied by UV melting (Fig. 1). Various mismatch targets were used and effects of the MGB on mismatch discrimination (ΔΔG) were calculated. A series of different length fluorogenic probes (±3′-MGB) were prepared and studied in the 5′-nuclease PCR assay (real-time PCR assay, TaqMan assay). The 5′-nuclease assay requires DNA probes that remain hybridized to the template during primer extension by Taq polymerase at 60–70°C. The 3′-MGB probes did not inhibit PCR, thus allowing robust performance in the 5′-nuclease assay.
Figure 1.

Structure of the DNA duplex formed with MGB-ODN1 and a 23mer complement. Effects of mismatched bases on stability of this duplex are shown in Figure 2. The putative MGB site is underlined in the complement. C→N polarity of the DPI3 peptide and structure of the N-terminal linker are shown below.
MATERIALS AND METHODS
Synthesis of MGB ODNs
The 3′-DPI3 ODN (Fig. 1) was prepared by automated DNA synthesis from a DPI3-modified glass support as described earlier (4). The 3′-CDPI3 probes (Fig. 5) were prepared from a trifunctional linker using methods described earlier (2). Oligonucleotide synthesis was performed on an ABI 394 synthesizer according to the protocol supplied by the manufacturer except that 0.015 M (instead of the standard 0.2 M) iodine solution was utilized in the oxidation step to avoid iodination of the MGB moiety. To prevent extension during PCR, probes without 3′-MGB were prepared with the 3′-hydroxyhexyl phosphate as previously described (9). 6-Carboxyfluorescein (6-FAM) or tetrachlorofluorescein (TET) phosphoramidites (Glen Research, Sterling, VA) were used to introduce the 5′-reporter dyes. Purine analogs containing aminopropyl linkers (Fig. 5) were incorporated for post-synthetic conjugation of the 3′-quencher dye tetramethylrhodamine (TAMRA). Synthesis of phosphoramidites derived from 3-(3-trifluroacetamidopropyl)-6-amino-4-hydroxy-1-β-d-2′deoxyribofuranosyl-pyrazolo[3,4-d]pyrimidine (aminopropyl-PPG) and 3-(3-tri-fluroacetamidopropyl)-4-amino-1-β-d-2′deoxyribofuranosyl-pyrazolo[3,4-d]pyrimidine (aminopropyl-PPA) will be described elsewhere. After ammonia deprotection, all oligonucleotides were reverse-phase HPLC purified as described earlier (2) and isolated as the sodium salts by butanol concentration/sodium perchlorate precipitation (10). Unmodified ODNs for melting studies and fluorogenic probes were purified by denaturing gel electrophoresis (PAGE) as described below.
Figure 5.

Structure of the DNA duplex formed with a fluorogenic 12mer MGB probe and a complementary DNA strand. Effects of probe length and mismatched bases on stability of this duplex are shown in Figure 6. The putative MGB site is underlined in the complement. N→C polarity of the CDPI3 peptide and structure of the C-terminal linker are shown. Fluorogenic probes were prepared using 5′-fluorescein (FAM) as the reporter dye. The quencher dye (TAMRA) was added using the pyrazolopyrimidine (PPA and PPG) linkers shown below.
Post-synthetic conjugation of ODNs with TAMRA
TAMRA NHS ester (Glen Research) was used to acylate the aminopropyl linkers in the fluorescent labeled ODNs according to the protocol supplied by the manufacturer. The resulting MGB-probes with two conjugated dyes were purified by PAGE using 8% polyacrylamide. The desired bands were excised and the gel slices were incubated overnight at 37°C in 10 ml of 100 mM Tris–HCl, 10 mM triethylammonium chloride and 1 mM EDTA (pH 7.8). The products were isolated from the extract by reverse phase HPLC, butanol concentration and sodium perchlorate precipitation. The pellets were dissolved in water and the concentrations were determined spectrophotometrically. A nearest-neighbor model (11) was applied to calculate extinction coefficients (ɛ260) of ODNs. A260 measurements were made in pH 7.2 PBS at ambient temperature and assumed a random coil DNA structure. For the conjugates and probes, extinction coefficients were calculated as a sum of ɛ260 for the ODN and the incorporated residues of DPI3 (68 000 M–1 cm–1), 6-FAM (22 800 M–1 cm–1) and TAMRA (34 000 M–1 cm–1).
UV melting studies
Hybrids formed between unmodified ODNs or MGB-probes and their complements were melted at a rate of 0.5°C/min on a Lambda 2S (Perkin-Elmer) spectrophotometer with a PTP-6 automatic multicell temperature programmer. Tm data reported in Figure 2 used 0.5× SSPE buffer (Sigma, pH 7.4). Tm data reported in Figure 6 used 20 mM Tris (pH 8.5) and 5 mM MgCl2. Complementary ODN strands (with or without mismatches) were designed for each probe and synthesized with short overhangs. Tm data reported in Figure 10 used 40 mM NaCl, 20 mM Tris (pH 8.7) and 5 mM MgCl2. Each ODN (1 µM of each strand) was mixed with its complement to give a 1:1 ratio. Prior to melting, samples were denatured at 100°C and then cooled to 10°C over a 10 min period. Melting temperatures of the hybrids were determined from the derivative maxima and are reported as Tm (±1°C). Mismatch discrimination for each type of duplex was calculated in terms of ΔΔG at 50°C using the equation:
Figure 2.

Effect of MGB on Tm of ODN1 with matched and mismatched targets. ODN1 was made with and without 3′-DPI3 ligand (±MGB). The minor groove binding region is underlined in the 23mer complement. The position of the mismatch is underlined. UV melting experiments with match or mismatch synthetic DNA complements were performed as described in Materials and Methods. Tm (match) and ΔTm (match–mismatch) are reported for each probe. A graphical representation of the UV melting experiments for the matched and mismatched duplex is shown below. The type and position of each mismatch is shown in relation to the MGB region.
Figure 6.

Sequence of PCR product, primers and fluorogenic probes. Inserts into pBK-CMV phagemid are shown in italics. Polymorphic base (match or mismatch) is in bold and underlined. The asterisk (*) indicates the modified base used to incorporate quenching dye. UV melting experiments with match or mismatch synthetic DNA complements were performed as described in Materials and Methods.
Figure 10.

Tm comparison of fluorogenic MGB probes and no-MGB ODNs. (A) Sequences of 10 fluorogenic 3′-MGB probes with variable G+C content and containing different types of mismatches. Location of the reporter dye (R) and TAMRA (*) are as indicated. ΔTm is shown for each MGB probe. (B) Tm of match and mismatch complements for sequences with representative (underlined) G+C content are plotted.
ΔΔG°50 = R × 323°K × ln(Kmatch/Kmismatch)
Kmatch and Kmismatch were determined using the relative fractions of duplex and single strands calculated from the melting curves at 50°C (12). A temperature of 50°C was chosen since it provided accurate values for both types of duplexes.
PCR
PCR assays with real-time fluorescence monitoring were performed in an Idaho Technology LC-24 LightCycler. The plasmid used in amplification was the LacZ gene (ATG:1183, TAA:799) in the 4518 bp pbk CMV phagemid (Stratagene), where the 1060–1083 bp region was substituted by TCTTTCTTCTTTTCTTTMAATTGCCC, where M was A (match) or C (mismatch). The PCR primer pair is shown in Figure 6. Each reaction contained PCR buffer (40 mM NaCl, 20 mM Tris–HCl, pH 8.9, 5 mM MgSO4, 0.05% bovine serum albumin), 125 µM each dNTP, 0.5 µM each primer, 0.1 µM fluorogenic MGB-probe, 0.5 U/10 µl Taq polymerase and 0.1 ng/10 µl plasmid DNA unless indicated otherwise. The cycling program was 50 cycles (or as indicated) of 2 s at 95°C, then 30 s at the indicated extension temperatures (55–70°C).
RESULTS AND DISCUSSION
Structure and synthesis of 3′-MGB ODNs and DNA duplex
3′-MGB ODNs with three different types of linker structures have been reported. Although post-synthetic modification of 3′-hexylamine ODNs has been used (2) we focus here on MGB-ODNs built from the convenient MGB CPG supports (2,4). The position of the linker affects the structure of the DNA duplex since the DPI3 tripeptide can be attached to the ODN at either the C- or N-terminus (Fig. 1). In one study (4) N-linked DPI3 had the same Tm as a C-linked CDPI3 and showed increased cooperativity during melting. The N-linked DPI3 structure was used for preparation of MGB-ODN1. The putative MGB site in the duplex was assigned based on analogy with the NMR structure (7).
Mismatch discrimination by 3′-MGB-ODNs
The 3′-MGB duplex system shown in Figure 1 was used for UV melting studies. A 15mer sequence (ODN1) was prepared ±3′-MGB and hybridized to a 23mer complement. As shown in Figure 2, Tm of the MGB duplex was 60°C whereas the unmodified DNA duplex melted at 42°C. This 18°C increase in Tm is not surprising since the A+T-rich region at the 3′-terminus of ODN1 provides a good MGB site. A series of mismatched 23mer targets were prepared which varied the type and position of mismatch. In general, mismatched duplexes formed with MGB-ODN1 were higher melting than the no-MGB control. For both types of duplexes (±MGB) ΔTm was affected by both the position and type of mismatch. Examination of the ΔTm data shows that mismatches within the minor groove binding region are especially destabilizing. For example, the T/G mismatch in the MGB region has ΔTm of 15°C in comparison to 6°C for the no-MGB control.
Since Tm is not a linear measure of thermodynamic stability (ΔG), we calculated the mismatch discrimination for each type duplex in terms of ΔΔG°50. The results are shown graphically in Figure 3. This analysis shows that mismatches under the MGB binding region were more easily discriminated (larger values of ΔΔG°50) for MGB-ODN1 in comparison to ODN1. The T/G mismatch under the MGB binding region gave almost a 3-fold increase in ΔΔG°50 (5.6 versus 2.0 kcal/mol). Molecular analysis showed that the 3′-linker to the ODN can allow the MGB to slide towards the 5′-end of the probe by an additional 1–2 bp to a better minor groove binding site. Thus it is not surprising that the A/C mismatch at the 3′-end of MGB-ODN1 gave smaller ΔΔG°50. It is not clear why the C/A mismatch at position 6 from the 3′-end of MGB-ODN1 gave smaller ΔΔG°50 or why the A/C mismatch at position 7 (just outside the putative MGB region) was so destabilizing. A detailed thermodynamic analysis of the position and type of mispairs in MGB ODNs is ongoing in our laboratory.
Figure 3.

Mismatch discrimination with ODN1 (±MGB). UV melting curves from the DNA duplexes shown in Figure 2 were used to calculate a free energy difference (ΔΔG°50) for each mismatch type and location. Mismatch discrimination for each duplex is shown graphically in relation to the MGB region.
5′-Nuclease PCR assay and the structure of fluorogenic MGB probes
Recently, quantitative ‘real-time’ PCR methods have been developed [5′-nuclease assay (13–15), hairpin probes (16) or primers (17) with native fluorescence quenching; hybridization-based resonance energy transfer probes (18); double strand-specific DNA dyes (19)]. These single tube methods eliminate post-PCR sample handling and the possibility of carry-over contamination, an important feature for high throughput testing. The use of fluorogenic MGB probes in the 5′-nuclease assay is shown in Figure 4.
Figure 4.
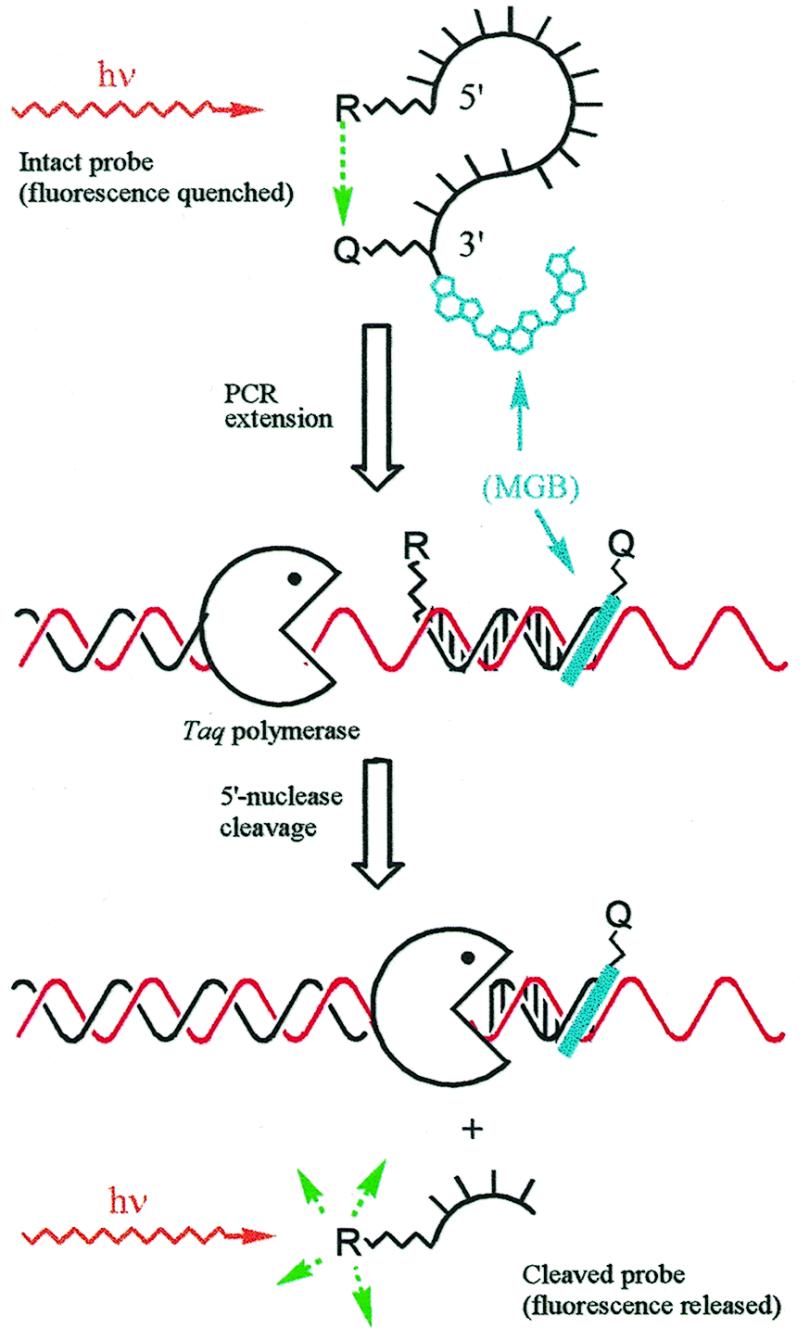
Real-time measurement of PCR using the 5′-nuclease fluorescence assay and MGB probes. MGB probes were prepared with 5′-reporter dye (R, fluorescein), internal quencher dye (Q, TAMRA) and 3′-MGB. Fluorescence of the intact probe is quenched by FRET. During the primer extension step, the hybridized probe is cleaved by the 5′ exonuclease activity of Taq polymerase and a fluorescent signal is released. Fluorescence of the cleaved probe during PCR is monitored in real time by instruments designed for this purpose. The MGB raises Tm, allowing shorter, more sequence specific probes to be used.
Fluorogenic probes for the 5′-nuclease assay have been developed using fluorescein (FAM) as a reporter dye and TAMRA as a fluorescent quencher dye (15,20). We used the same dyes for preparation of fluorogenic probes (±3′-MGB) as shown in Figure 5. The 12mer MGB duplex system illustrates the sequence used in the PCR model system (Fig. 6), and shows the putative MGB region and position of the mismatch. In contrast to the MGB duplex studied earlier (Fig. 1) the 3′-end of the probe was linked to the C-terminus of the DPI3 tripeptide and a carbamoyl group was added to the N-terminus (hence CDPI3). The CDPI3 linker system was used because the CPG intermediate was more efficiently prepared than the DPI3-CPG (prepared via peptide synthesis). Automated DNA synthesis used the MGB-CPG (or hexanol-CPG for no-MGB probes) and an aminopropyl-PPG or PPA phosphoramidite as indicated. The 5′-FAM was added as a phosphoramidite and the TAMRA quencher was added to the alkylamine linker in a post-synthetic conjugation reaction. All probes were purified by PAGE to ensure complete removal of free dye.
Model PCR system
Fluorogenic probes for the 5′-nuclease assay were designed to bind to the same strand in the center of an 81 bp PCR product. The model PCR system used two plasmid templates which varied by only a single base pair at the indicated probe position as shown in Figure 6. Probes with and without 3′-MGB were prepared. Each fully matched probe was designed to have a Tm value close to the optimal temperature (65–72°C) of the Taq polymerase extension step (21). MGB probes of length 12, 15 and 18 nt were synthesized with identical 3′-A+T-rich sequences and a mismatch five bases from the 3′-end. In order to function at PCR extension temperatures, no-MGB probes of length of 25 and 27 nt were required.
Probe hybridization properties
Melting temperatures of the duplexes formed from each probe and a complementary ODN strand are shown in Figure 6. As anticipated from our earlier research (2,6), addition of the MGB ligand to the 12–18mers gave significant enhancement in duplex stability with a Tm of 66–70°C. No-MGB probes with the same sequences had a Tm of 44–56°C. The shorter the probe, the greater the MGB contribution to the overall duplex stability. Interestingly, the no-MGB 27mer formed a duplex of about the same stability (Tm = 65°C) as the MGB 12mer (Tm = 66°C). For this 12mer sequence, coupling the MGB ligand near the A/T-rich 3′-end contributed as much stability as the addition of 15 bases to the 5′-end.
The most striking feature of the Tm data in Figure 6 is the increased mismatch discrimination by the MGB probes. The MGB 12mer probe had a 20°C difference between the matched and mismatched Tm values (ΔTm). No-MGB probes stable enough for the 5′-nuclease assay had a ΔTm of 4–6°C.
Comparison of probe performance in 5′-nuclease PCR assay
The probes shown in Figure 6 were compared for their ability to function in the 5′-nuclease assay. PCR was conducted with a two-step cycle, with annealing and extension at the same temperature. The relationship between probe Tm and primer extension temperature was studied by varying the extension temperature between 55 and 70°C. Ideally, the Tm of the probe with perfectly matched template should be slightly higher than the extension temperature, and the Tm of a probe with the single nucleotide mismatched template should be less. Because of the large ΔTm for the MGB probes, these criteria were easily met. Figure 7 compares the performance of the MGB 12mer and the no-MGB 27mer probes in the 5′-nuclease PCR assay. The real-time PCR fluorescence curves for the MGB 12mer probe showed excellent mismatch discrimination over a wide range of primer extension temperatures (55–70°C). In contrast, discrimination by the no-MGB 27mer probe was difficult to achieve. Some discrimination was seen at 67°C but the increase in fluorescence was low. At 65°C there was poor discrimination (data not shown) and at 70°C there was no signal.
Figure 7.

Effect of extension temperature on mismatch discrimination in 5′-nuclease PCR assay. Fluorogenic MGB 12mer and no-MGB 27mer probes were compared. PCR was performed as described in Materials and Methods. Extension temperature is indicated on top of the diagram columns. Each diagram shows two real-time PCR fluorescence curves with either match (upper curve) or mismatch (lower curve) plasmids as templates.
Another remarkable feature of the MGB probes is the low background fluorescence (at cycle 1). As seen in Figure 8, the background of the intact MGB probes slightly increased with probe length but was several times lower than either of the no-MGB probes. Quenching of FAM emission by fluorescence energy transfer (FRET) to TAMRA requires close proximity of the reporter and quencher dyes during the excited state lifetime of the reporter dye. The random coil structure of the probes in solution allows FRET despite the long distance between the dyes (15). The lower background observed for the MGB probes is presumably due to the short length but there may be other structural factors that contribute to more efficient quenching (22). As a result, the dynamic range of MGB probes in the real-time PCR fluorescence curves is much greater than with conventional no-MGB probes.
Figure 8.

Mismatch discrimination by various fluorogenic probes in 5′-nuclease PCR assay. Primer extension temperature was 65°C unless otherwise indicated. Each diagram shows two real-time PCR fluorescence curves with either match (upper curve) or mismatch (lower curve) plasmids as templates.
A useful term for discussion of probe performance in the 5′-nuclease assay is the signal-to-noise ratio (S/N) which we define as the fluorescent signal (at cycle 50) divided by the fluorescent background (at cycle 1). Clearly there are many variables which can affect the S/N of a probe, but for a specific PCR system S/N of a fully matched probe should be as large as possible. For example, the S/N for the MGB 15mer was >6 whereas optimized conditions for the no-MGB 25mer gave a S/N of ~1.5.
Sensitivity and quantitative performance of MGB probes
To determine the ultimate sensitivity of the MGB probes in the 5′-nuclease PCR assay, serial dilutions from one copy to 105 copies of the matched plasmid template were added to the PCR reactions, with a large excess of genomic DNA present. Figure 9 shows the results of this titration. Single copies of the target sequence could clearly be detected with absolute mismatch discrimination. The fluorescence curves are evenly spaced and the S/N is consistent, indicative of the quantitative nature of the assay. The data shown for one copy is typical for a positive result at this dilution. Since there is some probability that no target is present at this dilution, we repeated the experiment seven times and reproduced the same PCR curve five times (71% detection). This is close to the statistical estimate (Poisson distribution) of 67% for finding a single target at this dilution. The sensitivity is comparable to that reported for conventional fluorogenic probes in this assay (23).
Figure 9.

Titration of PCR template with genomic DNA background. 100 000 to 1 copies of the match plasmid per PCR reaction were detected using the MGB 15mer probe as described in Materials and Methods. 200 ng of herring sperm genomic DNA was added to each reaction. Fluorescence at cycle 1 was subtracted from each curve using the manufacturer’s software.
The presence of a large amount of genomic DNA (200 ng, or 4 × 104 copies) did not alter the sensitivity of the assay. Since the volume of sample in the Light Cycler was only 7 µl, this is equivalent, on a concentration basis, to the presence of almost 3 µg of genomic DNA in the larger 100 µl reaction volumes used by other thermal cyclers. This performance indicates there is no problem with non-specific interactions of MGB with highly complex DNA.
Design of MGB probes
To meet the high Tm requirements of PCR conditions, no-MGB probes vary substantially in length from 14 to 40mers depending on G+C content of the amplified DNA fragment. We found that for MGB probes this length variation is narrowed to a range of 12–20mers. To demonstrate this point, a group of MGB probes with G+C contents ranging from 17 to 69% were studied (Fig. 10). All of these 13–18mer MGB probes had a Tm of matched duplexes near 65°C. Consequently they showed excellent performance (S/N ratio) as well as mismatch discrimination in 5′ nuclease assays using the same temperature cycle (65°C annealing/extension temperature, data not shown).
No-MGB ODNs were also prepared for UV melting studies. They had good ΔTm values because of the length (13–18mers) but they are too short (Tm = 48–59°C) to satisfy PCR conditions. Addition of the MGB stabilized these ODNs, bringing them into a Tm range of 62–68°C. Addition of MGB to probes with an A+T-rich MGB site (#1 and #2) gave a larger increase in Tm than the probe with the G+C-rich MGB site (#8). This ‘Tm leveling effect’ provided by the MGB has been described earlier (6) and makes probe design easier. This in turn is critical for multiplexed PCR assays.
For the sequences shown in Figure 10, differences in ΔTm values for corresponding MGB probes and no-MGB ODNs did not exceed 20%. This was anticipated since many of these probes were designed to have mismatches located outside the MGB-binding region. However, enhanced mismatch discrimination was observed for probe #7 where the mismatched base was located within the MGB-binding region. ΔTm was increased from 6.5 to 11°C for a T/G mismatch. This mismatch was at the seventh position from the 3′-end of the probe. In contrast, the same mismatch at the eleventh position from the 3′-end of probe #2 showed no change in discrimination between MGB and no-MGB probes (ΔTm = 8.5°C). The MGB can slide towards the 5′-end of the probe by an additional 1–2 bp to a better minor groove binding site. Thus the mismatch at the seventh base pair from the 3′-end of the MGB probe could directly influence the structure of the MGB binding site. This is consistent with the results from the DPI3 model system (Fig. 2). In combination with the data from Figure 6, it seems clear that mismatches in or near the MGB site are more easily discriminated than mismatches in the ‘normal’ DNA duplex region of the probes.
CONCLUSIONS
The data presented here demonstrate improved hybridization properties of MGB-ODNs. The increased stability of DNA duplexes formed with MGB-ODNs allows use of shorter probes which are more sensitive to single base mismatches. Additional sequence specificity is provided when the mismatch is under the MGB site. To exploit these properties, fluorogenic 3′-MGB probes were prepared and studied in the 5′-nuclease PCR assay. The shorter length gave MGB probes better sequence specificity and lower fluorescent background in comparison to no-MGB probes. This makes MGB probes quite attractive for use in SNP detection and allelic discrimination. We are currently improving methods for synthesis of fluorogenic 3′-MGB probes, refining methods for Tm prediction of MGB-ODNs and determining the utility of MGB probes for other hybridization based assays such as DNA microarrays.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Dr Bill Andrews for preparation of the recombinant plasmids and to Mark Metcalf for technical assistance.
REFERENCES
- 1.Sinyakov A.N., Lokhov,S.G., Kutyavin,I.V., Gamper,H.B. and Meyer,R.B. (1995) J. Am. Chem. Soc., 117, 4995–4996. [Google Scholar]
- 2.Lukhtanov E.A., Kutyavin,I.V., Gamper,H.B. and Meyer,R.B.,Jr (1995) Bioconjug. Chem., 6, 418–426. [DOI] [PubMed] [Google Scholar]
- 3.Afonina I., Kutyavin,I., Lukhtanov,E., Meyer,R.B. and Gamper,H. (1996) Proc. Natl Acad. Sci. USA, 93, 3199–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lukhtanov E.A., Kutyavin,I.V. and Meyer,R.B. (1996) Bioconjug. Chem., 7, 564–567. [DOI] [PubMed] [Google Scholar]
- 5.Afonina I., Zivarts,M., Kutyavin,I., Lukhtanov,E., Gamper,H. and Meyer,R.B. (1997) Nucleic Acids Res., 25, 2657–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kutyavin I.V., Lukhtanov,E.A., Gamper,H.B. and Meyer,R.B. (1997) Nucleic Acids Res., 25, 3718–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar S., Reed,M.W., Gamper,H.B.,Jr, Gorn,V.V., Lukhtanov,E.A., Foti,M., West,J., Meyer,R.B.,Jr and Schweitzer,B.I. (1998) Nucleic Acids Res., 26, 831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boger D.L., Coleman,R.S. and Invergo,B.J. (1987) J. Org. Chem., 52, 1521–1530. [Google Scholar]
- 9.Gamper H.B., Reed,M.W., Cox,T., Virosco,J.S., Adams,A.D., Gall,A.A., Scholler,J.K. and Meyer,R.B. (1993) Nucleic Acids Res., 11, 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milesi D., Kutyavin,I., Lukhtanov,E.A., Gorn,V.V. and Reed,M.W. (1999) In Phillips,M.I. (ed.), Methods in Enzymology, Vol. 313. Academic Press, Orlando, FL, pp. 164–173. [DOI] [PubMed]
- 11.Cantor C.R., Warshaw,M.M. and Shapiro,H. (1970) Biopolymers, 9, 1059–1077. [DOI] [PubMed] [Google Scholar]
- 12.Lokhov S.G. and Pyshnyi,P.V. (1997) FEBS Lett., 420, 134–138. [DOI] [PubMed] [Google Scholar]
- 13.Holland P.M., Abramson,R.D., Watson,R. and Gelfand,D.H. (1991) Proc. Natl Acad. Sci. USA, 88, 7276–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee L.G., Connell,C.R. and Bloch,W. (1993) Nucleic Acids Res., 21, 3761–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Livak K.J., Flood,S.J.A., Marmaro,J., Giusti,W. and Deetz,K. (1995) PCR Methods Appl., 5, 357–362. [DOI] [PubMed] [Google Scholar]
- 16.Tyagi S., Bratu,D.P. and Kramer,F.R. (1998) Nat. Biotechnol., 16, 49–53. [DOI] [PubMed] [Google Scholar]
- 17.Nazerenko I.A., Bhatnagar,S.K. and Hohman,R.J. (1997) Nucleic Acids Res., 25, 2516–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bernard P.S., Lay,M.J. and Wittwer,C.T. (1998) Anal. Biochem., 255, 101–107. [DOI] [PubMed] [Google Scholar]
- 19.Ririe K.M., Rasmussen,R.P. and Wittwer,C.T. (1997) Anal. Biochem., 244, 1–7. [DOI] [PubMed] [Google Scholar]
- 20.Mullah B., Livak,K., Andrus,A. and Kenney,P. (1998) Nucleic Acids Res., 26, 1026–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barany F. (1991) Proc. Natl Acad. Sci. USA, 88, 189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonnet G., Tyagi,S., Libchaber,A. and Kramer,F.R. (1999) Proc. Natl Acad. Sci. USA, 96, 6171–6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lockey C., Otto,E. and Long,Z. (1998) Biotechniques, 24, 744–746. [DOI] [PubMed] [Google Scholar]