


Abstract
Here we describe a new procedure (cloning of polymorphisms, COP) for enrichment of single nucleotide polymorphisms (SNPs) that represent restriction fragment length polymorphisms (RFLPs). COP would be applicable to the isolation of SNPs from particular regions of the genome, e.g. CpG islands, chromosomal bands, YACs or PAC contigs. A combination of digestion with restriction enzymes, treatment with uracil-DNA glycosylase and mung bean nuclease, PCR amplification and purification with streptavidin magnetic beads was used to isolate polymorphic sequences from the genomes of two human samples. After only two cycles of enrichment, 80% of the isolated clones were found to contain RFLPs. A simple method for the PCR detection of these polymorphisms was also developed.
INTRODUCTION
Single nucleotide polymorphisms (SNPs) are the most frequent form of DNA polymorphism found in the human genome (1,2). Over the past years, microsatellite markers have almost completely replaced the use of restriction fragment length polymorphisms (RFLPs) (3), but with the identification of SNPs, the situation is now significantly different. SNPs, of which RFLPs represent a subclass, have a number of advantages over microsatellite markers (3,4). For example, they are more abundant, more evenly spaced and more stably inherited.
There is increasing agreement that SNPs will be a key element in finding disease genes involved in complex traits. They will also be extremely useful for understanding human evolution, population genetics, pharmacogenetics, etc. Many methods have been identified for the detection and analysis of SNPs (1–3,5,6), but an optimal method for the isolation of previously unknown SNPs has yet to be developed (7). The most useful and largely exploited method for SNP isolation uses the sequence data generated by the human genome and expressed sequence tag (EST) projects (1,2,7,8). However, this approach has some obvious limitations. For example, it cannot be applied to many non-human species.
Development of a simple, reliable and efficient method for isolation of unknown SNPs is therefore highly desirable (7). Here we describe a new procedure for simple and robust enrichment, cloning and testing of SNPs that represent RFLPs. In this procedure DNA A and DNA B, isolated from two individuals, were digested with BamHI and PCR amplified (DNA A in the presence of dUTP). After subtractive hybridization, homohybrids A, heterohybrids and single-stranded DNA were destroyed with uracil-DNA glycosylase (UDG) and mung bean nuclease. Homohybrids B representing RFLPs were purified and cloned. A PCR method was developed for detection of these RFLPs. This method is based on differential PCR amplification of RFLP DNA segments having different length and exploits agarose gel electrophoresis to detect polymorphisms.
MATERIALS AND METHODS
General methods
The isolation of genomic DNA, restriction enzyme digestion and DNA ligation procedures were performed as previously described (9).
Cloning, Southern blotting and hybridization were performed as previously described (9). Sequencing was undertaken using an ABI 310 sequencer (Perkin Elmer), according to the manufacturer’s instructions.
Oligonucleotides were from Life Technologies (Gibco BRL).
The cloning of polymorphisms (COP) procedure
DNA was extracted from blood samples taken from two individuals (A and B). Both DNA samples (2 µg) were digested with BamHI, BglII and BclI (20 U each) at 37°C for 5 h, followed by enzyme inactivation at 65°C for 20 min. Ligation of 0.5 µg of the digested DNA with 20 M excess of the linker, Blsubtr1/2, was performed at room temperature (see Table 1).
Table 1. List of primers and linker used.
| Primer/linker | Size (nt) | Primer and linker sequences |
|---|---|---|
| Blsubtr1/2 | 36 | 5′-GATCCGCGGCCGCGGTCCCAAAAGGGTCAGTGCTGG-3′ |
| 33 | 5′-GCGCCGGCGCCAGGGTTTTCCCAGTCACGACCC-3′ | |
| Antiuniv | 21 | 5′-CAGCACTGACCCTTTTGGGAC-3′ |
| PBsub | 21 | 5′-bio-CAGCACTGACCCTTTTGGGAC-3′ |
| SNP1F | 20 | 5′-TCCATGAAGTCAGGGGTTTG-3′ |
| SNP1R | 20 | 5′-TGAGGAACCTGGAGACCAAA-3′ |
| SNP2F | 20 | 5′-CCCATAAGGGTCAGTGCTGA-3′ |
| SNP2R | 20 | 5′-CCAAGTTGGTCATCCTCCCT-3′ |
| SNP3F | 20 | 5′-TTCTCACCTTGTCGAAAGCA-3′ |
| SNP3R | 20 | 5′-CGGTGTCGTGTCTGAAACAT-3′ |
Following ligation, PCR of DNA B was performed in a 100 µl solution containing 67 mM Tris–HCl (pH 9.1), 16.6 mM (NH4)2SO4, 1.0 mM MgCl2, 0.1% Tween 20, 200 µM each of the four dNTPs, 100 ng DNA, 400 nM biotinylated PBsub primer (Table 1) and 5 U of Taq polymerase. PCR of DNA A was performed in 20 tubes using the same PCR conditions as described for DNA B, with the following exceptions: the concentration of MgCl2 was increased to 2.5 mM, dUTP (300 µM) was used instead of dTTP and Antiuniv primer (as PBsub but without biotin) was used in place of PBsub.
The PCR cycling conditions were 72°C for 5 min, followed by 30 cycles of 95°C for 40 s, 60°C for 45 s and 72°C for 1.5 min, with a final extension at 72°C for 5 min. PCR-amplified DNA A (1000 µl) was mixed with 10 µl of PCR-amplified DNA B. This mixture was purified with a JETquick PCR Purification Spin Kit (Genomed Inc.), concentrated with ethanol and dissolved in 10 µl H2O.
After denaturation at 100°C for 8 min, the first hybridization was performed for 40 h in 18 µl buffer containing 0.4 mM NaCl, 100 mM Tris–HCl (pH 8.5) and 1 mM EDTA. The mixture was then diluted to 200 µl and extracted with an equal volume of chloroform:isoamyl alcohol (24:1) to remove the mineral oil. The mixture was treated with 30 U UDG (Boehringer Mannheim) for 4 h at 37°C in buffer containing 70 mM HEPES–KOH (pH 7.4), 1 mM EDTA and 1 mM dithiothreitol. The DNA was then concentrated with ethanol and dissolved in 25 µl TE buffer (10 mM Tris–HCl, pH 8.0, and 0.1 mM EDTA). Then, 3 µl 10× MBN buffer (30 mM sodium acetate, pH 4.6, 50 mM NaCl, 1 mM zinc acetate and 0.001% Triton X-100) and 20 U of mung bean nuclease (Boehringer Mannheim) were added and the sample was incubated at 37°C for 30 min. The reaction was terminated by the addition of EDTA to a final concentration of 1 mM.
The resulting product was purified with streptavidin-coupled Dynabeads M-280 (Dynal AS, Oslo, Norway), according to the manufacturer’s instructions, and dissolved in 20 µl TE. PCR amplification of 0.5 µl of this DNA sample was performed as described above for DNA B. After PCR, 0.1 µg of this DNA was mixed with 10 tubes of PCR-amplified DNA A and the hybridization and treatment with UDG and mung bean nuclease was repeated.
The final product was then PCR amplified, purified with a JETquick PCR Purification Spin Kit and cloned into pCR·4-TOPO using the TOPO™TA cloning kit (Invitrogen).
PCR detection of polymorphisms
Partial sequencing of clones 1–3 was performed using M13 reverse primer in an ABI 310 sequencer (Perkin Elmer) according to the manufacturers’ protocols.
Genomic DNA was completely digested with BamHI or BglII and self-ligated at low concentration (4 µg/ml) overnight.
PCR amplification was performed using 100 ng of the ligation product. The sequences of the PCR primers used for detection of the polymorphisms are shown in Table 1. The primers were designed using the Primer 3 program (http://www-genome. wi.mit.edu/cgi-bin/primer/primer3_www.cgi ) and the PCR conditions were selected using a program developed by Breslauer et al. (10; http://alces.med.umn.edu/rawtm.html ).
RESULTS AND DISCUSSION
An overview of the COP procedure is shown in Figure 1. DNA A and B obtained from two individuals were digested with BamHI, BglII and BclI, and Blsubtr1/2 linkers (Table 1) were ligated to the digested DNA fragments. DNA A was amplified using dUTP and an unmodified primer, and DNA B was amplified with a biotinylated primer, but with normal dNTPs. The PCR products (mainly in the range 0.5–2 kb) were denatured, hybridized at a ratio of 1:100 DNA B to A, and then treated with UDG to destroy all the DNA originating from sample A and with mung bean nuclease to digest the single-stranded DNA and all non-perfect hybrids. The resulting sample B homohybrids were purified and concentrated with streptavidin beads. The cycle was then repeated. The final DNA product was then repurified, amplified and cloned into an appropriate vector.
Figure 1.

Flow chart of the COP procedure for the analysis of DNA A and B from two individuals. R, recognition site of a restriction endonuclease; b, biotin.
This procedure was applied to DNA isolated from two different people of the same race, and a number of clones were obtained from the transformation. DNA from 10 random clones was isolated and the inserts were analyzed by Southern blot hybridization for the presence of polymorphisms (Fig. 2). Eight clones were clearly polymorphic and two clones were not. These results indicate a good selectivity of the COP procedure.
Figure 2.

Hybridization of the isolated recombinant clones to the Southern blots with DNA isolated from individual A (lanes 1, 3, 5, 7, 9, 11, 13, 15, 17 and 19) and B (lanes 2, 4, 6, 8, 10, 12, 14, 16, 18 and 20). DNA was digested with the restriction enzyme indicated in the figure.
SNPs that represent RFLPs can be identified by Southern blotting. However, genomic DNA is not always available in quantities sufficient for Southern blotting analysis and PCR is easier to perform. We wished to develop a straightforward procedure for testing for the presence or absence of SNPs in the clones generated in this study. The procedure we used was based on identification of the different sized polymorphic fragments generated by restriction enzymes. We selected three polymorphic clones, sequenced the inserts in these clones, and designed primers for detection of the polymorphisms. These primers worked in opposite directions.
Genomic DNA was digested with BamHI or BglII, and after heat inactivation of the enzyme, the solution was diluted 20 times and self-ligated. PCR amplification was performed as described in Materials and Methods. As shown in Figure 3, all three primers detected SNPs.
Figure 3.
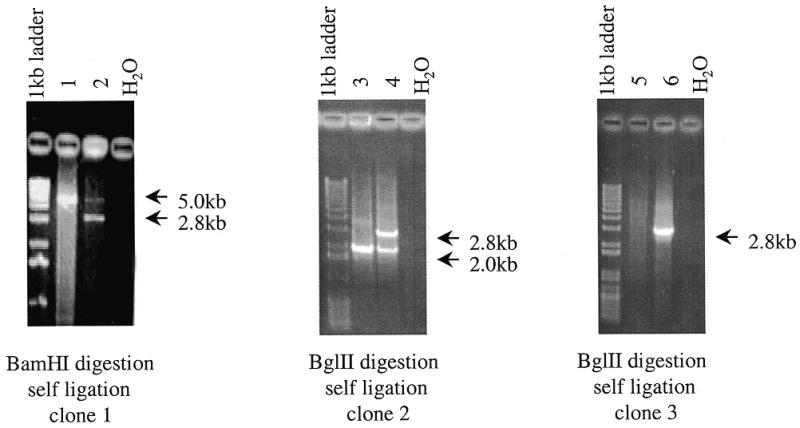
Detection of polymorphic sequences in DNA A and B using PCR. PCR conditions are described in Materials and Methods. Electrophoresis was performed in 1% agarose gels.
Figure 1 does not reflect all the complex reactions involved in COP that enrich for RFLP fragments. Importantly, polymorphic fragments are PCR enriched in COP because of their relatively small size. However, digestion with restriction enzymes generates two fragments from a single RFLP fragment. This may increase the probability of forming homodimers, partially from the replacement of a short plus strand with a longer plus strand in a duplex originally comprised of a long minus strand and a short plus strand. This duplex formed by two long strands will be more stable than the duplex formed by short and long strands.
Two scenarios probably play a major role in RFLP enrichment. Firstly, enzyme digestion of one long DNA fragment of A (e.g. 15 kb) may generate a long fragment (A1) (e.g. 14.5 kb) and a very short fragment (A2) (e.g. 0.5 kb). After the first step of PCR amplification, only fragment A2 would be present in amplified DNA (Fig. 4A). Clone 3 in Figure 3 demonstrates this case. Another situation would be if enzyme digestion generated two short fragments (e.g. 0.5 and 0.4 kb) from a medium DNA fragment (e.g. 0.9 kb). In this case, all three fragments would be PCR amplified on the first step and would therefore participate in the subsequent reactions (Fig. 4B). Clone 1 in Figure 3 is an example of this situation. Clone 2 in Figure 3 reflects the same situation but the polymorphic fragment is longer.
Figure 4.


General schemes (A and B) explaining PCR detection of polymorphic sequences. R, recognition site of a restriction endonuclease. Dashed lines denote sequences produced with reverse primer (see Materials and Methods), small arrows indicate localization of primers used for the PCR amplification.
In both scenarios, after the first PCR amplification, the complexity of the DNA would be significantly decreased and the proportion of polymorphic (mainly shorter) fragments would be dramatically increased.
In this study, three enzymes (BamHI, BglII and BclI) were used for the digestion of human DNA. These enzymes would generate fragments of an average size of ~1500 bp. Sau3A, which generates restriction fragments with an average size of 250–300 bp, could have been used in place of these enzymes.
If we assume that two equivalent chromosomes have on average one SNP per 1000 bp DNA (2), then at least 10 000 polymorphic fragments could be cloned using the COP procedure described in this study. In the calculation we assume that the size of the human genome is 2.5 × 109 bp and roughly 1500 bp of human DNA contain a site for one of the three enzymes used in the study, i.e. BamHI, BclI or BglII. Using Sau3A (one site per 250 bp), 40 000 fragments could be cloned. Using all combinations of restriction enzymes with a 4 bp recognition site (4 bp cutters), including enzymes recognizing multiple and non-palindromic sequences, practically all SNPs could be identified and cloned by this method. Using only two 4 bp cutters having CG pair(s) in the recognition site, cloning of at least 80 000 SNPs, located mostly in gene-rich regions, could be achieved. This effort would be comparable in productivity to SNP generation by EST cloning and sequencing programs. The importance of this type of SNP is that they may locate not only in expressed but also in promoter/enhancer regions.
It is worthwhile mentioning that the COP procedure itself results in enrichment of polymorphic sequences. Actually, to detect SNPs, another step, sequencing of the polymorphic clone, is needed.
We think COP will be used predominantly as a complementary method to other approaches for SNP detection. For example, the most practical method for detecting the most common polymorphisms in the human genome will continue to be sequencing of the whole human genome. Similarly, detection of SNPs in specific genes is likely to involve direct sequencing of genes from different individuals.
We suggest that this procedure could be used to isolate SNPs from particular regions of the genome. In this case, DNA B would originate from individual or contigs of YAC, PAC or BAC clones from the region of interest. Other obvious applications for the developed method would be generation of SNPs in CpG islands (11) using CG-recognizing enzymes and in different human populations and for non-human organisms. Use of the COP procedure to clone markers from regions that have lost heterozygosity could result in the isolation of tumor suppressor genes or could be used to detect rearranged immunoglobulin loci (12). Another application could be ‘chromosome landing’ (13,14) to facilitate positional cloning in organisms for which high resolution maps have not been developed. The efficiency of the COP procedure could be increased, if necessary, by adding one more cycle of enrichment or by omitting PCR amplification after the first cycle of enrichment.
The COP procedure resembles ‘RFLP subtraction’ in the cloning RFLP using subtractive procedure (12,13). Otherwise, however, these procedures are very different in the biochemical techniques used to clone the RFLPs and the results. RFLP subtraction is a significantly more complicated and laborious method. In addition to the multiple (three to four cycles) ‘classical’ sub-tractive hybridization steps it uses gel purification, a reassociation step to remove poorly hybridizing DNA, sub-traction based on representational difference analysis (15) and multiple combinations of linkers and PCR primers. The most effective enrichment steps in the COP procedure (with UDG and mung bean nuclease) are not used in RFLP subtraction at all. RFLP subtraction results in the cloning of RFLP segments that are present in one DNA sample and absent in the other. However, the COP procedure yields DNA fragments that are heterozygous in one DNA sample but homozygous in the other. Because of these distinctive properties, we think that the COP procedure will find wide application in studies of genetic diversity.
Acknowledgments
ACKNOWLEDGEMENTS
The authors are grateful to Dr A. J. Brookes for fruitful discussions and valuable advice. This work was supported by research grants from the Swedish Cancer Society, Pharmacia & Upjohn, Karolinska Institute, Ingabritt och Arne Lundbergs Forskningsstiftelse and the Åke Wiberg Foundation. V.Z. was the recipient of a fellowship from the Concern Foundation in Los Angeles and the Cancer Research Institute in New York.
REFERENCES
- 1.Gu W., Aguirre,G.D. and Ray,K. (1998) Biotechniques, 24, 836–837. [DOI] [PubMed] [Google Scholar]
- 2.Brookes A.J. (1999) Gene, 234, 177–186. [DOI] [PubMed] [Google Scholar]
- 3.Landegren U., Nilsson,M. and Kwok,P.Y. (1998) Genome Res., 8, 769–776. [DOI] [PubMed] [Google Scholar]
- 4.Weiss K.M. (1998) Genome Res., 8, 691–697. [DOI] [PubMed] [Google Scholar]
- 5.Lyamichev V., Mast,A.L., Hall,J.G., Prudent,J.R., Kaiser,M.W., Takova,T., Kwiatkowski,R.W., Sander,T.J., de Arruda,M., Arco,D.A., Neri,B.P. and Brow,M.A. (1999) Nature Biotechnol., 17, 292–296. [DOI] [PubMed] [Google Scholar]
- 6.Gilles P.N., Wu,D.J., Foster,C.B., Dillon,P.J. and Chanock,S.J. (1999) Nature Biotechnol., 17, 365–370. [DOI] [PubMed] [Google Scholar]
- 7.Kwok P.Y. and Chen,X. (1998) Genet. Eng., 20, 125–134. [DOI] [PubMed] [Google Scholar]
- 8.Taillon-Miller P., Gu,Z., Li,Q., Hillier,L. and Kwok,P.Y. (1998) Genome Res., 8, 748–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sambrook J. Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 10.Breslauer K.J., Frank,R., Blocker,H. and Marky,L.A. (1986) Proc. Natl Acad. Sci. USA, 83, 3746–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bird A.P. (1987) Trends Genet., 3, 342–347. [Google Scholar]
- 12.Rosenberg M., Przybylska,M. and Straus,D. (1994) Proc. Natl Acad. Sci. USA, 91, 6113–6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corrette-Bennett J., Rosenberg,M., Przybylska,M., Ananiev,E. and Straus,D. (1998) Nucleic Acids Res., 26, 1812–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young N.D. and Phillips,R.L. (1994) Plant Cell, 6, 1193–1195. [Google Scholar]
- 15.Lisitsyn N., Lisitsyn,N. and Wigler,M. (1993) Science, 259, 946–951. [DOI] [PubMed] [Google Scholar]