


Abstract
Protein–protein interactions have been widely used to study gene expression pathways and may be considered as a new approach to drug discovery. Here I report the development of a universal protein array (UPA) system that provides a sensitive, quantitative, multi-purpose, effective and easy technology to determine not only specific protein–protein interactions, but also specific interactions of proteins with DNA, RNA, ligands and other small chemicals. (i) Since purified proteins are used, the results can be easily interpreted. (ii) UPA can be used multiple times for different targets, making it economically affordable for most laboratories, hospitals and biotechnology companies. (iii) Unlike DNA chips or DNA microarrays, no additional instrumentation is required. (iv) Since the UPA uses active proteins (without denaturation and renaturation), it is more sensitive compared with most existing methods. (v) Because the UPA can analyze hundreds (even thousands on a protein microarray) of proteins in a single experiment, it is a very effective method to screen proteins as drug targets in cancer and other human diseases.
INTRODUCTION
Gene expression in eukaryotic cells is controlled by numerous fundamental and selective protein–protein, protein–DNA, protein–RNA and protein–ligand interactions. Cancer, as well as other genetic diseases, results from abnormal gene expression. Interactions of proteins with proteins and other biomolecules play a pivotal role in almost every aspect of gene expression. Therefore, factors involved in these interactions, including transcription factors, signal transduction factors, growth factors and the products of other oncogenes, tumor suppressor genes, viral genes and many cellular genes, have been implicated as potential targets for new drugs (1–4).
Use of transcription factors has proved to be a successful means to identify new drug targets in cancer and other human disease. The basal transcription machinery of class II genes consists of at least six general transcription factors, including TFIIB, TFIID, TFIIE, TFIIF, TFIIH and RNA polymerase II. However, an additional activator(s) and coactivator(s) are required for regulated (activated) transcription (5,6). Both basal and activated transcription are controlled largely through protein–protein interactions between transcription factors and through protein–DNA interactions. Thus, insight into factor communication holds not only the key to understanding mechanisms of gene regulation, but also provides a means of understanding mechanisms of pathogenesis and of identifying anticancer drugs. At present, in addition to the two-hybrid system and co-immunoprecipitation assays usually used to detect protein–protein interactions in vivo, the glutathione S-transferase (GST) pull-down assay is probably the most common method to determine specific protein–protein interactions in vitro. Cross-linking, gel mobility shift, footprinting and others have been often used to study protein–DNA and protein–RNA interactions (7,8). Recently, several methods, including serial analysis of gene expression (SAGE) (9), cDNA microarrays (10) and oligonucleotide-based DNA chips (11), have been employed to study the relationship between gene expression and cancer and have made significant contributions to our understanding of the mechanism of tumorigenesis. However, our knowledge of which trans-acting factors are involved and how they change gene expression patterns is still limiting due to the lack of efficient and reproducible techniques to examine intermolecular communications.
Based on the biological function of trans-acting factors, I have developed a universal protein array (UPA) system that can be used to effectively and quantitatively determine protein interactions with other biomolecules. It can be used in most molecular biology and biochemistry laboratories to study protein–protein, protein–DNA, protein–RNA and protein–ligand interactions involved in gene expression pathways, including transcription, RNA processing, replication, translation, signal transduction and others. It can also be used in biotechnology and pharmaceutical companies to screen new drugs and as a commercial product available to the market.
MATERIALS AND METHODS
Preparation of the UPA
General transcription factors, activators and coactivators were overexpressed either in bacteria, baculovirus or in mammalian cells and purified to near homogeneity as previously described (12–16). The serine–arginine (SR) protein fraction was prepared from HeLa cell nuclear extracts essentially according to Zahler et al. (17). GST–nucleolin fusion protein was prepared by overexpressing plasmid GST-HNB (provided by Dr M. Srivastava), which contains nucleolin coding sequence positions 290–707, in bacteria and purified on a glutathione–Sepharose column.
An average of 7.5 pmol (normalized by Bio-Rad protein assay) of each of the 48 highly purified proteins (or fractions) was spotted on a 12 × 8 cm nitrocellulose membrane using a 96-well dot blot apparatus (Bio-Rad). Each sample was diluted to 100 µl with buffer A100 (100 mM KCl, 10% glycerol, 20 mM HEPES Na pH 7.9, 0.2 mM EDTA, 10 mM 2-mercaptoethanol and 0.5 mM PMSF) and duplicated in two adjacent wells. Each well was rinsed with 2 × 500 µl buffer A100 and the vacuum kept for 3–5 min. The protein array was then rinsed with two changes of buffer A100.
Interaction with a protein probe
Purified GST-K–p52 protein (18) was labeled by heart muscle kinase (HMK) in a 50 µl reaction containing 10 µg of substrate protein, 40 µCi [γ-32P]ATP and 10 U of the catalytic subunit of Ca-independent protein kinase A from bovine heart (Sigma) at 30°C for 30 min. The 32P-labeled protein was purified through glutathione–Sepharose beads to separate uncoupled free nucleotide. In the case of the ASF/SF2 probe, pET11a-6H(K)-ASF/SF2 was created by inserting the ASF/SF2 coding region into the vector pET11a-6H(K) (18) and overexpressed in Escherichia coli cells. Recombinant protein was affinity purified and labeled by HMK in vitro as described above. Pre-treatment took place in buffer A100 containing 1% non-fat milk at room temperature for at least 30 min. The array was then incubated with 30–50 ng probe/ml buffer A100 (+1% milk) at 4°C for over 12 h. After incubation, the array was sequentially washed with three changes of buffer A100 (100 mM KCl), A500 (500 mM KCl) and A1000 (1000 mM KCl). The resulting signals were visualized by autoradiography (exposure from 30 min to 10 h) and quantified with a densitometer (Molecular Dynamics).
Interaction with a DNA probe
A double-stranded (ds) oligonucleotide (64 bp with plus strand 5′-AGGGGGGCTATAAAAGGGGGTGGGGGCGCGTTCGT-CCTCACTCTCTTCCGCATCGCTGTCTGCG and minus strand 5′-CCCTCGCAGACAGCGATGCGGAAGAGAG-TGAGGACGAACGCGCCCCCACCCCCTTTTATAGCCC) corresponding to the adenovirus major late promoter region from –39 to +29 was labeled at the 3′-end of the minus strand with Klenow fragment in the presence of [32P]dCTP. After labeling, the free nucleotides were separated from the probe by passing the labeling reaction through a G-50 nick column (Pharmacia). Pre-treatment took place with buffer A containing 60 mM KCl, 2× Denhardt’s solution and 25 µg/ml poly(dG·dC) (Sigma) at room temperature for 30 min. For interaction, 5 ng/ml of 32P-labeled double-stranded (ds)DNA was added to the same buffer and incubation was carried out at 4°C for >12 h. The array was then sequentially washed with three changes of buffer A100, A500 and A1000 followed by autoradiography and quantification. When the array was analyzed with a single-stranded (ss)DNA probe, the 64mer minus strand of the dsDNA probe was labeled at the 5′-end by T4 polynucleotide kinase in the presence of [γ-32P]ATP. Other conditions were exactly the same as those for the dsDNA probe.
Interaction with a RNA probe
An SV40 early pre-mRNA was synthesized in vitro from the plasmid pSVi66 by SP6 RNA polymerase as previously described (18). Interaction was carried out at 4°C for >12 h in the presence of 20 mM HEPES Na pH 7.9, 5% glycerol, 10 mM 2-mercaptoethanol, 0.2 mM EDTA Na pH 8.0, 60 mM KCl, 2 mM MgCl2, 0.5 mg/ml BSA, 25 µg/ml tRNA and ~5 ng/ml 32P-labeled SV40 early pre-mRNA. The array was then sequentially washed and visualized by autoradiography as described for the DNA probe.
Interaction with a ligand probe
l-3,5,3′-[125I]Triiodothyronine (T3) was purchased from NEN (catalog no. NEX110H). The interaction conditions were essentially the same as for the RNA probe except that tRNA was omitted and 0.3 µCi/ml [125I]T3 was added instead of the RNA probe.
Removal of the probe from the array
The same universal protein array was reused with a protein probe, a dsDNA probe, a ssDNA probe, a RNA probe and a ligand probe. After each use, the filter was stripped with buffer A containing 1 M (NH4)2SO4 and 1 M urea at room temperature for 30–60 min. Then the stripped array was equilibrated with buffer A100 before being incubated with another probe.
RESULTS
Design of a 96 protein array
A novel human protein, p52, has recently been identified as a general transcriptional coactivator capable of potentiating activated transcription of class II genes and as a splicing regulator capable of mediating the splicing of an SV40 early pre-mRNA in vitro (18,19). To identify target proteins for the transcriptional coactivator p52, I developed a protein array system to analyze a specific target(s). Forty-eight individual highly purified proteins, including general transcription factors, activators and coactivators, as well as several RNA processing factors (Tables 1 and 2), were analyzed simultaneously. Glutathione S-transferase fused to a HMK site (RRASV) (GST-K) (18) was used as a negative control in the experiments. Most proteins were overexpressed in either bacteria or in baculovirus and purified to near homogeneity. An average of 7.5 pmol of each protein (or protein fraction) was diluted to 100 µl with buffer A100 and spotted onto a nitrocellulose filter with a 96-well dot blot apparatus. Each sample was duplicated in two adjacent wells to provide a useful internal control.
Table 1. The name and position of each of 48 proteins arrayed on a nitrocellulose membrane with a 96-well dot blot apparatus.

Each sample was duplicated in two adjacent wells. The actual size of the membrane is 12 × 8 cm (height × width) with eight columns and 12 rows.
Interaction with a protein probe
GST-K–p52 was labeled in vitro with [γ-32P]ATP by HMK (18) and further purified through glutathione–Sepharose beads. The protein array was first treated with buffer A100 containing 1% non-fat milk and then incubated with 32P-labeled GST-K–p52 as described in Materials and Methods. The filter was extensively washed with buffer A containing 100, 500 and 1000 mM KCl prior to each autoradiographic analysis. A low salt wash (with 100 mM KCl) allowed the detection of most possible interactions (Fig. 1A), while a high salt wash (with 500–1000 mM KCl) allowed the detection of highly specific and high affinity interactions (Fig. 1B). No significant difference was found between the 500 and 1000 mM salt washes. The relative affinity of each tested protein for the probe could be measured with either a densitometer or a phosphorimager (Fig. 1C). Among all 48 proteins (or fractions), the SR protein fraction (12c/d) and the recombinant GST–nucleolin (12e/f) had the highest affinities for the transcriptional coactivator p52.
Figure 1.
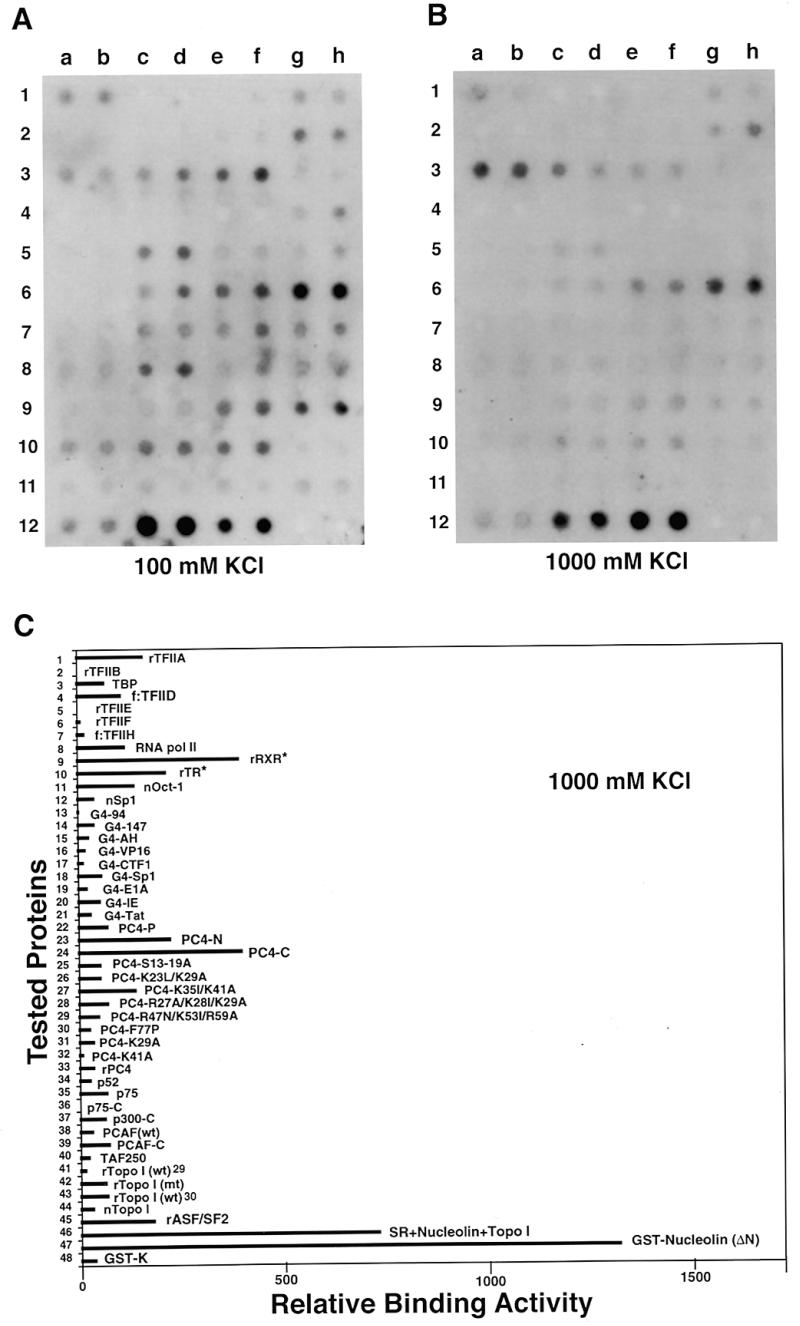
Protein–protein interactions monitored with the use of a universal protein array. (A) Autoradiographic signals detected from the array that was incubated with the 32P-labeled GST-K–p52 and washed with buffer A100. (B) Signals detected after washing the array (from A) with buffer A1000. (C) Quantitative representation of relative affinities (in reading units from a densitometer) of 48 proteins for the transcriptional coactivator p52 after washing with 1000 mM KCl.
We previously showed that in addition to the ability to interact specifically with a 34 kDa doublet corresponding to the splicing factor ASF/SF2, p52 could also interact strongly with a 100 kDa protein found to be present in the SR fraction by far-western blot analysis (18). Protein microsequence analysis indicated that the 100 kDa band isolated from the SR protein fraction contained two proteins, nucleolin and DNA topoisomerase I (topo I) (data not shown). In the present experiment, p52 strongly interacted with the recombinant GST–nucleolin but not with topo I, either recombinant proteins expressed in baculovirus (Fig. 1A, 11a–f) or naturally purified protein from mammalian cells (11g/h). This observation demonstrates that p52 interacts with the nucleolin rather than the topo I present in the SR protein fraction, which is consistent with our recent observation that nucleolin is a component of the multiprotein complex associated with p52 in HeLa cells (Y.Z.Si, Y.M.Zhao and H.Ge, unpublished data).
Nucleolin has been implicated in regulating pre-rRNA processing (20), pre-mRNA splicing (21), B cell-specific transcription (22), unwinding DNA, RNA or DNA–RNA duplexes (23) and mediating cell doubling time in human cancer cells (24). Like the splicing factor ASF/SF2, nucleolin also contains RNP type RNA-binding domains as well as RGG repeats (20,25). Its activity can be modulated through mitosis-specific phosphorylation by p34cdc2 kinase or casein kinase II (23). Therefore, it would be interesting to further examine the biological significance of nucleolin interaction with the general transcriptional coactivator and splicing regulator p52.
Interaction with other probes
To test whether this protein array system could also be used to detect interactions with other biological molecules, the same array was stripped (see Materials and Methods) and reprobed with a 32P-labeled double-stranded oligonucleotide (64 bp) containing the adenovirus major late core promoter elements (see Materials and Methods). The results shown in Figure 2A indicate that, after washing with 500 mM salt, phosphorylated PC4 (PC4-P, 6c/d), an inactive form of a previously described transcriptional coactivator (26), purified from HeLa cells had the highest affinity for the tested dsDNA probe among 48 samples (see quantification in Table 2). PC4-P had 3- to 5-fold higher affinity for dsDNA compared to other PC4 derivatives, including wild-type PC4 (9a/b). In contrast, a single amino acid change at position 77 (Phe→Pro) completely abolished the dsDNA binding ability of PC4 (8c/d). These results are in agreement with the observations reported recently using gel mobility shift assays that phosphorylated PC4 bound bubble DNA with higher affinity and the region around position 77 was critical for the DNA-binding activity of PC4 (27).
Figure 2.

Protein–DNA, protein–RNA and protein–ligand interactions monitored with the use of the universal protein array. Autoradiographic signals detected from the same array which was repeatedly stripped and incubated with the 32P-labeled dsDNA probe (A), the ssDNA probe (B), the RNA probe (C) and the 125I-labeled ligand T3 (D) after the array was washed with 500 mM KCl.
Table 2. Quantitative indication of relative affinities of 48 tested proteins in the UPA to the transcriptional coactivator p52, the dsDNA, the ssDNA and the SV40 early pre-mRNA.

The number, position, name/source (and related reference), affinities for each probe and known function of each of the 48 proteins are indicated. The highest affinities of the individual proteins for each probe [GST–nucleolin for p52 (12e/f), PC4-P for the dsDNA (6c/d), SR for the ssDNA (12c/d) and PC4-m4 for the RNA (8a/b)] were normalized to 100 and are indicated in bold.
Although it is known that TBP can specifically bind the present probe, the signal is relatively weak compared to other DNA-binding proteins. This result is consistent with the observation from gel mobility shift assays that the binding activity of TBP to TATA box-containing DNA was barely detectable. However, it can be significantly enhanced by the presence of another transcription factor, TFIIA (5). On the other hand, however, many other general (non-sequence-specific) DNA-binding proteins had much stronger signals than TBP, suggesting that the present system may not be suitable for determining the binding activity of sequence-specific DNA-binding (and/or RNA-binding) proteins. ASF/SF2 was identified as an RNA-binding protein playing an essential role(s) in pre-mRNA splicing. Both the recombinant ASF/SF2 (12a/b) and the native ASF/SF2-containing SR protein fraction (12c/d) bound dsDNA as well as ssDNA (see below) very strongly, even tighter than most of the DNA-binding proteins tested (see quantification in Table 2), indicating that ASF/SF2 is also a DNA-binding protein. After the array was analyzed with a ssDNA probe (Fig. 2B), although several differences were observed, the overall pattern of protein–ssDNA interactions was similar to that of protein–dsDNA interactions, suggesting that most DNA-binding proteins are capable of binding both dsDNA and ssDNA.
This protein array system was also used successfully to analyze interactions with an RNA probe transcribed from the SV40 early region-containing plasmid pSVi66 (18) and a 125I-labeled ligand, T3. In the case of the RNA probe, several interesting observations were revealed. At first, phosphorylation by casein kinase II in vivo apparently decreased the affinity of PC4 for the RNA probe (6c/d in Fig. 2C; see also Table 2), although it increased the affinity of PC4 for both the dsDNA and ssDNA probes (6c/d in Fig. 2A and B). Second, in contrast to the DNA-binding activity, the RNA-binding activity of PC4 was not significantly affected by the mutation at position 77 (8c/d in Fig. 2C). Third, both p52 and p75 could strongly bind the RNA probe (9c–h in Fig. 2C), but did not significantly bind either the dsDNA or ssDNA probe in this assay (9c–h in Fig. 2A and B). Finally, PCAF, a p300/CBP-associated factor that functions as a histone acetyltransferase (28), could bind the RNA probe very strongly (10c–f in Fig. 2C; see Table 2 for quantification), suggesting a possible role of PCAF in RNA metabolism. On the other hand, in the case of the ligand T3, only the recombinant thyroid hormone receptor bound 125I-labeled T3 strongly and specifically (3c/d in Fig. 2D).
In addition to GST-K–p52, I have also tested other protein probes in the UPA system. Figure 3 shows the binding activity of 32P-labeled splicing factor ASF/SF2, a member of the SR protein family, to 16 selected proteins (Fig. 3A). ASF/SF2 significantly bound to five of the 16 proteins, including the affinity-purified TFIID complex (Fig. 3B and C, 2d), retinoid-X receptor (3a), histone H1 (3c), co-histones (3d) and ASF/SF2 itself (4b). However, after washing the UPA with 500 mM KCl, ASF/SF2 appeared to have the highest affinity for itself (Fig. 3C, 4b), which is in agreement with the previous observation that in vitro translated ASF/SF2 could strongly bind to GST–ASF/ SF2 in a GST pull down assay (29). Interestingly, ASF/SF2 also showed high affinity for the TFIID complex. Since ASF/SF2 did not interact with TBP (2c), ASF/SF2 might interact directly with TBP-associated factors. Whether such an interaction reflects the function of TFIID or ASF/SF2 in transcription or pre-mRNA splicing or coupling of these two processes remains to be investigated. Taken together, these experiments indicate that UPA can be used to detect protein interactions with various targets.
Figure 3.
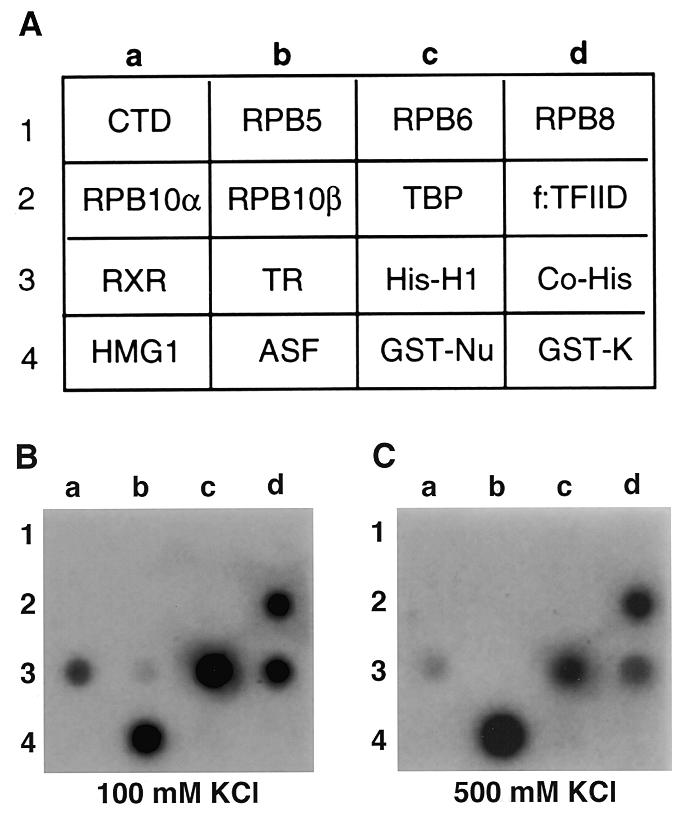
Use of UPA to detect ASF/SF2-interacting proteins. (A) Sixteen selected proteins (or fractions) were analyzed for interaction with 32P-labeled 6H(K)ASF/SF2. CTD, the C-terminal domain of RNA polymerase II fused to GST; RPB5, RPB6, RPB8, RPB10α and RPB10β correspond to individual subunits of RNA polymerase II fused to GST; TBP, TATA-binding protein; f:TFIID, affinity-purified flag-tagged TBP-containing TFIID complex from HeLa cells; RXR, retinoid-X receptor; TR, thyroid hormone receptor; His-H1, histone H1; Co-His, co-histones; HMG1, high mobility group protein 1; ASF, alternative splicing factor; GST–Nu, GST–nucleolin fusion; GST-K, GST fused with a synthetic heart muscle kinase site. (B) Autoradiographic detection of UPA after washing the membrane with 100 mM KCl. (C) Signals detected after washing the membrane with 500 mM KCl.
DISCUSSION
The targeting of proteins, via specific protein–protein interactions, has emerged as a major criterion to determine the function of proteins involved in gene transcription, replication, translation or in signal transduction and cell cycle pathways. The present report describes the development of a UPA system that may herald a new era in studying gene regulation and screening of targeted proteins as new drugs.
Although dot blot analysis has been used for the detection of specific antibody–antigen interactions, use of highly purified and fully active recombinant or native proteins to target a specific protein, or other biomolecules, has not been reported. Because the UPA assay is carried out under non-denaturing conditions, it represents a unique technology distinct from other related assay systems. (i) In the case of far-western blot analysis, protein fractions are usually analyzed by SDS–PAGE and electrotransferred to a membrane, followed by denaturation and renaturation before probing with a radiolabeled protein probe. On average only 1–10% of the activity (without considering the loss of protein during the transfer process) can be recovered for most proteins with such a procedure (18). UPA, however, simply uses fully active proteins by directly spotting them onto a membrane. Therefore, it is at least 10- to 100-fold more sensitive than the far-western blot assay. (ii) Since the amount of active protein assayed equals the amount of protein applied, the affinities of individual proteins for a specific probe, either a protein or another type of biomolecule, can be easily quantified and compared with each other. (iii) Most existing assay systems were designed for a single purpose. For example, the two hybrid system, co-immunoprecipitation, far-western blotting and GST assays are all used only for protein–protein interaction, the gel mobility shift, footprinting and cross-linking assays are used for protein–nucleic acid (DNA or RNA) interactions, and microarrays or DNA chips are used only for nucleic acid interactions. However, the same UPA has been successfully used for detection of protein–protein, protein–DNA, protein–RNA and protein–ligand interactions. It will probably also be useful for protein–metal ion interactions. Using the same UPA, I have shown that PC4 with a single point mutation (Phe→Pro) at position 77 lost both dsDNA- and ssDNA-binding activity (Fig. 2A and B, 8c/d), but still retained RNA-binding activity (Fig. 2C, 8c/d). In contrast, phosphorylation of PC4 by casein kinase II stimulated the DNA-binding activity (Fig. 2A and B, 6c/d), but reduced its RNA-binding activity (Fig. 2C, 6c/d). These observations demonstrate that UPA is an effective method to map protein interaction domains and DNA- or RNA-binding domains of a protein. In addition, once protein samples are available, it is possible to analyze hundreds or even thousands of proteins in a single experiment. (iv) Unlike DNA microarrays or DNA chips, no additional sophisticated equipment is required for the UPA. It can be carried out in most research laboratories and biotechnology companies.
Given that the major part of the genome sequence has been identified, that the entire genome sequence is expected to be completed by the year 2003 (30) and that most active proteins can be overexpressed in and purified from either bacteria, baculovirus or mammalian cells, it is thus feasible that the availability of 100 000 human gene products (30) will provide a rich source of proteins for such studies. Therefore, the UPA system will not only provide an alternative and efficient method to explore the mechanisms of gene expression pathways, but also a new pipeline to screen and to design new drugs, with the potential for disease diagnosis.
Acknowledgments
ACKNOWLEDGEMENTS
I am grateful to Drs A. Wolffe and I. Dawid for their support and encouragement, P. Wade and N. Kaludov for critical comments on the manuscript, Y. Nakatani and V. Ogryzko for recombinant p300, PCAF and TAFII250 proteins, Y. Pommier and Z. Wang for recombinant DNA topo I proteins, C. Chiang for the f:TFIIH cell line, M. Srivastava for the GST–nucleolin construct and M. Olson for anti-nucleolin antibodies. Protein sequence analysis was performed by the Protein/DNA Technology Center of the Rockefeller University.
REFERENCES
- 1.Hurst H.C. (1996) Eur. J. Cancer, 32A, 1857–1863. [DOI] [PubMed] [Google Scholar]
- 2.Bustin S.A. and McKay,I.A. (1994) Br. J. Biomed. Sci., 51, 147–157. [PubMed] [Google Scholar]
- 3.Powis G. (1994) Pharmac. Ther., 62, 57–95. [DOI] [PubMed] [Google Scholar]
- 4.Krantz A. (1998) Nature Biotechnol., 16, 1294. [DOI] [PubMed] [Google Scholar]
- 5.Orphanides G., Lagrange,T. and Reinberg,D. (1996) Genes Dev., 10, 2657–2683. [DOI] [PubMed] [Google Scholar]
- 6.Ptashne M. and Gann,A. (1997) Nature, 386, 569–577. [DOI] [PubMed] [Google Scholar]
- 7.Fields S. and Sternglanz,R. (1994) Trends Genet., 10, 286–292. [DOI] [PubMed] [Google Scholar]
- 8.Harris M. (1998) Methods Mol. Biol., 88, 87–99. [DOI] [PubMed] [Google Scholar]
- 9.Velculescu V.E., Zhang,L., Vogelstein,B. and Kinzler,K.W. (1995) Science, 270, 484–487. [DOI] [PubMed] [Google Scholar]
- 10.Schena M., Shalon,D., Davis,R.W. and Brown,P.O. (1995) Science, 270, 467–470. [DOI] [PubMed] [Google Scholar]
- 11.Chee M., Yang,R., Hubbell,E., Berno,A., Huang,X.C., Stern,D., Winkler,J., Lockhart,D.J., Morris,M.S. and Fodor,S.P.A. (1996) Science, 274, 610–614. [DOI] [PubMed] [Google Scholar]
- 12.Chiang C.M., Ge,H., Wang,Z., Hoffmann,A. and Roeder,R.G. (1993) EMBO J., 12, 2749–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kershnar E., Wu,S.Y. and Chiang,C.M. (1998) J. Biol. Chem., 18, 34444–34453. [DOI] [PubMed] [Google Scholar]
- 14.Luo Y., Fujii,H., Gester,T. and Roeder,R.G. (1992) Cell, 71, 231–241. [DOI] [PubMed] [Google Scholar]
- 15.Jackson S.P. and Tjian,R. (1989) Proc. Natl Acad. Sci. USA, 86, 1781–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ge H., Martinez,E., Chiang,C.M. and Roeder,R.G. (1996) Methods Enzymol., 274, 57–71. [DOI] [PubMed] [Google Scholar]
- 17.Zahler A.M., Lane,W.S., Stolk,J.A. and Roth,M.B. (1992) Genes Dev., 6, 837–847. [DOI] [PubMed] [Google Scholar]
- 18.Ge H., Si,Y.Z. and Wolffe,A.P. (1998) Mol. Cell, 2, 751–759. [DOI] [PubMed] [Google Scholar]
- 19.Ge H., Si,Y.Z. and Roeder,R.G. (1998) EMBO J., 17, 6723–6729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouvet P., Jain,C., Belasco,J.G., Amalric,F. and Erard,M. (1997) EMBO J., 16, 5235–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishikawa F., Matunis,M.J., Dreyfuss,G. and Cech,T.R. (1993) Mol. Cell. Biol., 13, 4301–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanakahi L.A., Dempsey,L.A., Li,M.J. and Maizels,N. (1997) Proc. Natl Acad. Sci. USA, 94, 3605–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tuteja N., Huang,N.W., Skopac,D., Tuteja,R., Hrvatic,S., Zhang,J., Pongor,S., Joseph,G., Faucher,C., Amalric,F. et al. (1995) Gene, 28, 143–148. [DOI] [PubMed] [Google Scholar]
- 24.Derenzini M., Sirri,V., Trere,D. and Ochs,R.L. (1995) Lab. Invest., 73, 497–502. [PubMed] [Google Scholar]
- 25.Valdez B.C., Henning,D., Busch,R.K., Srivastava,M. and Busch,H. (1995) Mol. Immunol., 32, 1207–1213. [DOI] [PubMed] [Google Scholar]
- 26.Ge H., Zhao,Y., Chait,B.T. and Roeder,R.G. (1994) Proc. Natl Acad. Sci. USA, 91, 12691–12695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Werten S., Stelzer,G., Goppelt,A., Langen,F.W.M., Gros,P., Timmers,H.Th.M., Van der Vliet,P.C. and Meisterernst,M. (1998) EMBO J., 17, 5103–5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogryzko V.V., Schiltz,R.L., Russanova,V., Howard,B.H. and Nakatani,Y. (1996) Cell, 87, 953–959. [DOI] [PubMed] [Google Scholar]
- 29.Xiao S.H. and Manley,J.L. (1998) EMBO J., 17, 6359–6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collins F.S., Patrinos,A., Jordan,E., Chakravarti,A., Gesteland,R. and Walters,L. (1998) Science, 282, 682–689. [DOI] [PubMed] [Google Scholar]
- 31.Mizzen C.A., Yang,X.-J., Kokubo,T., Brownell,J.E., Bannister,A.J., Owen-Hughes,T., Workman,J., Wang,L., Berger,S.L., Kouzarides,T., Nakatani,Y. and Allis,C.D. (1996) Cell, 87, 1261–1270. [DOI] [PubMed] [Google Scholar]
- 32.Wang Z. and Roeder,R.G. (1998) Mol. Cell, 1, 749–757. [DOI] [PubMed] [Google Scholar]
- 33.Pourquier P., Pilon,A.A., Kohlhagen,G., Mazumder,A., Sharma,A. and Pommier,Y. (1997) J. Biol. Chem., 272, 26441–26447. [DOI] [PubMed] [Google Scholar]