


Abstract
Formaldehyde crosslinking has been widely used to study binding of specific proteins to DNA elements in intact cells. However, previous studies have not determined if this crosslinker preserves the bona fide pattern of DNA binding. Here we show that formaldehyde crosslinking of Drosophila embryos maps an interaction of the transcription factor Zeste to a known target element in the Ultrabithorax promoter. This data agrees broadly with previous mapping of the same Zeste binding sites by in vivo UV crosslinking, though the formaldehyde method does give a low, possibly artifactual signal on other DNA fragments that is not detected by the UV method. We also demonstrate, using an in vitro assay, that formaldehyde crosslinking accurately reflects the DNA binding specificities of both Zeste and a second transcription factor, Eve. The crosslinking reagent methylene blue is shown to preserve DNA binding specificity in vitro as well. Our results suggest that crosslinking by formaldehyde, and possibly also by methylene blue, provide an accurate guide to the interaction of proteins with their high affinity target sites in cells.
INTRODUCTION
Many sequence-specific DNA-binding proteins have been identified, and their interactions with DNA have been extensively studied in vitro (1–4; http://transfac.gbf.de/transfac ). To understand how these proteins function, however, it is crucial to know which DNA sites they bind to in living cells and to measure the level of occupancy on these sites. The most direct way of accomplishing this is by use of in vivo crosslinking. Several methods have been published that use UV light or formaldehyde to covalently attach proteins to their natural DNA sites in vivo (5–9). These methods then identify the DNA sequences crosslinked by extracting the crosslinked protein–DNA complexes from cells, immunoprecipitating protein–DNA adducts with antibodies that recognize the protein of interest, and analyzing the co-precipitated DNAs by PCR, Southern blot or other hybridization method.
Using this crosslinking strategy, we previously showed that in Drosophila embryos, the transcription factor Zeste is UV crosslinked to discrete regions within the Ultrabithorax (Ubx) gene and is not crosslinked to non-target genes, whereas the homeoproteins Eve, Ftz, Bicoid and Paired crosslink to DNA sequences throughout the length of most genes (8,10,11). These results, together with the subsequent experiments they have spawned, have significantly altered our understanding of how Zeste and homeoproteins act (11–15). However, the UV crosslinking data is limited by the low yield of protein–DNA crosslinks induced in vivo, permitting convincing data to be obtained only for large DNA fragments that contain multiple binding sites. Even in the most favorable cases, UV crosslinking can only map binding to regions of 0.2–1 kb that contain 5–10 binding sites. For this reason, it is crucial to find a crosslinking method that can detect binding at higher resolution, preferably one able to detect binding of a single transcription factor molecule.
To this end, in this paper we have modified an existing in vivo formaldehyde crosslinking protocol for use in Drosophila embryos. The chief advantage of formaldehyde is that it crosslinks protein to DNA much more efficiently than UV light (16). In principle then, it should allow detection of binding by single molecules. However, formaldehyde also induces protein–protein crosslinks (17,18) and thus has the potential to connect proteins to DNAs that they do not directly contact. This property can be useful to study chromatin proteins that don’t themselves bind DNA, but it could also give a misleading indication of which genes or promoter regions a protein interacts with. Therefore, to assess the accuracy and specificity of formaldehyde crosslinking, we have sought to compare our formaldehyde crosslinking results with our earlier UV crosslinking data. In addition, previous in vitro experiments have shown that UV crosslinking gives a quantitative measure of DNA binding that accurately reflects the specificities of all transcription factors tested (10,11,19). Therefore, we have sought to determine if this is also true for formaldehyde crosslinking.
MATERIALS AND METHODS
Cloned DNAs and proteins
The Zeste C-terminal expression construct, pETzIII, consists of a NdeI fragment encoding the C-terminal 140 amino acids of Zeste (PCR amplified from pETzeste provided by V. Pirrotta) cloned into pET15b from Novagen. This construct expresses a His-tagged Zeste C-terminal polypeptide. All other plasmids have been described previously (8). The C-terminal Zeste polypeptide was purified according to Novagen’s protocols, and Eve and Zeste proteins were prepared as before (10).
Antibodies
Polyclonal goat anti-Drosophila RNA polymerase II large subunit was a gift from A. Greenleaf. For the in vivo formaldehyde crosslinking experiments, polyclonal rabbit anti-Zeste antibody was affinity purified from crude serum (7386, kindly provided by V. Pirrotta) using the His-tagged C-terminal Zeste polypeptide (pETzIII). This affinity-purified antibody recognizes amino acids 470–574 of Zeste. For the in vitro DNA binding and crosslinking experiments with Zeste, rabbit polyclonal antibodies against a Zeste β-galactosidase fusion were used (20). Antibodies to the N-terminal 246 amino acids of Eve were affinity purified from a polyclonal rabbit serum kindly provided by M. Frasch.
In vivo formaldehyde crosslinking of embryos and chromatin purification
Staged embryos were collected from population cages of adult Drosophila maintained according to standard techniques. Three to five grams of embryos were dechorionated for 2 min in 50% Clorox bleach and then rinsed. Residual water that will interfere with permeabilization by hexane was removed by briefly dispersing embryos in 300 ml of isopropanol and then by blotting the embryos dry. One volume of hexanes (a mixture of isomers) was equilibrated for 30 min against 0.175 vol of 37% formaldehyde and 0.130 vol of 10× phosphate-buffered saline (PBS) (1.37 M NaCl, 27 mM KCl, 43 mM Na2HPO4 and 14 mM KH2PO4), pH 7.3, to make the mixture 5% formaldehyde and 1× PBS. Only the upper organic phase was used for crosslinking. Embryos were fixed in 10 ml of buffered 5% formaldehyde/hexanes per gram of embryos by vigorous shaking for 5 min at room temperature. The embryos were allowed to settle and the formaldehyde/hexanes solution was poured off. The embryos were washed twice in a solution containing 1× PBS and 0.5% Triton X-100, using the same volume as used for fixing. They were then blotted dry, frozen in liquid nitrogen and stored at –70°C.
Chromatin was prepared by quick thawing frozen embryos and resuspending them in nuclei irradiation buffer [0.3 M sucrose, 15 mM NaCl, 5 mM MgCl2, 15 mM Tris pH 7.5, 60 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 0.5 mM dithiothreitol (DTT) and 1 mM phenylmethylsulfonyl fluoride (PMSF); 19]. They were homogenized with two strokes of a motorized Dounce homogenizer (Glas-col model GKH) and five strokes of a hand-held type B Dounce homogenizer. Triton X-100 was added to 0.3% and the homogenate was centrifuged (2000 g for 15 min in an SS-34 rotor). The pellet was resuspended in nuclei lysis buffer (100 mM NaCl, 10 mM Tris pH 7.9, 1 mM EDTA, 0.1% v/v NP-40 and 1 mM PMSF; 19) and homogenized with five strokes of a hand-held type B Dounce homogenizer. SDS was added to a final concentration of 3% and the material was vortexed. The released chromatin was then sheared by two passes through a 23 gauge needle to an average size of >20 kb. Triton X-100 and Sarkosyl were each added to a final concentration of 1% and the mixture was layered onto a step gradient (8.5 ml each of 1.5, 1.4 and 1.3 g/ml CsCl in 2% Sarkosyl, 1 mM EDTA and 1 mM PMSF). Gradients were centrifuged at 25 000 r.p.m. in a Beckman SW28 rotor for 40 h at 20°C. At the end of the spin, a fluffy white band containing the majority of the crosslinked DNA was located ~3 cm from the bottom of the tube. This material was recovered in ~3 ml of solution using an 18 gauge needle and a 5 ml syringe. Consistent with earlier reports, the density of the crosslinked chromatin isolated was ~1.38 g/ml (7,21). This contrasts with UV crosslinked chromatin, which has the density of uncross-linked DNA, i.e. 1.66 g/ml. The lower density of formaldehyde crosslinked chromatin indicates that it contains a much higher degree of covalently bound protein than UV crosslinked material (N.B. the density of free protein is 1.3 g/ml).
The crosslinked chromatin (usually ~3 ml) was dialyzed to remove the CsCl as described previously, except that the first buffer also contained 0.1% Sarkosyl and 0.1 mM PMSF (9,19). After 2 h, SDS was added to the chromatin in the dialysis bag to a final concentration of 0.1%, and the dialysis was then continued as described previously except that all of the dialysis buffers contained 0.1% SDS and 0.1 mM PMSF (9,19). The DNA concentration was quantitated spectrophotometrically, and the chromatin was frozen in liquid nitrogen and stored at –70°C.
Immunoprecipitation and Southern blotting
After thawing formaldehyde crosslinked chromatin at room temperature, restriction digests were set up that contained 50 µg chromatin, 0.1% SDS, 0.2% dodecylmaltoside, 0.4% Zwittergent 3-12, 1× the appropriate restriction enzyme buffer, 0.1 mg/ml BSA, 1 mM PMSF and 5 U of each restriction enzyme/µg chromatin. After 3 h at 37°C, the digests were centrifuged at 16 000 r.p.m. in a microfuge for 10 min. Then sodium deoxycholate and Triton X-100 were added to final concentrations of 0.1 and 1%, respectively, followed by the addition of 1 µg of the appropriate affinity-purified antibody. The samples were incubated at room temperature for 3 h, then immunoprecipitated using Staph A cells (9,19). The precipitated DNA was then released from the crosslinked protein, purified, resolved by agarose gel electrophoresis and analyzed by Southern blotting using standard techniques (9,19).
In vitro crosslinking and immunoprecipitation assays
A plasmid containing 3.5 kb of the Ubx gene (p3102) was restriction digested with ScaI, BsaJI and BssHII, and the resulting fragments were end-labeled with 32P by Klenow enzyme. This labeled DNA was used for in vitro DNA binding reactions performed as described previously except that 20 mM HEPES (pH 7.6) was substituted for Tris buffer (pH 7.5) (10). For ‘standard in vitro DNA binding assays’, purified protein was incubated with the labeled Ubx DNA on ice. The resulting protein–DNA complexes were then immunoprecipitated and washed with a mild buffer (20 mM Tris pH 7.5, 0.25 mM EDTA, 10% glycerol, 6.25 mM MgCl2, 0.05% NP-40) that does not disrupt the interaction of protein with high and moderate affinity DNA binding sites. Then the bound DNA was eluted from the washed complexes using 1% SDS, 1.5 mg/ml calf thymus DNA and analyzed on an acrylamide gel (8). For in vitro formaldehyde crosslinking assays, formaldehyde (freshly diluted from a stock solution of 37% formaldehyde/10% methanol) was added to a final concentration of either 0.74 or 0.074% after the DNA binding reactions had reached equilibrium. These reactions were incubated for a further 30 min on ice, then crosslinked protein–DNA complexes were immunoprecipitated and washed with a stringent buffer (10 mM Tris pH 7.5, 100 mM MgCl2, 150 mM NaCl, 0.2 mg/ml calf thymus DNA, 1% Triton X-100, 0.2% Sarkosyl, 0.1 mg/ml bovine serum albumin) that elutes DNA fragments that are not covalently attached to protein. Then, separately, the covalently crosslinked DNA fragments were eluted by incubation with proteinase K overnight at 65°C and analyzed on an acrylamide gel (10). The crosslinking efficiency of each protein was measured by comparing the signal intensities (quantitated by MacBas with data from a Fuji 2000 phosphorimager) of each DNA fragment from the ‘standard in vitro binding reaction’ lane versus the crosslinking lanes. For the quenching experiments described in Figure 5B, 2 µl of a 1.85% formaldehyde solution was incubated on ice with a 10-fold molar excess of Tris (pH 7.5) for 30 min prior to addition to the binding reactions. For the experiments described in Figure 7, methylene blue was added to a final concentration of 20 µM and the reactions were transferred to a parafilm-covered depression-well dish on ice and exposed to white light from a 100 W incandescent bulb at a distance of 3 cm for 30 min. (A glass plate was placed between the dish and bulb to prevent heating of the samples.) After methylene blue crosslinking, reactions were analyzed by the same protocol used to study formaldehyde crosslinking.
Figure 5.
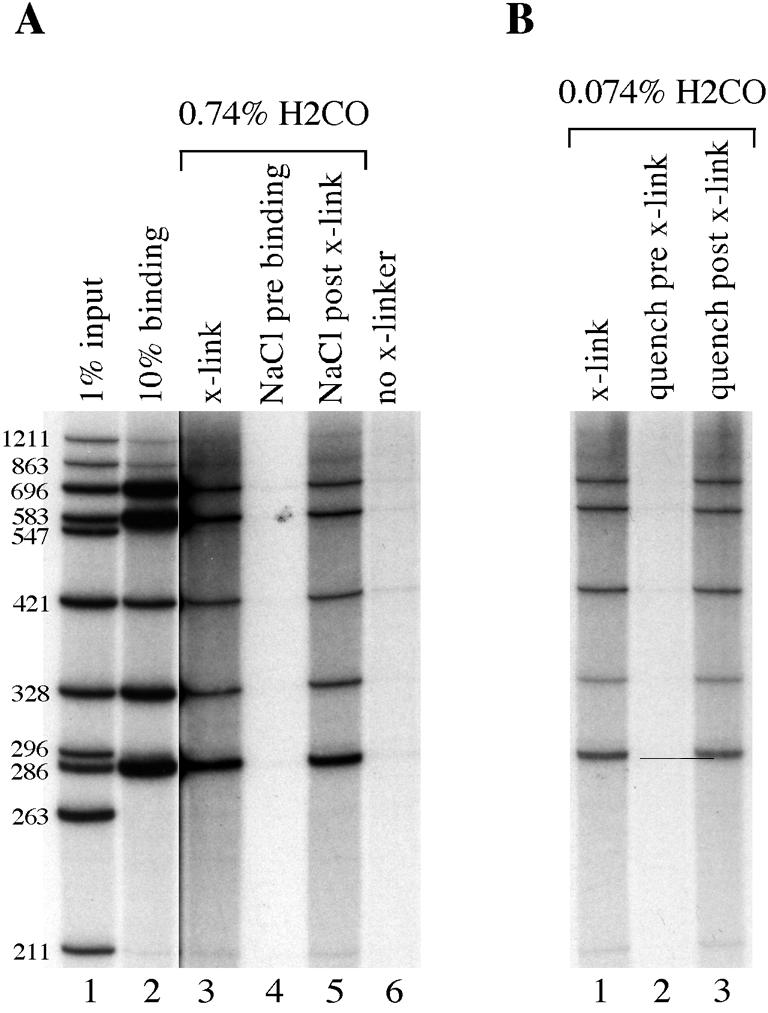
In vitro formaldehyde crosslinking of Eve protein reproduces the same pattern of DNA fragments as an in vitro binding reaction. Autoradiograms of 6% denaturing polyacrylamide gels. (A) Lane 1, 1% of the DNA used in all DNA binding and crosslinking reactions; lane 2, 10% of the DNA immunoprecipitated from a standard in vitro DNA binding reaction containing Eve protein; lanes 3–6, all of the DNA recovered from various formaldehyde crosslinking experiments containing Eve protein and either 0.74% (lanes 3–5) or no formaldehyde (lane 6); lanes 4 and 5, DNA fragments recovered when NaCl was added to a final concentration of 500 mM to DNA binding reactions prior to (lane 4) or after (lane 5) formaldehyde crosslinking. The DNA fragments used in this and all other in vitro crosslinking experiments are from a BsaJI + BssHII + ScaI digest of a 3.5 kb Ubx proximal promoter from plasmid p3102. The sizes of the DNA fragments in base pairs are indicated along the left. The autoradiogram of lanes 3–6 was exposed for 12 times longer than the autoradiogram of lanes 1 and 2. (B) Lanes 1–3, all of the DNA recovered from formaldehyde crosslinking experiments containing Eve protein and 0.074% formaldehyde; lanes 2 and 3, DNAs recovered when Tris quencher was added before (lane 2) or after crosslinking (lane 3). The DNAs used in this experiment are the same as those in (A). The autoradiogram of this gel was exposed for the same length of time as the autoradiogram of lanes 3–6 in (A).
Figure 7.
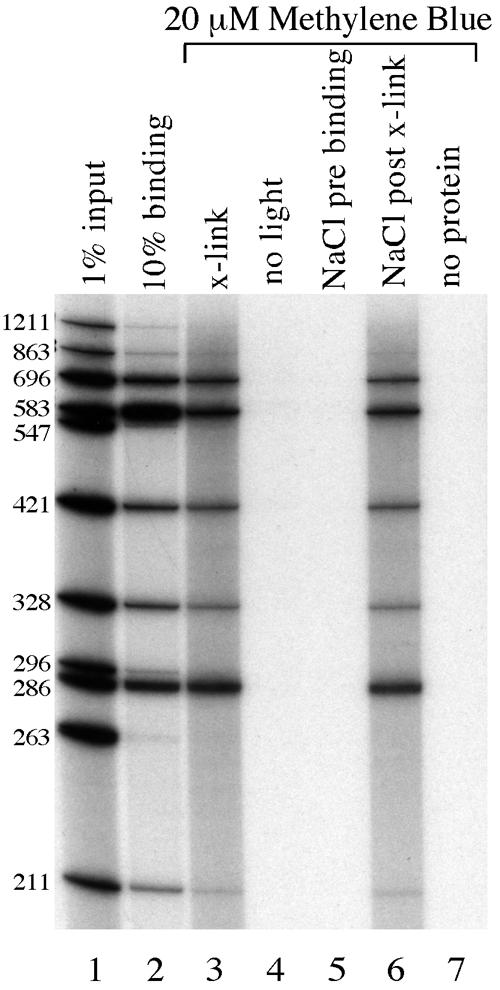
Methylene blue efficiently crosslinks Eve protein to DNA in vitro. Lane 1, 1% of the input DNA used in the binding and crosslinking reactions; lane 2, 10% of the DNA isolated in a standard DNA binding reaction containing Eve protein; lanes 3–7, all of the DNA recovered from a series of in vitro crosslinking reactions with Eve protein; lanes 3 and 4, DNA immunoprecipitated from reactions containing 20 µM methylene blue that had either been irradiated with white light for 30 min (lane 3) or that had been kept in the dark (lane 4); lanes 5 and 6, methylene blue crosslinking experiments in which 500 mM NaCl had been added either prior to (lane 5) or after (lane 6) crosslinking; lane 7, DNAs recovered when protein was not included in the reaction. The DNAs used in this experiment are the same as those used in Figure 5, and the autoradiogram of this gel was exposed for the same length of time as lanes 3–6 of Figure 5.
RESULTS
We have designed an in vivo formaldehyde crosslinking method that can localize transcription factor DNA binding by restriction enzyme mapping (Fig. 1; Materials and Methods). Previous formaldehyde crosslinking methods make it difficult to map binding sites in this way as they extract crosslinked chromatin from cells by sonicating it into pieces of <1 kb, preventing subsequent detection of immunoprecipitated DNA fragments by Southern blotting. Our method overcomes this problem by extracting crosslinked chromatin as fragments of at least 20 kb in length. Because subsequent restriction digestion of this material yields coherent, full-length restriction fragments, DNAs that are crosslinked to specific proteins can then be identified by Southern blotting.
Figure 1.

Outline of our in vivo formaldehyde crosslinking protocol.
We were able to eliminate the normally employed sonication step by extracting large chromatin fragments using buffers containing 0.1% SDS, thus preventing these otherwise insoluble complexes from aggregating. However, 0.1% SDS in the absence of a countering detergent inhibits restriction enzymes. Therefore, once the chromatin has been purified by buoyant density ultracentrifugation, we add 0.4% Zwittergent 3-12 and 0.2% dodecylmaltoside to the chromatin solution just prior to restriction digestion. We tested many combinations of detergents and found this to be optimal for allowing restriction digestion while maintaining the solubility of large chromatin fragments (unpublished data).
Formaldehyde crosslinking maps the interaction of Zeste with the Ubx promoter in Drosophila embryos
To demonstrate that our method detects binding of sequence-specific transcription factors in Drosophila embryos, we first examined crosslinking of Zeste. Zeste activates Ubx transcription via a cluster of high affinity sites just upstream of the Ubx proximal promoter (10,12,22). Formaldehyde crosslinked chromatin from 0–12 h old embryos was digested with EcoRI and subsequently immunoprecipitated with either affinity-purified anti-Zeste antibodies or non-specific immunoglobulin, then the immunoprecipitated DNAs were analyzed by Southern blotting. Compared to a dilution series of the total amount of DNA in the reaction, the anti-Zeste antibody immunoprecipitated 0.2% of a 3.9 kb EcoR1 Ubx promoter fragment that includes the Zeste binding sites (Fig. 2A), whereas the control IgG only precipitated 0.005% of this fragment. (The faint band appearing just above the 3.9 kb fragment is thought to be due to cross-reaction of the radioactive probe with bacterial DNA from the Staph A cells, which elutes variably in different immunoprecipitation experiments; unpublished data.) In contrast, when this same blot was stripped and reprobed with DNA sequences that hybridize to an 8.7 kb EcoRI fragment from the actin 5C gene, the anti-Zeste antibodies brought down only 0.005% of this gene fragment and the non-specific antibodies precipitated only 0.001% (Fig. 2B). Because signals between 0.005 and 0.001% are commonly observed in immunoprecipitations using non-specific IgG antibodies, it cannot be assumed from the data in Figure 2B that Zeste specifically crosslinks to the actin 5C gene. It is quite possible that Zeste does not bind at all to actin 5C in vivo and that the signal in Figure 2B represents the background of the assay.
Figure 2.
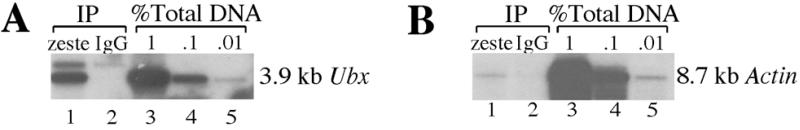
Zeste binds specifically to the Ubx proximal promoter. (A) Immunoprecipitation of EcoRI digested, formaldehyde crosslinked chromatin from 0–12 h embryos with either anti-Zeste antibody (lane 1) or with non-specific IgG (lane 2). A dilution series of the total amount of input DNA in the immunoprecipitation is also shown (lanes 3–5). The Southern blot was probed with a 3.5 kb fragment of the Ubx proximal promoter that spans from –3.1 to +0.4 kb: the size of the EcoRI fragment detected is indicated. (B) The same Southern blot used in (A) was stripped and reprobed with an 8.7 kb EcoRI fragment from the actin 5C gene (35). The lane designations are the same as in (A).
The above formaldehyde crosslinking results are largely consistent with our earlier in vivo UV crosslinking data. Zeste UV crosslinks to the 3.9 kb Ubx fragment, which contains the Zeste recognition elements, but cannot be detected on the actin 5C gene, suggesting that if Zeste interacts with actin 5C, it must do so at >100-fold lower levels than it does to Ubx (8,13).
To localize the site of Zeste interaction with the Ubx proximal promoter, crosslinked chromatin was digested with various restriction enzymes that cut the Ubx promoter near the cluster of high affinity Zeste sites (Fig. 3A). Digestion of chromatin with EcoRI and StuI separates the Ubx proximal promoter region into a 1 kb StuI–EcoRI fragment that contains the high affinity Zeste binding sites (Fig. 3A, fragment b) and a 2.8 kb EcoRI–StuI fragment that lacks these sites (Fig. 3A, fragment a). The 1 kb fragment is immunoprecipitated by anti-Zeste antibodies at 0.8% of total DNA in the reaction (Fig. 3A, lane b), while the 2.8 kb fragment is precipitated at only 0.02% of input DNA (Fig. 3B, lane a). Thus, the 1 kb StuI–EcoRI fragment, containing the high affinity Zeste sites, is enriched 40-fold relative to the 2.8 kb fragment. When chromatin is digested with EcoRI and EheI, two hybridizing fragments are generated: a 3.4 kb EcoRI–EheI fragment lacking the high affinity Zeste sites (Fig. 3A, fragment c) and a 542 bp EheI–EcoRI fragment containing them (Fig. 3A, fragment d). In immunoprecipitations of EcoRI + EheI digested chromatin, the 3.4 kb fragment was recovered at only 0.03% of total DNA, while the 542 bp fragment was recovered at 0.6% (Fig. 3B, lanes c and d). Thus, these experiments map the major site of Zeste interaction to the region 3′ of the EheI site at –177 bp, a result that is consistent with the in vitro mapping of Zeste sites to nucleotides between –160 and –31 bp.
Figure 3.
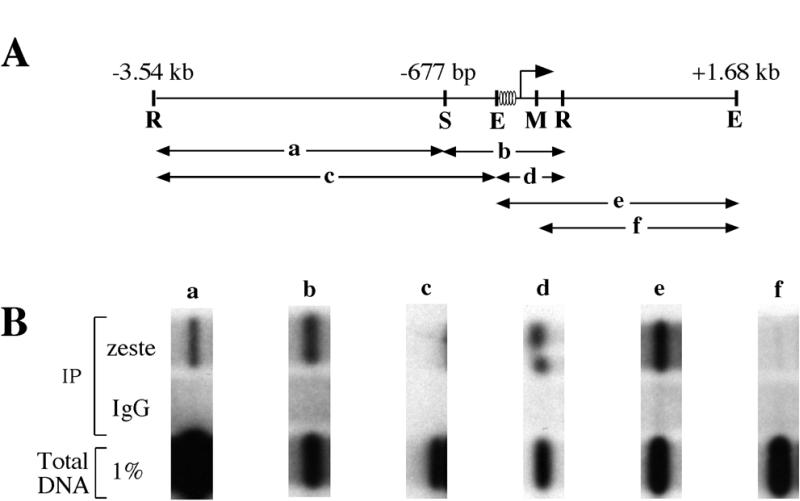
Zeste is crosslinked most strongly to a 297 bp region of the Ubx promoter. (A) Diagram of the restriction fragments produced by various digests of the Ubx proximal promoter. The positions of EcoRI (R), EheI (E), MluI (M) and StuI (S) restriction sites are marked, and the RNA start site at +1 is indicated by an arrow. The restriction fragments to which binding has been examined are a 2.8 kb EcoRI–StuI fragment that spans nucleotides –3.54 kb to –677 bp (a), a 1 kb EcoRI–StuI fragment from –677 to +365 bp (b), a 3.4 kb EcoRI–EheI fragment from –3.54 kb to –177 bp (c), a 540 bp EcoRI–EheI fragment from –177 to +365 bp (d), a 1.8 kb EheI–EheI fragment from –177 bp to +1.63 kb (e) and a 1.5 kb MluI–EheI fragment from +120 bp to +1.63 kb (f). Ovals denote the positions of high affinity Zeste binding sites. (B) Results of formaldehyde crosslinking experiments examining binding to the DNA fragments shown in (A). Vertical columns show blots of immunoprecipitations of crosslinked chromatin from wild-type 0–12 h embryos (a–f). The top row contains DNA fragments immunoprecipitated with anti-Zeste antibodies, the middle row contains the result obtained with non-specific antibody and the bottom row contains 1% of the input DNA. The Southern blots examining binding to fragments a–d were probed with the same 3.5 kb fragment of the Ubx proximal promoter as used in Figure 2A. The blots examining binding to fragments e and f were probed with an adjacent EcoRI DNA fragment that lies between nucleotides +365 bp and +3.68 kb.
To map the downstream boundary of this major site of Zeste crosslinking, EheI or EheI + MluI digested chromatin was immunoprecipitated with anti-Zeste antibodies, and the resulting blots were hybridized with an EcoRI DNA fragment spanning nucleotides +365 bp to +3.68 kb. An EheI digest of crosslinked chromatin produces a 1.8 kb fragment that includes the Zeste high affinity sites (Fig. 3A, fragment e). Immunoprecipitation reactions recovered 0.3% of this fragment using anti-Zeste antibodies (Fig. 3B, lane e). In contrast, an EheI + MluI digest produces a 1.5 kb fragment that lacks the high affinity sites (Fig. 3A, fragment f). Only 0.005% of this fragment was recovered by the anti-Zeste antibody (Fig. 3B, lane f). Thus the presence of a 297 bp region (from nucleotides –177 to +120 bp), which contains the high affinity Zeste binding sites, correlates with efficient immunoprecipitation of crosslinked chromatin fragments by anti-Zeste antibodies.
This mapping of Zeste binding sites agrees broadly with the in vivo UV crosslinking data, which shows that Zeste specifically crosslinks to sequences between nucleotides –200 and –31 bp in the Ubx promoter (8,13). However, the formaldehyde mapping data does differ from the UV data in one regard: we reproducibly detect low but significant crosslinking to the region between nucleotides –677 bp and –3.54 kb with formaldehyde, but not with UV. Possible explanations for this difference are suggested in the discussion.
In vivo formaldehyde crosslinking fails to convincingly detect the interaction of Eve with DNA
Having shown that the formaldehyde method detects DNA binding of Zeste in vivo, we set out to detect binding of the selector homeoprotein Eve. Eve is expressed at high levels only in 4–5 h old embryos. Consequently, crosslinked chromatin was purified from 4–5 h embryos, restriction digested, then immunoprecipitated using either affinity-purified anti-Eve antibodies or control IgG. The recovered DNAs were then analyzed on a Southern blot that was probed with eve upstream sequences (Fig. 4A). Compared to a dilution series of the input DNA, the anti-Eve antibodies precipitated only 0.004% of the total eve promoter sequences present in the reaction. Because this is within the range of signals observed in immunoprecipitations using non-specific IgG, this may represent a non-specific background, or it could represent specific but weak crosslinking of Eve.
Figure 4.
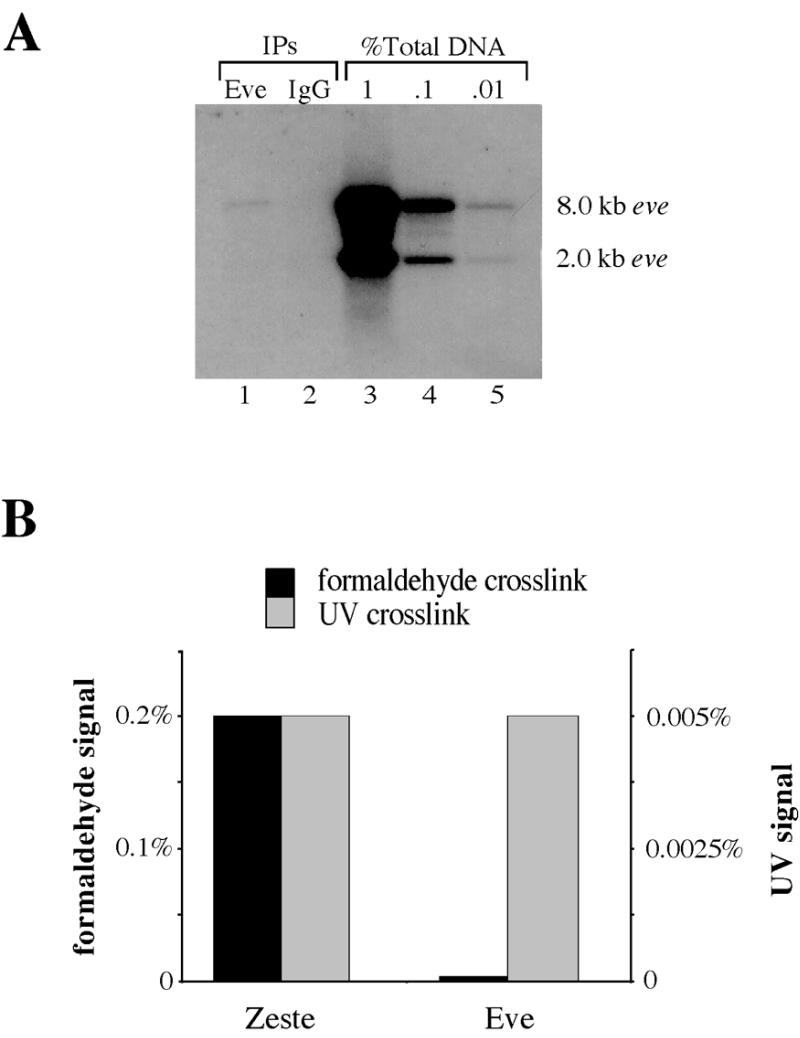
In vivo formaldehyde crosslinking does not convincingly detect DNA binding by Eve. (A) Southern blot of EcoRI digested, formaldehyde crosslinked chromatin from 4–5 h embryos immunoprecipitated either with anti-Eve antibodies (lane 1) or with non-specific IgG (lane 2). The blot was probed with eve gene sequence spanning nucleotides –6.4 kb to –400 bp. A dilution series of input DNA is shown on the left (lanes 3–5) and the size of the EcoRI fragment detected is indicated. (B) Normalized crosslinking efficiency of Zeste and Eve detected by the in vivo UV and formaldehyde techniques. The percent formaldehyde crosslinking signals for Eve and Zeste are shown by the gray bars and indicated by the scale on the left. The percent UV crosslinking is shown by the black bars and the values are indicated on the right hand scale. The values for Zeste are for crosslinking to the 3.5 kb proximal promoter fragment of the Ubx gene and for Eve are for the eve upstream promoter fragment used in (A). The data for UV crosslinking are from Solomon et al. (7).
This inability to convincingly detect binding of the homeoprotein Eve with the formaldehyde method contrasts with the results obtained by in vivo UV crosslinking: in vivo UV crosslinking detects Eve and Zeste equally well, at levels at least 100-fold above any background in the assay (Fig. 4B).
In vitro formaldehyde crosslinking accurately reflects relative levels of DNA binding in vitro
To investigate why, in some cases, these two crosslinking methods give different results and to evaluate the accuracy with which formaldehyde reproduces the correct pattern of DNA binding, we have employed an in vitro formaldehyde crosslinking assay. In this method, a purified transcription factor is first incubated with a mixture of radioactively labeled DNA fragments, the bound protein is then crosslinked to DNA, the protein is immunoprecipitated, non-covalently linked DNA fragments are removed by washing the immunoprecipitated material with stringent buffers, then covalently attached DNA fragments are eluted from the antibody–protein complexes by proteinase K digestion, the purified DNA fragments are analyzed on denaturing polyacrylamide gels, and finally the amount of specific DNA fragments crosslinked is quantitated. The advantage of this method is that it allows direct comparison between the relative levels of DNA binding and crosslinking and a range of different affinity DNAs under the same experimental conditions (10).
First, we wanted to determine if formaldehyde can crosslink Eve protein to DNA in vitro, particularly as previous in vitro experiments suggested that formaldehyde may not crosslink some proteins to DNA (16). Figure 5 compares the results of ‘standard DNA binding reactions’ with the results of in vitro formaldehyde crosslinking reactions. In the ‘standard DNA binding assay’, immunoprecipitated protein–DNA complexes are first washed with a much milder buffer than that used in the crosslinking assay, prior to the elution of bound DNA fragments (see Materials and Methods). This milder buffer does not disrupt binding of proteins to high and moderate affinity DNA sites, and thus this experiment measures the affinity of binding to different DNA fragments. The ‘standard DNA binding assay’ shows that Eve protein binds with different affinities to DNA fragments from the Ubx proximal promoter (Fig. 5A, lane 2). Although Eve binds to many DNA fragments, the binding is sequence specific, as opposed to non-specific, since DNA fragments lacking homeoprotein recognition sites are not bound (Fig. 5A, lane 2). In the in vitro formaldehyde crosslinking assay, which only detects that minority of DNA fragments that have become covalently attached to Eve (see Materials and Methods), DNAs crosslinked to Eve give a very similar pattern of band intensities to the ‘standard DNA binding reaction’ (Fig. 5A, compare lanes 2 and 3). (Note that the autoradiogram of lane 3 in Fig. 5A has been exposed for 12 times longer than the autoradiogram in lane 2.) Thus, the in vitro crosslinking assay faithfully reflects the DNA binding specificity of Eve.
To ensure that the in vitro formaldehyde crosslinking assay only detects molecules that are covalently coupled to DNA, the following controls were performed. No DNA was recovered from reactions in which DNA binding was disrupted by high concentrations of NaCl prior to crosslinking (Fig. 5A, compare lanes 3 and 4), whereas increasing the concentration of salt after crosslinking does not affect DNA recovery (Fig. 5A, compare lanes 3–5). Thus the formaldehyde crosslinking assay only recovers DNA that is bound by protein during the crosslinking reaction. If an excess of Tris is preincubated with the formaldehyde prior to the crosslinking step, the Tris reacts with the formaldehyde and inactivates it, and no DNA is recovered (Fig. 5B, compare lanes 1 and 2). However, if Tris is added after the crosslinking reaction, DNA is efficiently recovered (Fig. 5B, compare lanes 1 and 3). Therefore, the recovery of DNA is dependent on the chemical reactivity of formaldehyde. Finally, in the absence of formaldehyde, only an extremely weak residual pattern of DNA fragments is recovered, which resembles the input DNA rather than the Eve pattern (Fig. 5A, compare lanes 6, 3 and 1). Thus the high salt washes employed in the in vitro formaldehyde crosslinking protocol ensure that only protein molecules that are chemically crosslinked to DNA are detected. We conclude that the crosslinking assay is a good measure of DNA occupancy by Eve.
Figure 6 compares the results of Zeste assayed in a standard DNA binding reaction with the results of Zeste assayed in an in vitro formaldehyde crosslinking reaction. Lane 1 shows a series of DNA fragments from the Ubx proximal promoter, one of which is a 263 bp fragment that contains the high affinity Zeste binding sites. In the standard DNA binding assay (Fig. 6, lane 2) the 263 bp fragment is bound most strongly. In the in vitro formaldehyde crosslinking assay (Fig. 6, lane 3) the same 263 bp fragment is the only DNA recovered at significant levels. Thus, formaldehyde crosslinking reproduces the DNA binding preferences for Zeste protein in vitro, just as it does for Eve.
Figure 6.
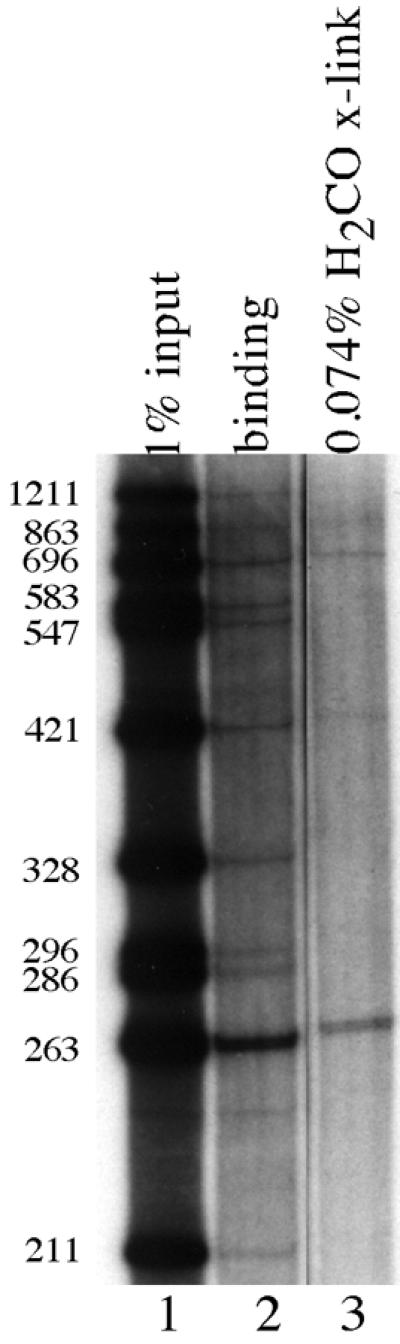
In vitro formaldehyde crosslinking localizes the binding of Zeste protein to the same region of the Ubx gene to which it crosslinks in vivo. Autoradiogram of a 6% denaturing polyacrylamide gel. Lane 1, 1% of the DNA from the same digest of Ubx DNA used in Figure 5; lane 2, all of the DNA immunoprecipitated from an in vitro DNA binding assay containing purified Zeste protein; lane 3, all of the DNA recovered from an in vitro formaldehyde crosslinking reaction with Zeste protein. The sizes of the DNA fragments in base pairs are indicated on the left. These DNA fragments are the same as those used in Figure 5, and the autoradiogram of this gel was exposed for the same length of time as lanes 3–6 of Figure 5.
Interestingly, in light of the inability of the in vivo formaldehyde crosslinking assay to convincingly detect DNA binding by Eve in embryos, formaldehyde crosslinks Eve to DNA in vitro much less efficiently than it crosslinks Zeste. Approximately 33% of the Zeste bound to the Ubx promoter element in vitro is crosslinked to DNA, whereas only 0.25–0.70% of Eve bound to DNA becomes crosslinked: a 47- to 132-fold difference in crosslinking efficiency. This result is consistent with the at least 50-fold difference in formaldehyde crosslinking efficiencies seen in vivo. One possibility is that the amino acid side chains within the Eve DNA-binding domain do not react as favorably with formaldehyde as the side chains in the Zeste DNA-binding domain, causing lower crosslinking efficiencies.
Phenothiazinium compounds crosslink proteins to DNA in a manner that preserves the pattern of DNA binding
Because of this variation in the degree of crosslinking by formaldehyde, we wished to explore other crosslinking reagents that might react more efficiently with Eve and with other homeoproteins. Using an in vitro assay, we explored a number of compounds that had previously been reported to induce protein–DNA crosslinks, including dimethylarsinic acid, cisplatin, potassium chromate, methylene blue and acridine orange (23–26). In preliminary trials, only phenothiazinium compounds were effective in crosslinking Eve to DNA, the most effective being methylene blue (Fig. 7, lane 3). This compound forms protein–DNA crosslinks only in the presence of white light (Fig. 7, compare lanes 3 and 4) and, like UV light and formaldehyde, gives a very similar pattern of DNA fragments as an in vitro binding reaction (Fig. 7, compare lanes 2 and 3), indicating that methylene blue should be a useful reagent for quantitative studies of DNA binding. The efficiency of crosslinking is 2- to 10-fold higher than that obtained using formaldehyde, so methylene blue may be a better crosslinking reagent than formaldehyde for studying homeoproteins. However, because the increase in efficiency is modest, further experiments may be needed to develop a crosslinking method that efficiently detects homeoprotein DNA binding at high resolution in vivo.
DISCUSSION
We have used formaldehyde crosslinking to map DNA elements bound by Zeste in Drosophila embryos and have compared our results to those obtained by UV crosslinking. The two methods give similar results in that they both most strongly detect crosslinking to a short region of the Ubx promoter that contains a well-characterized Zeste target element. We have also shown that, like UV crosslinking, formaldehyde crosslinking accurately preserves the DNA binding specificities of proteins in vitro. Therefore, our results strongly suggest that UV and formaldehyde crosslinking both faithfully reflect the binding of Zeste to its high affinity sites in vivo.
However, the formaldehyde method also detects lower levels of Zeste crosslinking to a fragment just 5′ of the Ubx target element that the UV method does not detect. The amount of Zeste crosslinked by formaldehyde to the 5′-fragment is 34-fold less than that to the target element but is well above the background of the assay. In UV crosslinking experiments, Zeste crosslinks to the Ubx target element at at least 100 times higher levels than it does to the adjacent 5′-region. Thus, the UV method is sufficiently sensitive to detect the interaction with this 5′-region. The fact that it does not indicates that the difference between the two assays is significant.
There could be several reasons for this discrepancy. Only proteins bound within a few angstroms of DNA are coupled to DNA by UV crosslinking (19,27,28). However, because formaldehyde induces high levels of protein–protein crosslinking, it can crosslink proteins to DNA sequences that they do not directly contact (29). Thus, one possibility is that Zeste does not directly bind to the DNA 5′ of the Ubx target element but becomes indirectly attached to this DNA region in the formaldehyde assay via intermediary proteins, at least one of which does directly contact the 5′-region. Another possibility is that Zeste does directly contact the 5′-region but does so by using a different means of interaction than used at high affinity specific sites. In this case, the difference between the two assays could be explained if formaldehyde can crosslink molecules that make such interactions and UV light cannot. Whatever the reason, this discrepancy does not detract from the clear agreement between the UV and formaldehyde methods at elements crosslinked strongly by Zeste.
Our results also indicate a second difference between UV and formaldehyde crosslinking. Both in vitro and in vivo, UV crosslinking detects DNA binding of Zeste as efficiently as it detects binding by a second protein Eve, whereas formaldehyde crosslinking detects Zeste at least 50-fold more efficiently than it detects Eve, again both in vitro and in vivo. To explain this, we suggest that there must be an innate difference in the reactivities of the DNA-binding domains of Eve and Zeste to formaldehyde. Because formaldehyde is thought to crosslink lysine, arginine, glutamine, asparagine and, perhaps, histidine residues to DNA (21), and because these amino acids are abundant in the DNA-binding domains of both Eve and Zeste (30,31), we suggest that differences between the two proteins reactivities are due to differences in the precise geometry or alignment of these amino acids with specific bases on the DNA. Other workers have also noted differences in the efficiency with which distinct proteins are coupled to DNA by formaldehyde (16), so formaldehyde may only be useful for examining a subset of proteins. However, the same is probably also true for UV crosslinking, since proteins whose binding sites do not contain a thymine are not UV crosslinked to DNA effectively (27,28). Thus, neither method is likely to be universally applicable.
Because one of our goals was to develop a high efficiency crosslinking method that can map binding of homeoproteins to single sequence elements in vivo, we have also explored the effectiveness and specificity of other chemical crosslinking reagents in vitro. A survey of a number of compounds known to crosslink proteins to DNA suggested that the most effective were phenothiazinium biological stains, including methylene blue and acridine orange. These compounds are believed to couple proteins to DNA because, when exposed to visible light, they form highly reactive singlet oxygen free radicals (32–34). These oxygen radicals diffuse away and catalyze the formation of covalent bonds between molecules that are in close contact.
Our in vitro results suggest that, like UV and formaldehyde, methylene blue crosslinking accurately reproduces the DNA binding specificity of transcription factors. However, this compound is only modestly more efficient than formaldehyde in crosslinking Eve to DNA in vitro, and it remains to be determined if this reagent will be effective in mapping Eve DNA binding sites in vivo. In preliminary trials, we found that methylene blue does crosslink high levels of protein to DNA in embryos, as indicated by the altered buoyant density of purified chromatin (data not shown). Given that our studies indicate that different crosslinking reagents show different preferences for the proteins they react with, it will be important to identify a range of crosslinking reagents so that all classes of DNA-binding proteins can be studied.
REFERENCES
- 1.Johnson P.F. and McKnight,S.L. (1989) Annu. Rev. Biochem., 58, 799–839. [DOI] [PubMed] [Google Scholar]
- 2.Faisst S. and Meyer,S. (1992) Nucleic Acids Res., 20, 3–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heinemeyer T., Wingender,E., Reuter,I., Hermjakob,H., Kel,A.E., Kel,O.V., Ignatieva,E.V., Ananko,E.A., Podkolodnaya,O.A., Kolpakov,F.A., Podkolodny,N.L. and Kolchanov,N.A. (1998) Nucleic Acids Res., 26, 362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pabo C.O. and Sauer,R.T. (1992) Annu. Rev. Biochem., 61, 1053–1095. [DOI] [PubMed] [Google Scholar]
- 5.Gilmour D.S. and Lis,J.T. (1985) Mol. Cell. Biol., 5, 2009–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orlando V. and Paro,R. (1993) Cell, 75, 1187–1198. [DOI] [PubMed] [Google Scholar]
- 7.Solomon M.J., Larsen,P.L. and Varshavsky,A. (1988) Cell, 53, 937–947. [DOI] [PubMed] [Google Scholar]
- 8.Walter J., Dever,C.A. and Biggin,M.D. (1994) Genes Dev., 8, 1678–1692. [DOI] [PubMed] [Google Scholar]
- 9.Biggin M.D. (1999) Methods Enzymol., 304, 496–515. [DOI] [PubMed] [Google Scholar]
- 10.Walter J. and Biggin,M.D. (1996) Proc. Natl Acad. Sci. USA, 93, 2680–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carr A. and Biggin,M.D. (1999) EMBO J., 18, 1598–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laney J.D. and Biggin,M.D. (1996) Development, 122, 2303–2311. [DOI] [PubMed] [Google Scholar]
- 13.Laney J.D. and Biggin,M.D. (1997) Proc. Natl Acad. Sci. USA, 94, 3602–3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biggin M.D. and McGinnis,W. (1997) Development, 124, 4425–4433. [DOI] [PubMed] [Google Scholar]
- 15.Liang Z. and Biggin,M.D. (1998) Development, 125, 4471–4482. [DOI] [PubMed] [Google Scholar]
- 16.Solomon M.J. and Varshavsky,A. (1985) Proc. Natl Acad. Sci. USA, 82, 6470–6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meluh P.B. and Koshland,D. (1997) Genes Dev., 11, 3401–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong S.S. (1993) Chemistry of Protein Conjugation and Cross Linking. CRC Press, Boca Raton, FL.
- 19.Walter J. and Biggin,M.D. (1997) Methods, 11, 215–224. [DOI] [PubMed] [Google Scholar]
- 20.Mansukhani A., Crickmore,A., Sherwood,P.W. and Goldberg,M.L. (1988) Mol. Cell. Biol., 8, 615–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orlando V., Strutt,H. and Paro,R. (1997) Methods, 11, 205–214. [DOI] [PubMed] [Google Scholar]
- 22.Benson M. and Pirrotta,V. (1988) EMBO J., 7, 3907–3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamanaka K., Tezuka,M., Kato,K., Hasegawa,A. and Okada,S. (1993) Biochem. Biophys. Res. Commun., 191, 1184–1191. [DOI] [PubMed] [Google Scholar]
- 24.Miller C.A., Cohen,M.D. and Costa,M. (1991) Carcinogenesis, 12, 269–276. [DOI] [PubMed] [Google Scholar]
- 25.Lin X., Zhuang,Z. and Costa,M. (1992) Carcinogenesis, 13, 1763–1768. [DOI] [PubMed] [Google Scholar]
- 26.Villanueva A., Canete,M., Trigueros,C., Rodriguez-Borlado,L. and Juarranz,A. (1993) Biopolymers, 33, 239–244. [DOI] [PubMed] [Google Scholar]
- 27.Hockensmith J.W., Kubasek,W.L., Vorachek,W.R., Evertsz,E.M. and von Hippel,P.H. (1991) Methods Enzymol., 208, 211–236. [DOI] [PubMed] [Google Scholar]
- 28.Blatter E.E., Ebright,Y.W. and Ebright,R.H. (1992) Nature, 359, 650–652. [DOI] [PubMed] [Google Scholar]
- 29.Strutt H., Cavalli,G. and Paro,R. (1997) EMBO J., 16, 3621–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott M.P., Tamkun,J.W. and Hartzell,G.W. (1989) Biochim. Biophys. Acta, 989, 25–48. [DOI] [PubMed] [Google Scholar]
- 31.Pirrotta V. (1991) Adv. Genet., 29, 301–348. [DOI] [PubMed] [Google Scholar]
- 32.Dunn D.D., Lin,V.H. and Kochevar,I.E. (1991) Photochem. Photobiol., 53, 47–56. [DOI] [PubMed] [Google Scholar]
- 33.Hagmar P., Pierrou,S., Nielsen,P., Norden,B. and Kubista,M. (1992) J. Biomol. Struct. Dyn., 9, 667–679. [DOI] [PubMed] [Google Scholar]
- 34.Tuite E.M. and Kelley,J.M. (1993) J. Photochem. Photobiol. B, 21, 103–124. [DOI] [PubMed] [Google Scholar]
- 35.Fyrberg E.A., Kindle,K.L. and Davidson,N. (1980) Cell, 19, 365–378. [DOI] [PubMed] [Google Scholar]