


Abstract
We have developed a novel biochemical method to simultaneously amplify and immobilize a target gene onto insoluble particles using PCR. This method employs the covalent attachment of one of two PCR primers to a particle surface either directly during DNA synthesis of the primer or post-DNA synthesis, through the use of chemical crosslinkers. Immobilization of the target gene can be achieved directly during PCR amplification, with one bead-bound primer and one soluble primer. Alternatively, this can be achieved post-PCR, through covalent attachment of a chemically modified primer incorporated into the amplicon to an activated particle. All of the immobilized DNA templates containing appropriate regulatory regions were fully competent for transcription and translation reactions and several could be re-used in serial reactions. The most successful strategy utilized amino-silanized controlled pore glass beads, which were coupled to phosphorylated primers using carbodiimide chemistry. These bead-bound primers were used during PCR to generate attached DNA templates that could be collected and re-used for at least seven sequential transcription reactions without significant loss in efficiency. This method has also been successfully applied to the amplification, transcription and translation of multiple DNA templates using a single, immobilized primer. The combined PCR-based amplification/immobilization method was shown to be more durable than post-PCR chemical immobilization and affords the convenience of performing sequential PCR amplification, transcription and translation reactions in a single tube.
INTRODUCTION
PCR is a powerful tool that enables logarithmic amplification of DNA sequences, resulting in a 106 to 108-fold enhancement of a target sequence which can be used for a variety of downstream experimental or diagnostic purposes. As efficient as the PCR process can be, recovery of amplification products still involves tedious purification procedures such as gel electrophoresis, organic extraction, centrifugation and/or column purification (1).
The attachment of PCR amplicons to a variety of solid phases has enabled the utility of PCR to be harnessed while eliminating the necessity for extensive PCR product purification. Several methods, including UV irradiation (2,3), biotin–avidin/ streptavidin and covalent chemical attachment, have been utilized to coat DNA and PCR products onto glass or plastic tubes, nitrocellulose or nylon filters, microtiter wells, agarose bead gels and magnetic particles (2–18). Assays utilizing post-PCR immobilized DNA have been developed for a myriad of applications, including: the detection of single or multiple nucleotide polymorphisms (3,19), the identification of bacterial agents (20,21), genetic phylogeny analysis and hybridization assays for gene detection (22–25), in vitro transcription (10,11) and the development of cDNA microarrays for analysis of gene expression (26,27). Thus, minute amounts of DNA can be amplified and by immobilizing the PCR product onto a surface, assays that utilize colorimetric or fluorescent signal generation can be designed to facilitate the identification and quantitation of hybridization events. These advances have promoted the development of microfabrication and automation strategies for high throughput, compact DNA diagnostic tools.
Although the utility of these techniques is clear, they still involve initial PCR amplification and subsequent immobilization of the PCR products. To improve the efficiency of this technology, PCR amplification and immobilization can be combined into a single step. Rasmussen and co-workers have developed a method in which 5′-phosphorylated DNA primers are bound to secondary amines on microtiter well surfaces through carbodiimide condensation (20,21). In this report, we analyze a variety of covalent chemical attachment methodologies to immobilize one of two PCR primers onto CPG and/or polymer supports. Using our methodology, bead-bound primers are used to simultaneously amplify and covalently immobilize one or more DNA amplicons, which we show to be competent templates for in vitro transcription and subsequent translation reactions. This novel methodology has the advantages of previous technologies (amplification of starting material, ease of sequence manipulation, elimination of extensive PCR product purification), and now introduces the ability to use the amplicon beyond the confines of a test tube, glass slide or microtiter plate. The immobilization of amplified DNA to particles has far-reaching potential in nucleic acid vaccine and gene therapy research and provides the versatility of mixing and matching beads, to which specific DNA molecules are bound, to suit a specific diagnostic assay or experimental need.
MATERIALS AND METHODS
DNA synthesis
DNA oligomers were prepared using a PE Applied Biosystems model 394 DNA/RNA synthesizer (Foster City, CA) using conventional cyanoethyl (CE) phosphoramidites; an exception to this is described below under Oligo affinity supports. All synthesis chemicals, phosphoramidites and chemical modifiers were obtained from Glen Research (Sterling, VA). Phosphate or disulfide groups were introduced at the 5′-end of oligos using Chemical Phosphorylation Reagent I or Thiol Modifier C6 S-S, respectively (Glen Research), following the manufacturer’s protocols. Specific details for the coupling of modified oligomers to various supports are given below. Primers for the T7 promoter region (T7Prom, 5′-TAG GGC GTG AGT CGT ATT AAA ATT AAT ACG ACT CAC TAT AGG GAG A-3′) and the T7 termination site (T7Term, 5′-CAA GGG GTT ATG CTA GTT ATT GCT CAG CGG-3′) were prepared and used as either soluble or bead-immobilized PCR primers. Table 1 details the specific modifications made to these basic sequences. The DNA oligomers were purified and detritylated using C-18 SPE cartridges (Supelco, Bellefonte, PA), and then quantified as described previously (8).
Table 1. Summary of primer modificationsa.
| Primer | 5′ Chemical modification | Crosslinker | Bead type | Text abbreviation |
|---|---|---|---|---|
| T7Term | None | None | None | T7 Term |
| T7Term | biotin | None | Avidin–MPG | B-MPG |
| T7Term | phosphorylation | None | EDA-modified CPGb | PC-E |
| T7Term | phosphorylation | None | HDA-modified CPGb | PC-H |
| T7Term | C6-thiol | SIAB | EDA-modified CPGb | E-SIAB |
| T7Term | C6-thiol | SIAB | HDA-modified CPGb | H-SIAB |
| T7Term | C6-thiol | NHS-PEG-MAL | EDA-modified CPGb | E-PEG |
| T7Term | C6-thiol | NHS-PEG-MAL | HDA-modified CPGb | H-PEG |
| T7Term | Uncleavable support | None | OAS-PS | T-PS |
| T7Term | Uncleavable support | None | OAS-CPGb | T-CPG |
| T7Prom | None | None | None | T7Prom |
| T7Prom | Uncleavable support | None | OAS-PS | Pr-PS |
| T7Prom | Uncleavable support | None | OAS-CPGb | Pr-CPG |
aT7Prom and T7Term, PCR primers for T7 promoter and T7 terminator regions, respectively, as described in Materials and Methods; SIAB, N-succinimidyl-(4-iodoacetyl)aminobenzoate; NHS-PEG-MAL, N-hydroxysuccinimidyl polyethyleneglycol maleimide, MW2000; MPG, magnetic porous glass; CPG, controlled pore glass; EDA, N-(2-aminoethyl)-3-aminopropylmethyl dimethoxy silane; HDA, N-(6-aminohexyl)aminopropyl trimethoxysilane; OAS, oligo affinity supports; OAS-PS, polystyrene–polyethyleneglycol co-polymer.
bOAS-CPG is a commercially available support (Glen Research, Sterling, VA) and is chemically unrelated to the EDA- and HDA-modified CPG which was amino-silanized in our laboratory at NRL.
Bead silanization
For bead silanization, dH2O refers to water obtained from a Nanopore purification system which was >18 MΩ, UV-sterilized and 0.22 µm filtered prior to use. CPG beads (CPG Inc., Lincoln Park, NJ) having a mean pore diameter of 3.1 µm on a particle size 80/120 mesh, pore volume of 95 cc/g and surface area of 8.41 m/g were cleaned by immersion in 1:1 concentrated HCl:MeOH for 45 min, rinsed three times with dH2O, immersed in concentrated H2SO4 for 45 min, and rinsed in dH2O until neutral pH was achieved. The beads were heated in 65°C dH2O for 45 min prior to silanization. Beads were then silanized using N-(2-aminoethyl)-3-aminopropylmethyl dimethoxy silane (EDA) (Gelest, Tullytown, PA) or N-(6-aminohexyl)-aminopropyl trimethoxysilane (HDA) (Gelest). Acid-cleaned beads (typically 50–100 mg) were shaken with 3–5 ml of a 2% solution of EDA or HDA in 25 mM aqueous CH3COOH for 1 h at 25°C. (Care must be taken to avoid exposure of the stock silanes to moisture, thus the silanes were opened, dispensed and re-sealed in a glovebag under N2.) The beads were then rinsed 5–10 times with dH2O (until a pH of ~5 was attained for the wash solution) and dried at 120°C to ensure completion of silane film crosslinking (dehydration). The dried beads were then ready for use in carbodiimide crosslinking or treatment with thiolated DNA as described below.
Carbodiimide crosslinking
Reactions containing EDA-modified (15.5 mg, 1 ml) or HDA-modified beads (12.5 mg, 0.8 ml) were incubated on an orbital shaker with 5 µM 5′-phosphorylated T7Term and 10 mM 1-ethyl- 3-(3-dimethylaminopropyl)carbodiimide-HCl (EDC) (Sigma Chemical Co., St Louis, MO) in 10 mM 1-methylimidazole (Sigma), pH 7.0, for 3 h at 50°C, then 70 h at 25°C. The beads were then washed briefly twice with dH2O, then washed twice with 1 ml of 1 M NaCl for 1 h at 25°C (to remove non-covalently bound DNA; 8). 5′-Phosphorylated DNA primers attached to EDA- or HDA-modified CPG by carbodiimide crosslinking are referred to in the text as PC-E and PC-H, respectively (Table 1).
Treatment with thiolated DNA
EDA- or HDA-silanized beads (3.5–4.0 mg) were treated with one of two heterobifunctional crosslinkers: N-succinimidyl- (4-iodoacteyl)aminobenzoate (SIAB) (Pierce, Rockford, IL) or N-hydroxysuccinimidyl polyethyleneglycol maleimide, MW 2000 (NHS-PEG-MAL) (Shearwater Polymers, Huntsville, AL). Crosslinkers were prepared as a 10 mM solution in 1:4 DMSO:MeOH, adding the DMSO first to solubilize the crosslinker. Crosslinkers were opened under N2 in a glovebag to minimize hydrolysis of the NHS ester. Beads were agitated with crosslinker solution (1.5 ml/3.5–4.0 mg beads) for 2.5 h at 25°C, then rinsed five times with 1 ml portions of MeOH immediately before coupling to post-PCR thiolated DNA. DNA primers which carried a 5′-disulfide modifier were used to generate PCR amplicons (see PCR, below). PCR reactions (100 µl) were purified using Qiaquick PCR Product Purification Spin Columns (Qiagen, Valencia, CA) yielding 50 µl amplicon in Qiagen EB buffer per reaction. For DNA attachment reactions, the thiol-modified amplicon was reacted with EDA- or HDA-modified beads via a SIAB crosslinker (E-SIAB and H-SIAB, respectively) or via a NHS-PEG-MAL crosslinker (E-PEG and H-PEG, respectively). Reactions containing 25 µl thiolated amplicon, 1.5 mg beads in HEPES buffer (10 mM HEPES, 1 mM EDTA, pH 6.5), and 200 µM tris-(2-carboxyethyl)phosphine (TCEP) (Molecular Probes, Eugene, OR) were agitated for 2 h at 25°C. TCEP rapidly reduces the disulfide modifier on the amplicon to yield free thiol that can react with iodoacetamide or maleimide groups on the crosslinker-modified beads). The beads were then washed with dH2O several times, followed by incubation in 1 M NaCl for 15 h to remove non-covalently attached DNA (8).
Oligo affinity supports (OAS)
Two different types of oligo affinity supports were obtained from Glen Research: OAS-PS (polystyrene–polyethyleneglycol co-polymer) and OAS-CPG. These supports were used with 5′-CE-phosphoramidites (Glen Research) to synthesize oligos in the 5′→3′ direction, which leaves a free OH group at the 3′-end (the 5′-end remains attached to the OAS support). The OAS oligos are not susceptible to cleavage during deprotection but remain tethered to the support. T7Term and T7Prom primers synthesized on OAS-PS and OAS-CPG supports are referred to in the text as T-PS and T-CPG and Pr-PS and Pr-CPG, respectively, where T indicates T7Term and Pr indicates T7Prom primers (Table 1). The oligo-modified supports are used to prime PCR reactions as described below.
DNA templates
Plasmids used as templates in this work include pET3a-derived (Novagen, Madison, WI) pET23 (28) and pETGFP (this work). These plasmids all utilize the bacteriophage T7 expression system, which was selected because of its relatively short (17 bp) consensus promoter sequence and high level of transcription. pET23 contains a 1.56 kb gene encoding the 57 kDa bacteriophage T4 major capsid protein (T4 MCP). pETGFP was constructed by isolating the green fluorescent protein (GFP) gene from p6XHis-GFP (Clontech, Palo Alto, CA) via PCR and using primer-inserted NdeI and BamHI restriction enzyme sites to clone the GFP gene into the pET3a expression vector cleaved with the same restriction enzymes. A third plasmid, pTEI, and soluble primers complementary to its sequence were used in amplification experiments designed to quantitate non-specific binding to T7-derived immobilized primers (a generous gift of Dr J.M. Mauro, Geo-Centers Inc.).
PCR
The procedure used is a modification of previously published work (19,20). Standard PCR reaction conditions were as follows: 0.1 µg each DNA template (pETHISGFP and/or pET23), 1 µM soluble primer A, 50 nM soluble primer B ‘spike’, 30–352 µg bead-immobilized primer B (as indicated), 20 µM each dNTP, 0.01% BSA, 0.1% Tween 20, 1 U Taq polymerase (Gibco BRL, Gaithersburg, MD), 10 mM Tris–HCl pH 8.3 at 25°C, 50 mM KCl, 1.5 mM MgCl2, 1 mM DTT, 1 mM EDTA and 0.1% gelatin. In this context, primer A refers to unmodified T7Prom or T7Term oligomers and primer B refers to the bead-immobilized versions of these primers as described in Table 1. Primer B ‘spike’ refers to the small concentration (50 nM) of soluble primer B added to reactions containing mainly an immobilized form of primer B. Control reactions in which neither primer was immobilized and those in which a thiolated primer was used for generation of thiolated amplicons contained 1 µm of each soluble primer.
PCR was initiated under hot start conditions in a PE thermocycler (1). PCR was conducted for 35 cycles (each cycle consisting of 1 min at 94°C, 45 s at 66°C, 1 min at 72°C). The final cycle was followed by an additional 10 min at 72°C to ensure complete extension. For some experiments, tubes were removed during the denaturation step and vortexed to resuspend the beads. Subsequent to PCR, the supernatant was removed and the beads were washed for 2 × 30 min in 150 mM NaCl and then in nuclease-free dH2O three times (initially for 1 h, then for 2 × 15 min) with gentle shaking at room temperature. The bead-immobilized DNA templates were then resuspended in 6 µl nuclease-free dH2O.
Quantitation of bead-bound DNA
The amount of 1 kb DNA (pETGFP template) specifically attached to beads during simultaneous amplification/immobilization was determined using T4 polynucleotide kinase (PNK) (Boehringer Mannheim, Indianapolis, IN) catalyzed incorporation of [γ-32P]dATP (3000 Ci/mmol; NEN Life Science Products, Boston, MA) according to the manufacturer’s 5′-end-labeling protocol. PCR with a soluble and a bead-immobilized primer was performed; the soluble control reaction used a 5′-biotinylated primer and an unmodified primer. After the completion of PCR amplification/immobilization, the soluble amplicon from each PCR reaction was purified via Qiaquick PCR Purification Spin Columns (Qiagen) and the bead-bound amplicon was washed in 150 mM NaCl twice and rinsed three times with dH2O. All samples were resuspended in 25 µl dH2O final volume. These reactions were subsequently treated with T4 PNK (37°C, 1 h, with gentle shaking) and unincorporated [γ-32P]dATP removed through Qiaquick PCR Purification Spin Columns. Radiolabeled DNA was then eluted with 50 µl dH2O. In a parallel preparation, bead-bound DNA was treated with T4 PNK, under the same conditions described above, and washed twice with 150 mM NaCl and rinsed three times with dH2O. Samples were placed in 10 ml Ecoscint H (National Diagnostics, Atlanta, GA) and a Packard 1500 Liquid Scintillation Counter (Packard, Downers Grove, IL) was utilized to determine [γ-32P]dATP incorporation in the samples. Non-specific binding to CPG or PS beads was determined by 5′-end-labeling a target gene (pTEI template, target gene ~0.7 kb) whose sequence was non-complementary to the T7-derived immobilized primer. The quantities of both soluble and bead-immobilized DNA bound were calculated using the specific activity of the [γ-32P]dATP. The percentage of total amplicon immobilized during solid phase PCR was determined as the ratio of bead-associated c.p.m. to the total c.p.m. (c.p.m. of bead-bound DNA plus c.p.m. of soluble DNA from a given PCR reaction). The data reported are the results of triplicate experiments.
In vitro transcription/translation reactions
The Linked in vitro SP6/T7–Transcription/Translation Kit (non-radioactive) (Boehringer Mannheim, Indianapolis, IN) was used, according to the manufacturer’s protocol, to obtain mRNA transcripts and protein products using soluble or bead-immobilized DNA. The mMessage mMachine in vitro transcription system (Ambion, Austin, TX) was used to determine whether bead-immobilized DNA templates could be recycled or re-used. Briefly, immobilized DNA templates were incubated with transcription reagents for 80 min at 37°C with periodic gentle shaking or continuous agitation. Samples were pelleted briefly (700 g, 2 min, 25°C) and the mRNA transcript-containing supernatant was removed and analyzed electrophoretically (see Electrophoretic analysis). Bead-immobilized DNA templates were then washed twice with 150 mM NaCl and twice with nuclease-free dH2O (30 min each with gentle shaking) and fresh transcription reaction reagents added. This was repeated for a total of seven cycles.
Electrophoretic analysis
mRNA transcripts were analyzed on 1–2% agarose gels prepared in 1× MOPS and 6% formaldehyde under RNase-free conditions and electrophoresed at 100 V (100–150 mA) for 1–1.5 h (1). RNA was visualized using ethidium bromide and photographed under UV light. Band intensities of the experimental relative to the control transcript were determined by scanning the photo into CorelDraw 7 (Corel Corp.) and using NIH Image 1.61 (W.Rasband, NIH, Bethesda, MD). For western blot analysis, protein products were electrophoresed in 12% precast Novex (San Diego, CA) acrylamide gels, electroblotted onto Immobilon-P membranes (Millipore, Billerica, MA), and probed with a 1:1000 dilution of streptavidin–horseradish peroxidase (HRP) conjugate (Gibco BRL, Gaithersburg, MD). Stabilized TMB substrate (Promega, WI) was used to visualize the biotinylated protein–streptavidin–HRP complexes.
RESULTS AND DISCUSSION
Immobilization of DNA primers
Primers synthesized on OAS supports linked to PS or CPG beads were synthesized in the 5′→3′ direction and remained attached to the PS or CPG support at the 5′-end. Thus, primers synthesized using OAS supports can be used directly as immobilized primers in PCR reactions without further manipulation. A potential disadvantage to the use of OAS supports is an inability to remove failure sequences generated during DNA synthesis. All other primers were purified after synthesis before coupling to beads, to remove failure sequences.
In order to be useful for a PCR reaction, bead-bound primers must remain intact during the thermal cycling process. We initially tested 5′-thiolated primers attached via heterobifunctional crosslinkers to amine-functionalized CPG, and, as expected (8), this chemistry proved to be heat labile (data not shown). Thus, a soluble 5′-disulfide-modified primer was used to generate thiolated PCR product and immobilization took place as a separate step, subsequent to amplification. The remainder of the immobilized primer chemistries tested were heat stable and thus suitable for solid phase PCR experiments. Table 1 summarizes the various combinations of primers, attachment chemistries and beads or polymeric supports which were used in this study.
Several amplification conditions were analyzed for the generation of a bead-bound DNA template that was functional for transcription by bacteriophage T7 polymerase. The primers used in the amplification reactions were as follows (see Table 1): (i) soluble T7Prom and soluble T7Term; (ii) soluble T7Prom and soluble 5′-thiolated T7Term; (iii) soluble T7Prom and immobilized T7Term (PC-E, PC-H, T-PS or T-CPG); (iv) immobilized T7Prom (Pr-PS or Pr-CPG) and soluble T7Term. All PCR and transcription reactions yielded soluble DNA or RNA fragments of the expected size, as verified by electrophoretic analysis and exemplified in Figure 1A and B.
Figure 1.
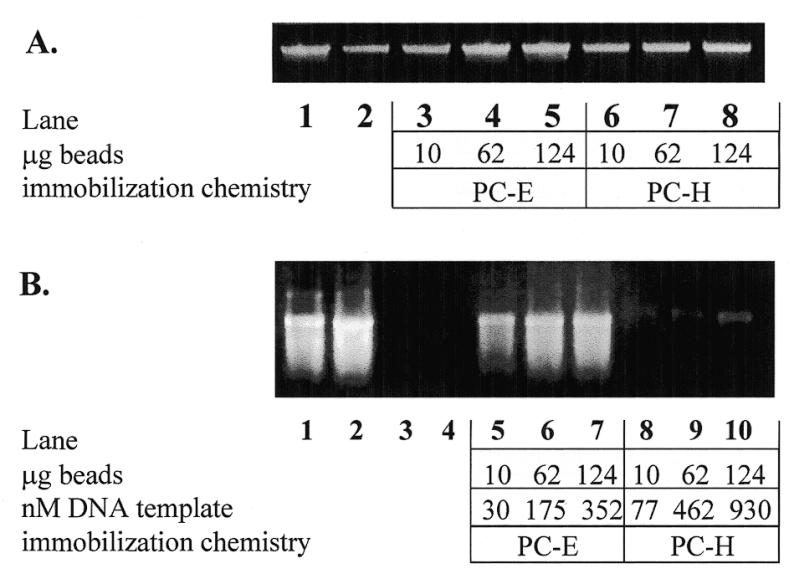
Soluble PCR amplification (A) and in vitro transcription (B) products from reactions containing indicated amounts of PC-E and PC-H immobilized primers. (A) Lane 1, 1 µM each soluble primers T7Prom and T7Term; lane 2, 1 µM and 50 nM primers T7Prom and T7Term, respectively; lanes 3–8, 1 µM T7Prom with 10, 62 or 124 µg PC-E (lanes 3–5) or 10, 62 or 124 µg PC-H (lanes 6–8), respectively. (B) mRNA transcripts from soluble or immobilized DNA. DNA templates used for in vitro transcript production shown in (B) are as follows: lane 1, 0.5 µg soluble Xenopus EF1 control DNA; lane 2, 0.5 µg soluble 1.0 kb PCR-amplified gene encoding T4 MCP from pET23; lanes 3 and 4, washed PCR reaction tubes originally containing soluble PCR amplification products shown in lanes 1 and 2 of (A); lanes 5–10, immobilized DNA templates corresponding to lanes 3–8 of (A).
The amount of DNA covalently bound to the CPG (PC-E or PC-H), OAS-CPG (T-CPG or Pr-CPG) or PS (T-PS or Pr-PS) beads was determined by 5′-end-radiolabeling DNA amplicons obtained following PCR with or without an immobilized primer. A 5′-biotin-labeled primer was used as one of two primers in the soluble control reaction to limit radiolabeling to just one of the 5′-ends of the amplicon and, thus, to more closely simulate the situation in which one primer is bead bound. Briefly, the gene for GFP was amplified from pETGFP using one of the following conditions: (i) two soluble primers (T7Prom and 5′-biotinylated T7Term); (ii) one soluble and one 5′-thiol-modified primer (T7Prom and 5′-SH-modified T7Term); (iii) one soluble and one immobilized primer [(T7Term and PC-E, PC-H, T-PS or T-CPG) or (Pr-PS or Pr-CPG and T7Term)]. The thiolated PCR amplicon generated under condition (ii) above was then immobilized to heterobifunctional crosslinker-modified CPG beads. All bead-immobilized templates and soluble PCR fractions were 5′-end-labeled as described in Materials and Methods. Non-specific DNA attachment was determined by amplifying a DNA template without any complementarity to the above soluble or immobilized primers and attempting to radiolabel the bead-associated product. Specific covalent attachment to the immobilized primers was determined by the amount of 32P incorporation associated with the beads minus any non-specific attachment. The specific activity of the [γ-32P]dATP was used to determine nmol DNA bound/g beads. Table 2 compares the yield of DNA PCR amplicons on CPG, OAS-CPG or OAS-PS beads using each type of immobilized primer, or post-PCR attachment of thiolated amplicons. The results indicate a wide range of DNA attachment in the nmol DNA/g bead range for the immobilization chemistries surveyed. It is interesting to note that HDA silanized beads (H-SIAB, H-PEG and PC-H) consistently yielded higher quantities of covalently attached DNA than EDA silanized beads for all three chemical attachment methods tested. HDA (six carbons) is a longer silane than EDA (three carbons) and may be less densely packed on the surface than the EDA. A lower density of silane may provide a greater proportion of silanes that react with the heterobifunctional crosslinkers. DNA template attachment is most successful on beads to which primers were coupled prior to PCR via carbodiimide chemistry (PC-E and PC-H) and those which underwent post-PCR immobilization onto HDA crosslinker-modified CPG (see PC-E, PC-H, H-SIAB and H-PEG, Table 2). The percentage of total PCR product that was bound to the immobilized beads during solid phase PCR is shown in Table 3. The mean bead-bound amplicon generated by PC-E and PC-H immobilized primers (expressed as a percentage of total amplicon) was 30.9 ± 0.73 and 60.4 ± 2.2%, respectively. Amplification using T-PS and T-CPG solid phase primers resulted in 51.8 ± 2.7 and 15.6 ± 3.5% of total amplicon bound to PS and CPG beads, respectively. Analysis of the four OAS supports indicated a comparatively low mean percentage of total amplicon bound to P-PS and P-CPG immobilized primers (3.2 ± 0.54 and 3.0 ± 0.53%, respectively). This result may be sequence-dependent as these primer–bead combinations used the T7Prom primer which contains a potential hairpin loop sequence that may have interfered with annealing of the primer to the target molecule.
Table 2. Quantitation of bead-immobilized DNA.
| Primer used for immobilization of amplicon | Specific binding (nmol DNA/g bead) |
|---|---|
| PC-E | 141 ± 0.48 |
| PC-H | 374 ± 0.39 |
| 5′-Thiol-modified T7Term (→E-SIAB)a | 77 ± 0.75 |
| 5′-Thiol-modified T7Term(→H-SIAB)a | 154 ± 2.5 |
| 5′-Thiol-modified T7Term(→E-PEG)a | 15.4 ± 0.63 |
| 5′-Thiol-modified T7Term(→H-PEG)a | 75.3 ± 0.28 |
| T-PS | 25.2 ± 0.6 |
| T-CPG | 10.8 ± 0.28 |
| P-PS | 18.9 ± 0.84 |
| P-CPG | 41.8 ± 1.2 |
Experimental details are given in Materials and Methods. Bead-bound primers used during the PCR amplification/immobilization protocol are indicated in the far left column using the abbreviations listed in the legend to Table 1. After PCR amplification, soluble and immobilized DNAs were purified separately and labeled with [γ-32P]dATP using T4 polynucleotide kinase at 37°C for 1 h. Bound DNA was determined by the specific activities of the oligomers. Non-specific binding was determined by PCR amplification of an ~1 kb target gene, whose sequence is non-compatible to the bead-bound primer.
aThe 5′-thiol-modified T7Term primers were used to generate soluble PCR amplicons which were subsequently crosslinked to CPG beads via the chemistry indicated in parentheses. The total soluble thiolated PCR product and the immobilized DNA were radiolabeled as described above to quantitate the amount of PCR product immobilized versus total soluble PCR product. Amplification of a 1 kb target sequence, compatible with the soluble and bead-bound primer, was used to determine specific binding. The data are reported as the means of triplicate experiments ± SD. Non-specific binding is calculated as the total binding minus non-specific binding for a particular immobilized primer.
Table 3. Percentage of total PCR amplicon immobilized.
| Solid phase primer | Percent of total |
|---|---|
| Non-specific PC | 0.45 ± 0.25 |
| PC-E | 30.9 ± 0.73 |
| PC-H | 60.4 ± 2.2 |
| Non-specific OAS-PS | 0.44 ± 0.16 |
| P-PS | 3.21 ± 0.54 |
| T-PS | 51.8 ± 2.7 |
| Non-specific OAS-CPG | 0.38 ± 0.08 |
| P-CPG | 3.04 ± 0.53 |
| T-CPG | 16.7 ± 3.5 |
Experimental details are given in Materials and Methods. Non-specific PC, PS and CPG refer to non-specific binding to those specific beads. Percentage of total PCR amplicon immobilized was determined by dividing bead-bound c.p.m. by total c.p.m. (bead-bound c.p.m. + soluble DNA c.p.m.) and multiplying by 100%. Experiments were conducted in triplicate and the data are given as the means ± SEM.
Solid phase PCR and in vitro transcription of bead-bound amplicons
In designing the solid phase PCR experiments, we selected the bacteriophage T7 transcriptional promoter and termination sequences as primers since the T7 promoter allows for high levels of tightly regulated transcription and, thus, would be useful for recombinant cloning and protein production applications (30). We wished to create a solid phase PCR system that offered widespread utility for amplification and immobilization of a wide array of gene targets. With design of appropriate primers, however, we anticipate that this method would work equally well for other bacterial, viral or mammalian expression systems.
The solid phase PCR protocol was a modification of the procedure established by Saiki et al. (31). In the present study, solid phase PCR was performed using minimal, or ‘spike’, concentrations of the soluble equivalent to the immobilized primer. This was critical in the initial stages of PCR amplification to increase the ability of the DNA template to interact with bead-immobilized primers that settled in the bottom of the PCR reaction tube. Rassmussen and co-workers similarly used asymmetric PCR to immobilize PCR amplicons using PCR primers attached to microtiter plate wells (20,21). Figure 1 shows a representative result for solid phase PCR and in vitro transcription with bead-immobilized PCR products using pET23 as the template DNA, which yields a 1.56 kb PCR product (gene encoding T4 MCP). For PCR and subsequent in vitro transcription reactions, 10, 62 and 124 µg of beads attached to the indicated primer (PC-E or PC-H) were used. From our radiolabeling experiments, we have calculated the bead-bound primer concentrations in each PCR reaction to be 30, 175 and 352 nM (PC-E, Fig. 1A, lanes 3–5) and 77, 462 and 930 nM (PC-H, Fig. 1A, lanes 6–8). Figure 1A, lane 1, shows the soluble PCR product produced from two soluble primers T7Prom and T7Term (1 µM each). Lane 2 shows the soluble amplicon produced from two soluble primers T7Prom and T7Term (at 1 µM and 50 nM, respectively), under conditions which mimic the solid phase PCR conditions except that the immobilized primer is not present. The soluble PCR product from solid phase PCR reactions using 1 µM T7Prom, 50 nM soluble T7Term and the indicated amounts of PC-E and PC-H immobilized primers is shown in Figure 1A, lanes 3–8. The PCR reactions yielded a single soluble DNA band of the expected molecular weight (1.56 kb) for the gene encoding T4 MCP. The band intensity of the soluble PCR product shown in Figure 1A, lanes 3–8 (the same reaction conditions as used in lane 2 plus the indicated amount of immobilized T7Term primer), is of equal or greater intensity than the PCR product shown in lane 2. This is an expected result because the enzymatic extension of the immobilized primer creates additional template (antisense) strand that can be used to create more soluble product by the soluble primer present in the reaction. PCR reactions that contained soluble T7Prom and immobilized T7Term primers, but no soluble ‘spike’ of T7Term primer, resulted in undetectable soluble PCR product formation (data not shown). These results indicate that the small increase in soluble PCR product observed in lanes 3–8 is not due to dissociation of primer from the bead during amplification. The addition of a ‘spike’ of soluble T7Term primer was necessary to increase the likelihood of an interaction between the soluble coding strand and the bead-bound T7Term primer, which settles to the bottom of the PCR tube during amplification. To determine whether settlement of the beads during PCR was inhibiting efficient coding strand capture, we repeated select reactions but altered the protocol such that the reaction tubes were vortexed vigorously during the denaturation stage of several cycles (2, 4, 6, 8, 12 and 24). Contrary to our expectations, this resulted in a diminished yield of lower quality transcript (which reached a maximum of 66% of the non-vortexed reaction).
All bead-immobilized DNAs tested were functional as templates for in vitro transcription (representative data are shown in Fig. 1B). Following the PCR reactions, the 1.56 kb gene encoding T4 MCP attached to PC-E and PC-H beads was subjected to in vitro transcription. Electrophoretic analysis of the mRNA transcripts produced from these reactions is shown in Figure 1B. Lanes 1 and 2 show mRNA transcript production from two soluble DNA templates. As negative controls, the PCR reaction tubes that originally contained the soluble PCR products shown in Figure 1A, lanes 1 and 2, were washed (in parallel with the bead-containing PCR reactions, Fig. 1A, lanes 3–8) and used for in vitro transcription reactions. The lack of mRNA product in Figure 1B, lanes 3 and 4, demonstrated that the observed mRNA transcription observed in lanes 5–10 was not due to carry-over of soluble DNA non-specifically bound to the reaction tube. A comparison of Figure 1B, lanes 5–7 and 8–9 shows that although mRNA is produced with each bead type, the PC-E-immobilized PCR amplicon is more readily transcribed than DNA attached through PC-H primers. This result was also consistent with in vitro transcription results for amplicons produced from 5′-thiol-modified primers and subsequently attached to EDA- or HDA-modified CPG beads (E-SIAB, H-SIAB, E-PEG and H-PEG) in that more mRNA was produced when EDA-silanized beads were used. Paradoxically, the observed higher levels of amplicon attachment to HDA silanized beads may be detrimental to efficient in vitro transcription of the immobilized template DNA. This may be due to the inability of T7 polymerase and other required transcriptional components to efficiently access and transcribe because of tight DNA packing on and inside the CPG bead pores.
We examined a range of concentrations of bead-bound DNA template to establish whether or not a linear relationship exists between the quantity of bead-bound DNA template added to transcription reactions and the mRNA produced. A comparison of band intensities of mRNA transcribed from bead-immobilized DNA versus soluble DNA template showed a negligible difference in transcript production above 62 µg beads. The results relative to the Xenopus control (1.00) (Fig. 1B, lane 1) were 0.65, 0.80 and 0.84 for 10, 62 and 124 µg PC-E-immobilized DNA, respectively. The results for PC-H relative to the same control were 0.32, 0.28 and 0.32 for 10, 62 and 124 µg beads. These observations indicated a near saturated state with little change with increasing bead-bound amplicon concentration. For nearly all of the transcription experiments, reactions were periodically gently agitated during incubation. To determine whether the observed limitation in product formation was due to the restricted accessibility to the immobilized DNA of the transcription reactants as the beads settled to the bottom of the reaction tube, we repeated select experiments with continuous agitation during the incubation period. This resulted in an average increase of 19.5% product mRNA (when >50 µg beads were used per transcription reaction), verifying that exposure of the beads to fresh transcription reagents through mixing improved yield.
It is important to note that the relative intensities of the mRNA bands (Fig. 1B, lanes 5–7) were ~80% of the soluble Xenopus DNA transcriptional control (Fig. 1B, lane 1), demonstrating the efficiency of the solid phase templates in in vitro transcription reactions.
Our primary interest in developing a solid phase PCR amplification system using bead-bound primers was their potential use in repetitive in vitro transcription reactions. In vivo protein synthesis of polymeric proteins and certain mammalian proteins can be problematic due to the rearrangement or modification of highly repetitive DNA sequences, improper protein folding and cellular toxicity. Although in vitro transcription/translation systems could provide an alternative to in vivo protein synthesis, these methodologies can be impractical because of the laborious and often expensive necessity to resynthesize DNA templates after each transcription reaction. Conversely, automated solid phase peptide synthesis can be quite economical but may be limited by the length and sometimes the sequences of the desired protein. Bead-immobilized DNA templates would provide a convenient way to circumvent these problems by enabling the bead-bound DNA templates to be collected and recycled after each transcription reaction. This approach permits the synthesis of large proteins not available by automated synthesis and is more economical than conventional in vitro transcription/translation reactions. Figure 2 shows the results of an experiment designed to determine whether DNA templates, coding for GFP (1.0 kb), immobilized via PC-E and E-PEG crosslinked supports, could be efficient templates for multiple sequential in vitro transcription reactions. In these experiments, bead-immobilized PCR amplicons were used as templates for in vitro transcription, then the immobilized DNA primers were collected by brief centrifugation, rewashed and exposed to fresh in vitro transcription reagents. Significant differences in sequential mRNA production were observed for each type of immobilized DNA template. Although E-PEG yielded high levels of transcript initially, these templates were unable to survive the recycling process beyond two or three cycles of repetitive rounds of in vitro transcription. This result proved to be the case for most immobilized DNA templates tested with the exception of PC-E-immobilized DNA. In this case, our findings indicated that PC-E-bound templates can be collected and recycled effectively for use in at least seven repetitive rounds of in vitro transcription.
Figure 2.

Repetitive in vitro transcription using solid phase DNA templates. mRNA electrophoresed on a 1% agarose–formaldehyde gel. Lane 1, mRNA markers in kb as indicated; lanes 2–4 and 5–7, mRNA from the first, fifth and seventh sequential rounds of in vitro transcription using GFP DNA templates crosslinked to CPG beads via PC-E or E-PEG chemistry.
Protein production from solid phase DNA templates
To determine whether mRNA transcripts produced from solid phase DNA templates were able to initiate translation, reagents for non-radioactive in vitro translation were added to the supernatants from in vitro transcription reactions. The biotinylated protein products were detected using a streptavidin–HRP conjugate with TMB as substrate. Figure 3A shows a western blot of biotinylated products produced from solid phase templates through a coupled in vitro transcription/translation system. All solid phase DNA templates produced the desired protein product (57 kDa T4 MCP). A useful feature of the bead immobilization methodology is the ability to amplify and to immobilize either single or multiple DNA target sequences. The results of such a mixing experiment are shown in Figure 3B. In this experiment, the genes for GFP and T4 MCP were amplified via solid phase PCR using the immobilized PC-E primer and plasmids pETGFP and pET23 as templates (either separately or mixed together). The bead-immobilized amplicons from each solid phase PCR reaction (GFP, MCP and GFP + MCP) were collected, washed and used as templates for in vitro transcription/translation reactions. Figure 3B, lanes 3 and 4, shows the separate production of GFP and T4 MCP and lane 5 shows in vitro translation of both proteins simultaneously from a mixture of immobilized templates. Thus, the addition of multiple DNA gene templates to solid phase PCR reactions yields efficient immobilization of both genes and the subsequent production of multiple protein products.
Figure 3.
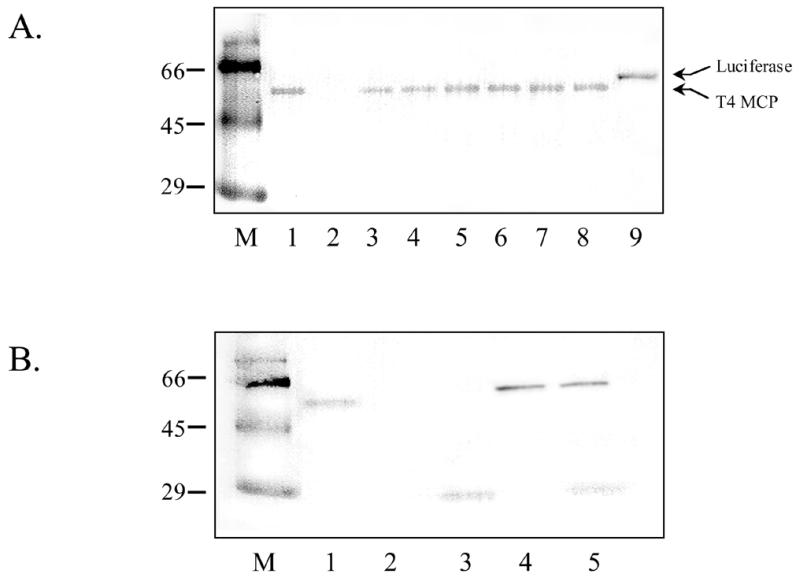
Western blot showing production of T4 MCP from immobilized DNA templates. (A) DNA templates used for in vitro transcription product production and subsequent protein translation are as follows: lane 1, soluble DNA encoding MCP; lane 2, no DNA control; lane 9, soluble DNA encoding luciferase protein. MCP DNA templates used to obtain protein products in lanes 3–8 were immobilized using soluble T7Prom and the following immobilized primers: lane 3, P-PS; lane 4, P-CPG; lane 5, T-PS; lane 6, T-CPG; lane 7, 5′-thiol-modified T7Term (attached to CPG post-PCR to generate E-SIAB immobilized DNA); lane 8, 5′-thiol-modified T7Term (attached to CPG post-PCR to generate E-PEG immobilized DNA). (B) Western blot showing the results of a mixing experiment using templates immobilized via PC-E primer during solid phase PCR singly or together. DNA templates used to obtain protein shown in (B) are as follows: lane 1, soluble gene encoding MCP; lane 2, no DNA; lane 3, GFP + PC-E; lane 4, MCP + PC-E; lane 5, GFP + PC-E and MCP+PC-E.
CONCLUSIONS
Two solid phase DNA templates, one coding for the 27 kDa GFP and the other coding for the 57 kDa bacteriophage T4 major capsid protein (gp23) were covalently immobilized to CPG, OAS-CPG or OAS-PS supports using seven different chemistries via solid phase PCR amplification or post-PCR reactions (Tables 1 and 2). All DNA templates immobilized by this approach were shown to be competent for in vitro transcription and subsequent protein production (Fig. 3), and multiple templates could be simultaneously amplified and immobilized using a single immobilized primer/soluble primer pair. Although other methods utilizing post-PCR immobilization have been used to attach DNA amplicons to a variety of surfaces, these methodologies have critical limitations. For example, we also generated solid phase PCR protocols using a single biotinylated primer bound to avidin-coated MPG particles (Table 1), but found the biotin–avidin interaction to be unsuitable for repetitive in vitro transcription reactions (data not shown). Our finding, which concurs with other work, suggests that success in using avidin–biotin-immobilized DNAs as recyclable templates for in vitro transcription reactions may be dependent on the location of the biotin modifier (31). In contrast to this, our covalent immobilization methodology allows for repetitive collection and re-use of bead-bound DNA templates (Fig. 2), which addresses a significant problem with current extracellular protein production methods: the inability to easily recover and re-use soluble nucleic acid templates.
The contribution of the solid phase PCR DNA immobilization methodology we present in this paper, however, goes beyond DNA hybridization assays and in vitro protein production. The ease of amplification and simultaneous covalent immobilization onto particles enables these amplicons to be utilized directly in a variety of downstream applications. One can envision, for example, simultaneous amplification, immobilization and subsequent transcription/translation of numerous DNA templates under the control of various transcriptional promoters, by including appropriate mixtures of immobilized priming sequences (e.g. plasmids under the control of T7, Sp6 and retroviral promoters). Bead-immobilized gene targets attached to mammalian transcriptional and translational regulatory regions can also be created using our solid phase PCR approach and may be used to characterize transcriptional complexes or possibly employed directly as DNA vaccines (32–34). Covalently immobilized templates on particles may also be potentially useful as recyclable and mobile elements in microfabricated diagnostic devices or biosensors. In addition, our approach provides the unique flexibility of mixing preamplified bead-bound DNA sequences for immediately realizable diagnostic assays tailored to a specific need.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Dr J. M. Mauro (Geo-Centers, Inc.) for kindly supplying us with pTE1 and its respective primers and for helpful suggestions. We would also like to thank Drs J.M. Mauro and J. Pancrazio for review of this manuscript. A National Research Council Post-doctoral Fellowship to J.D.A. supported this work.
REFERENCES
- 1.Maniatis T., Fritsch,E.F. and Sambrook,J. (1982) Molecular Cloning: A Laboratory Manual, 1st Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 2.Conner B.J., Reyes,A.A., Morin,C., Itakura,K., Teplitz,R.L. and Wallace,R.B. (1983) Proc. Natl Acad. Sci. USA, 80, 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lockley A.K., Jones,C.G., Bruce,J.S., Franklin,S.J. and Bardsley,R.G. (1997) Nucleic Acids Res., 25, 1313–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joos B., Kuster,H. and Cone,R. (1997) Anal. Biochem., 247, 96–101. [DOI] [PubMed] [Google Scholar]
- 5.Cohen G., Deutsch,J., Fineberg,J. and Levine,A. (1997) Nucleic Acids Res., 25, 911–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang M.S., Kong,R.Y.C., Kazmi,N. and Leung,A.K.C. (1998) Chem. Lett., 3, 257–258. [Google Scholar]
- 7.Maskos U. and Southern,E.M. (1992) Nucleic Acids Res., 20, 1679–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chrisey L.A., Lee,G.U. and O’Ferrall,C.E. (1996) Nucleic Acids Res., 24, 3131–3039. [Google Scholar]
- 9.Chrisey L.A., O’Ferrall,C.E., Spargo,B.J., Dulcey,C.S. and Calvert,J.M. (1996) Nucleic Acids Res., 24, 3040–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marble H.A. and Davis,R.H. (1995) Biotechnol. Prog., 11, 393–396. [DOI] [PubMed] [Google Scholar]
- 11.Liu M. and Price,D.H. (1997) Promega Notes Mag., 64, 21–25. [Google Scholar]
- 12.Weiler J. and Hoheisel J.D. (1996) Anal. Biochem., 243, 218–227. [DOI] [PubMed] [Google Scholar]
- 13.Beattie W.G., Meng,L., Turner,S.L., Varma,R.S., Dao,D.D. and Beattie,K.L. (1995) Mol. Biotechnol., 4, 213–225. [DOI] [PubMed] [Google Scholar]
- 14.Rasmussen S.R., Larsen,M.R. and Rasmussen,S.E. (1991) Anal. Biochem., 198, 138–142. [DOI] [PubMed] [Google Scholar]
- 15.Timofeev E., Kochetkova,S.V., Mirzabekov,A.D. and Florentiev,V.L. (1996) Nucleic Acids Res., 24, 3142–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yershov G., Zaslavsky,A., Gemmell,A., Shick,V., Proudnikov,D., Arenkov,P. and Mirzabekov,A. (1997) Anal. Biochem., 250, 203–211. [DOI] [PubMed] [Google Scholar]
- 17.DeAngelis M.M., Wang,D.G. and Hawkins,T.L. (1995) Nucleic Acids Res., 23, 4742–4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haukanes B.I. and Kvam,C. (1993) Biotechnology, 11, 60–63. [DOI] [PubMed] [Google Scholar]
- 19.Saiki R.K., Walsh,P.S., Lenvenson,C.H. and Erlich,H.A. (1989) Proc. Natl Acad. Sci. USA, 86, 6230–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rasmussen S.R., Rasmussen,H.B., Larsen,M.R., Hoff-Jorgensen,R. and Cano,R.J. (1994) Clin. Chem., 40, 200–205. [PubMed] [Google Scholar]
- 21.Oroskar A., Rasmussen,S.R., Rasmussen,H.B., Rasmusssen,S.R., Sullivan,B.M. and Johansson,A. (1996) Clin. Chem., 42, 1547–1555. [Google Scholar]
- 22.Drobyshev A., Shik,M.N., Pobedimskaya,D., Yershov,G. and Mirzabekov,A. (1997) Gene, 188, 45–52. [DOI] [PubMed] [Google Scholar]
- 23.Keller G.H., Huang,d.-P. and Manak,M.M. (1991) J. Clin. Microbiol., 29, 638–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chevrier D., Popoff,M.Y., Diaon,M.P., Hermant,D. and Guesdon,J.L. (1995) FEMS Immunol. Med. Microbiol., 10, 245–501. [DOI] [PubMed] [Google Scholar]
- 25.Kohsaka H. and Carson,D.A. (1994) J. Clin. Lab. Anal., 8, 452–455. [DOI] [PubMed] [Google Scholar]
- 26.Schena M., Shalon,D., Davis,R.W. and Brown,P.O. (1995) Science, 270, 467–470. [DOI] [PubMed] [Google Scholar]
- 27.Schena M., Shalon,D., Heller,R., Chai,A., Brown,P.O. and Davis,R.W. (1996) Proc. Natl Acad. Sci. USA, 93, 10614–10619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andreadis J.D. and Black,L. (1998) J. Biol. Chem., 273, 34075–34086. [DOI] [PubMed] [Google Scholar]
- 29.Ujvari A. and Martin,C.T. (1997) J. Mol. Biol., 273, 775–781. [DOI] [PubMed] [Google Scholar]
- 30.Fujita K. and Silver,J. (1993) Biotechniques, 14, 608–617. [PubMed] [Google Scholar]
- 31.Saiki R.K., Gelfand,D.H., Stoffel,S., Scharf,S.J., Higuche,R., Horn,G.T., Mullis,K.B. and Erlich,H.A. (1988) Science, 239, 487–491. [DOI] [PubMed] [Google Scholar]
- 32.Wang Z.Y. and Rana,T.M. (1997) Proc. Natl Acad. Sci. USA, 94, 6688–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ulmer J.B., Donnelly,J.J., Parker,S.E., Rhodes,G.H., Felgner,P.L., Dwarki,V.J., Gromkowski,S.H., Deck,R.R., DeWitt,C.M. and Friedman,A. (1993) Science, 259, 1745–1749. [DOI] [PubMed] [Google Scholar]
- 34.Raz E., Carson,D.A., Parker,S.E., Parr,T.B., Abai,A.M., Aichinger,G., Gromkowski,S.H., Singh,M., Lew,D. and Yankauckas,M.A. (1994) Proc. Natl Acad. Sci. USA, 91, 9519–9523. [DOI] [PMC free article] [PubMed] [Google Scholar]