


Abstract
The Holliday junction is a central intermediate in genetic recombination. This four-stranded DNA structure is capable of spontaneous branch migration, and is lost during standard DNA extraction protocols. In order to isolate and characterize recombination intermediates that contain Holliday junctions, we have developed a rapid protocol that restrains branch migration of four-way DNA junctions. The cationic detergent hex-adecyltrimethylammonium bromide is used to lyse cells and precipitate DNA. Manipulations are performed in the presence of the cations hexamine cobalt(III) or magnesium, which stabilize Holliday junctions in a stacked-X configuration that branch migrates very slowly. This protocol was evaluated using a sensitive assay for spontaneous branch migration, and was shown to preserve both artificial Holliday junctions and meiotic recombination intermediates containing four-way junctions.
INTRODUCTION
Genetic recombination involves the exchange of DNA strands between duplex DNA molecules. The point of strand exchange is called a Holliday junction (1,2), a four-stranded intermediate that is central to most models of homologous recombination (3–5). Four-way junctions have been observed in studies of both homologous (6–8) and site-specific recombination (9). The Holliday junction is capable of movement by branch migration, where DNA strands are exchanged by the stepwise breakage and reformation of base pairs, generating a hybrid joint between the homologous chromosomes (2). Enzymes that catalyze this reaction have been identified in Escherichia coli (10).
Testing models for genetic recombination often requires the physical detection and analysis of relevant intermediates. However, branch migration presents a problem for the isolation of recombination intermediates that contain Holliday junctions. The rate of spontaneous branch migration is determined by the structure of the junction, which is acutely sensitive to metal ions (11,12). DNA extraction protocols generally use EDTA to chelate divalent cations, thereby inhibiting nuclease activity. In the absence of cations, the four-way DNA junction exists in an unstacked square planar conformation that branch migrates very rapidly (12–14), and will therefore be lost during standard DNA extraction procedures. In contrast, the rate of spontaneous branch migration is very slow in the presence of multivalent cations such as Mg2+ (12), which allow the junction to adopt a folded X structure where base stacking is maintained through the crossover point (15). Nevertheless, using Mg2+ to stabilize Holliday junctions during DNA extraction is impractical on account of accompanying nuclease activities.
Studies of site-specific recombination have trapped Holliday junction intermediates with flanking heterology or by use of recombinase step-arrest mutants (16,17). This approach is not practical in studies of homologous recombination between linear DNA molecules; instead, recombination intermediates have been stabilized by psoralen crosslinking before purification (7,18). However, this treatment renders the DNA recalcitrant to analysis of individual strands, since crosslinks must be removed by a lengthy process before the DNA can be denatured (19).
We have developed a rapid procedure for DNA extraction in the presence of hexamine cobalt(III) [Co3+(NH3)6], a polyvalent cation that efficiently folds four-way DNA junctions into a stacked X structure (15). Cells are lysed and DNA is extracted using the cationic detergent hexadecyltrimethylammonium bromide (CTAB). In developing this protocol, we used a sensitive assay to monitor spontaneous branch migration (11,12). We demonstrate that four-way DNA junctions can be preserved during this extraction procedure without the need for crosslinking.
MATERIALS AND METHODS
Preparation of branch migration substrates
Two sets of tailed duplex substrates (Fig. 1) were used. S1 and S2, with homologous duplex lengths of 300 bp, were a gift from Mikhail Grigoriev and Peggy Hsieh (NIH, Bethesda, MD) (11). To generate R1 and R2, with homologous duplex lengths of 273 bp, a 281 bp fragment of the ARG4 gene of Saccharomyces cerevisiae was amplified by PCR using the primers 5′-ACTTGGTCAGAAAGGGTGTTCC-3′ and 5′-TGC-GCATAACGTCGCCATCTGC-3′. The PCR product was purified by Microcon 100 spin filtration (Amicon), digested with BsmAI, and a 257 bp fragment purified from an agarose gel. Partial duplexes (D1 and D2) were obtained by annealing 5′-ACCATGCTCGAGATTACGAGATATCGATGCATGCG-3′ with 5′-CTGACGCATGCATCGATATAATACGTGAGGC-CTAGGATC-3′ for D1, and 5′-GATCCTAGGCCTCACGT-ATTATATCGATGCATGCG-3′ with 5′-CTGACGCATGCAT-CGATATCTCGTAATCTCGAGCATGGT-3′ for D2, followed by two rounds of purification from native polyacrylamide gels (20). The second oligonucleotide in each pair was 5′-phosphorylated during synthesis. Partial duplexes were ligated to the 257 bp ARG4 fragment, and the products were purified from a native polyacrylamide gel. One of the pair of substrates was 5′-32P-end-labeled using T4 polynucleotide kinase and [γ-32P]ATP, and purified by extraction with phenol:chloroform followed by ethanol precipitation.
Figure 1.

Branch migration assay. Two homologous duplexes (hatched) each having heterologous single-strand tails (black and white) are rapidly annealed to form a four-stranded structure with a Holliday junction. Two sets of substrates were used: S1 and S2 (300 bp homologous duplex) and R1 and R2 (273 bp homologous duplex). Spontaneous branch migration proceeds as a random walk until the junction reaches the distal end of the homologous region, leading to the irreversible dissociation of two duplex products. Asterisks indicate 5′-32P-label.
Branch migration assays
Branch migration assays (Fig. 1) were performed essentially as described previously (12). A 32P-labeled duplex substrate (5–10 µg/ml final concentration) was mixed on ice with a 10-fold molar excess of a second unlabeled substrate, in a 2–4 µl reaction volume containing TNM buffer (10 mM Tris–HCl, pH 7.5, 0.1 mM EDTA, 50 mM NaCl, 10 mM MgCl2). Substrates were incubated at 37°C for 3 min to promote annealing, and then diluted on ice in 20–30 µl TNM buffer. For branch migration in the absence of free Mg2+, EDTA was added on ice to 20 mM, followed by Co3+(NH3)6Cl–3 (Fluka) or spermidine (Sigma) as appropriate. Samples were then incubated at 37°C (unless indicated otherwise), and the reaction stopped by adding 2.5 vol of ice-cold stop buffer (10 mM Tris–HCl pH 7.5, 0.1 mM EDTA, 10 mM MgCl2, 10% v/v glycerol, 0.1% w/v bromophenol blue, 0.1% w/v xylene cyanol FF, 1 µg/ml ethidium bromide). Samples were stored on ice, and resolved on native 4.75% (w/v) polyacrylamide (40:1) gels in 89 mM Tris–borate pH 8.3, 1 mM EDTA, 3 mM MgCl2, 0.5 µg/ml ethidium bromide, at 8 V/cm for 4.5 h at 4°C. Gels were vacuum dried for 2 h at 80°C, and quantification performed using a Fuji BAS2000 phosphorimager. The step time for branch migration was calculated as T1/2/0.76L2, where T1/2 is the time required for half-maximal dissociation of the Holliday junction by branch migration and L is the length (in base pairs) of the duplex fragment (21).
Branch migration during ‘standard’ DNA extraction with SDS
Individual steps of a yeast DNA extraction protocol using SDS were tested as follows. 32P-labeled S1 and excess unlabeled S2 were annealed and diluted on ice in 20 µl TNM buffer. An aliquot of 1 µl of annealed substrate was added to 10 µl spheroplasting solution [1.2 M sorbitol, 20 mM EDTA, 1% β-mercaptoethanol ± 10 mM Co3+(NH3)6Cl–3] or 10 µl SDS lysis solution [100 mM NaCl, 50 mM Tris–HCl pH 8.5, 20 mM EDTA ± 10 mM Co3+(NH3)6Cl–3] and incubated at 37°C for 30 min. Alcohol precipitation was tested using 1 µl annealed substrate in 10 µl 0.3 M sodium acetate ± 10 mM Co3+(NH3)6Cl–3. To this were added 10 µl isopropanol or 20 µl ethanol, and the DNA was pelleted and resuspended in 10 µl ice-cold TNM buffer. To test extraction protocols, 1 µl annealed substrate was added to 10 µl TNM buffer and extracted with 10 µl chloroform or 10 µl phenol:chloroform [1:1 v/v phenol equilibrated with either 10 mM Tris–HCl pH 7.5, 1 mM EDTA, or 50 mM NaCl, 10 mM Tris–HCl pH 7.5, 20 mM EDTA, 10 mM Co3+(NH3)6Cl–3]. Reactions were stopped by the addition of 25 µl of ice-cold stop buffer, and analyzed as described above.
DNA precipitation assay
Bacteriophage λ DNA was digested with XmaI, end-labeled using Klenow enzyme, dGTP and [α-32P]dCTP, and purified by passage through a Nuctrap column (Stratagene). An aliquot of 50 ng of 32P-labeled λ DNA was mixed with 20 µg carrier salmon sperm DNA in a 20 µl reaction volume containing TNM buffer, supplemented as appropriate. Samples were incubated at 37°C for 2 h, centrifuged at 16 000 g for 15 min at 4°C, and the amount of 32P present in the supernatant and pellet was measured separately by liquid scintillation counting.
DNA extraction using CTAB
The following solutions were used. Spheroplasting buffer: 1 M sorbitol, 50 mM potassium phosphate pH 7, 10 mM EDTA, 5 mM Co3+(NH3)6Cl–3; Extraction buffer: 3% (w/v) CTAB (Sigma), 2 M NaCl, 100 mM Tris–HCl pH 7.5, 25 mM EDTA, 2% (w/v) polyvinyl pyrrolidone 40 (Sigma), 20 mM Co3+(NH3)6Cl–3. Extraction buffer was filter sterilized and mixed in the following order: 10 ml of 200 mM Co3+(NH3)6Cl–3, followed by 60 ml of 3.33 M NaCl, 167 mM Tris–HCl pH 7.5, 41.7 mM EDTA, 3.33% (w/v) polyvinyl pyrrolidone 40, and, finally, 30 ml of 10% (w/v) CTAB. Extraction buffer was stored at 37°C to prevent precipitation. Dilution buffer: 1% (w/v) CTAB, 50 mM Tris–HCl pH 7.5, 10 mM EDTA, 4 mM Co3+(NH3)6Cl–3. Wash buffer: 0.4 M NaCl, 10 mM Tris–HCl pH 7.5, 1 mM EDTA, 1 mM Co3+(NH3)6Cl–3. NaCoHex solution: 1.42 M NaCl, 10 mM Tris–HCl pH 7.5, 1 mM EDTA, 1 mM Co3+(NH3)6Cl–3. TMNa solution: 10 mM Tris–HCl pH 7.5, 10 mM MgCl2, 200 mM NaCl. TMSpe buffer: 10 mM Tris–HCl pH 7.5, 2 mM MgCl2, 50 µM spermidine.
A saturated overnight culture (~1.6 × 109 haploid cells in 8 ml) of S.cerevisiae strain S1007 [MATa ura3 lys2 ho::LYS2 arg4Δ(eco47III-hpa1) cyh2-z], a derivative of SK1 (22), was pelleted at 3000 g for 2 min, washed in 2 ml ice-cold spheroplasting buffer, and pelleted again. Cells were resuspended in 500 µl spheroplasting buffer plus 1% (v/v) β-mercaptoethanol, 0.5 mg/ml Zymolyase 100T (ICN), and incubated at 37°C for 5 min. Spheroplasts were pelleted and the supernatant removed completely. The pellet was resuspended gently in 500 µl of extraction buffer, followed by the stepwise addition of β-mercaptoethanol to 0.16% (v/v), proteinase K (Sigma) to 0.5 mg/ml, and RNase (Boehringer Mannheim) to 20 µg/ml. The homogenate was incubated at 37°C for 15 min with occasional mixing, extracted with 200 µl chloroform:isoamyl alcohol (24:1 v/v) by vigorous vortexing, and centrifuged at 16 000 g for 3 min. All subsequent precipitations and washes were performed without centrifugation. The aqueous phase was transferred to a 5 ml round-bottomed tube, and 1.5 ml of dilution buffer was added. The tube was inverted gently about five times until a diffuse precipitate formed, incubated for 10 min at room temperature, and was then inverted a further 20 times until a discrete precipitate appeared; this procedure reduces RNA co-precipitation. The supernatant was removed and the precipitate was washed twice with 2 ml of ice-cold wash buffer. An aliquot of 0.5 ml of ice-cold NaCoHex solution was added and the tube agitated gently until the DNA precipitate became translucent. The suspension (including the DNA) was transferred to a 1.5 ml tube, 1 ml of ethanol (at room temperature) was added, and the tube inverted until a discrete precipitate formed. The supernatant was removed and the precipitate was washed with 1 ml of 70% (v/v) ethanol + 30% (v/v) 1 mM Co3+(NH3)6Cl–3. The DNA was resuspended on ice in NaCoHex solution and precipitated with ethanol as before. The DNA was resuspended on ice in 100 µl of ice-cold TMNa solution and precipitated with 200 µl of ethanol (at room temperature). The DNA was washed with 200 µl of 70% (v/v) ethanol + 30% (v/v) 10 mM MgCl2, and resuspended on ice in 100 µl of ice-cold TMSpe buffer.
To test branch migration during DNA extraction with CTAB, 32P-labeled R1 and unlabeled R2 were annealed, and diluted on ice in 20 µl TNM buffer. An aliquot of 3 ml of a saturated overnight culture of S.cerevisiae was spheroplasted in 150 µl spheroplasting buffer, and the cell pellet resuspended in 200 µl extraction buffer. A sample of 3.5 µl of annealed branch migration substrate was added, and DNA extraction performed as described above (volumes were scaled proportionately). After extraction with chloroform:isoamyl alcohol, a 20 µl aliquot was added to 50 µl stop buffer on ice. DNA pellets obtained after precipitation with NaCoHex solution and ethanol, and after precipitation with TMNa and ethanol, were resuspended in 20 µl ice-cold TMSpe buffer, and 50 µl of ice-cold stop buffer was added. Samples were kept on ice prior to electrophoresis.
Isolation of joint molecules from meiotic cultures
A 400 ml synchronous meiotic culture of S.cerevisiae SK1 strain MJL2444 [MATa/α ura3/ura3 lys2/lys2 ho::LYS2/ho::LYS2 arg4Δ(eco47III-hpa1)/arg4Δ(eco47III-hpa1) cyh2-z/CYH2 LEU2/leu2-R his4Δ(Sal1-Cla1)::URA3-Δ(Sma1-Eco47III)-arg4-EcPal(1691)/his4Δ(Sal1-Cla1)::URA3-Δ(Sma1-Eco47III)-ARG4] was prepared as described (23). Samples (50 ml) were taken and DNA extracted using CTAB as described above. DNA (2 µg) was digested with XmnI and resolved on a native/native 2D agarose gel essentially as described (18,24), except that electrophoresis was performed at 4°C in 89 mM Tris–borate pH 8.3, 1 mM EDTA, 3 mM MgCl2 to limit branch migration. DNA was transferred to Zetaprobe GT membrane (Bio-Rad) by alkaline transfer (20), and hybridized with a radioactive probe prepared by random priming of a HpaI fragment containing the ARG4 gene of S.cerevisiae.
RESULTS AND DISCUSSION
Hexamine cobalt(III) chloride reduces the rate of branch migration
We assayed spontaneous branch migration as outlined in Figure 1 (11,12). Two substrates consisting of homologous duplexes with single-stranded tails were annealed to form a four-stranded structure containing a Holliday junction that is capable of branch migration. Spontaneous branch migration terminates when the Holliday junction reaches the distal end of the homologous region and the two duplex products dissociate.
Spontaneous branch migration in the presence of 10 mM Mg2+ is exceedingly slow (11). However, DNA extraction in the presence of Mg2+ is impractical, owing to the presence of intracellular nucleases. We therefore assayed branch migration under conditions found in typical DNA extraction protocols, where intracellular Mg2+ is chelated by the addition of excess EDTA. In order to retard branch migration in the presence of EDTA, we examined the effect of spermidine and hexamine cobalt(III) (Fig. 2). Both of these polyvalent cations efficiently fold four-way junctions into a stacked X conformation (15). Branch migration of a Holliday junction intermediate at 37°C occurred very slowly in 10 mM Mg2+ (Fig. 2A). When Mg2+ was chelated by EDTA, branch migration proceeded very rapidly, and was almost complete after 5 min at 37°C. Supplementing this reaction with 2 mM spermidine or 2 mM Co3+(NH3)6Cl–3 significantly retarded branch migration. We calculated the 1 bp step time of branch migration to be 145 ms in 10 mM Mg2+, <0.9 ms after addition of EDTA, and 30 and 112 ms, respectively, when the reaction was supplemented with 2 mM spermidine or 2 mM Co3+(NH3)6Cl–3. These values are in good agreement with published data (12). We subsequently determined that under the conditions tested, 10 mM is the optimum concentration of Co3+(NH3)6Cl–3 for restraining branch migration (Fig. 2B). After 2 h incubation at 37°C, only 5% of the Holliday junction-containing intermediate had dissociated by branch migration into duplex products.
Figure 2.

Effect of polyvalent cations and temperature on branch migration. (A) Effect of polyvalent cations. 32P-labeled S1 and excess unlabeled S2 substrates were annealed in TNM buffer (10 mM MgCl2) and aliquoted to reactions containing the indicated supplements. Reactions were incubated at 37°C for the indicated time, and dissociation of the Holliday junction (upper band) to duplex products (lower band) by branch migration was monitored by gel electrophoresis. The middle band (→) is 32P-labeled duplex DNA lacking single-stranded tails, which does not participate in the reaction. Net product refers to the fraction of total 32P label migrating as monomer duplex, after subtraction of residual unannealed substrate (in this case, duplex remaining after 1 min in 10 mM MgCl2). (B) Effect of Co3+(NH3)6Cl–3 concentration. R1 and R2 were annealed in TNM buffer and aliquoted to reactions containing 10 mM MgCl2, 10 mM MgCl2 + 20 mM EDTA or 10 mM MgCl2 + 20 mM EDTA + Co3+(NH3)6Cl–3 at the indicated concentrations. Reactions were incubated at 37°C for 2 h, and branch migration was determined as described in (A). (C) Effect of temperature. S1 and S2 were annealed in TNM buffer, which was then supplemented with 20 mM EDTA and 10 mM Co3+(NH3)6Cl–3. Reactions were incubated for 1 h at the indicated temperatures, and branch migration was determined as described in (A).
We found it important to balance the concentrations of Co3+(NH3)6Cl–3 and EDTA. Hexamine cobalt(III) can condense and precipitate DNA (25,26), which would have the effect of reducing the concentration of free Co3+(NH3)6Cl–3 in solution. Consistent with this, we observed an increased rate of branch migration at Co3+(NH3)6Cl–3 concentrations associated with DNA precipitation (Table 1 and Fig. 2B). Increasing the concentration of EDTA reduced DNA precipitation but also led to rapid branch migration (Table 1). This is probably due to the chelation of Co3+(NH3)6Cl–3 by EDTA; chelation of other polyvalent cations by EDTA has been observed (27). Intermediate concentrations, such as 10 mM Co3+(NH3)6Cl–3, 20 mM EDTA, minimized both DNA precipitation and branch migration. Varying the concentration of NaCl from 50 to 150 mM did not affect DNA precipitation by Co3+(NH3)6Cl–3 significantly, and had only a modest effect on branch migration (data not shown).
Table 1. DNA precipitation and branch migration as a function of Co3+(NH3)6Cl–3 and EDTA concentration.
| [EDTA] (mM) | DNA precipitation (%)a |
Branch migration (%)b |
|||||
|---|---|---|---|---|---|---|---|
| [Co3+(NH3)6Cl–3] (mM) |
[Co3+(NH3)6Cl–3] (mM) |
||||||
| 2 | 5 | 10 | 25 | 2 | 5 | 10 | |
| 0 | 12 | 56 | 55 | 58 | 0 | 12 | 22 |
| 10 | 0 | 1 | 65 | 65 | 1 | 0 | 17 |
| 20 | 0 | 0 | 1 | 65 | 7 | 0 | 2 |
| 50 | 0 | 0 | 0 | 1 | 63 | 29 | 8 |
aDNA precipitation assays using 32P-labeled λ DNA were performed in TNM buffer supplemented as indicated, for 2 h at 37°C. Precipitation was calculated as the fraction of total 32P label found in the pellet after centrifugation.
bBranch migration assays using 300 bp substrates S1 and S2 were performed in TNM buffer supplemented as indicated, for 1 h at 37°C. Branch migration was determined as in Figure 2.
The rate of branch migration increases ~10-fold with every 10°C increase in temperature (11). We confirmed that this temperature dependence also exists in 10 mM Co3+(NH3)6Cl–3 (Fig. 2C). Incubation at 45°C or greater dramatically increased the rate of branch migration. Similar results were obtained at lower concentrations of Co3+(NH3)6Cl–3 (data not shown).
In summary, a DNA extraction protocol using Co3+(NH3)6Cl–3 to reduce branch migration of Holliday junctions should be performed at 37°C or less, and the concentrations of EDTA and Co3+(NH3)6Cl–3 should be in a ratio of ~2:1.
DNA extraction with CTAB in the presence of hexamine cobalt(III) chloride
We attempted to incorporate Co3+(NH3)6Cl–3 into a standard yeast DNA extraction protocol (see for example 23), in which cells are spheroplasted, lysed with SDS, and DNA is extracted with phenol:chloroform and precipitated with alcohol (Table 2). Addition of 10 mM Co3+(NH3)6Cl–3 restrained branch migration in spheroplasting solution. No branch migration was observed during chloroform extraction, or during precipitation with ethanol or isopropanol (Table 2). Presumably, alcohol-induced DNA condensation folds the Holliday junction into a conformation that is recalcitrant to branch migration. However, phenol:chloroform extraction led to rapid branch migration, even in the presence of Co3+(NH3)6Cl–3. This is to be expected, as phenol decreases the thermal stability of DNA (28). Branch migration also occurred in the SDS lysis solution; addition of Co3+(NH3)6Cl–3 had little effect, and precipitation of hexamine cobalt(III):dodecyl sulfate complexes occurred. Substitution of sarkosyl for SDS led to similar results (data not shown).
Table 2. Branch migration during a yeast DNA extraction protocol using SDS.
| Treatmenta | Branch migration (%)b | |
|---|---|---|
| No addition | + Co3+(NH3)6Cl–3c | |
| Spheroplasting | 85 | 0 |
| SDS lysis | 85 | 68 |
| Phenol:chloroform | 73 | 50 |
| Chloroform | 0 | n.d. |
| Isopropanol | 0 | 0 |
| Ethanol | 0 | 0 |
n.d., not determined.
aAnnealed substrates were aliquoted into the following: spheroplasting buffer or SDS lysis buffer, and incubated at 37°C for 30 min; TNM buffer, and extracted with phenol:chloroform or chloroform; 0.3 M sodium acetate, and precipitated using isopropanol or ethanol.
bBranch migration using 300 bp substrates S1 and S2 was measured as in Figure 2.
cSolutions were supplemented with 10 mM Co3+(NH3)6Cl–3 as indicated. Phenol, but not chloroform, was equilibrated with buffer containing 10 mM Co3+(NH3)6Cl–3.
As Co3+(NH3)6Cl–3 is incompatible with anionic detergents, we developed a DNA extraction protocol that uses the cationic detergent CTAB (29–32). This protocol is outlined in Figure 3A, and is described in detail in Materials and Methods. CTAB performs all of the core DNA extraction functions: it lyses cells, solubilizes proteins and precipitates nucleic acids. Initial lysis is performed in ~1.4 M NaCl to prevent DNA condensation and the subsequent co-precipitation with cellular debris (26). After chloroform extraction to remove protein and debris, the Na+ concentration is lowered to ~0.4 M, bringing about DNA condensation and precipitation by CTA+. The precipitate is washed to remove excess CTAB, and is resuspended in 1.4 M NaCl to dissociate the CTA+ from the DNA. Finally, the DNA is precipitated with ethanol, thereby removing residual CTAB (which is highly soluble in ethanol).
Figure 3.
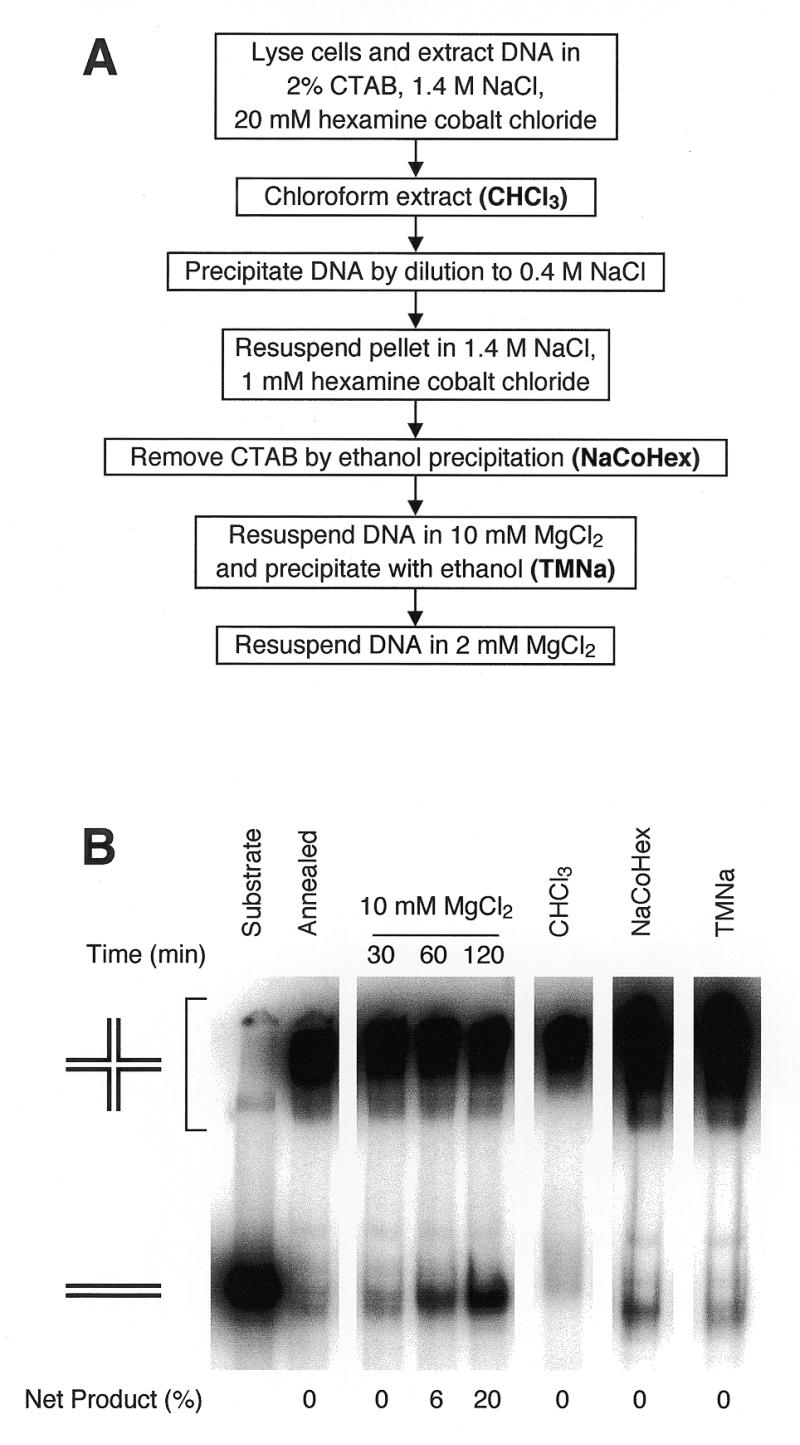
DNA extraction with CTAB in the presence of Co3+(NH3)6Cl–3. (A) Outline of the protocol. Experimental details are given in Materials and Methods. (B) Branch migration during yeast DNA extraction using the protocol. R1 and R2 were annealed in TNM buffer. As a positive control for branch migration, an aliquot was incubated at 37°C in TNM buffer (10 mM MgCl2) for the indicated times. Remaining annealed substrate was added to yeast spheroplasts, and DNA was extracted as described in Materials and Methods. A sample was taken after chloroform:isoamyl alcohol extraction (CHCl3). DNA pellets obtained after ethanol precipitation from NaCoHex solution (NaCoHex) and after ethanol precipitation from TMNa solution (TMNa) were resuspended in TMSpe buffer on ice. Branch migration was measured as described in Figure 2; net product refers to the fraction of total 32P label migrating as monomer duplex, after subtraction of residual unannealed substrate.
The optimal concentration of Co3+(NH3)6Cl–3 differed at each step. Extraction buffer could be supplemented with 20 mM Co3+(NH3)6Cl–3, since 1.4 M Na+ prevents DNA precipitation (25). However, CTA+:DNA precipitation by dilution to 0.4 M NaCl is inefficient if the final concentration of Co3+(NH3)6Cl–3 is >8 mM, possibly because Co3+(NH3)6 competes with CTA+ for DNA binding. Ethanol precipitation of DNA from solutions containing >1 mM Co3+(NH3)6Cl–3 results in an insoluble precipitate. The final steps in the protocol substitute MgCl2 for Co3+(NH3)6Cl–3, as Mg2+ limits branch migration without the risk of DNA precipitation in solutions of low ionic strength.
Several other points should be emphasized. Efficient precipitation of CTA+:DNA complexes depends somewhat on the concentration of DNA in the lysate. This protocol was optimized for 8 ml of a saturated culture of S.cerevisiae (~40 µg genomic DNA) lysed in 500 µl of extraction buffer, but should be transferable to other cell types provided that volumes are scaled appropriately. Addition of proteinase K and RNase to the lysate improved the yield and quality of the DNA. The lysate was incubated at 37°C, as higher temperatures led to rapid branch migration (Fig. 2C), and have been shown to reduce the yield of DNA isolated by CTAB (33).
We tested the DNA extraction protocol presented here for its effect on branch migration (Fig. 3B). Annealed Holliday junction substrates were added to yeast spheroplasts, and subjected to the protocol described. Samples were taken after chloroform:isoamyl alcohol extraction, ethanol precipitation from the 1.4 M NaCl solution, and ethanol precipitation from the MgCl2 solution. There was no detectable Holliday junction dissociation at any step of the protocol, indicating that the total extent of branch migration during DNA extraction must have been less than 273 bp, the duplex length of the substrates. In fact, CTA+ ions alone can stabilize Holliday junctions, as we observed very little branch migration during extraction with CTAB in the absence of Co3+(NH3)6Cl–3 (data not shown). Nevertheless, Co3+(NH3)63+ or Mg2+ are necessary to restrain branch migration before the formation of CTA+:DNA complexes and after CTAB has been dissociated from DNA.
The method presented here meets all the requirements of a DNA extraction protocol that restrains spontaneous branch migration. DNA is always in the presence of multivalent cations, specifically hexamine cobalt(III) and magnesium, that efficiently fold four-way junctions into a stacked X structure. Furthermore, the use of CTAB ensures that the DNA is partially or fully condensed for virtually all of the extraction protocol, thereby greatly stabilizing any perishable intermediates such as Holliday junctions. In addition, the method is rapid (<2 h) and does not use incubation temperatures that would result in significant branch migration. This protocol was used to isolate joint molecules from meiotic yeast cultures (Fig. 4); these structures are recovered very poorly by conventional extraction methods (8), unless the DNA is crosslinked with psoralen (7). We are currently analyzing meiotic recombination intermediates isolated using the method presented here.
Figure 4.
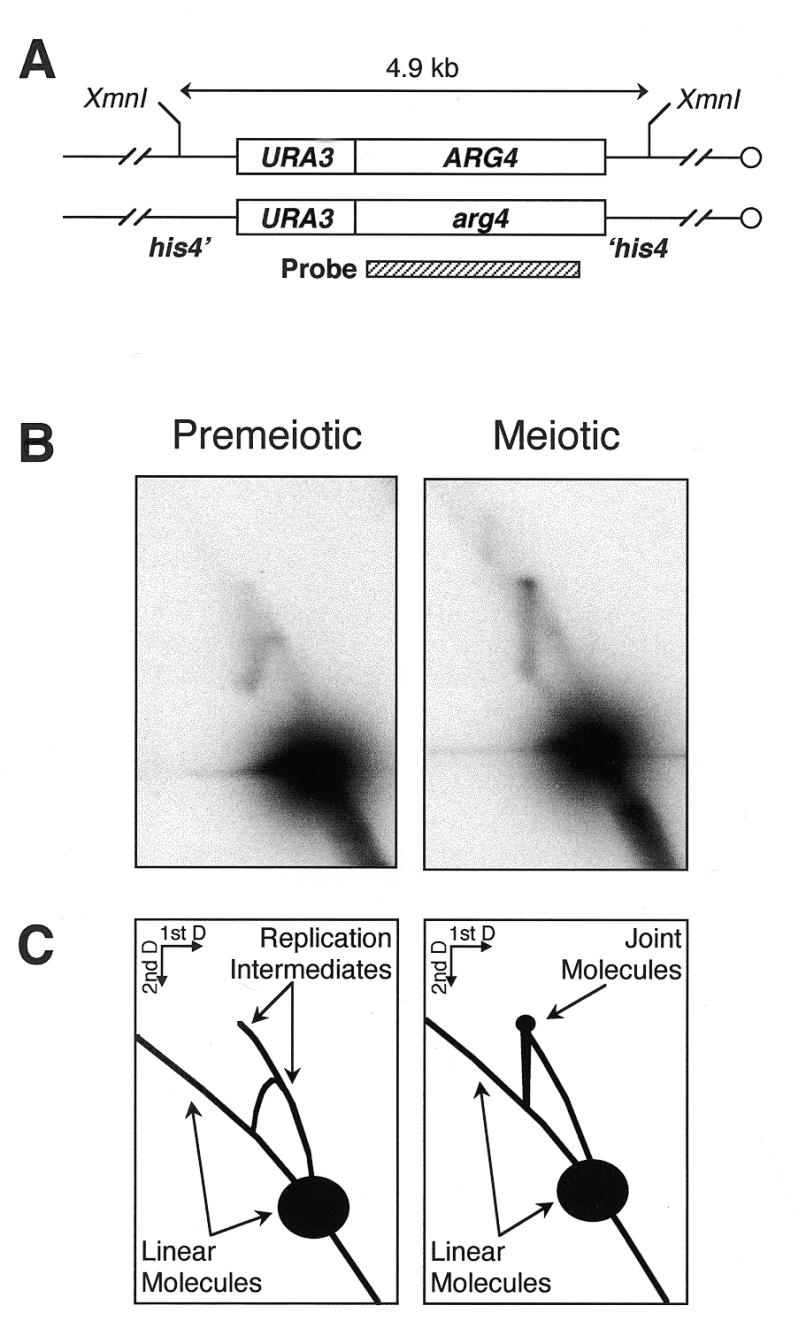
Joint molecules observed during yeast meiosis. (A) Structure of the locus examined. The his4::URA3-ARG4 insert in MJL2444 consists of a 1.1 kb HindIII–SmaI fragment of URA3 and a 2.3 kb Eco47III–PstI fragment of ARG4 inserted in HIS4 coding sequences. (B) DNA was extracted with CTAB, either from a premeiotic culture or 4 h after the onset of meiosis, digested with XmnI, resolved on a 2D agarose gel and hybridized with the indicated probe as described in Materials and Methods. (C) Interpretation of the results. While only DNA replication intermediates are seen in the premeiotic sample, a distinct off-arc spot is observed in the meiotic sample, at the position expected for joint molecules formed between the homologs illustrated in (A) (7).
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Mikhail Grigoriev and Peggy Hsieh for generously providing branch migration substrates and invaluable technical advice. We thank Valérie Borde, Robert Shroff, Carol Wu, Claude Klee and Dhruba Chattoraj for their helpful discussions.
REFERENCES
- 1.Holliday R. (1964) Genet. Res., 5, 282–304. [Google Scholar]
- 2.Sigal N. and Alberts,B. (1972) J. Mol. Biol., 71, 789–793. [DOI] [PubMed] [Google Scholar]
- 3.Meselson M.S. and Radding,C.M. (1975) Proc. Natl Acad. Sci. USA, 72, 358–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Resnick M.A. (1976) J. Theor. Biol., 59, 97–106. [DOI] [PubMed] [Google Scholar]
- 5.Orr-Weaver T.L., Nicolas,A. and Szostak,J.W. (1988) Mol. Cell. Biol., 8, 5292–5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bell L. and Byers,B. (1979) Proc. Natl Acad. Sci. USA, 76, 3445–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwacha A. and Kleckner,N. (1994) Cell, 76, 51–63. [DOI] [PubMed] [Google Scholar]
- 8.Collins I. and Newlon,C.S. (1994) Cell, 76, 65–75. [DOI] [PubMed] [Google Scholar]
- 9.Stark W.M., Boocock,M.R. and Sherratt,D.J. (1992) Trends Genet., 8, 432–439. [PubMed] [Google Scholar]
- 10.West S.C. (1997) Annu. Rev. Genet., 31, 213–244. [DOI] [PubMed] [Google Scholar]
- 11.Panyutin I.G. and Hsieh,P. (1994) Proc. Natl Acad. Sci. USA, 91, 2021–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Panyutin I.G., Biswas,I. and Hsieh,P. (1995) EMBO J., 14, 1819–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lilley D.M. and Clegg,R.M. (1993) Annu. Rev. Biophys. Biomol. Struct., 22, 299–328. [DOI] [PubMed] [Google Scholar]
- 14.Seeman N.C. and Kallenbach,N.R. (1994) Annu. Rev. Biophys. Biomol. Struct., 23, 53–86. [DOI] [PubMed] [Google Scholar]
- 15.Duckett D.R., Murchie,A.I. and Lilley,D.M. (1990) EMBO J., 9, 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitts P.A. and Nash,H.A. (1987) Nature, 329, 346–348. [DOI] [PubMed] [Google Scholar]
- 17.Nunes-Duby S.E., Matsumoto,L. and Landy,A. (1987) Cell, 50, 779–788. [DOI] [PubMed] [Google Scholar]
- 18.Bell L.R. and Byers,B. (1983) Cold Spring Harbor Symp. Quant. Biol., 47, 829–840. [DOI] [PubMed] [Google Scholar]
- 19.Shi Y.B., Spielmann,H.P. and Hearst,J.E. (1988) Biochemistry, 27, 5174–5178. [DOI] [PubMed] [Google Scholar]
- 20.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 21.Hsieh P. and Panyutin,I.G. (1995) In Eckstein,F. and Lilley,D.M.J. (eds), Nucleic Acids and Molecular Biology. Springer-Verlag, Berlin, Germany, Vol. 9, pp. 42–65.
- 22.Kane S.M. and Roth,R. (1974) J. Bacteriol., 118, 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goyon C. and Lichten,M. (1993) Mol. Cell. Biol., 13, 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bell L. and Byers,B. (1983) Anal. Biochem., 130, 527–535. [DOI] [PubMed] [Google Scholar]
- 25.Widom J. and Baldwin,R.L. (1980) J. Mol. Biol., 144, 431–453. [DOI] [PubMed] [Google Scholar]
- 26.Manning G.S. (1978) Q. Rev. Biophys., 11, 179–246. [DOI] [PubMed] [Google Scholar]
- 27.Hoopes B.C. and McClure,W.R. (1981) Nucleic Acids Res., 9, 5493–5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leng M., Drocourt,J.L., Helene,C. and Ramstein,J. (1974) Biochimie, 56, 887–891. [DOI] [PubMed] [Google Scholar]
- 29.Jones A.S. (1953) Biochim. Biophys. Acta, 10, 607–612. [DOI] [PubMed] [Google Scholar]
- 30.Dutta S.K., Jones,A.S. and Stacey,M. (1953) Biochim. Biophys. Acta, 10, 613–622. [DOI] [PubMed] [Google Scholar]
- 31.Jones A.S. (1963) Nature, 199, 280–282. [DOI] [PubMed] [Google Scholar]
- 32.Murray M.G. and Thompson,W.F. (1980) Nucleic Acids Res., 8, 4321–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shahjahan R.M., Hughes,K.J., Leopold,R.A. and DeVault,J.D. (1995) Biotechniques, 19, 332–334. [PubMed] [Google Scholar]