

Abstract
The proto-oncogenic protein, c-KIT, plays a crucial role in regulating cellular transformation and differentiation processes, such as proliferation, survival, adhesion, and chemotaxis. The overexpression of, and mutations, in c-KIT can lead to its dysregulation and promote various human cancers, particularly gastrointestinal stromal tumors (GISTs); approximately 80–85% of cases are associated with oncogenic mutations in the KIT gene. Inhibition of c-KIT has emerged as a promising therapeutic target for GISTs. However, the currently approved drugs are associated with resistance and significant side effects, highlighting the urgent need to develop highly selective c-KIT inhibitors that are not affected by these mutations for GISTs. Herein, the recent research efforts in medicinal chemistry aimed at developing potent small-molecule c-KIT inhibitors with high kinase selectivity for GISTs are discussed from a structure–activity relationship perspective. Moreover, the synthetic pathways, pharmacokinetic properties, and binding patterns of the inhibitors are also discussed to facilitate future development of more potent and pharmacokinetically stable small-molecule c-KIT inhibitors.
Keywords: c-KIT, GISTs, stem cell growth factor, c-KIT inhibitors, SAR, SCFR
1. Introduction
The majority of mesenchymal neoplasms of the digestive system are gastrointestinal stromal tumors (GISTs), which develop from interstitial Cajal cells [1,2]. GISTs typically manifest as a sharply demarcated submucosal or subserosal mass in the small intestine (25%) and stomach (60%), but can also occur less commonly in the colon, rectum, esophagus, mesentery, and omentum [3,4]. They often have spindled (70%), epithelioid (20%), or mixed (10%) cytomorphology characteristics. GISTs associated with neurofibromatosis type 1 are typically found in the stomach, have a distinctive multilobular or plexiform architecture, and exhibit either an epithelioid or mixed morphology, but almost never a pure spindle-cell morphology [5]. They are also more likely to occur in multiple locations and to have a spindle-shaped morphology. Anemia, indigestion, bleeding, and abdominal pain brought on by stressful situations are some of the typical clinical signs and symptoms of GISTs [6].
The KIT receptor tyrosine kinase gene, which codes for c-KIT (also known as mast/stem cell growth factor receptor, SCFR, or CD117), has been found to have primary activating mutations in approximately 90% of GISTs. c-KIT is a type III receptor tyrosine kinase that is present on the surface of hematopoietic stem cells and other cell types [7,8]. When it interacts to its cognate ligand stem cell factor (SCF), it becomes activated. A role in the control of cell survival, proliferation, and differentiation is played by this binding, which leads to the production of receptor dimers, which, in turn, phosphorylate and activate signal transduction molecules in the cell [9,10,11]. Exon 11, which codes for the JM region and destroys its autoinhibitory function, is where c-KIT mutations most frequently occur [12]. This results in continual activation of c-KIT. One of the hotspots of the c-kit gene, codons 550 and 560, is where the majority of these exon 11 mutations occur as deletions and clusters [13]. Other, less frequent mutations in exon 9, which codes for the EC region of c-KIT, include an internal tandem duplication of Ala502-Tyr503, which simulates the conformational change that occurs when c-KIT dimerizes after binding to SCF [13,14]. With a combined frequency of 1–2% across all GISTs, mutations in exons 13 (encoding the ATP-binding region of c-KIT) and 17 (encoding the activation loop of the kinase) are extremely uncommon (Figure 1) [15].
Figure 1.

Structure of the KIT receptor. The main mutation sites and phosphorylation sites of KIT in GISTs. Reprinted and modified from Ref. [15].
Cancer stemness, also known as the cancer stem cell (CSC) phenotype, is described as a subset of cancer cells that have the capacity to self-renew, specialize into specific progenies, initiate tumor growth, and promote metastasis, recurrence, and therapy resistance [16,17,18]. In several malignancies, stemness has been connected to c-KIT. Research using spheroid cultures obtained from tumor cells from patients with colon cancer grown in serum-free and non-adherent plates—a technique frequently used to study CSCs—have linked c-KIT expression to cancer stemness, and mounting evidence implies a role of c-KIT in colon cancer stemness [19,20]. Recent investigations have shown that more differentiated colon tumor cells release SCF, which affects the proliferation of CSC-like colon tumor cells that express c-KIT [21]. This suggests the possibility of a paracrine mechanism wherein SCF can activate CSC-like cells present in colonospheres. Recent research has also shown that c-KIT increases CSC characteristics in colorectal cancer cells, including the expression of CD44 and other stem cell markers [22,23].
Imatinib, a c-KIT inhibitor, has been shown to be effective in treating GISTs, but it has also been linked to primary resistance in some oncogenic mutations, and secondary resistance has frequently emerged [24,25]. Imatinib therapy has a success rate of roughly 70% in the treatment of GISTs with main mutations [26]. After an average of two years, however, acquired resistance is seen in 40–50% of cases [27,28]. Patients with imatinib-resistant GISTs most frequently have the V654A exon 13 resistance mutation [29]. Sunitinib, regorafenib, and ripretinib, which have been approved for the treatment of GISTs, only partially target these resistance mutations and do not improve the overall response rate, as evidenced by their limited therapeutic benefit. Furthermore, these therapies are associated with significant adverse effects (Figure 2) [30,31,32,33]. Thus, there is a critical need for the creation of effective inhibitors for the resistance mutation KIT V654A. Herein, small-molecule c-KIT inhibitors that have been reported to be effective against GISTs are reviewed from a structure–activity relationship (SAR) perspective. It is important to note that only reported c-kit inhibitors with a sufficient number of derivatives to derive a SAR were discussed. Moreover, for each discussed series, the synthetic route as well as the binding pattern (if available) are discussed in detail in an effort to accelerate the discovery of novel c-KIT inhibitors for the treatment of GISTs. The synthesis and Schemes S1–S25 for all the compounds are available in the supplementary information (ref. [34,35,36,37,38,39]).
Figure 2.

Chemical structures of KIT inhibitors approved for the treatment of GISTs.
2. c-KIT Inhibitors Targeting GIST
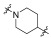
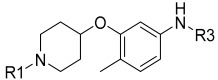
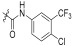
Wang et al. designed and synthesized a novel series of substituted N-(4-methyl-3-(piperidin-4-yloxy)phenyl) amide derivatives, and the 27 synthesized compounds were screened for type-II c-KIT inhibition activity for GISTs against the Tel-c-KIT-BaF3, Parental BaF3, K562 cell lines [40]. EL-c-KIT-BaF3 and parental BaF3 cells were employed to monitor the activity, while K562 cells were used to monitor BCR-ABL inhibitory activity. The CF3 group substitution at the meta-position of the benzene as R3 was selected as a beginning point for the SAR study. Replacement of the aromatic amine linker of I to a flexible 2-carbon aliphatic ether linker at the R2 fragment (6a and 6b) showed a 20–25-fold loss of growth inhibitory activity against TEL-c-KIT-BaF3 cells. After replacement of the aliphatic chain with the piperidine ring (6c), the c-KIT activity was improved by 3–4-fold compared with compound I (GI50 = 0.11 vs. 0.40 μM) and demonstrated good selectivity between TEL-c-KIT-BaF3 and the parental BaF3 cells (GI50 ≥ 10 μM). In addition, this compound showed moderate BCR-ABL inhibitory activity (GI50 = 2.94 μM). Relocation of the R1 fragment nitrogen position (6d and 6e) led to the complete loss of BCR-ABL inhibitory activity but maintained c-KIT inhibitory activity (GI50 = 0.33 and 0.19 μM, respectively) and selectivity over the parental BaF3 cells (GI50 ≥ 10 μM). Replacing pyridine with a five-membered heterocyclic group (6f) in R1 drastically improved the BCR-ABL activity (GI50 = 0.16 μM). Loss of c-KIT activity was observed when the aromatic ring in the R1 fragment was replaced with aliphatic chains, such as ethyl (6g) and ethylene (6h). The c-KIT inhibitory activity was abolished when the ether linkage was shifted to the three-position from the four-position of the piperidine (6i and 6j) in R2, along with the replacement of the methyl group to a hydrogen atom in R4. Furthermore, a 4–5-fold loss of c-KIT activity (GI50 = 0.49 vs. 0.11 μM) was observed when the methyl group in R4 of 6c was replaced with a hydrogen atom (6k). When the methyl group was substituted with an electron-withdrawing group (-Cl), compound 6l displayed a nine-fold loss of activity against c-KIT (GI50 = 0.96 μM), and when it was substituted with an electron-donating group (-Ome), compound 6m showed complete loss of c-KIT activity (Table 1).
Table 1.
SAR exploration of the R1/R2/R4 positions.
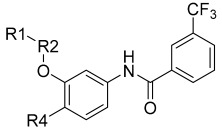
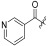
| ||||||
|---|---|---|---|---|---|---|
| Compd. | R1 | R2 | R4 | Tel-c-KIT-BaF3 (GI50: μM) |
Parental BaF3 (GI50: μM) |
K562 (GI50: μM) |
| I |
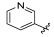
|
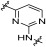
|
–Me | 0.40 | >10 | 0.12 |
| 6a |
|
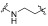
|
–Me | 7.94 | >10 | >10 |
| 6b |
|
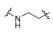
|
–Me | 9.97 | >10 | >10 |
| 6c |
|
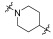
|
–Me | 0.11 | >10 | 2.94 |
| 6d |
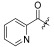
|
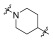
|
–Me | 0.33 | >10 | >10 |
| 6e |
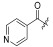
|
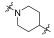
|
–Me | 0.19 | >10 | >10 |
| 6f |
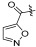
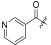
|
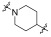
|
–Me | 0.22 | >10 | 0.16 |
| 6g |
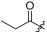
|
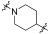
|
–Me | 1.55 | >10 | >10 |
| 6h |
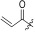
|
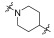
|
–Me | 0.62 | >10 | 8.36 |
| 6i |
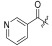
|
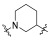
|
–H | 8.07 | 8.07 | >10 |
| 6j |
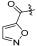
|
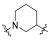
|
–H | >10 | >10 | >10 |
| 6k |
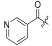
|
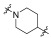
|
–H | 0.49 | >10 | >10 |
| 6l |
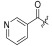
|
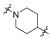
|
–Cl | 0.96 | >10 | 2.8 |
| 6m |
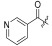
|
|
–OMe | >10 | >10 | >10 |
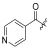
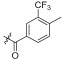
These results demonstrate that the piperidine ring in the R2 fragment and the methyl ring in the R4 fragment play important roles in achieving high inhibitory activity. By keeping intact the ether linked piperidine at R2, the introduction of a methyl group at the four-position of the benzene ring (6n) led to a six-fold improvement in the c-KIT activity (GI50 = 0.031 vs. 0.11 μM) through a loss of selectivity against K562 (GI50 = 2.32 μM) and BaF3 cells (GI50 = 6.39 μM). Replacement of the R1 fragment with other heterocyclic groups (6o, 6p, and 6q) and the introduction of a –Cl (6r, 6s) or –F (6t) atom at the five-position of the benzene ring in the R3 fragment led to improved BCR-ABL inhibitory activity and c-KIT inhibitory activity, respectively. The pyridine ring in R3 (6u and 6v) resulted in complete loss of activity against c-KIT and BCR-ABL, and the replacement of the CF3 group in R3 with a methoxy group (10a) led to 15-fold activity loss against c-KIT compared with 6c (GI50 = 1.48 vs. 0.11 μM). Alteration of the benzene ring in R3 to a tert-butyl isoxazole moiety (10b) led to three-fold activity loss against c-KIT (GI50 = 0.34 vs. 0.11 μM), whereas alterations to benzodioxole (10c) and quinolone (10d) moieties led to a 50-fold loss of c-KIT activity. The introduction of a urea linkage (13) between the R3 fragment and the linker moiety in place of an amide increased the BCR-ABL activity and decreased the c-KIT activity (Table 2). Of the tested compounds, 6e displayed selectivity over ABL kinase and showed potent inhibition against c-KIT kinase. SAR study of this series disclosed that the introduction of a methyl group at the four-position of the benzene ring and piperidine ring in the R2 position was responsible for the c-KIT inhibitory activity. In addition, the terminal pyridine moiety is important to gain selectivity against c-KIT over ABL (Figure 3).
Table 2.
SAR exploration of the R1/R3 positions.
| |||||
|---|---|---|---|---|---|
| Compd. | R1 | R3 | Tel-c-KIT-BaF3 (GI50: μM) |
Parental BaF3 (GI50: μM) |
K562 (GI50: μM) |
| 6n |
|
|
0.031 | 6.39 | 2.32 |
| 6o |
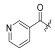
|
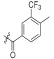
|
0.083 | 5.47 | 5.85 |
| 6p |
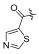
|
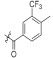
|
0.10 | >10 | 5.24 |
| 6q |
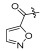
|
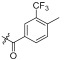
|
0.065 | 8.94 | 1.96 |
| 6r |
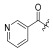
|
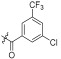
|
0.16 | >10 | 3.66 |
| 6s |
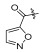
|
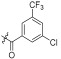
|
0.15 | >10 | 1.95 |
| 6t |
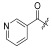
|
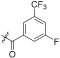
|
0.15 | >10 | 4.56 |
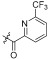
| 6u |
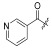
|
|
>10 | >10 | >10 |
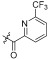
| 6v |
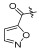
|
|
>10 | >10 | >10 |
| 10a |
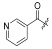
|
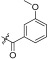
|
1.48 | >10 | >10 |
| 10b |
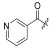
|
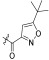
|
0.34 | >10 | 9.92 |
| 10c |
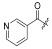
|
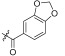
|
4.8 | >10 | >10 |
| 10d |
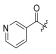
|
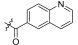
|
5.55 | >10 | >10 |
| 13 |
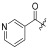
|
|
2.6 | 3.3 | 0.78 |
Figure 3.

SAR summary and pharmacophore description of substituted N-(4-methyl-3-(piperidin-4-yloxy)phenyl) analogs.
Compound 6e inhibited c-KIT kinase (IC50 = 99 nM) and PDGFRβ (IC50 = 120 nM) in the ADP-Glo assay and DDR1 kinase (IC50 = 126 nM) and CSF1R (IC50 = 133 nM) in the Z’lyte assay. In addition, 6e showed a strong inhibition in the growth of KIT-dependent GIST cancer cells, such as GIST-T1 (GI50 = 0.021 μM) and GIST-882 (GI50 = 0.043 μM). Surprisingly, it did not show potent inhibitory activity against the c-KIT-independent GIST cell line GIST48B (GI50 ≥ 10 μM), FLT3 kinase activity, and did not exhibit antiproliferative activity against BCR-ABL-driven CML cell lines, such as K562 (GI50 ≥ 10 μM), MEG-01 (GI50 = 7.43 μM), and KU812 cells (GI50 = 6.71 μM). Moreover, 6e showed a good safety profile over normal Chinese hamster ovary cells, CHO and CHL cells, and leukemic cell lines, such as U937, HL-60, and REC-1 cells. Pharmacokinetics analysis of compound 6e demonstrated a half-life of 4.11 h and satisfactory bioavailability (F = 36%) when administered orally. In contrast, 6e exhibited fast clearance (CLz, 4.988 L/h/kg) and a short half-life (T1/2, 0.45 h) when administered intravenously. The antitumor effectiveness of 6e at 50 and 100 mg/kg doses in a GIST-T1 cell-inoculated xenograft mice model greatly inhibited tumor development.
Molecular docking of the potent derivative 6e into c-KIT kinase and ABL kinase revealed that 6e might adopt the DFG-out conformation of c-KIT kinase (PDB ID: 1T46) with a typical type II binding mode, as well as a similar type II binding mode, in the ABL kinase (PDB ID: 2HYY). The amide carbonyl linking the terminal pyridine and piperidine moiety of 6e forms an O-H-N hydrogen bond with the Cys673 at the hinge-binding region; in contrast, compound I uses the terminal pyridine as a hinge binder. The amide moiety in the tail section of 6e forms two usual hydrogen bonds with Glu640 and Asp810 (Figure 4A). The hydrogen bond established in the hinge-binding region of the ABL kinase (PDB ID: 2HYY) is somewhat longer than that of the c-KIT kinase (3.6 vs. 2.9 Å). Furthermore, the adjacent Tyr253 in the P-loop (3.1 Å to the pyridine moiety) creates a potential steric hindrance that hinders 6e from binding to the ABL kinase, which was clearly illustrated when superimposing 6e and I in the X-ray crystal structure of 1-ABL kinase. The pyridine moiety in I establishes a hydrogen bond with a distance of 2.9 Å in the hinge-binding region, and this aromatic moiety moves further away from Tyr253 than the pyridine moiety in 6e, probably to prevent potential steric hindrance. Tyr253 was replaced by Gly596 in the c-KIT kinase to accommodate the terminal pyridine (Figure 4).
Figure 4.

(A) docking of 6e into c-KIT kinase (PDB ID: 1T46) (B) docking of 6e into ABL kinase (PDB ID: 2HYY) (C) Superimposition of docking model of 6e with ABL kinase (PDB ID: 2HYY) (in white) and 1-ABL kinase X-ray crystal structure (in purple). (D) Superimposition of 6e with c-KIT kinase (PDB ID: 1T46) (in white) and docked model of 6e with ABL kinase (PDB ID: 2HYY) (in purple). Reprinted with permission from Ref. [40].
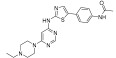
Li et al. designed and synthesized a series of 6,7-dimethoxy-4-phenoxyquinoline derivatives, and the 30 synthesized compounds were tested for their kinase inhibitory activity against c-KIT wt and c-KIT-T670I using BaF3-TEL-c-KIT, BaF3-TEL-c-KIT-T670I, and parental BaF3 cells [41]. Compound 22a, which contains a urea group instead of the amide group in compound I, displayed enhanced activity for c-KIT wt (GI50 = 0.12 μM) and c-KIT-T670I (GI50 = 0.071 μM) and maintained a 12-fold selectivity against parental BaF3 cells (GI50 = 1.5 μM). However, compound 33, which had a much larger group (N,N′-dimethylcyclopropane-1,1-dicarboxamide) in place of the amide group, showed no kinase inhibitory activity (GI50 ≥ 10 μM). The R2 moiety was investigated with R1 fixed as urea. With the trifluoromethyl group removed, Compound 22b completely lost activity against c-KIT wt and c-KIT-T670I (GI50 ≥ 10 μM), indicating the importance of the hydrophobic interaction between the CF3 group and the hydrophobic environment for binding. In contrast, substituting the phenyl group with a piperidine group (27) resulted in a significant loss of activity in BaF3-TEL-c-KIT-T670I cells (GI50 ≥ 10 μM) (Table 3). According to these findings, the (trifluoromethyl)benzyl group at position R2 and the urea group at position R1 are favored for better activity. They focused on exploring the R3 position, which is known to enhance the binding affinity and selectivity for type II inhibitors (Table 4). Shifting the propionyl group to position three (22c) from position four (22a) decreased activity against BaF3-TEL-c-KIT-T670I cells by 13-fold (GI50 = 0.92 μM). However, substituting the propionyl group in 22a with an acetyl group (22d) maintained activity against BaF3-TEL-c-KIT (GI50 = 0.11 μM) and BaF3-TEL-c-KIT-T670I cells (GI50 = 0.046 μM) with improved selectivity against parental BaF3 cells (GI50 = 7.1 μM). A ten-fold decreased activity against the c-KIT T670I mutant was observed for larger group substitutions, such as dimethylbutyl (22e), glycine (22f), and alanine (22g).
Table 3.
SAR exploration of the R1/R2 positions.
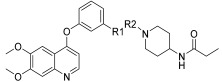
| |||||
|---|---|---|---|---|---|
| Compd. | R1 | R2 | BaF3-TEL-c-KIT (GI50: μM) |
BaF3-TEL-c-KIT-T670I (GI50: μM) |
BaF3 (GI50: μM) |
| I |
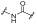
|
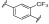
|
0.4 ± 0.011 | 2.7 ± 0.058 | >10 |
| 22a |
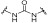
|
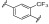
|
0.12 ± 0.016 | 0.071 ± 0.0011 | 1.5 ± 0.17 |
| 33 |
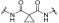
|
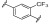
|
>10 | >10 | >10 |
| 22b |
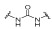
|
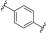
|
>10 | >10 | >10 |
| 27 |
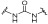
|
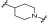
|
0.34 ± 0.01 | >10 | >10 |
Table 4.
SAR of the R3 position.
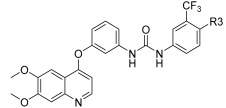
| ||||
|---|---|---|---|---|
| Compd. | R3 | BaF3-TEL-c-KIT (GI50: μM) |
BaF3-TEL-c-KIT-T670I (GI50: μM) |
BaF3 (GI50: μM) |
| 22c |
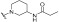
|
0.28 ± 0.025 | 0.92 ± 0.04 | 1.3 ± 0.17 |
| 22d |
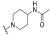
|
0.11 ± 0.016 | 0.046 ± 0.001 | 7.1 ± 0.38 |
| 22e |
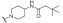
|
0.43 ± 0.01 | 0.34 ± 0.025 | >10 |
| 22f |
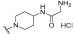
|
0.13 ± 0.01 | 0.58 ± 0.02 | 5 ± 1.0 |
| 22g |
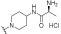
|
0.12 ± 0.011 | 0.48 ± 0.011 | 3 ± 0.15 |
| 22h |
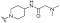
|
0.038 ± 0.001 | 0.053 ± 0.001 | 2.9 ± 0.17 |
| 22i |
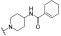
|
0.76 ± 0.0057 | 0.76 ± 0.1 | 1.1 ± 0.47 |
| 22j |
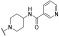
|
0.02 ± 0.001 | 0.32 ± 0.025 | 7 ± 0.74 |
| 22k |
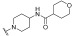
|
0.087 ± 0.0015 | 0.083 ± 0.021 | 2.9 ± 0.056 |
| 22l |
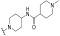
|
0.16 ± 0.01 | 0.081 ± 0.006 | 1.7 ± 01 |
| 22m |
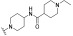
|
0.039 ± 0.041 | 0.087 ± 0.008 | 1.3 ± 0.11 |
| 22n |
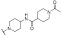
|
0.059 ± 0.001 | 0.23 ± 0.0001 | 3.4 ± 1.2 |
| 22o |
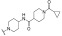
|
0.13 ± 0.0057 | 0.12 ± 0.02 | 0.8 ± 0.084 |
| 22p |
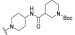
|
0.13 ± 0.001 | 0.17 ± 0.05 | 0.51 ± 0.15 |
| 22q |
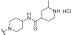
|
0.46 ± 0.021 | 0.65 ± 0.076 | 4.0 ± 0.32 |
| 22r |
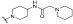
|
0.12 ± 0.0001 | 0.037 ± 0.009 | 0.48 ± 0.006 |
| 22s |
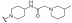
|
0.33 ± 0.015 | 0.2 ± 0.011 | 0.87 ± 0.068 |
| 22t |
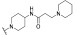
|
0.44 ± 0.03 | 0.26 ± 0.052 | 7.6 ± 0.61 |
| 22u |
|
0.19 ± 0.021 | 0.2 ± 0.02 | 1.2 ± 0.11 |
| 22v |
|
0.042 ± 0.003 | 0.059 ± 0.006 | 3.6 ± 0.89 |
| 22w |
|
3.9 ± 0.023 | 3.8 ± 0.02 | >10 |
| 22x |
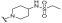
|
0.034 ± 0.002 | 0.33 ± 0.029 | 6.2 ± 0.38 |
| 22y |
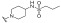
|
0.059 ± 0.003 | 0.95 ± 0.04 | 4.2 ± 0.36 |
| 22z |
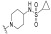
|
0.14 ± 0.021 | 0.063 ± 0.002 | 1.9 ± 0.058 |
| 22aa |
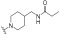
|
0.057 ± 0.0021 | 0.063 ± 0.001 | >10 |
| 22ab |
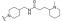
|
0.25 ± 0.06 | 0.46 ± 0.01 | 4.4 ± 1.37 |
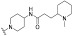
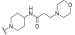
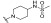
However, N,N-dimethylglycine (22h) exhibited improved activity against c-KIT wt (GI50 = 0.038 μM) and regained activity against the c-KIT T670I mutant (GI50 = 0.053 μM) while maintaining good selectivity toward parental BaF3 cells (GI50 = 2.9 μM), compared with 22a. The addition of cyclohexene (22i) and pyridine groups (22j) decreased the activity five- to ten-fold, while the addition of tetrahydropyran (22k) led to the regained activity against both c-KIT wt (GI50 = 0.087 μM) and c-KIT T670I (GI50 = 0.083 μM). N-Methylpiperidine (22l) and N-ethylpiperidine (22m) showed a similar potency. N-Acyl (22n), N-cyclopropanecarbonyl (22o), and Boc (22p) led to a two- to three-fold reduction in activity, and 2-methylpiperidine (22q) showed a ten-fold decreased activity against c-KIT T670I. Selectivity window to parental BaF3 cells (22r and 22s) or reduced activity against c-KIT T670I (22t and 22u) was observed by increasing the size of the piperidine-derived substituents. However, ethyl-linked morpholine (22v) displayed impressive activity against c-KIT wt (GI50 = 0.042 μM) and c-KIT T670I (GI50 = 0.059 μM), with good selectivity for parental BaF3 cells (GI50 = 3.6 μM). Switching the amide moiety to sulfonamide derivatives (22w–22y) with different aliphatic chains did not improve antiproliferative efficacy against c-KIT T670I mutant. However, introducing the cyclopropyl group (22z) regained the activity against BaF3-TEL-c-KIT-T670I cells (GI50 = 0.063 μM). N-(piperidin-4-ylmethyl)propionamide (22aa) improved activity against c-KIT wt (GI50 = 0.057 μM), maintained activity against c-KIT T670I, and improved selectivity to parental BaF3 cells (GI50 > 10 μM), whereas further enlargement (22ab) showed significantly reduced activity, compared with 22aa. A SAR study of this series revealed that by switching the amide linkage to the urea linkage and increasing the size of the substituent that is coupled to the urea, moiety improved the activity and selectivity against the c-KIT and c-KIT T670I mutant (Figure 5).
Figure 5.

Discovery, SAR summary, and pharmacophore description of quinoline-based urea derivatives as c-KIT wt and c-KIT T670I inhibitors.
Compound 22aa exhibited potent antiproliferative activity against both c-KIT wt-expressing GIST cancer cell line GIST-T1 (GI50 = 0.004 μM) and the c-KIT T670I-expressing GIST cancer cell line GIST-5R (GI50 = 0.026 μM). The pharmacokinetics of 22aa demonstrated a half-life of 2.04 h and clearance of 2.76 L/h/kg when administered via intravenous injection. However, oral administration of 22aa resulted in negligible absorption, which precluded its use via the oral route in the animal model. The antitumor effect of 22aa at 25, 50 and 100 mg/kg dosages in a BaF3-TEL-c-KIT-T670I cell-inoculated xenograft mouse model was evaluated. Over a period of 10 days of continuous treatment, the growth of BaF3-TEL-c-KIT-T670I tumors was found to be dose-dependently inhibited by compound 22aa. At a dosage of 100 mg/kg, the compound exhibited a tumor growth inhibition of 47.7%.
Molecular modeling showed that compound 22aa adopted a type II binding mode. In c-KIT, the nitrogen atom of the quinoline moiety established a hydrogen bond with Cys673 of the c-KIT domain at the hinge-binding region, and the urea moiety NHs interacted with Glu640 via a hydrogen bond formation. The hydrophobic pocket was occupied by the piperidine moiety and amide NH to form an additional hydrogen bond with Ile789 (Figure 6A). A similar type II binding mechanism was adopted by compound 22aa in the c-KIT T670I mutant homology model. The O-bridged phenyl moiety in 22aa was oriented at an angle that allowed room for the large residue isoleucine due to the three hydrogen bonds created by the urea moiety (Figure 6B).
Figure 6.

(A) Molecular modeling of compound 22aa with c-KIT wt (PDB code 1T46); (B) Molecular modeling of compound 22aa with the c-KIT T670I homology model (generated based on PDB code 1T46). Reprinted with permission from Ref. [41].
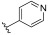
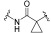
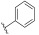
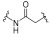
In continuation to their previously developed c-KIT inhibitor CHMFL-KIT-8140, Wu et al. designed and synthesized a series of substituted N-(4-((6,7-dimethoxyquinolin-4-yl)oxy)phenyl)acetamide derivatives [42]. The 35 synthesized compounds were screened for their inhibitory activity against IL-3-independent BaF3 cells expressing c-KIT wt (BaF3-tel-c-KIT) and c-KIT T670I (BaF3-tel-c-KIT-T670I). The inhibitory activity of CHMFL-KIT-8140 was significantly reduced by replacing the urea fragment (L2) with acetamide and the R1 tail moiety with a phenyl ring (35a) or pyridine (35b). Modification of L2 in 35a to cyclopropanecarboxamide (36) did not increase the potency. Nevertheless, introducing a CF3 group at the para-position of R1 (35c) significantly increased the activity against c-KIT wt and c-KIT T670I with strong selectivity against parental BaF3 cells (GI50 = 2.16 μM) and GI50 values of 0.022 μM and 0.011 μM, respectively. Similar efficacy against c-KIT wt and c-KIT T670I was demonstrated by an analogous molecule with an m-CF3 substituent (35d) (GI50 = 0.020 and 0.001 μM, respectively). Significant activity loss occurred when the R1 substituent was changed from m-CF3 to m-F (35e) or m-OMe (35f). The p-F substitution added to R1 in 35d (35g) resulted in a 13-fold decrease in activity against c-KIT T670I. Cl substitution (35h), however, increased the selectivity for parental BaF3 cells (GI50 = 5.97 μM) and efficacy against c-KIT wt and c-KIT T670I. The activity against c-KIT wt and c-KIT T670I was slightly decreased by installing a larger substituent, such as a methyl group, at R1 (35i). The effects of various substituents at the meta- and para-positions of the benzene ring (35k–m) were further investigated, and this led to a considerable loss of activity against c-KIT wt. In addition, activity loss was observed when a F or CF3 group was installed to the benzene ring ortho-position (35n–p). Larger groups at R1, such as benzodioxole (35q), naphthyl (35r), and aliphatic rings such as cyclohexyl (35s) and N-methyl piperazine (35t), led to a loss of activity. When the amide of L2 in 35c (43) and 35d (44) was changed to a reversed amide, activity loss occurred (Table 5).
Table 5.
SAR exploration focused on the L2/R1 moieties.
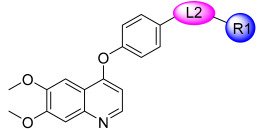
| |||||
|---|---|---|---|---|---|
| Compd. | L2 | R1 | Parental BaF3 (GI50: μM) |
BaF3-Tel-c-KIT (GI50: μM) |
BaF3-Tel-c-KIT-T670I (GI50: μM) |
| CHMFL-KIT-8140 | - | - | >10 | 0.057 | 0.063 |
| 35a |
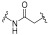
|
|
>10 | >10 | >10 |
| 35b |
|
|
5.13 | 4.17 | 6.99 |
| 36 |
|
|
>10 | >10 | >10 |
| 35c |
|
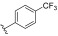
|
2.16 | 0.022 | 0.001 |
| 35d |
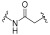
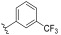
|
|
1.89 | 0.020 | 0.001 |
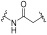
| 35e |
|
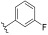
|
1.0 | 0.923 | 0.342 |
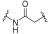
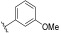
| 35f |
|
|
2.78 | 0.293 | 0.339 |
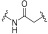
| 35g |
|
|
1.03 | 0.024 | 0.013 |
| 35h |
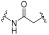
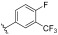
|
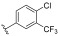
|
5.97 | 0.001 | 0.004 |
| 35i |
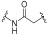
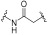
|
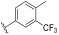
|
7.26 | 0.017 | 0.014 |
| 35j |
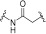
|
|
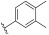
6.42 | 0.027 | 0.015 |
| 35k |
|
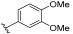
|
4.13 | 0.319 | 0.179 |
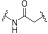
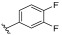
| 35l |
|
|
0.67 | 0.312 | 0.309 |
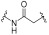
| 35m |
|
|
2.03 | 0.342 | 0.037 |
| 35n |
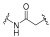
|
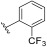
|
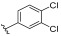
6.70 | 0.424 | 0.309 |
| 35o |
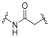
|
|
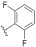
1.93 | 2.78 | 1.70 |
| 35p |
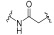
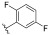
|
|
0.704 | 0.040 | 0.285 |
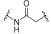
| 35q |
|
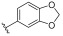
|
3.10 | 0.11 | 0.167 |
| 35r |
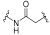
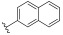
|
|
6.23 | 0.112 | 0.034 |
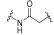
| 35s |
|
|
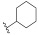
2.47 | 0.635 | 0.362 |
| 35t |
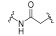
|
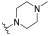
|
8.70 | 7.33 | 8.52 |
| 43 |
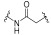
|
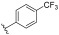
|
6.18 | 2.90 | 0.668 |
| 44 |
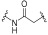
|
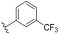
|
10 | 2.35 | 4.61 |
To further explore the SAR of the compound series, they focused on the L1 moiety while fixing the L2 and R1 moieties as phenoxyacetamide and 4-chloro-3-(trifluoromethyl)phenyl, respectively (Table 6). Replacement of the oxygen atom in the L1 linker of compound 35h with nitrogen (48), N-methyl (50), or sulfur (52) resulted in a significant loss of activity against both c-KIT wt and c-KIT T670I. The effects of various substituents on the benzene ring of the L1 moiety was investigated while the phenoxy group was kept intact. The addition of an F atom to the acetamide ortho-position (39) improved the activity against both c-KIT wt and c-KIT T670I (GI50 = 0.049 and 0.018 μM, respectively). Replacement of the F atom with a chlorine atom (35a) or a methyl group (40a) displayed similar activity compared to 39. However, shifting the F atom from the ortho- to the meta-position (40) decreased the activity against c-KIT wt (GI50 = 0.116 μM). Substituting the meta-position with various groups, such as chloro (40b), methyl (40c), methoxy (40d), trifluoromethyl (40e), and nitrile (40f), all resulted in a loss of activity compared with compound 35h. SAR study of this series disclosed that changing the urea linkage to a phenylacetamide linker and substituting the tail phenyl ring with a chloro group in combination with the trifluoromethyl group para to the acetamide linker improved the activity and selectivity (Figure 7).
Table 6.
SAR exploration focused on the substitution (L1) moiety.
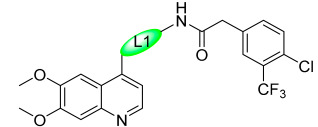
| ||||
|---|---|---|---|---|
| Compd. | L1 | Parental BaF3 (GI50: μM) |
BaF3-Tel-c-KIT (GI50: μM) |
BaF3-Tel-c-KIT-T670I (GI50: μM) |
| 48 |
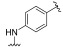
|
2.21 | 0.742 | 0.4 |
| 50 |
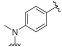
|
2.67 | 4.28 | 8.46 |
| 52 |
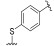
|
3.09 | 0.154 | 0.11 |
| 39 |
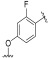
|
3.21 | 0.049 | 0.018 |
| 35u |
|
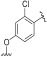
8.22 | 0.081 | 0.070 |
| 40a |
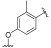
|
4.86 | 0.061 | 0.075 |
| 40 |
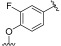
|
1.99 | 0.116 | 0.021 |
| 40b |
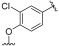
|
9.45 | 0.078 | 0.056 |
| 40c |
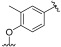
|
7.05 | 0.105 | 0.009 |
| 40d |
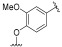
|
8.48 | 0.034 | 1.56 |
| 40e |
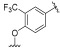
|
2.92 | 3.0 | 2.84 |
| 40f |
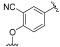
|
9.97 | 0.989 | 0.21 |
Figure 7.

Discovery, SAR summary, and pharmacophore description of compound 35h from 22aa to improve the pharmacokinetic profile.
Compound 35h showed a strong growth inhibition in KIT-dependent GIST cancer cells, such as GIST-T1 and GIST-882 (GI50 = 0.013 and 0.006 μM) and maintained a selectivity window against the GIST-48B cancer cell line (GI50 = 1.37 μM). When administered orally, the pharmacokinetics of compound 35h exhibited half-lives of 4.5 h, 6.4 h, and 19.4 h in mice, rats and dogs, respectively. Compound 35h has an acceptable bioavailability in mice (F = 43%), rats (F = 50%), and dogs (F = 81%). The antitumor effect of 35h at dosages of 20, 40, 80 and 100 mg/kg in a BaF3-tel-c-KIT-T670I cell-inoculated xenograft mouse model was evaluated. Over an 11-day course of treatment, compound 35h showed a dose-dependent inhibition of BaF3-tel-c-KIT-T670I tumor progression with almost 100% tumor growth inhibition at a dosage of 100 mg/kg/day.
The docking results revealed that the potent derivative 35h adopted a type II binding mode in the c-KIT wt (Figure 8A) and c-KIT T670I mutant. At the hinge-binding region, the nitrogen atom of the quinoline moiety forms a hydrogen bond with Cys673. Two more hydrogen bonds were established by the amide group with Asp810 and Glu640, and the tail phenyl group fit into the hydrophobic pocket in the c-KIT wt model (Figure 8B).
Figure 8.

Molecular docking analysis of 35h (A) 35h with the c-KIT wt (PDB code: 6GQK); (B) 35h with the c-KIT V654A homology model (PDB code: 6GQK); Reprinted with permission from Ref. [42].
Liu et al. designed and synthesized structurally modified derivatives of the FDA-approved drug axitinib and tested them for their inhibitory activity [43]. The inhibitory activity of the 29 synthesized compounds was evaluated against IL-3-independent BaF3 cells (GI50) expressing c-KIT wt (BaF3-tel-c-KIT) and c-KIT T670I (BaF3-tel-c-KIT-T670I). When the phenyl thiol ether linkage was replaced with a malonamide linkage (57a), it showed an improved potency against c-KIT wt and c-KIT T670I (GI50 = 0.025 and 0.002 μM, respectively). 57a also displayed selectivity against parental BaF3 cells (GI50 = 7.4 μM), which indicates effective antiproliferative inhibition. Shorter linkers, such as amide 58a, ethyleneamide 58b, and urea 59, showed a decreased activity against c-KIT T670I. Increasing the linker size (57b) decreased the activity against both c-KIT wt and c-KIT T670I (Table 7).
Table 7.
SAR exploration of the linker moiety (L).
| ||||
|---|---|---|---|---|
| Compd. | Linker (L) | BaF3 (GI50: μM) |
BaF3-Tel-c-KIT (GI50: μM) |
BaF3-Tel-c-KIT-T670I (GI50: μM) |
| Axitinib | - | 1.64 | 0.105 | 0.108 |
| 57a |
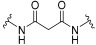
|
7.4 | 0.025 | 0.002 |
| 58a |
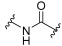
|
1.71 | 2.97 | 1.71 |
| 58b |
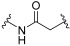
|
3.79 | 5.5 | 3.18 |
| 59 |
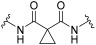
|
1.9 | 1.6 | 1.15 |
| 57b |
|
>10 | 1 | 0.23 |
Removal of vinylpyridine in 57a with a hydrogen atom (67a) or methyl group (67b) led to a substantial loss of activity. However, 1-methyl-1H-pyrazole, pyridine, and 2-methylpyridine (67c–e) displayed potent inhibitory activity against c-KIT T670I (GI50 = 0.023, 0.045, and 0.059 μM, respectively) and had 7- to 15-fold increased selectivity over c-KIT, indicating the importance of the aromatic head moiety for c-KIT kinase binding. Altering the pyridine in 67d to 5-(methylcarbamoyl)pyridin (67f), fluorophenyl (67g), or 3-carbamoylphenyl (67h) led to a loss of activity against c-KIT wt and c-KIT T670I. N-Methyl formamyl (67i) showed a six-fold selectivity over c-KIT wt and good activity against c-KIT T670I (GI50 = 0.057 μM). Larger groups, such as 4-N-methyl piperazine (67j) and N-methyl piperazinyl methylene (67k), showed more potent activity against c-KIT T670I and c-KIT wt, but also exhibited inhibitory activity against parental BaF3 cells (Table 8).
Table 8.
SAR exploration of the R1moiety.
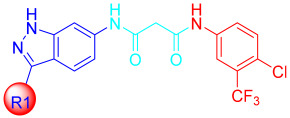
| ||||
|---|---|---|---|---|
| Compd. | R1 | BaF3 (GI50: μM) |
BaF3-Tel-c-KIT (GI50: μM) |
BaF3-Tel-c-KIT-T670I (GI50: μM) |
| 67a | H | >10 | >10 | 2.67 |
| 67b | Me | >10 | >10 | 1.88 |
| 67c |
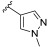
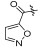
|
>10 | 0.35 | 0.023 |
| 67d |
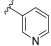
|
>10 | 0.32 | 0.045 |
| 67e |
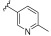
|
4.91 | 0.183 | 0.059 |
| 67f |
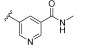
|
>10 | >10 | 4.48 |
| 67g |
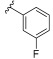
|
>10 | 1.12 | 0.179 |
| 67h |
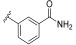
|
>10 | 1.55 | 0.339 |
| 67i |
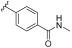
|
>10 | 0.338 | 0.057 |
| 67j |
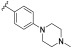
|
1.35 | 0.025 | 0.012 |
| 67k |
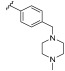
|
0.911 | 0.019 | 0.012 |
According to these findings, vinylpyridine in 57a was preferred for its increased efficacy against c-KIT T670I and for its higher selectivity for c-KIT wt. The impact of the R2 tail moiety was tested while keeping the head and linker the same (Table 9). Compound 57c, which has a simple phenyl group at R2, displayed a 19-fold selectivity over c-KIT wt, but showed a reduced activity against c-KIT T670I (GI50 = 0.117 μM). Meta-halogen-substituted phenyl groups (57d–f) were tested at R2 to increase the hydrophobicity and m-fluoro (57d), showing the best selectivity over c-KIT wt (26–fold) and highest potency against c-KIT T670I (GI50 = 0.044 μM). Larger substituent groups, such as m-methyl (57g), m-methoxy (57h), m-N,N-dimethyl (57i), and m-trifluoromethyl (57j), showed a reduced activity against c-KIT T670I and poorer selectivity for c-KIT wt. The m-N-methyl piperazinyl group (57k) caused significant activity loss. Fluoro-containing groups, such as o-fluoro (57l), p-fluoro (57m), and various multifluoro substituents (57n–p), resulted in loss a of activity against c-KIT T670I, compared with compound 57d.
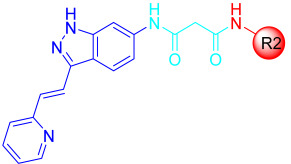
Table 9.
SAR exploration of the R2 position.
| ||||
|---|---|---|---|---|
| Compd. | Tail (R2) | BaF3 (GI50: μM) |
BaF3-Tel-c-KIT (GI50: μM) |
BaF3-Tel-c-KIT-T670I (GI50: μM) |
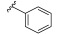
| 57c |
|
6.72 | 2.22 | 0.117 |
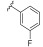
| 57d |
|
3.97 | 1.15 | 0.044 |
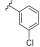
| 57e |
|
3.61 | 0.923 | 0.111 |
| 57f |
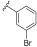
|
6.09 | 0.943 | 0.113 |
| 57g |
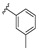
|
>10 | 1.43 | 0.17 |
| 57h |
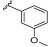
|
>10 | 2.74 | 0.234 |
| 57i |
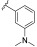
|
>10 | 3.87 | 0.594 |
| 57j |
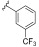
|
>10 | 0.216 | 0.086 |
| 57k |
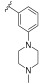
|
>10 | >10 | 6.17 |
| 57l |
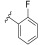
|
5.09 | 2.16 | 0.312 |
| 57m |
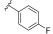
|
5.86 | 0.554 | 0.111 |
| 57n |
|
4.24 | 1.87 | 0.399 |
| 57o |
|
5.08 | 3.46 | 0.847 |
| 57p |
|
3.68 | 2.95 | 1.3 |
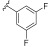
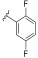
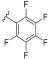
A SAR study of this series revealed that the replacement of the phenyl thioether linker with a malonamide moiety and by substituting the tail phenyl ring with meta halogen groups improved the activity and selectivity against c-KIT T670I over c-KIT wt (Figure 9).
Figure 9.

Design, optimization strategy, SAR summary, and pharmacophore description for the discovery of compound 57d.
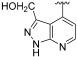
The pharmacokinetics of active compound 57d in a rat model showed 27.5% bioavailability and a half-life of 4.9 h at a dosage of 10 mg/kg when administered orally. In mice, it exhibited a 16.4% bioavailability with a moderate half-life of 1.6 h. The compound had a clearance rate of 12.4 L/h/kg and 6.8 L/h/kg in rats and mice, respectively. The antitumor efficacy of 57d at a dose of 100 mg/kg in a GIST-5R cell-inoculated xenograft model displayed 83% tumor growth inhibition.
A docking study of the potent compound 57d revealed that the compound adopted a canonical type II binding mode in the c-KIT wt and c-KIT T670I mutant. In the c-KIT wt model, the indazol nitrogen atoms established two hydrogen bonds with Cys673 and Glu671 in the hinge-binding region. Two additional hydrogen bonds formed between the amide moiety and Glu640 and Asp810 at the DFG motif. Furthermore, the hydrophobic pocket was occupied by the tail part. The modeling study was unable to explain the selectivity of compound 57d against c-KIT T670I over c-KIT wt (Figure 10).
Figure 10.

(A) Binding mode of 57d with c-KIT wt (PDB code: 1T46). (B) Binding mode of 57d with c-KIT T670I (PDB code: 1T46). Reprinted with permission from Ref. [43].
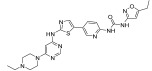
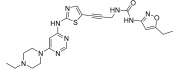
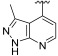
Kaitsiotou et al. designed and synthesized trisubstituted 3-ethynyl-N-(4-((4-methylpiperazin-1-yl)methyl)phenyl)benzamide derivatives and screened them against various c-KIT mutants, such as V654A, T670I, and D816H along with wild-type KIT [44]. The design of compounds was started by maintaining the potency of ponatinib and modifying the substitutions in the R1–R4 regions, while the alkyne linker, benzoic acid moiety, and N-methylpiperazine moiety of ponatinib were all kept intact throughout the SAR optimization.
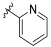
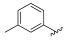
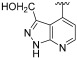
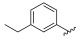
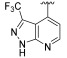
The 24 synthesized derivatives were tested against various c-KIT mutants, as mentioned above. Similar to the potent precursor of ponatinib (70a, 6.8 nM), the alkyne precursor (71a) displayed a loss of activity (231 nM) against the wild-type KIT. Unsubstituted pyridine analogues (71b, 3.5 nM) and (71e, 2.1 nM) exhibited similar activity against the wild-type KIT and other KIT mutants. Substitution of an electron-donating group, such as a benzyloxy group at the five-position on the pyridine ring (71c), decreased the potency compared with 71b, whereas methoxy group substitution at the three-position (71d) led to a retained potency. Replacement of the pyridine heterocycle with 2-aminopyrimidine (71g) increased the potency against the wild-type KIT and the tested mutants (2–247 nM), whereas 3-aminopyridazine (71f) retained similar potency against the wild-type KIT but lost activity against all KIT mutants. Substitution of the amine group of 2-aminopyrimidine with an aliphatic chain (71h) exhibited an IC50 value of 3.3 nM, but lost activity in all KIT mutants compared with 71g. Further replacements of pyridine, such as 2-aminopyridin-5-yl (71l), 2-amino-3-methylpyridin-5-yl (71j), and isoquinolin-1-amine (71m), resulted in a loss of potency against the D816H and T670I mutant forms. These results suggest that the 2-aminopyrimidine moiety is crucial for potency. Replacing the trifluoromethyl group from the most active derivatives 71a–lb, with a hydrogen atom (71n–q) and F atom (71r–s), resulted in a significant loss of inhibitory activity against KIT mutants. These results indicate that the trifluoromethyl group was necessary for inhibitory activity against KIT mutant forms. Shifting the methyl group from the four-position of 71g to the two-position on the phenyl (71t), resulted in a loss of activity against V654A and T670I mutants (Table 10).
Table 10.
IC50 determination of trisubstituted derivatives.
| Compd. | R1 | R2 | R3 | R4 | IC50 [nM] | |||
|---|---|---|---|---|---|---|---|---|
| KITWT | KITV559D/T670I | KITV559D/V654A | KITD816H | |||||
| 70a | I | CF3 | H | CH3 | 6.8 ± 21 | a | a | a |
| 70b | H | CF3 | F | CH3 | 555.7 ± 41.6 | a | a | a |
| 70c | I | CF3 | Et | CH3 | 1039.3 ± 513.9 | a | a | a |
| 70d | I | CF3 | F | CH3 | 183.7 ± 186.5 | a | a | a |
| 71a | H | CF3 | H | CH3 | 230.7 ± 1.2 | a | a | a |
| 71b |
|
CF3 | H | CH3 | 3.5 ± 0.6 | 116 ± 20.1 | a | a |
| 71c |
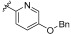
|
CF3 | H | CH3 | 120.9 ± 154.7 | a | a | a |
| 71d |
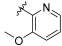
|
CF3 | H | CH3 | 6.7 ± 1.9 | 158.7 ± 43 | a | a |
| 71e |
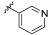
|
CF3 | H | CH3 | 2.1 ± 0.6 | 24 ± 3.1 | 1101.6 ± 47.8 | 137.6 ± 2.3 |
| 71f |
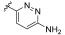
|
CF3 | H | CH3 | 5.8 ± 0.2 | 150.2 ± 35.3 | a | 2267.8 ± 105.3 |
| 71g |
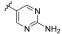
|
CF3 | H | CH3 | 1.9 ± 0.6 | 21.4 ± 1.2 | 246.6 ± 71.2 | 42.2 ± 13.0 |
| 71h |
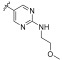
|
CF3 | H | CH3 | 3.3 ± 0.9 | 106.8 ± 7.2 | 252.5 ± 13.2 | 2614.3 ± 745.4 |
| 71i |
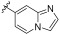
|
CF3 | H | CH3 | 1.6 ± 0.9 | 72.0 ± 5.2 | a | a |
| 71j |
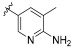
|
CF3 | H | CH3 | 2 ± 0.5 | 29.8 ± 1.4 | 777.2 ± 88.6 | 99.3 ± 43.3 |
| 71k |
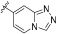
|
CF3 | H | CH3 | 2.5 ± 0.9 | 2147.2 ± 213.1 | a | a |
| 71l |
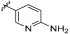
|
CF3 | H | CH3 | 2.0 ± 0.6 | 28.8 ± 1.3 | 908.3 ± 165.0 | 140 ± 13.4 |
| 71m |
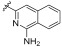
|
CF3 | H | CH3 | 4 ± 0 | 18.2 ± 6.8 | 313.5 ± 132.2 | 104.3 ± 8.7 |
| 71n |
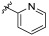
|
H | H | CH3 | 239.6 ± 307.6 | a | a | a |
| 71o |
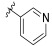
|
H | H | CH3 | 3.3 ± 1.8 | a | a | a |
| 71p |
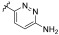
|
H | H | CH3 | 100.3 ± 84.2 | a | a | a |
| 71q |
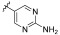
|
H | H | CH3 | 2.6 ± 1.0 | a | a | a |
| 71r |
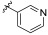
|
F | H | CH3 | 2.3 ± 0.5 | a | a | a |
| 71s |
|
F | H | CH3 | 6.5 ± 1.1 | a | a | 1706.6 ± 803.4 |
| 71t |
|
CF3 | CH3 | H | 6.2 ± 2.3 | 21 ± 3.6 | 1084.7 ± 304.0 | 325.8 ± 111.1 |
| Ponatinib | 1.7 ± 0.7 | 17.4 ± 9.8 | 136.0 ± 39.9 | 20 ± 2.2 | ||||
a No inhibition.
These derivatives were further screened for their inhibitory activity against GIST-T1, GIST-T1-T670I, GIST430-V654A, and GIST-T1-D816E cells. The results revealed that compounds 71b, 71d–j, and 71l–m exhibited excellent activity against imatinib-resistant GIST-T1-T670I cells. Compounds 71g, 71j, and 71l demonstrated potent antiproliferative activity (GI50 = 141 nM, 474 nM, and 221 nM, respectively) against GIST-T1-D816E cells. In addition, it exhibited improved inhibitory potency against GIST430-V654A cells compared with ponatinib (51 vs. 149 nM). The precursor compounds 70a–d and 71a did not reduce the cell viability in all tested GIST cell lines. The tested compounds did not exhibit inhibitory activity against KIT-negative GIST-48B cell lines. Compounds 71n–s demonstrated weak inhibitory activity over all KIT-positive and KIT-negative cells. Interestingly, compound 71t retained an inhibitory activity against GIST430-V654A (308 nM) and GIST-T1-D816E cells (381 nM) and showed a decreased activity against KIT-negative GIST cells. A SAR analysis of this series revealed that the trifluoromethyl group at the R2 position was an important structural feature for activity. Additionally, the replacement of the imidazo[1,2-b]pyridazine heterocycle with 2-aminopyrimidine significantly improved the activity against all tested KIT mutants, and also against the wild type c-KIT. In addition, the repositioning the methyl group of the phenylcarboxamide moiety resulted in a significant loss of activity against the secondary mutant V654A and T670I mutant (Figure 11).
Figure 11.

Design strategy, SAR summary, and pharmacophore description of ponatinib structure-modified derivatives.
Western blot analysis demonstrated that 71g was the most effective compound for inhibiting autophosphorylation in GIST430-V654A and GIST-T1-D816E cells. A pharmacokinetics study of the potent compound 71g displayed satisfactory clearance in human liver microsomes (CLint, 1 μL min−1 mg−1), 91% human plasma stability, 98.4% to 99.6% plasma protein binding, and good permeability in Caco-2 cells. These data are in the same range as ponatinib and the FDA-approved KIT inhibitors (Table 11).
Table 11.
GI50 determination of trisubstituted derivatives.
| Compd. | GI50 [nM] | ||||
|---|---|---|---|---|---|
| GIST-48B | GIST-T1 | GIST-T1-T670I | GIST-T1-D816E | GIST430-V654A | |
| 70a | 4877 ± 358 | 896 ± 175 | 3342 ± 282 | 9891 ± 1741 | 3905 ± 1799 |
| 70b | a | 10,019 ± 1870 | 15,291 ± 2665 | a | a |
| 70c | 9942 ± 1049 | 11,006 ± 825 | 7609 ± 286 | a | 15,858 ± 498 |
| 70d | 3627 ± 1346 | 5729 ± 705 | 9512 ± 403 | a | a |
| 71a | a | 7840 ± 891 | 10,136 ± 2058 | a | a |
| 71b | 3627 ± 532 | 55 ± 9 | 97 ± 19 | 1673 ± 328 | 697 ± 76 |
| 71c | 1860 ± 250 | 121 ± 320 | 4375 ± 1985 | a | 2005 ± 890 |
| 71d | 3793 ± 525 | 82 ± 115 | 165 ± 33 | 991 ± 95 | 564 ± 32 |
| 71e | 2705 ± 296 | 51 ± 13 | 76 ± 18 | 332 ± 77 | 333 ± 1 |
| 71f | 6247 ± 242 | 54 ± 11 | 89 ± 17 | 580 ± 141 | 459 ± 47 |
| 71g | 2024 ± 545 | 23 ± 6 | 44 ± 8 | 141 ± 12 | 51 ± 5 |
| 71h | 5132 ± 511 | 119 ± 21 | 198 ± 25 | 704 ± 110 | 481 ± 6 |
| 71i | 3037 ± 620 | 59 ± 12 | 184 ± 26 | 2289 ± 382 | 723 ± 45 |
| 71j | 2087 ± 147 | 59 ± 11 | 117 ± 29 | 474 ± 37 | 307 ± 2 |
| 71k | 4146 ± 1116 | 282 ± 78 | 2971 ± 1150 | a | 4145 ± 94 |
| 71l | 2237 ± 156 | 42 ± 8 | 67 ± 12 | 221 ± 42 | 404 ± 335 |
| 71m | 2065 ± 288 | 191 ± 64 | 301 ± 68 | 645 ± 744 | 926 ± 385 |
| 71n | a | 7097 ± 522 | 12,473 ± 3527 | a | a |
| 71o | a | 150 ± 37 | 6352 ± 1825 | 9610 ± 1089 | 4032 ± 97 |
| 71p | a | 4189 ± 727 | a | a | a |
| 71q | 6877 ± 1409 | 33 ± 6 | 1224 ± 758 | 1269 ± 53 | 2063 ± 1294 |
| 71r | 11,966 ± 2280 | 57 ± 15 | 3294 ± 314 | 5358 ± 1309 | 1639 ± 176 |
| 71s | 8285 ± 2983 | 31 ± 16 | 1362 ± 576 | 904 ± 490 | 920 ± 105 |
| 71t | 15,832 ± 390 | 35 ± 3 | 51 ± 1 | 381 ± 33 | 308 ± 26 |
| Ponatinib | 2000 ± 450 | 17 ± 8 | 40 ± 19 | 106 ± 60 | 149 ± 36 |
a No inhibition.
The molecular docking of potent compound 71g and SAR analysis demonstrated that the main hinge binder was a pyrimidine heterocycle. Compound 71g established two hydrogen bond interactions with Cys673 and Glu640. The amine group of the pyrimidine moiety formed an additional hydrogen bond to the hinge region of the kinases. The trifluoromethyl group was anchored in the back subpocket. The nitrogen atom of the N-methylpiperazine moiety displayed H-bond interactions with the carbonyl groups of Ile789/His790 residues (Figure 12).
Figure 12.

(A) Docking of 71g in complex with wild-type KIT. (B) Close-up view of the back pocket. Reprinted with permission from Ref. [44].
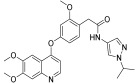
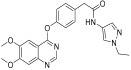
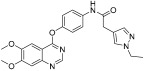
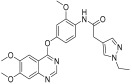
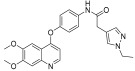
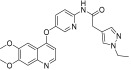
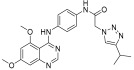
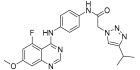
Kettle et al. designed and synthesized a potent derivative and tested it against the KIT mutant Ba/F3 and PDGFR cell lines to treat GISTs [45]. Screening of previously developed quinazoline-based compounds as PDGFR and VEGFR inhibitors against KIT mutant Ba/F3 cell lines, along with the KDR cell line, resulted in the identification of the lead compound AZD2932 [46], which exhibited excellent inhibitory activity in this panel along with the KDR cell line. A selective PDGFR inhibitor, compound I-a [47], which has a central phenoxy ring, displayed less inhibitory activity against KIT mutants and the KDR cell line but showed activity against the parental cell line. Compound I-b, which has an amine linker instead of a phenoxy linker (I-a), showed a decreased potency against all the tested cell lines. Methoxy substitution at the meta position on the phenoxy ring (I-c) led to retained potency, as well as improved KDR selectivity. Replacement of the phenyl ring with meta-pyridine (I-d) displayed a high selectivity against KDR and an excellent potency against KIT-mutant Ba/F3 cell lines. Quinazoline to quinoline modification (I-e) led to a retained potency against KIT mutants, but a decreased selectivity against KDR. The combination of quinoline with a meta-methoxyphenol linker (I-f) resulted in nanomolar activity in all tested cell lines. Compound II and its reverse amide (II-a) (ref. [16,26]) from the PDGFR program demonstrated similar activity against V654A and D816H, but the reverse amide showed a decreased activity against T6701 and improved selectivity against KDR. Methoxy substitution on the central ring (II-b) did not improve the activity. Quinoline analogues (III and III-a) showed a significant loss of activity in KIT mutants (Table 12; Figure 1).
Table 12.
SAR of the literature PDGFR inhibitors in KIT-mutant Ba/F3 cell lines and their effect on KDR.
| Compd. | Structure | Ba/F3 GI50 (μM) | ||||
|---|---|---|---|---|---|---|
| Parental | Exon 11 Del + V654A | Exon 11 Del + D816H | Exon 11 Del + T670I | KDR | ||
| AZD2932 |
|
>10 | 0.012 | 0.070 | 0.004 | 0.093 |
| I-a |
|
4.289 | 0.685 | 0.786 | 1.918 | 2.674 |
| I-b |
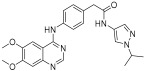
|
>10 | 0.337 | 2.592 | 0.078 | 3.595 |
| I-c |
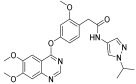
|
>10 | 0.008 | 0.026 | 0.012 | 0.442 |
| I-d |
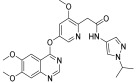
|
>10 | 0.021 | 0.176 | 0.060 | 8.030 |
| I-e |
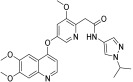
|
>10 | 0.013 | 0.027 | 0.061 | 3.924 |
| I-f |
|
>10 | 0.006 | 0.008 | 0.007 | 0.077 |
| II |
|
>10 | 0.044 | 0.488 | 0.007 | 0.680 |
| II-a |
|
>10 | 0.021 | 0.250 | 0.141 | 1.615 |
| II-b |
|
>10 | 0.078 | 0.251 | 0.086 | 2.551 |
| III |
|
>10 | 0.026 | 0.047 | 0.007 | 0.082 |
| III-a |
|
>10 | 0.150 | 0.518 | 0.008 | 0.761 |
| IV |
|
>10 | 0.003 | 0.019 | 0.017 | 0.612 |
| V |
|
>10 | 0.007 | 0.050 | 0.055 | 1.222 |
| V-a |
|
>10 | 0.003 | 0.048 | 0.107 | 5.408 |
| V-b |
|
>10 | 0.002 | 0.025 | 0.068 | >10 |
| 75 |
|
>10 | 0.003 | 0.009 | 0.016 | 1.378 |
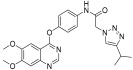
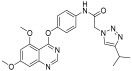
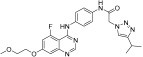
By screening various heterocyclic portions for the modification of isopropyl pyrazole, the N-linked triazole compound (IV) was found to exhibit potent inhibition. When the methoxy group was relocated from the six-position to the five-position (V), a three-fold loss of activity was observed. Replacement of the ether-linked aromatic ring with the amine-linked aromatic ring (V-a) led to potent inhibition against KIT mutants along with a 113-fold improved selectivity against KDR. When the five-position methoxy group was replaced with an F atom (V-b), the activity against KIT mutants was retained, and the selectivity against KDR was improved to a greater extent, but the compound had a 99.7% protein binding. The addition of a methoxyethoxy group (75), rather than a methoxy group, retained KIT mutant inhibitory activity and selectivity over KDR. The lead compound, AZD2932, which was identified from their previous work as a PDGFR and VEGFR inhibitor, was utilized to design a potent c-KIT inhibitor. A SAR study of this series revealed that the incorporation of the fluoro group at the five-position of the quinoxaline core improved the potency. Additionally, modifying the methoxy group with an methoxyethoxy group at the seven-position of the core improved the activity against mutant c-KIT. In addition, replacing the quinoxaline ether linker with an amine linker resulted in the retention of the mutant potency and a significant improvement in selectivity over KDR (Figure 13).
Figure 13.

Schematic design, SAR summary, and pharmacophore description for the discovery of compound 75.
The ADME results of 75 indicate that it has good bioavailability (mouse, rat, dog, human: 4.9, 2.2, 6.8, 3.5%), low clearance (mouse, rat, dog, human: 5, 17, <1, <1 μL/min/10–6 cells) and low hERG activity (IC50 = 33.3 μM). Compound 75 showed excellent growth inhibition in all KIT-mutant Ba/F3 cell lines and PDGFR-driven cell lines relevant to the subsets of GISTs, including the clinically GIST-relevant D842V mutant compared with clinically approved and unapproved KIT inhibitors. The in vivo results of compound 75 in a Ba/F3 KIT-exon 11 del/D816H mouse allograft tumor model revealed that there was a 5% inhibition, and tumor size was not affected. However, at a dosage of 20 mg/kg b.i.d., 75 showed excellent regression of tumor volume (75%), and the data are encouraging when compared with regorafenib (39%) at 100 mg/kg q.d. In a Ba/F3 KIT-exon 11 del/V654A mouse allograft tumor model, 75 displayed strong regression (85%) at a dosage of 20 mg/kg b.i.d., but sunitinib showed similar regression (87%) at a dosage of 80 mg/kg q.d.
The cocrystal structure of 75 indicates that the triazolo group binds in the DFG pocket, and there is a water-mediated interaction at the gatekeeper, encouraging selectivity. The C7 side chain was oriented outside of the active site and into the solvent pocket (Figure 14).
Figure 14.

Cocrystal structure of 75 bound to c-KIT (PDB code: 6GQM). Reprinted with permission from Ref. [45].
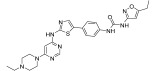
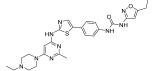
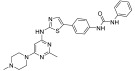
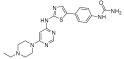
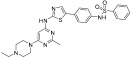
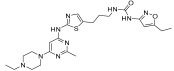
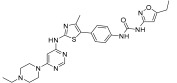
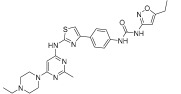
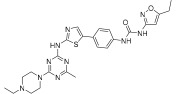
Wu et al. designed and synthesized a series of 5-phenyl-thiazol-2-ylamine derivatives, and the 14 synthesized compounds as well as compound 81a were evaluated for c-KIT kinase activity and GIST-T1 cell proliferation [48]. This demonstrated that 2-methylpyrimidine 81b had comparable inhibitory effects on both the wild type c-KIT kinase and GIST-T1 cells to compound 81a (Table 13), while phenyl-substituted urea 81 displayed strong inhibition against c-KIT kinase. Modifications, such as the removal of the 5-ethylisoxazole moiety in 81a (81d) and replacement of the urea component of 81a with a sulfonamide (81e) or an amide (81f), led to a reduced cellular potency, but there was no significant impact on c-KIT kinase inhibitory activity. Insertion of a nitrogen atom (81g) on the phenyl ring in 81a, saturation of the phenyl ring (90), and replacement of the aromatic ring with an aliphatic chain (93 and 95) with the urea component intact did not result in a significant change in inhibitory activity against both c-KIT kinase and GIST-T1 cells. Methyl group substitution at the four-position on the thiazole ring in compound 81h resulted in a slight decrease in activity against c-KIT kinase and GIST-T1 cells. Shifting the phenylurea tail four-position of the thiazole ring (84) led to loss of enzymatic and cellular activity. When the pyrimidine component of 81a was changed to 2-methyl-1,3,5-triazine (81i), it resulted in a six-fold decrease in activity against GIST-T1 cells, revealing the significance of the pyrimidine component in 81a. Compound 81j, which has an N-(2-hydroxyethyl)piperazine group, demonstrated comparable potency to 81a in both assays, while compound 81k, which has a pyrrolidin-3-ylamine group, enhanced enzymatic inhibition against c-KIT but showed lower cellular activity than 81a. Among the tested compounds, compound 81g only exhibited slightly improved activity, compared with 81a, in both enzymatic and cellular assays, but it had a lower synthetic yield (Figure 15).
Table 13.
c-KIT enzymatic inhibitory activity and GIST-T1 cell proliferation with the synthesized compounds.
| Compd. | Structure | IC50 (nM) | GI50 (nM) |
|---|---|---|---|
| c-KIT | GIST-T1 | ||
| 81a |
|
82 | 2.2 |
| 81b |
|
97 | 3.2 |
| 81c |
|
20 | 5.4 |
| 81d |
|
98 | 86 |
| 81e |
|
54 | 42 |
| 81f |
|
130 | 25 |
| 81g |
|
43 | 1.0 |
| 90 |
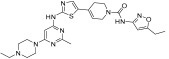
|
82 | 4.9 |
| 93 |
|
38 | 9.9 |
| 95 |
|
50 | 10 |
| 81h |
|
122 | 3.3 |
| 84 |
|
950 | >1000 |
| 81i |
|
86 | 13 |
| 81j |
|
71 | 3.3 |
| 81k |
|
15 | 6.4 |
| Sunitinib |
|
48 | 38 |
Figure 15.

SAR summary and pharmacophore description of 81a.
5-Phenyl-thiazol-2-ylamine pyrimidine template was utilized to design and develop a potent c-KIT inhibitor. A SAR study of this series revealed that substituted phenyl urea was an important structural feature for activity. Additionally, the pyrimidine component was necessary for maintaining the inhibitory activity. In addition, modification of the ethylisoxazole or urea moiety did not affect the c-KIT inhibitory activity (Figure 15).
Notably, compounds 81c and 81k exhibited potent inhibition against c-KIT but had unfavorable pharmacokinetic profiles. Thus, compound 81a was selected for its biological activity and in vivo efficacy. Compound 81a exhibited potent antiproliferative activity against GIST882, GIST430, and GIST48 (GI50 = 3, 1, and 2 nM, respectively).
The pharmacokinetics of compound 81a in a mouse model demonstrated moderate bioavailability (F = 38%), AUC (2796 ng/mL∙h) and a short half-life (t1/2 = 2.8 h) with oral administration. Compound 81a exhibited a 7.1 L/kg volume of distribution (Vss) and a 20.2 mL/min/kg plasma clearance rate (Cl) with intravenous administration. The antitumor efficacy of 81a at 40 and 25 mg/kg dosages in a GIST430 tumor xenograft model was superior to the FDA-approved sunitinib and showed 59% reduction in tumor size on day 14.
The molecular interactions of 81a in the unactivated c-KIT kinase domain displayed type II binding mode (Figure 16). The thiazolylamine formed two hydrogen bonds in the hinge-binding area of c-KIT with the backbone of Cys673. The back pocket of the ATP-binding site was occupied by the tail group, which formed three more H-bonds with the urea group, backbone of Asp810, and side chain of Glu640. The phenyl ring of 81a formed an aromatic interaction with Phe811, while the ethylpiperazine was oriented toward the solvent region.
Figure 16.

Chemical structure of 81A and in complex with the unactivated c-KIT kinase domain (PDB 6ITT). Reprinted with permission from Ref. [48].
Lin et al. continued their research from their previous compound 81a by rational design, and the team synthesized a series of five-aromatic substituted thiazol-2-ylamine pyrimidine derivatives [49]. The 14 synthesized compounds were evaluated against c-KIT and wt-FLT3 kinases GIST-T1 and MOLM-13 cell lines (Table 14). Compound 102 had a higher inhibitory effect against c-KIT (IC50 = 24 nM) than I (GI50 = 35 nM), but lost its antiproliferative activity against MOLM-13 cells. Changing the phenyl ring in 102 to a pyridine ring, led to compound 103, which had a similar inhibitory potency against c-KIT and FLT3, but decreased cellular potencies (GI50 = 42 and 36 nM for GIST-T1 and MOLM-13, respectively) relative to I. When the nitrogen atom was shifted to the third position, compounds 104a (R2 = CH3) and 104b (R2 = H) exhibited similar inhibitory effects against the kinases as compound I but were less active in the cellular assays (GI50 values of 26–140 nM). Compound 105a (R2 = CH3) had single-digit nanomolar activity against GIST-T1 (GI50 = 7.1 nM) and MOLM-13 (GI50 = 9.4 nM) cell lines and exhibited similar inhibitory activities against c-KIT and FLT3 compared with I. As compared with 105a, 105b (R2 = H) displayed no improvement in cellular potency (GI50 > 10 nM); however, it exhibited potent inhibitory activities against c-KIT and FLT3 (IC50 < 30 nM). Next, the effect of water-solubilizing substituents on the pyrimidine ring four-position was evaluated, and the potency of compounds 105c–f was compared to that of the N-ethylpiperazine analog 105a. Limited water-solubilizing groups were considered in this study. Analogs 105g and 106 were produced by inserting a methyl group on the pyridine ring of 105a and changing the pyridine ring of 105a to a pyrimidine ring, respectively. N-(2-fluoroethyl)piperazine, N-(2-hydroxyethyl)piperazine, N,N-dimethylpiperidin-4-amine, and 4-(2-hydroxyethyl)morpholine derivatives (105c, 105d, 105e, and 105f, respectively) did not affect the activity against the kinases c-KIT and FLT3. The N,N-dimethylpiperidin-4-amine group in 105e increased the cellular activities slightly (GIST-T1 GI50 = 1.5, MOLM-13 GI50 = 3.5 nM), whereas the morpholine group linked by a two-carbon ether in 105f led to decreased cellular potency (GIST-T1 GI50 = 27 nM, MOLM-13 GI50 = 42 nM). The study considered only a few water-soluble groups since the impact of some groups on biological activities, in vivo toxicities, and pharmacokinetics had been well-established during the development of 5-phenylthiazol-2-ylamine-based inhibitors. Analogs 105g and 106, with a single methyl substituent on the pyridine ring of 105a and a pyrimidine moiety instead of pyridine, respectively, were produced. The 2-methylpyridine compound 105g had a two- to four-fold decrease in potency against c-KIT, FLT3, and MOLM-13 cells compared with 105a, but retained activity (GI50 = 7.7 nM) against GIST-T1 cells. Pyrimidine 106 had a moderate inhibitory activity for c-KIT (IC50 = 123 nM) and FLT3 (IC50 = 163 nM), but a significantly reduced cellular potency (GI50 > 100 nM). Optimization of the 5-pyridin-4-yl-thiazol-2-yl series of pyrimidines (compound 105) was performed by investigating the effects of substitutions on 2-aminothiazole. When the pyridine ring in 104a was moved from the five- to the four-position on the thiazole ring (109), compounds 109 and 111 showed a significant decrease in both enzymatic activities (IC50 > 1000 nM) and cellular potencies (GI50 > 1000 nM), compared with 104a. In response to previous studies that demonstrated the ability of 2-aminothiazole to form hydrogen bonds with the hinge region of the ATP pocket, benzamide 111 was produced by replacing the pyrimidine ring with a solubilized para-substituted benzamide ring, which is a recognized scaffold for FLT3 kinase inhibitors. Benzamide 111 was found to have submicromolar activities against MOLM-13 cells (GI50 = 544 nM) and GIST-T1 (GI50 = 647 nM), but showed moderate inhibitory activity against c-KIT/FLT3. Therefore, no further modification of lead 111 was conducted. The pyridine-substituted 2-aminothiazole analogs 105a and 150c–e exhibited potent dual inhibition of c-KIT and FLT3 with cellular antiproliferative activities of less than 10 nM (Figure 17).
Table 14.
Thiazole analogs for enzyme inhibition and cell proliferation.
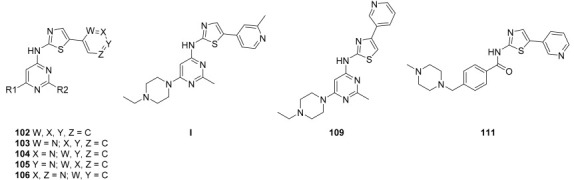
| ||||||
|---|---|---|---|---|---|---|
| Compd. | R1 | R2 | IC50 (nM) | GI50 (nM) | ||
| c- KIT | wt-FLT3 | GIST-T1 | AML MOLM-13 |
|||
| I | - | - | 97 | 38 | 3.2 | 2.0 |
| 102 |
|
-CH3 | 24 | 38 | 8.0 | 35 |
| 103 |
|
-CH3 | 100 | 24 | 42 | 36 |
| 104a |
|
-CH3 | 69 | 63 | 26 | 140 |
| 104b |
|
-H | 91 | 60 | 80 | 81 |
| 105a |
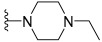
|
-CH3 | 56 | 30 | 7.1 | 9.4 |
| 105b |
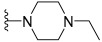
|
-H | 29 | 20 | 15 | 12 |
| 105c |
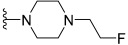
|
-CH3 | 47 | 37 | 3.8 | 7.7 |
| 105d |
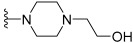
|
-CH3 | 49 | 43 | 2.8 | 6.5 |
| 105e |
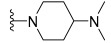
|
-CH3 | 53 | 35 | 1.5 | 3.5 |
| 105f |
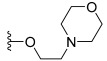
|
-CH3 | 49 | 35 | 27 | 42 |
| 105g | - | - | 100 | 64 | 7.7 | 39 |
| 106 |
|
-CH3 | 123 | 163 | 120 | 280 |
| 109 | - | - | >1000 | >1000 | >1000 | >1000 |
| 111 | - | - | 107 | 103 | 647 | 544 |
| imatinib | 53 | 131 | 40 | >1000 | ||
| sunitinib | 48 | 31 | 38 | 54 | ||
| regorafenib | 116 | 82 | 119 | 887 | ||
| midostaurin | 109 | 40 | 235 | 68 | ||
Figure 17.

Rational design, SAR summary, and pharmacophore description of compound 105a.
A SAR study of this series suggested that altering the phenyl urea moiety with a pyridine ring, with its four-position to thiazole, led to the identification of the most potent derivative with a favorable pharmacokinetic profile (Figure 17).
The pharmacokinetics, antitumor activities, and toxicities of 105a and 150c–e in SCID mouse xenografts or normal mice showed that 105a has a greater impact on GIST430 xenografts than 105c and 105d, as well as a lower toxicity and more favorable pharmacokinetic profile than 105c and 105e.
The pharmacokinetics of the potent compound 105a in male Sprague–Dawley rats and ICR mice demonstrated a bioavailability of 36% in rats and 68% in mice, and a moderate half-life (t1/2) of 3.9 h in rats and 4.1 h in mice when administered orally. When used via intravenous administration, 105a displayed high volumes of distribution (Vss = 14.7 L/kg in mice and 10.1 L/kg in rats) and plasma clearances (Cl = 70.7 mL/min/kg in mice and 393.6 mL/min/kg in rats). The antitumor effect of 105a at 10, 20, and 40 mg/kg dosages in NOD/SCID mice bearing GIST430 tumors showed rapid tumor regression.
The crystal structure of c-KIT, in complex with compound 105a (Figure 18), revealed that thiazolylamine formed two hydrogen bonds with Cys673 and established hydrophobic interactions with Leu595, Tyr672, Cys673, and Leu799. The ethylpiperazine group was oriented toward the solvent exposed region. The pyridine moiety formed additional hydrophobic interactions with Leu799 and Ala621.
Figure 18.

Interactions of 105a in complex with c-KIT and the chemical structure of 105a. Reprinted with permission from Ref. [49].
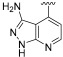
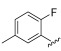
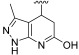
Lu et al. designed structural modifications to linifanib and synthesized a series of 3-methyl-1H-pyrazolo[3,4-b]pyridine derivatives [50]. The structural modification of a linifanib (I) resulted in the generation of 58 derivatives, which were screened for their inhibitory potency (IC50) against PDGFRα, VEGFR2, and FGFR1. Based on their initial optimization (Table 15), compound 122d, the most potent PDGFRα inhibitor, was selected to explore its activity against other RTKs as it showed potent inhibitory activity against c-KIT with an IC50 value of 2.1 nM (Table 16).
Table 15.
Structures and kinase inhibitory activities of compounds 116a–122b.
| ||||||
|---|---|---|---|---|---|---|
| Compd. | R1 | R2 | R3 | IC50 (nM) | ||
| PDGFRα | VEGFR2 | FGFR1 | ||||
| I |
|
|
O | 15 | 250 | 5479 |
| 116a |
|
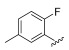
|
O | >50,000 | >50,000 | 33,849 |
| 116b |
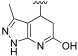
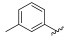
|
|
O | >50,000 | >50,000 | 18,325 |
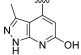
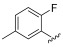
| 119a |
|
|
O | 1179 | 1856 | 4635 |
| 119b |
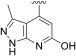
|
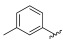
|
O | 560 | 1224 | 2264 |
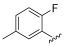
| 122c |
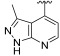
|
|
O | 87 | 4293 | >50,000 |
| 122d |
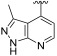
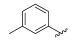
|
|
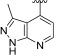
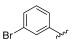
O | 40 | 1225 | >50,000 |
| 122e |
|
|
O | 137 | 3145 | >50,000 |
| 122f |
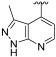
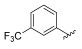
|
|
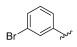
O | 106 | 1921 | >50,000 |
| 122a |
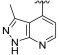
|
|
S | 1081 | 6235 | 19,999 |
| 122b |
|
|
S | 1572 | 8008 | 42,859 |
Table 16.
Kinase spectrum of 122d against RTK.
| Compd. | IC50 (nM) | |||||||
|---|---|---|---|---|---|---|---|---|
| PDGFRα | VEGFR2 | FGFR1 | c-KIT | PDGFRβ | FLT3 | VEGFR1 | VEGFR3 | |
| 122d | 40 | 1225 | >50,000 | 2.1 | 197 | 1880 | 3493 | 376 |
This led to the development of a dual inhibitor against c-KIT and PDGFRα, and VEGFR2 kinase was used as a reference to monitor selectivity. To evaluate the impact of various three-position substituents of the pyrazolo[3,4-b]pyridine scaffold, the 3-methyl group was replaced with hydroxymethyl (126a, 126b) and trifluoromethyl groups (122g, 122h). The replacement of the 3-methyl group with a hydroxymethyl group (126a vs. 122d) resulted in a two-fold activity reduction in PDGFRα (IC50 = 87 vs. 40 nM), while the c-KIT inhibition remained relatively potent (IC50 = 2.4 vs. 2.1 nM) (Table 17).
Table 17.
Inhibitory activities of compounds 126a–122i against kinases.
| |||||
|---|---|---|---|---|---|
| Compd. | R1 | R2 | IC50 (nM) | ||
| c-KIT | PDGFRα | VEGFR2 | |||
| 126a |
|
|
2.4 | 87 | 4920 |
| 126b |
|
|
3.7 | 32 | 3360 |
| 122g |
|
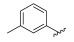
|
>50,000 | >50,000 | >50,000 |
| 122h |
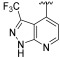
|
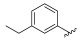
|
6768 | >50,000 | >50,000 |
| 122i |
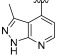
|
|
3.9 | 22 | 1208 |
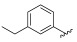
The trifluoromethyl group (122g) resulted in complete loss of activity in all kinases (IC50 > 50,000 nM). These results revealed that for the dual inhibition of c-KIT and PDGFRα, the optimal pharmacophore was the 3-methyl-pyrazolo[3,4-b]pyridine moiety. As a potential template, the urea linker and the 3-methyl-1H-pyrazolo[3,4-b]pyridine scaffold were used to identify a dual c-KIT/PDGFRα inhibitor. Next, the substitution effect (R2) on the phenyl ring linked to terminal nitrogen of urea was investigated. Shifting the methyl group from the three-position (122d) to the two- (122j) or four-position (122k) resulted in a significant activity loss against c-KIT/PDGFRα. The 3-methyl group was found to be the most effective substituent among several 3-substituents (112d–f, 122l–n, and 130a) for inhibiting c-KIT/PDGFRα. Interestingly, when an N-methyl piperazinyl group was substituted at the three-position, the inhibitory activity against c-KIT/PDGFRα was moderately reduced. Compounds 130b–d were synthesized with a combination of a 3-methyl group and either an N-methyl piperazinyl or a morpholinyl group on the terminal phenyl ring. Compound 130d showed the most potent inhibitory activity against both c-KIT and PDGFRα kinases (IC50 = 2.4 and 7.2 nM, respectively) and maintained selectivity for VEGFR2, suggesting that compound 130d was a promising candidate for further development as a dual inhibitor. Changing the combination of the N-methyl piperazinyl or the morpholinyl or methyl groups (130e–h) resulted in a significant loss of activity against all three kinases. The introduction of a diethylamino group (130i and 130j) or a dimethylamino group (130k and 130l) on the terminal phenyl ring partially restored this dual activity, but the potency was lower than that achieved with compound 130d. The replacement of the 3-dimethylamino group with a 3-dimethylaminomethyl group (130m) displayed a complete loss of activity against all three kinases. Further screening of the R2 group (127a–y) was performed to identify more potent compounds (Table 18). While increasing the length of the linear alkyl groups (127a–e) improved the c-KIT/PDGFRα inhibition activity, replacing the linear alkyl group with a branched (127f and 127g) or cycloalkyl (127h–k) group did not result in an improved inhibitory activity compared with the original compound (122d). Heteroarylmethyl groups (127l–n) exhibited better c-KIT/PDGFRα inhibitory activity than the phenylmethyl group (127o), but the inhibitory activity was lower than that which was advised with 122d. By varying the chain length of phenylalkyl groups (127o–s), the potency against c-KIT/PDGFRα increased in the following order: phenylpropyl (122q) > phenylethyl (127p) > 2,3-dihydro-indene (127s) > phenylbutyl (127r). Compound 127q had better c-KIT inhibitory activity than 122d and 130d but showed weaker PDGFRα inhibitory activity than 130d (IC50 = 24 nM vs. 7.2 nM). Heteroaryl groups (127t–v) reduced the kinase potency by over six-fold when replacing the aryl group (R2). Fused aryl rings (127w–y) improved c-KIT/PDGFRα inhibition compared with 122d but not to single-digit nanomolar concentration. The structural optimization generated several novel compounds with an improved potency against the tested kinases. Compound 130d was particularly notable for its 17-fold improvement in potency, compared with the benchmark imatinib for c-KIT and for its dual potent activity against both c-KIT (IC50 = 2.4 nM) and PDGFRα (IC50 = 7.2 nM). Other promising compounds include 127e, 127q, and 127w, which also exhibited a high dual-target potency and appropriate selectivity against VEGFR2.
Table 18.
Inhibitory activities of compounds 122j–n, 130a–m, and 127a–y against kinases.
| Compd. | R2 | IC50 (nM) | Compd. | R2 | IC50 (nM) | ||||
|---|---|---|---|---|---|---|---|---|---|
| c-KIT | PDGFRα | VEGFR2 | c-KIT | PDGFRα | VEGFR2 | ||||
| 122j |
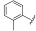
|
10,890 | 305 | 4322 | 127e |
|
1.4 | 27 | 10,462 |
| 122k |
|
1831 | 65 | 5613 | 127f |
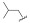
|
320 | 1123 | 8634 |
| 122l |
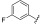
|
4794 | 262 | 6228 | 127g |
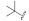
|
1416 | 1465 | 11,382 |
| 122m |
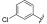
|
2402 | 182 | 4050 | 127h |
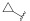
|
553 | 1133 | 4523 |
| 122n |
|
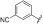
201 | 21,400 | >50,000 | 127i |
|
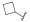
318 | 732 | 5002 |
| 130a |
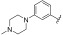
|
19 | 215 | 6235 | 127j |
|
144 | 576 | 6229 |
| 130b |
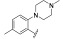
|
1949 | 1779 | 3940 | 127k |
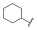
|
>50,000 | >50,000 | >50,000 |
| 130c |
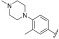
|
7.2 | 127 | 3695 | 127l |
|
51 | 387 | 3503 |
| 130d |
|
2.4 | 7.2 | 2280 | 127m |
|
26 | 157 | 1791 |
| 130e |
|
2225 | 1101 | 6816 | 127n |
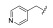
|
78 | 472 | 4079 |
| 130f |
|
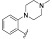
4581 | 928 | 4164 | 127o |
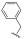
|
46,064 | >50,000 | >50,000 |
| 130g |
|
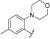
397 | 18,925 | 26,163 | 127p |
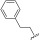
|
3.7 | 82 | 9760 |
| 130h |
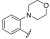
|
6438 | 4077 | 36,890 | 127q |
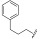
|
1.7 | 24 | 3857 |
| 130i |
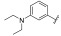
|
12 | 136 | 41,966 | 127r |
|
6.2 | 135 | 10,917 |
| 130j |
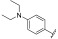
|
12 | 176 | 5352 | 127s |
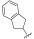
|
8.7 | 83 | >50,000 |
| 130k |
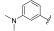
|
2.6 | 83 | 6920 | 127t |
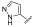
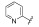
|
120 | 411 | 2391 |
| 130l |
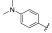
|
9.3 | 309 | 9046 | 127u |
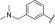
|
20 | 401 | >50,000 |
| 130m |
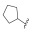
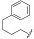
|
22,056 | 4608 | 15,935 | 127v |
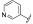
|
14 | 313 | 3109 |
| 127a |
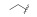
|
38,667 | >50,000 | >50,000 | 127w |
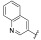
|
1.9 | 22 | 1845 |
| 127b |
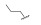
|
21,432 | >50,000 | >50,000 | 127x |
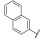
|
20 | 124 | >50,000 |
| 127c |
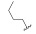
|
20,556 | 43,877 | >50,000 | 127y |
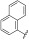
|
3.6 | 97 | >50,000 |
| 127d |
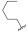
|
3.5 | 164 | >50,000 | |||||
A SAR study of this series revealed that the 3-methyl-1H-pyrazolo[3,4-b]pyridine pharmacophore was an important structural feature for the dual inhibition of c-KIT and PDGFRα. Additionally, it was observed that there was a correlation between the activity and the terminal amines. In addition, the introduction of the morpholine group para to the phenyl urea moiety improved activity by retaining selectivity over VEGFR2 (Figure 19).
Figure 19.

Design strategy, SAR summary, and pharmacophore description of compound 130d.
The antiproliferation activity of 130d showed significant improvement against the GIST-T1 (GI50 < 0.003 μM) and GIST-882 cell lines (GI50 < 0.020 μM).The pharmacokinetics of compound 130d in a rat model displayed a short half-life (t1/2) of 0.69 h, a small volume distribution (Vss) of 0.97 L/kg, and a moderate plasma clearance (Cl) of 0.95 L/h/kg at a dosage of 2 mg/kg via intravenous administration, while intraperitoneal injection at a dosage of 100 mg/kg improved the plasma exposure and half-life (T1/2 = 10.97 h). In contrast, the lack of absorption at the 10 mg/kg dosage during oral administration prevented 130d from being utilized for oral administration in the animal model. The in vivo efficacy of 130d at a dosage of 100 mg/kg in a BaF3-TEL-c-KIT-T670I cell-inoculated xenograft mouse model displayed 41.9% tumor growth inhibition and a good safety profile.
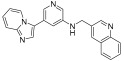
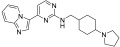
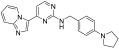
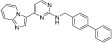
Andreas et al. designed and synthesized 3-(pyrimidin-4-yl)imidazo[1,2-a]pyridine derivatives as selective c-KIT inhibitors for the treatment of GISTs [51]. A high-throughput screening of derivatives supplied by the European Lead Factory resulted in the identification of an imidazopyridine derivative as a hit against KIT (V654A). The hit compound displayed high selectivity against KIT autophosphorylation at Y703 in a GIST430 cancer cell line over 28 kinases. Except for its microsomal stability, the hit compound had good solubility and permeability. The pyrrolidine and benzylic positions were the reason for the high metabolism. At first, hit-to-lead optimization was performed, and the SAR exploration was mainly concentrated on improving cellular potency and metabolic stability. The introduction of 4,6-substituted pyrimidine (III) in place of a three-position substitution on the pyridine ring improved the cellular potency, and it was identified as an ideal replacement. Linker region modification (II and IV) resulted in a three-fold decreased potency. When the pyrrolidine substituted pyridine moiety was replaced with a fused ring system and an aliphatic moiety (V and VI), there was a complete loss of potency, but the phenyl ring (VII) showed a slight increase in cellular potency (Table 19).
Table 19.
SAR overview for 3-position imidazopyridine modifications.
| Compd. | Structure | IC50 [nm] | Cell IC50 [nm] | Clint HLM/MLM |
Solubility [µM] |
|---|---|---|---|---|---|
| I |
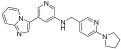
|
12 | 20 | h/h | 101 |
| II |
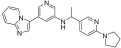
|
83 | 2600 | h/h | 124 |
| III |
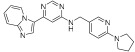
|
5.4 | 429 | m/h | <3 |
| IV |
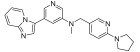
|
240 | - | - | 8 |
| V |
|
>10,000 | - | m/m | - |
| VI |
|
>10,000 | - | m/m | - |
| VII |
|
5.6 | 680 | - | 7 |
| VIII |
|
15 | 340 | m/m | <3 |
Modification of the pyrrolidine moiety with a phenyl ring (VIII) led to a further increase in both cellular potency and metabolic stability. Substitution at the six-position of imidazopyridine with electronic-donating groups for ether (IX) resulted in increased cellular potency. Increasing the length of the ether linker along with methyl ether substitution in the tail part further increased the cellular potency (X). Basic substitutions at the tail part of the extended ether linker increased the water solubility by maintaining the potency (XI). Bipheylmethanamine in the benzylic part, in combination with 6-methoxy substitution and 4,6-pyrimidine in the central core, resulted in the identification of compound (XII), which had an improved cellular potency. When the terminal part of the phenyl ring was replaced with a five-membered heterocycle (XIII), the metabolic stability was retained, and the cellular potency was further improved. The extended ether linker (135a) maintained metabolic stability and cellular potency (Table 20). Based on the SAR results, 135a was identified as a lead compound.
Table 20.
SAR overview for six-position imidazopyridine modifications.
| Compd. | Structure | IC50 [nm] | Cell IC50 [nm] | Clint HLM/MLM |
Solubility [µM] |
|---|---|---|---|---|---|
| IX |
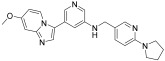
|
13 | 300 | h/h | 81 |
| X |
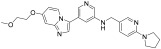
|
12 | 110 | h/h | 9 |
| XI |
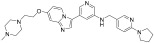
|
11 | 290 | m/h | 100 |
| XII |
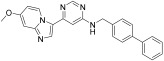
|
4.6 | 160 | m/L | <2 |
| XIII |
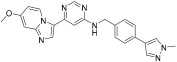
|
8.6 | 55 | m/L | <2 |
| 135a |
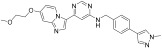
|
7.3 | 48 | m/h | <2 |
The lead optimization of 135a was focused on improving the solubility, permeability, and hERG inhibition. Terminal pyrazole ring replacement with additional five-membered heterocycles, such as triazole (136a and 136b) and oxazole (137), led to a retained cellular potency. The introduction of polar groups at the tail part of the extended ether linker (135b and 135c) reduced the permeability and increased the efflux; however, for 135d and 135e, the hERG activity was improved. All the derivatives displayed superior cell potency and kinase selectivity (Figure 20) (Table 21).
Figure 20.

Design strategy, SAR summary, and pharmacophore description of compound 135j.
Table 21.
SAR overview of advanced c-KIT inhibitors.
| Compd. | Cell IC50 (nM) |
hHEP [µL/min/106 cells] | CACO-2 [106 cm/s] (Efflux) |
FaSSiF [µg/mL] |
hERG [%inh] at 10 µM |
LogD | pKa |
|---|---|---|---|---|---|---|---|
| 135a | 48 | 18 | 12 (0.9) | 2 | 1% | 3.8 | - |
| 136a | 78 | 8 | 9.5 (0.7) | 4 | - | 4.0 | - |
| 136b | 46 | 4 | 6.4 (0.8) | - | - | 3.0 | - |
| 135b | 93 | 16 | 3.1 (4.7) | 16 | −15% | 4.4 | 6.9 |
| 135c | 61 | <4 | 5.7 (57) | 97 | −59% | 1.9 | 9.5 |
| 135d | 43 | - | - | - | −86% | 2.5 | nd |
| 135e | 70 | 18 | 12 (5.4) | - | −78% | 2.9 | 9.1 |
| 135f | 75 | 9 | 21 (5.0) | 13 | −23% | 3.0 | - |
| 135g | 76 | <4 | 7.6 (18) | 10 | −8% | 2.8 | 5.8 |
| 135h | 56 | 10 | 9.5 (15) | 16 | −19% | 1.5 | 7.3 |
| 135i | 73 | 10 | 1.8 (1.3) | 2 | −4% | 4.3 | 8.3 |
| 137 | 54 | 6 | 7.6 (7.5) | 10 | −7% | 3.2 | 6.3 |
| 135j | 59 | 21 | 5.7 (14) | 374 | −86% | 2.6 | 9.7 |
The HTS screening of the European Lead Factory derivatives resulted in the identification of an imidazopyridine derivative as a hit against KIT. A SAR study of this series revealed that the 4,6-pyrimide moiety was an important structural feature for cellular potency. In addition, the introduction of an ethylene or propylene ether linker at the six-position of the imidazopyridine core and the replacement of the pyrrolidine ring on the terminal phenyl ring with a N-methyl pyrazole improved the cellular potency. Furthermore, it was observed that the pyrrolidin-1-yl propylene ether linker resulted in improved solubility and strong hERG inhibition (Figure 20).
Across all preclinical species, the pharmacokinetics of 135j demonstrated great metabolic stability, a good half-life, and a high volume of distribution. Compound 135j displayed excellent cellular potency (IC50 = 4 ± 1 nM) against the imatinib-sensitive GIST430 cell line. In addition, it exhibited good cellular potency in the imatinib-resistant cell line GIST430/654 and the AML cell line Kasumi-1 (IC50 = 48 ± 21 and 4 ± 1 nM, respectively). The in vivo results on GIST430/654 xenografts indicated that 132j strongly inhibited tumor growth. The compound was further evaluated in in vivo studies in dogs and guinea pigs, which revealed its long half-life and lack of accumulation. The compound was nominated for a phase 1 clinical study in humans with metastatic and surgically unresectable GISTs.
The molecular docking study of 135j revealed that it could adopt a DFG-out conformation of c-KIT kinase with typical type II binding. The nitrogen of the imidazopyridine established a hydrogen bond with Cys673 in the kinase hinge region. The side chain of the ether linker was oriented toward the solvent exposure region. The benzylic NH formed an interaction with the Thr670 side chain, and the benzylic substituent was deeply embedded via π-interactions with Trp557. One of the pyrimidine nitrogen atoms was seen to engage with the Asp810 backbone carbonyl and the terminal amino group of the Lys623 side chain via the water-mediated process (Figure 21).
Figure 21.

X-ray structure of 135j (2.1 A resolution, PDB-ID: 7ZW8) in complex with the kinase domain of c-KIT. Reprinted with permission from Ref. [51].
Nam et al. designed and reported the synthesis of thiazolo[5,4-b]pyridine-based derivatives targeting antiproliferative activities [52].The 31 synthesized compounds were evaluated for their enzymatic inhibitory activity against c-KIT (Table 22). Imatinib and sunitinib were used as standard references with IC50 values of 0.27 µM and 0.14 µM, respectively. SAR analysis showed that among the compounds 142a–j, the 3-(trifluoromethyl)phenyl group (142h) only exhibited moderate enzymatic inhibitory activity (IC50 = 9.87 µM). Extension of the amide and introduction of a urea linkage led to a loss of activity. The addition of polar moieties in the para position to the amide group on the 3-(trifluoromethyl)phenyl group resulted in a two- to six-fold improved inhibitory activity, whereas addition of the meta position led to decreased inhibitory activity. By further exploring para substitution with various moieties, the (4-methylpiperazin-1-yl)methyl analog 142r was found to be the most potent derivative (IC50 = 0.14 µM). Modification of the amino group of 142r with cyclohexyl (143a) and phenyl amide (143b) led to decreased inhibitory activity compared with 142r (IC50 = 0.14 µM vs. 1.51 µM and 0.74 µM, respectively). The acetamide analog (143c) displayed little improvement in activity with an IC50 value of 0.10 µM. Additional acetamide derivatives (143f–h) with various para substitutions on the 3-trifluoromethyl phenyl amide moiety were more potent than their precursor compounds (142p, 142v, and 142w) (Figure 22).
Table 22.
c-KIT enzymatic inhibitory activities of thiazolo[5,4-b]pyridine derivatives.
| Compd. | R1 | R2 | IC50 (µM) a | Entry | R1 | R2 | IC50 (µM) a |
|---|---|---|---|---|---|---|---|
| imatinib | - | - | 0.27 | 142p |
|
- | 5.72 |
| sunitinib | - | - | 0.14 | 142q |
|
- | 3.23 |
| 142a |
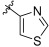
|
- | inactive b | 142r |
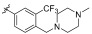
|
- | 0.14 |
| 142b |
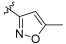
|
- | inactive b | 142s |
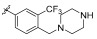
|
- | 0.37 |
| 142c |
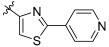
|
- | inactive b | 142t |
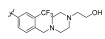
|
- | 1.25 |
| 142d |
|
- | inactive b | 142u |
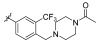
|
- | 4.56 |
| 142e |
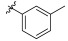
|
- | inactive b | 142v |
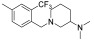
|
- | 0.39 |
| 142f |
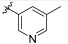
|
- | inactive b | 142w |
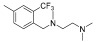
|
- | 0.25 |
| 142g |
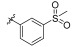
|
- | inactive b | 143a |
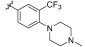
|
cyclohexyl | 1.51 |
| 142h |
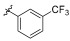
|
- | 9.87 | 143b |
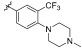
|
phenyl | 0.74 |
| 142i |
|
- | inactive b | 143c |
|
methyl | 0.1 |
| 142j |
|
- | inactive b | 143d |
|
cyclohexyl | inactive b |
| 142k |
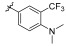
|
- | 4.31 | 143e |
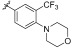
|
methyl | 0.88 |
| 142l |
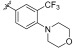
|
- | 1.76 | 143f |
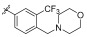
|
methyl | 3.63 |
| 142m |
|
- | 2.17 | 143g |
|
methyl | 0.18 |
| 142n |
|
- | inactive b | 143h |
|
methyl | 0.10 |
| 142o |
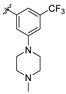
|
- | 5.03 |
a Radiometric biochemical kinase assay results. b less than 50% inhibition at a concentration of 10 µM.
Figure 22.

SAR summary and chemical structures of compounds 142r and 143c.
A SAR study of this series revealed that the thiazolo[5,4-b]pyridine core was an important structural feature for enzymatic and anti-proliferative activities. Additionally, the introduction of polar groups to the para-position of phenyl amide improved activity. Similarly, the substitution of an acetyl group on thiazolo[5,4-b]pyridine amine further improved the activity (Figure 22).
The compounds were further screened for their antiproliferative activity against c-KIT-dependent GIST-T1 and HMC1.2 cancer cell lines (Table 23). The results showed that compounds 142r (GI50 = 0.01 ± 0.00 µM), 142s (GI50 = 0.02 ± 0.00 µM), and 143c (GI50 = 0.01 ± 0.00 µM) exhibited similar antiproliferative activity against GIST-T1 cells compared with sunitinib (GI50 = 0.01 ± 0.00), but 142s (GI50 = 1.33 ± 0.43 µM), and 143c (GI50 = 1.52 ± 0.43 µM) displayed a potent antiproliferative activity against HMC1.2 cells (GI50 = 1.15 ± 0.96 µM) compared with imatinib (GI50 = 27.10 ± 3.36 µM). The enzymatic inhibitory activities revealed that the active compounds showed a five- to eight-fold improved activity against c-KIT V560G/D816V double mutant compared with imatinib (IC50 = 37.93 µM). On the other hand, these compounds displayed similar activity to sunitinib (IC50 = 3.98 µM). In addition, they exhibited similar activity against c-KIT D816V Ba/F3 cells, while having a higher cytotoxicity window for c-KIT D816V Ba/F3 cells compared with parental Ba/F3 cells than sunitinib.
Table 23.
Antiproliferative activities of thiazolo[5,4-b]pyridine derivatives.
| Compd. | GI50 (µM) a | Entry | GI50 (µM) a | ||
|---|---|---|---|---|---|
| GIST-T1 | HMC1.2 | GIST-T1 | HMC1.2 | ||
| imatinib | 0.02 ± 0.01 | 27.10 ± 3.36 | 142p | 0.66 ± 0.10 | 8.55 ± 2.40 |
| sunitinib | 0.01 ± 0.00 | 2.53 ± 0.35 | 142q | 0.42 ± 0.07 | 15.53 ± 3.97 |
| 142a | >50 | Inactive b | 142r | 0.01 ± 0.00 | 1.15 ± 0.96 |
| 142b | 33.05 ± 7.09 | Inactive b | 142s | 0.02 ± 0.00 | 1.33 ± 0.43 |
| 142c | 33.69 ± 3.45 | Inactive b | 142t | 0.12 ± 0.03 | 6.62 ± 0.75 |
| 142d | 16.36 ± 2.73 | 25.18 ± 2.21 | 142u | 0.66 ± 0.08 | 11.31 ± 0.34 |
| 142e | 3.45 ± 0.41 | 11.53 ± 0.62 | 142v | 0.05 ± 0.01 | 6.81 ± 0.67 |
| 142f | 20.94 ± 2.00 | Inactive b | 142w | 0.02 ± 0.01 | 4.99 ± 0.43 |
| 142g | 10.84 ± 1.06 | 36.77 ± 10.9 | 143a | 0.08 ± 0.01 | 4.58 ± 0.32 |
| 142h | 0.96 ± 0.10 | 7.26 ± 4.13 | 143b | 0.10 ± 0.01 | 5.22 ± 0.59 |
| 142i | 7.16 ± 0.87 | 9.65 ± 1.06 | 143c | 0.01 ± 0.00 | 1.52 ± 0.43 |
| 142j | 3.31 ± 0.73 | 6.35 ± 0.32 | 143d | 8.13 ±1.81 | Inactive b |
| 142k | 0.18 ± 0.03 | 2.27 ± 0.84 | 143e | 0.23 ± 0.07 | 14.04 ± 10.2 |
| 142l | 0.08 ± 0.01 | 4.34 ± 1.31 | 143f | 0.30 ± 0.06 | 28.39 ± 13.3 |
| 142m | 0.27 ± 0.17 | 3.41 ± 1.49 | 143g | 0.07 ± 0.01 | 4.88 ± 1.18 |
| 142n | 0.55 ± 0.09 | 7.00 ± 2.68 | 143h | 0.02 ± 0.00 | 4.98 ± 0.32 |
| 142o | 0.28 ± 0.08 | 2.36 ± 0.58 | |||
a Radiometric biochemical kinase assay results. b less than 50% inhibition at a concentration of 10 µM.
A molecular docking study was performed to elucidate the binding mode of the potent compounds 142r and 143c. Both compounds established hydrogen bonding at the hinge region with a Cys673 backbone, as well as with a Glu640/Asp810 and an Ile789/His790 backbone. The thiazolo[5,4-b]pyridine fragment was involved in hydrophobic interactions with Leu799, Val603, Ala621, and Val654, and the hydrophobic pocket was occupied by the 3-trifluoromethyl group (Figure 23).
Figure 23.

(A,B) Docking model of compound of 142r and 143c on c-KIT (PDB code: 1T46). Reprinted from Ref. [52].
3. Conclusions
In conclusion, c-KIT has emerged as a promising target in drug development for the treatment of GISTs, and the emergence of resistance to imatinib has highlighted the need for more potent and selective inhibitors. Various sub-structural modifications have been explored to discover a potent inhibitor, and in this review, we focused on the synthesis and the SAR optimization of various scaffolds for c-KIT inhibitors for GISTs. Several compounds have demonstrated potent c-KIT inhibitory activity and antitumor efficacy, including compounds 6e, 22aa, 35h, 57d, 71g, 75, and 130d. Compounds 81a and 105a have also shown promise as next-generation therapeutic candidates for GISTs. In addition, compound 135j (IDRX-42) has been designated as a clinical candidate and is undergoing testing in a phase 1 trial. The results of these studies provide insight for medicinal chemists to design new scaffolds targeting c-KIT-driven GISTs.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms24119450/s1. References [34,35,36,37,38,39] are cited in the Supplementary Materials.
Author Contributions
Conceptualization, S.G. and K.L.; literature review, S.G. and H.N.; draft preparation, S.G.; writing—review and editing, S.G., J.L., H.N., A.E., G.Q., Y.C. and K.L.; supervision, K.L.; project administration K.L.; funding acquisition, K.L. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and Supplementary Material.
Conflicts of Interest
The authors have declared no conflict of interest.
Funding Statement
This study was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) [No. 2018R1A5A2023127] & [No. 2023R1A2C3004599]. This work is also supported by the BK21 FOUR program, which was funded by the Ministry of Education of Korea through NRF.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Candelaria M., de la Garza J., Duenas-Gonzalez A. A clinical and biological overview of gastrointestinal stromal tumors. Med. Oncol. 2005;22:1–10. doi: 10.1385/MO:22:1:001. [DOI] [PubMed] [Google Scholar]
- 2.Schaefer I.M., Mariño-Enríquez A., Fletcher J.A. What is new in gastrointestinal stromal tumor? Adv. Anat. Pathol. 2017;24:259–267. doi: 10.1097/PAP.0000000000000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Søreide K., Sandvik O.M., Søreide J.A., Giljaca V., Jureckova A., Bulusu V.R. Global epidemiology of gastrointestinal stromal tumours (GIST): A systematic review of population-based cohort studies. Cancer Epidemiol. 2016;40:39–46. doi: 10.1016/j.canep.2015.10.031. [DOI] [PubMed] [Google Scholar]
- 4.Bardales R.H., Mallery S. EUS and EUS-FNA of intramural masses of the esophagus, stomach, and proximal intestinal tract. In: Bardales R.H., editor. Cytology of the Mediastinum and Gut Via Endoscopic Ultrasound-Guided Aspiration. Springer International Publishing; Cham, Switzerland: 2015. pp. 53–110. [Google Scholar]
- 5.Schaefer I.-M., DeMatteo R.P., Serrano C. The GIST of advances in treatment of advanced gastrointestinal stromal tumor. Am. Soc. Clin. Oncol. Educ. Book. 2022;42:885–899. doi: 10.1200/EDBK_351231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joensuu H., Hohenberger P., Corless C.L. Gastrointestinal stromal tumour. Lancet. 2013;382:973–983. doi: 10.1016/S0140-6736(13)60106-3. [DOI] [PubMed] [Google Scholar]
- 7.Miettinen M., Lasota J. KIT (CD117): A review on expression in normal and neoplastic tissues, and mutations and their clinicopathologic correlation. Appl. Immunohistochem. Mol. Morphol. 2005;13:205–220. doi: 10.1097/01.pai.0000173054.83414.22. [DOI] [PubMed] [Google Scholar]
- 8.Abbaspour Babaei M., Kamalidehghan B., Saleem M., Huri H.Z., Ahmadipour F. Receptor tyrosine kinase (c-Kit) inhibitors: A potential therapeutic target in cancer cells. Drug Des. Dev. Ther. 2016;10:2443–2459. doi: 10.2147/DDDT.S89114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pixley F.J., Stanley E.R. Chapter 319—Cytokines and cytokine receptors regulating cell survival, proliferation, and differentiation in hematopoiesis. In: Bradshaw R.A., Dennis E.A., editors. Handbook of Cell Signaling. 2nd ed. Academic Press; San Diego, CA, USA: 2010. pp. 2733–2742. [Google Scholar]
- 10.Tilayov T., Hingaly T., Greenshpan Y., Cohen S., Akabayov B., Gazit R., Papo N. Engineering stem cell factor ligands with different c-Kit agonistic potencies. Molecules. 2020;25:4850. doi: 10.3390/molecules25204850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lennartsson J., Rönnstrand L. Stem cell factor receptor/c-Kit: From basic science to clinical implications. Physiol. Rev. 2012;92:1619–1649. doi: 10.1152/physrev.00046.2011. [DOI] [PubMed] [Google Scholar]
- 12.Heinrich M.C., Rubin B.P., Longley B.J., Fletcher J.A. Biology and genetic aspects of gastrointestinal stromal tumors: KIT activation and cytogenetic alterations. Hum. Pathol. 2002;33:484–495. doi: 10.1053/hupa.2002.124124. [DOI] [PubMed] [Google Scholar]
- 13.Lasota J., Jasinski M., Sarlomo-Rikala M., Miettinen M. Mutations in exon 11 of c-Kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am. J. Pathol. 1999;154:53–60. doi: 10.1016/S0002-9440(10)65250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheikh E., Tran T., Vranic S., Levy A., Bonfil R.D. Role and significance of c-KIT receptor tyrosine kinase in cancer: A review. Biomol. Biomed. 2022;22:683–698. doi: 10.17305/bjbms.2021.7399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding H., Yu X., Yu Y., Lao X., Hang C., Gao K., Jia Y., Yan Z. Clinical significance of the molecular heterogeneity of gastrointestinal stromal tumors and related research: A systematic review. Oncol. Rep. 2020;43:751–764. doi: 10.3892/or.2020.7470. [DOI] [PubMed] [Google Scholar]
- 16.Oshimori N., Guo Y., Taniguchi S. An emerging role for cellular crosstalk in the cancer stem cell niche. J. Pathol. 2021;254:384–394. doi: 10.1002/path.5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ajani J.A., Song S., Hochster H.S., Steinberg I.B. Cancer stem cells: The promise and the potential. Semin. Oncol. 2015;42:S3–S17. doi: 10.1053/j.seminoncol.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 18.Nada H., Elkamhawy A., Lee K. Structure activity relationship of key heterocyclic anti-angiogenic leads of promising potential in the fight against cancer. Molecules. 2021;26:553. doi: 10.3390/molecules26030553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao J., Qian F., Tchabo N., Mhawech-Fauceglia P., Beck A., Qian Z., Wang X., Huss W.J., Lele S.B., Morrison C.D., et al. Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through Hypoxia-resistant metabolism. PLoS ONE. 2014;9:e84941. doi: 10.1371/journal.pone.0084941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warrier S., Bhuvanalakshmi G., Arfuso F., Rajan G., Millward M., Dharmarajan A. Cancer stem-like cells from head and neck cancers are chemosensitized by the Wnt antagonist, sFRP4, by inducing apoptosis, decreasing stemness, drug resistance and epithelial to mesenchymal transition. Cancer Gene Ther. 2014;21:381–388. doi: 10.1038/cgt.2014.42. [DOI] [PubMed] [Google Scholar]
- 21.Vermeulen L., de Sousa e Melo F., Richel D.J., Medema J.P. The developing cancer stem-cell model: Clinical challenges and opportunities. Lancet Oncol. 2012;13:e83–e89. doi: 10.1016/S1470-2045(11)70257-1. [DOI] [PubMed] [Google Scholar]
- 22.Zhao Y., Zhang W., Guo Z., Ma F., Wu Y., Bai Y., Gong W., Chen Y., Cheng T., Zhi F., et al. Inhibition of the transcription factor Sp1 suppresses colon cancer stem cell growth and induces apoptosis in vitro and in nude mouse xenografts. Oncol. Rep. 2013;30:1782–1792. doi: 10.3892/or.2013.2627. [DOI] [PubMed] [Google Scholar]
- 23.Tomizawa F., Jang M.-K., Mashima T., Seimiya H. c-KIT regulates stability of cancer stemness in CD44-positive colorectal cancer cells. Biochem. Biophys. Res. Commun. 2020;527:1014–1020. doi: 10.1016/j.bbrc.2020.05.024. [DOI] [PubMed] [Google Scholar]
- 24.Nada H., Kim S., Godesi S., Lee J., Lee K. Discovery and optimization of natural-based nanomolar c-Kit inhibitors via in silico and in vitro studies. J. Biomol. Struct. Dyn. 2023:1–12. doi: 10.1080/07391102.2022.2164061. [DOI] [PubMed] [Google Scholar]
- 25.Daub H., Specht K., Ullrich A. Strategies to overcome resistance to targeted protein kinase inhibitors. Nat. Rev. Drug Discov. 2004;3:1001–1010. doi: 10.1038/nrd1579. [DOI] [PubMed] [Google Scholar]
- 26.Oppelt P.J., Hirbe A.C., Van Tine B.A. Gastrointestinal stromal tumors (GISTs): Point mutations matter in management, a review. J. Gastrointest. Oncol. 2017;8:466–473. doi: 10.21037/jgo.2016.09.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oxnard G.R., Arcila M.E., Chmielecki J., Ladanyi M., Miller V.A., Pao W. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin. Cancer Res. 2011;17:5530–5537. doi: 10.1158/1078-0432.CCR-10-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rutkowski P., Nowecki Z., Nyckowski P., Dziewirski W., Grzesiakowska U., Nasierowska-Guttmejer A., Krawczyk M., Ruka W. Surgical treatment of patients with initially inoperable and/or metastatic gastrointestinal stromal tumors (GIST) during therapy with imatinib mesylate. J. Surg. Oncol. 2006;93:304–311. doi: 10.1002/jso.20466. [DOI] [PubMed] [Google Scholar]
- 29.Debiec-Rychter M., Cools J., Dumez H., Sciot R., Stul M., Mentens N., Vranckx H., Wasag B., Prenen H., Roesel J., et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128:270–279. doi: 10.1053/j.gastro.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 30.Demetri G.D., van Oosterom A.T., Garrett C.R., Blackstein M.E., Shah M.H., Verweij J., McArthur G., Judson I.R., Heinrich M.C., Morgan J.A., et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet. 2006;368:1329–1338. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 31.Demetri G.D., Reichardt P., Kang Y.K., Blay J.Y., Rutkowski P., Gelderblom H., Hohenberger P., Leahy M., von Mehren M., Joensuu H., et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295–302. doi: 10.1016/S0140-6736(12)61857-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blay J.Y., Serrano C., Heinrich M.C., Zalcberg J., Bauer S., Gelderblom H., Schöffski P., Jones R.L., Attia S., D’Amato G., et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21:923–934. doi: 10.1016/S1470-2045(20)30168-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farag S., Smith M.J., Fotiadis N., Constantinidou A., Jones R.L. Revolutions in treatment options in gastrointestinal stromal tumours (GISTs): The latest updates. Curr. Treat. Options Oncol. 2020;21:55. doi: 10.1007/s11864-020-00754-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jung F.H., Morgentin R.R., Ple P. Quinoline Derivatives. WO/2007/099323. 2007 September 7;
- 35.Jung F.H., Ple P. Quinazoline Derivatives. WO/2007/099317. 2007 September 7;
- 36.Chen C.-T., Hsu J.T.A., Lin W.-H., Lu C.-T., Yen S.-C., Hsu T., Huang Y.-L., Song J.-S., Chen C.-H., Chou L.-H., et al. Identification of a potent 5-phenyl-thiazol-2-ylamine-based inhibitor of FLT3 with activity against drug resistance-conferring point mutations. Eur. J. Med. Chem. 2015;100:151–161. doi: 10.1016/j.ejmech.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 37.Lee B.W., Lee S.D. [5,5]Sigmatropic shift of N-phenyl-N′-(2-thiazolyl)hydrazines and N,N′-bis(2-thiazolyl)hydrazines into 2-amino-5-(p-aminophenyl)thiazoles and 5,5′-bis(2-aminothiazole) derivatives. Tetrahedron Lett. 2000;41:3883–3886. doi: 10.1016/S0040-4039(00)00493-7. [DOI] [Google Scholar]
- 38.Décor A., Grand-Maître C., Hucke O., O’Meara J., Kuhn C., Forget L.C., Brochu C., Malenfant E., Bertrand-Laperle M., Bordeleau J., et al. Design, synthesis and biological evaluation of novel aminothiazoles as antiviral compounds acting against human rhinovirus. Bioorg. Med. Chem. Lett. 2013;23:3841–3847. doi: 10.1016/j.bmcl.2013.04.077. [DOI] [PubMed] [Google Scholar]
- 39.Cáceres-Castillo D., Carballo R.M., Tzec-Interián J.A., Mena-Rejón G.J. Solvent-free synthesis of 2-amino-4-arylthiazoles under microwave irradiation. Tetrahedron Lett. 2012;53:3934–3936. doi: 10.1016/j.tetlet.2012.05.093. [DOI] [Google Scholar]
- 40.Wang Q., Liu F., Wang B., Zou F., Chen C., Liu X., Wang A., Qi S., Wang W., Qi Z., et al. Discovery of N-(3-((1-Isonicotinoylpiperidin-4-yl)oxy)-4-methylphenyl)-3-(trifluoromethyl)benzamide (CHMFL-KIT-110) as a selective, potent, and orally available type II c-KIT kinase inhibitor for gastrointestinal stromal tumors (GISTs) J. Med. Chem. 2016;59:3964–3979. doi: 10.1021/acs.jmedchem.6b00200. [DOI] [PubMed] [Google Scholar]
- 41.Li B., Wang A., Liu J., Qi Z., Liu X., Yu K., Wu H., Chen C., Hu C., Wang W., et al. Discovery of N-((1-(4-(3-(3-((6,7-Dimethoxyquinolin-3-yl)oxy)phenyl)ureido)-2-(trifluoromethyl)phenyl)piperidin-4-yl)methyl)propionamide (CHMFL-KIT-8140) as a highly potent type II inhibitor capable of inhibiting the T670I “Gatekeeper” mutant of cKIT kinase. J. Med. Chem. 2016;59:8456–8472. doi: 10.1021/acs.jmedchem.6b00902. [DOI] [PubMed] [Google Scholar]
- 42.Wu Y., Wang B., Wang J., Qi S., Zou F., Qi Z., Liu F., Liu Q., Chen C., Hu C., et al. Discovery of 2-(4-Chloro-3-(trifluoromethyl)phenyl)-N-(4-((6,7-dimethoxyquinolin-4-yl)oxy)phenyl)acetamide (CHMFL-KIT-64) as a novel orally available potent inhibitor against broad-spectrum mutants of c-KIT kinase for gastrointestinal stromal tumors. J. Med. Chem. 2019;62:6083–6101. doi: 10.1021/acs.jmedchem.9b00280. [DOI] [PubMed] [Google Scholar]
- 43.Liu X., Wang B., Chen C., Qi Z., Zou F., Wang J., Hu C., Wang A., Ge J., Liu Q., et al. Discovery of (E)-N1-(3-Fluorophenyl)-N3-(3-(2-(pyridin-2-yl)vinyl)-1H-indazol-6-yl)malonamide (CHMFL-KIT-033) as a novel c-KIT T670I mutant selective kinase inhibitor for gastrointestinal stromal tumors (GISTs) J. Med. Chem. 2019;62:5006–5024. doi: 10.1021/acs.jmedchem.9b00176. [DOI] [PubMed] [Google Scholar]
- 44.Kaitsiotou H., Keul M., Hardick J., Mühlenberg T., Ketzer J., Ehrt C., Krüll J., Medda F., Koch O., Giordanetto F., et al. Inhibitors to overcome secondary mutations in the stem cell factor receptor KIT. J. Med. Chem. 2017;60:8801–8815. doi: 10.1021/acs.jmedchem.7b00841. [DOI] [PubMed] [Google Scholar]
- 45.Kettle J.G., Anjum R., Barry E., Bhavsar D., Brown C., Boyd S., Campbell A., Goldberg K., Grondine M., Guichard S., et al. Discovery of N-(4-{[5-Fluoro-7-(2-methoxyethoxy)quinazolin-4-yl]amino}phenyl)-2-[4-(propan-2-yl)-1H-1,2,3-triazol-1-yl]acetamide (AZD3229), a potent pan-KIT mutant inhibitor for the treatment of gastrointestinal stromal tumors. J. Med. Chem. 2018;61:8797–8810. doi: 10.1021/acs.jmedchem.8b00938. [DOI] [PubMed] [Google Scholar]
- 46.Plé P.A., Jung F., Ashton S., Hennequin L., Laine R., Morgentin R., Pasquet G., Taylor S. Discovery of AZD2932, a new Quinazoline Ether Inhibitor with high affinity for VEGFR-2 and PDGFR tyrosine kinases. Bioorg. Med. Chem. Lett. 2012;22:262–266. doi: 10.1016/j.bmcl.2011.11.019. [DOI] [PubMed] [Google Scholar]
- 47.Plé P.A., Jung F., Ashton S., Hennequin L., Laine R., Lambert-van der Brempt C., Morgentin R., Pasquet G., Taylor S. Discovery of new quinoline ether inhibitors with high affinity and selectivity for PDGFR tyrosine kinases. Bioorg. Med. Chem. Lett. 2012;22:3050–3055. doi: 10.1016/j.bmcl.2012.03.074. [DOI] [PubMed] [Google Scholar]
- 48.Wu T.-S., Lin W.-H., Tsai H.-J., Hsueh C.-C., Hsu T., Wang P.-C., Lin H.-Y., Peng Y.-H., Lu C.-T., Lee L.-C., et al. Discovery of conformational control inhibitors switching off the activated c-KIT and targeting a broad range of clinically relevant c-KIT mutants. J. Med. Chem. 2019;62:3940–3957. doi: 10.1021/acs.jmedchem.8b01845. [DOI] [PubMed] [Google Scholar]
- 49.Lin W.-H., Wu S.-Y., Yeh T.-K., Chen C.-T., Song J.-S., Shiao H.-Y., Kuo C.-C., Hsu T., Lu C.-T., Wang P.-C., et al. Identification of a multitargeted tyrosine kinase inhibitor for the treatment of gastrointestinal stromal tumors and acute myeloid leukemia. J. Med. Chem. 2019;62:11135–11150. doi: 10.1021/acs.jmedchem.9b01229. [DOI] [PubMed] [Google Scholar]
- 50.Lu Y., Mao F., Li X., Zheng X., Wang M., Xu Q., Zhu J., Li J. Discovery of potent, selective stem cell factor receptor/platelet derived growth factor receptor alpha (c-KIT/PDGFRα) dual inhibitor for the treatment of imatinib-resistant gastrointestinal stromal tumors (GISTs) J. Med. Chem. 2017;60:5099–5119. doi: 10.1021/acs.jmedchem.7b00468. [DOI] [PubMed] [Google Scholar]
- 51.Blum A., Dorsch D., Linde N., Brandstetter S., Buchstaller H.-P., Busch M., Glaser N., Grädler U., Ruff A., Petersson C., et al. Identification of M4205─A highly selective inhibitor of KIT mutations for treatment of unresectable metastatic or recurrent gastrointestinal stromal tumors. J. Med. Chem. 2023;66:2386–2395. doi: 10.1021/acs.jmedchem.2c00851. [DOI] [PubMed] [Google Scholar]
- 52.Nam Y., Kim C., Han J., Ryu S., Cho H., Song C., Kim N.D., Kim N., Sim T. Identification of thiazolo[5,4-b]pyridine derivatives as c-KIT inhibitors for overcoming imatinib resistance. Cancers. 2023;15:143. doi: 10.3390/cancers15010143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is contained within the article and Supplementary Material.