

Abstract
1,3-butadiynamides—the ethynylogous variants of ynamides—receive considerable attention as precursors of complex molecular scaffolds for organic and heterocyclic chemistry. The synthetic potential of these C4-building blocks reveals itself in sophisticated transition-metal catalyzed annulation reactions and in metal-free or silver-mediated HDDA (Hexa-dehydro-Diels–Alder) cycloadditions. 1,3-Butadiynamides also gain significance as optoelectronic materials and in less explored views on their unique helical twisted frontier molecular orbitals (Hel-FMOs). The present account summarizes different methodologies for the synthesis of 1,3-butadiynamides followed by the description of their molecular structure and electronic properties. Finally, the surprisingly rich chemistry of 1,3-butadiynamides as versatile C4-building blocks in heterocyclic chemistry is reviewed by compiling their exciting reactivity, specificity and opportunities for organic synthesis. Besides chemical transformations and use in synthesis, a focus is set on the mechanistic understanding of the chemistry of 1,3-butadiynamides—suggesting that 1,3-butadiynamides are not just simple alkynes. These ethynylogous variants of ynamides have their own molecular character and chemical reactivity and reflect a new class of remarkably useful compounds.
Keywords: 1,3-diynamides; ynamides; alkynes; homogeneous catalysis; heterocycles; annulation reactions; cycloaddition cascade reactions; helical twisted frontier molecular orbitals (Hel-FMO); Hexa-dehydro-Diels–Alder (HDDA) reaction; quinolines; indoles; carbazoles
1. Introduction
Since the disclosure of a first reliable and widely applicable synthesis of ynamides (N-alkynylamides) in 1998 [1], these functionalized acetylenes combining the diverse reactivity of a carbon–carbon triple bond with one of the most important functional groups of organic chemistry—the amino function—found widespread interest within the chemical community. Ynamides distinguish from their long-time known electron-rich variants—the ynamines (N-alkynylamines)—by having at least one electron-withdrawing group (EWG) attached to the nitrogen heteroatom. This EWG is capable of serving as a protective group in further chemical manipulations—but most importantly—it guarantees a decent chemical stability of the otherwise electron-rich acetylenic unit by lowering its HOMO. As such, the nitrogen bound EWG decreases the electron density of the adjacent alkyne moiety by inducing a more or less—but significantly—polarized carbon–carbon triple bond (Figure 1). Consequently, this results in tuning reactivity and selectivity. Ynamides are habitually bench-stable functionalized acetylenes that withstand an aqueous work-up procedure as well as purification by column chromatography on silica gel—both important features that are required for being useful molecular building blocks in organic synthesis. Over the years, the chemistry of ynamides became well-recognized. Ynamides nowadays serve as multipurpose substrates in organic synthesis including transition metal, ionic, radical and photoredox-mediated processes leading to N-heterocyclic compounds, natural products and other molecular targets for life and material sciences. Their exceptional rich and diverse chemistry covers several review articles [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16].
Figure 1.

Mesomeric resonance structures of ynamines, ynamides and 1,3-butadiynamides.
More recently, the ethynylogous variants of ynamides—the 1,3-butadiynamides—likewise received considerable attention in organic and heterocyclic chemistry. They found interesting use in transition metal-catalyzed annulation reactions, like the Hexa-dehydro-Diels–Alder (HDDA) reaction [17,18,19,20,21], and as optoelectronic materials due to their underlying push–pull character. Notably, the intrinsic polarization of ynamides is not only preserved in 1,3-butadiynamides. It is furthermore extendable via the ethynylogous principle from a 1,2- to a formal 1,4-polarization. Therefore, 1,3-butadiynamides are appealing for nitrogen-functionalized C4-building blocks for the construction of complex structural motifs. As both triple bonds can be simultaneously or consecutively functionalized via an electrophilic and/or nucleophilic reaction step, the selective introduction of more than one substituent might be achievable selectively. Other important features of molecular 1,3-butadiynamide scaffolds are their facile synthesis and their often superb stability. This not only accounts for 1,4-diynamides, but also for the higher conjugated oligoynamides. Some chemistry associated with 1,3-butadiynamides has been briefly mentioned in reviews covering synthesis and use of ynamines and ynamides [3,4,5,12,13,14,15,16]. However, there is so far no comprehensive article focusing on the ethynylogous variant of ynamides—the 1,3-butadiynamides. Highlighting the chemistry of 1,3-butadiynamides and compiling their reactivity, specificity and miscellaneous synthetic potential is the purpose of our present review.
2. Synthesis of 1,3-Butadiynamides
The majority of described 1,3-butadiynamides are derived from arylsulfonamides (EWG = Toluenesulfonyl (Ts), Methanesulfonyl (Ms), etc.), and only a few examples rely on carbamates (EWG = CO2Me) or oxazolidinones. Syntheses of 1,3-butadiynamides follow the basic three retrosynthesis disconnections [a–c] as shortlisted in Scheme 1.
Scheme 1.

Retrosynthesis of 1,3-butadiynamide.
2.1. Disconnection [a]: Cu-Catalyzed Cross-Coupling Reactions
Disconnection [a] to 1,3-butadiynamides follows common strategies for the assembly of 1,3-bisacetylenes starting from various terminal acetylenes (Glaser–Hay coupling) or cross-couplings of terminal acetylenes with 1-bromoalkynes (Cadiot–Chodkiewicz reaction). The use of terminal ynamides in copper-catalyzed cross-coupling reactions is the major strategy to access both symmetrical and unsymmetrical 1,3-butadiynamides. The required terminal ynamides are readily available by rendering the synthetic strategies outlined in Scheme 1 [13].
Application of the Glaser–Hay reaction to terminal ynamides proceeds readily affording various sets of symmetrical substituted 1,3-butadiynamides 2a–e (Scheme 2) [22,23].
Scheme 2.

Symmetrical substituted 1,3-diynamides via oxidative Glaser–Hay coupling.
The oxidative homocoupling of terminal ynamides is usually carried out in acetone at room temperature with CuI-TMEDA (tetramethylethylenediamine) as the catalyst and by exposure to atmospheric oxygen. It is also applicable to chiral amino acid-derived terminal ynamides to deliver 1,3-diynamide 2e [24].
Unsymmetrical 1,3-butadiynamides 3a–h are available via the Cadiot–Chodkiewicz cross-coupling between terminal ynamides and 1-bromoalkynes (Scheme 3) [25,26]. Reactions are usually performed in methanol at 40 °C and require copper(I) iodide as the catalyst, n-butylamine as the base and sub-stoichiometric quantities of hydroxylamine hydrochloride. The use of the latter is crucial to avoid ynamide homocoupling by reducing any copper(II) salts present in the reaction medium. Notably, the complementary approach utilizing bromine- or iodine-terminated ynamides with terminal acetylenes was unsuccessful so far. This is reasoned by the fact that bromine- or iodine-terminated ynamides are difficult to obtain and are quite unstable [27].
Scheme 3.

1,3-Butadiynamides via Cadiot–Chodkiewicz cross-coupling reaction.
The Cadiot–Chodkiewicz reaction with N-ethynylated oxazolidine-2-one 4 is also suitable for the synthesis of oxazolidine-2-one-derived 1,3-butadiynamides, which, for example, delivers the chiral push–pull 1,3-butadiynamides 5 in high yields (Scheme 4, (1)) [25].
Scheme 4.

Synthesis of oxazolidine-2-one-derived 1,3-butadiynamides.
Furthermore, but limited in scope, the palladium-mediated cross-coupling of ynamide 6 with 1-bromohexyne in (i-Pr)2NH/toluene (2:1 (v/v)) gives the corresponding 1,3-butadiynamide 7 via a Sonogashira type reaction (Scheme 4, (2)) [28].
2.2. Disconnection [b]: Amide N-Alkynylations
In analogy to related ynamide approaches, the synthesis of 1,3-butadiynamides via a copper-mediated C-N bond formation between suitable amides and 1-bromo-1,3-butadiynes was described, though only a few examples have been realized so far. Here, the stabilization of the acting copper amide complex prior to the coupling with the 1-bromo-1,3-diyne is mandatory. This is necessary to overpower the quite facile homocoupling of 1-bromodiynes (Scheme 5) [29,30]. C-N bond formation takes place at room temperature, but the drawbacks of requiring stoichiometric amounts of copper iodide and the need of high-quality pyridine should be taken into consideration. Indeed, the use of dry pyridine, freshly distilled over CaH2 under inert atmosphere, is required. The reaction conditions are also suitable for and were applied in the synthesis of the N-methanesulfonyl 1,3-butadiynamide 9c (EWG = Ms) [31].
Scheme 5.

Synthesis of 1,3-butadiynamide via Cu-mediated C-N bond formation.
Ynamide syntheses relying on sub-stoichiometric amounts of copper sulfate/1,10-phenanthroline as the catalyst in toluene at 60–95 °C [32] were extended to gain access to 1,3-butadiynamides. Efficiency, however, is lower compared to the protocol leading to carbamate-derived 1,3-butadiyne 9a via stoichiometric amounts of copper salt (36 vs. 80% yield) [29]. Notably, N-alkyl N-tosylamides are better substrates, and the synthesis of N-tosyl-1,3-butadiynamide 9d proceeds with high yields under copper-catalyzed cross-coupling conditions (Scheme 6, (1)) [33].
Scheme 6.

Copper-catalyzed synthesis of 1,3-butadiynamides.
It is worth noting that the synthesis of N-aryl N-toluenesulfonyl ynamides (R1 = Ar) via copper-catalyzed C-N bond-forming protocols with 1-bromo alkynes does not proceed at all, or proceeds in very low yields after several days of heating [34]. Probably, this is due to the weaker nucleophilicity of N-aryl N-toluenesulfonylamides. Therefore, the Cadiot–Chodkiewicz reaction with terminal ynamides remains the state of the art to obtain 1,3-diynamides having an N-aryl group. However, more recently, a copper-catalyzed electrophilic N-1,3-diynylation with triisopropylsilyl 1,3-diynyl benziodoxolone (10) was described to give straightforward TIPS protected 1,3-butadiynamides 11 including examples with N-aryl tosylamides (Scheme 6, (2)) [35].
2.3. Disconnection [c]: Functional Group Interconversion (FGI) at the C-Terminus
Chemical modifications of ynamides without affecting the triple bond have been scarcely explored until recently [13]. In the case of 1,3-butadiynamides, examples of modifications of the C-terminus are even fewer.
The synthesis of a terminal 1,3-butadiynamide through desilylation of the corresponding trimethylsilyl protected precursor with tributylammonium fluoride (TBAF) at −78 °C in dry THF proceeds in high yield (Scheme 7, (1)) [36]. Alternatively, access to terminal 1,3-butadiynamides is provided under basic conditions starting from the parent 1,3-butadiynamide terminated by the acetone adduct (Scheme 7, (2)) [25].
Scheme 7.

Modification of the 1,3-butadiynamide C-terminus.
1,3-Butadiynamide synthesis followed by transformation to a terminal 1,3-butadiynamide via protective group strategies allows for the development of iterative Cadiot–Chodkiewicz couplings to higher order ethynylogous ynamides. Such iterative cross-couplings offer a straightforward entry to conjugated tri- and tetraynamides 13 and 14 using as coupling partners diynamide 12e and a 1-bromoalkyne or a 1-bromo-1,3-diyne, respectively (Scheme 7, (3) and (4)) [25].
3. Molecular Structure and Electronic Properties
1,3-Butadiynamides and their higher homologues not only gain interest as molecular building blocks in organic synthesis, but they also receive considerable attention for their unique conformation on basis of principal helical twisted molecular orbitals delocalized over the entire conjugated carbon rod, with their optoelectronic properties including NLO performance, and their possible function as molecular wire or as monomer in solid-state topo-chemical polymerizations.
Whilst axial chirality in certain odd numbered cumulenes like allenes is well-documented, the understanding of chirality based on helical twisted frontier molecular orbitals (Hel-FMO) and its impact on chemical, optical and physical properties is still in its early infancy. Markedly, it is mainly limited to the theoretical and computational description of conjugated cumulenes [37,38], spiroconjugated systems [39] and suitable extended conjugated oligoynes (ECOs) [40,41,42,43]. The latter favor a twisted near orthogonal conformation caused by the combination of repulsive interactions between the terminating functional groups and the constructive orbital overlap upon twisting. Nitrogen atom-terminated oligoynes are predicted of being important members of the ECO family because of both being stable and having helical molecular orbitals delocalized over the entire conjugated polyyne carbon rod (Figure 2).
Figure 2.

Helical twisted molecular orbitals in extended conjugated oligoynamides.
For example, the high-level density functional theory (DFT) calculation of the still unknown—but synthetically feasible—diynamide 15 predict helical twisted molecular frontier orbitals with axial chiral geometry [41]. For the 1,3-diynamide 15, a torsion angle (TA) of TA = 66.74° was calculated together with a right-handed helical HOMO (Figure 2). Notably, similar values of TAs can be found in the single crystal structure data of the ynamine 16 (TA = 78.5°) [44], the 1,3-diynamines 17a (TA = 60.2°) [45] and 17b (TA = 85.2°) [46], the 1,3-diynamide 2d (TA = 76.6°) [23] and the 1,3,5-triynamide 14 (TA = 65.8°) [25]. Although these data are taken from solid-state structures, and conformational changes due to crystal structure-packing effects cannot be neglected, all TAs are of similar magnitudes and underline the influence of helical twisted molecular orbitals on the conformational output of 1,3-diynamides and their higher homologues.
A set of donor-π-spacer-acceptor “push–pull” ynamides end-capped with nitrogen donor and 4-nitro or 4-cyanophenyl acceptor units was investigated by the electro-optical absorption method (EOAM). The aim of the study was to examine their intramolecular charge transfer (ICT) properties in relation to the extent of the spacing-conjugated oligoyne units [25]. Dipole moments in solution of the ground (μg), the Franck–Condon excited state (μεFC) and their respective change of dipole moment (Δaμ) were obtained. Values of dipole moments for measurements in 1,4-dioxane of selected para-nitro-phenyl substituted ynamide derivatives are compiled in Table 1. The observed very high values for the change of dipole moments after transition from the ground to the Franck–Condon excited state show effective ICTs and consequently potential non-linear optical properties for these electronically tunable, extended, conjugated oligoynamides. Notably, the increase of change of dipole moments reaches a maximum for push–pull 1,3-diynamides with two spacing-conjugated alkyne units. Elongation of the conjugated alkyne units from one to two triple bonds results in an increase of the Δaμ values, while the ground-state dipole moments μg are almost unaffected. Increasing to three conjugated alkyne units, however, leads to a decrease of Δaμ (Figure 3).
Table 1.
Electrooptical absorption measurement (EOAM) of selected “push–pull” ynamides end-capped with a nitrogen donor and a 4-nitro-phenyl acceptor unit [25].
| Ynamide | μ g | Δaμ | μ a FC |
|---|---|---|---|
| [10−30 Cm] in 1,4-dioxane | |||

|
9.7 | 74.6 | 84.3 |
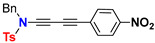
|
9.5 | 92.3 | 101.8 |

|
10.9 | 30.8 | 41.7 |

|
11.8 | 69.7 | 81.5 |
Figure 3.

Correlation of the number of alkyne units in ynamides with the magnitude of change of dipole moment from ground to Franck–Condon excited state Δaμ.
Topochemical solid-state polymerization of 1,3-diacetylenes in single crystals gives conjugated polymers that have attracted attention on their physical properties such as conductivity, optical nonlinearity and mechanical strength. Consequently, 1,3-butadiynamides that are often crystalline materials were also investigated in topochemical polymerizations [23,47,48].
4. Addition, Cycloaddition and Oxidation Reactions
Non-symmetrical 1,3-butadiynamides—unlike their symmetrical counterparts—often act as internal ynamides. In many examples of addition and cycloaddition reactions, the 1,2-activation of the 1,3-diyne is superior to the 1,4-activation, and the second triple bond remains unaffected in the underlying transformation. However, in view of the richness of acetylene chemistry, the obtained products can often undergo further chemical transformations, broadening the array of available molecular and heterocyclic structures.
4.1. Addition Reactions
Stereodefined 1,3-dienes bearing N-substituents are important building blocks in organic synthesis. Simple access to versatile 1,4-dihalogenated E,E-1,4-dienamides was achieved from readily available symmetrical 1,3-butadiynamides 2a–b in regio- and stereoselective hydrohalogenations (Scheme 8, (1)) [49].
Scheme 8.

Regio- and stereoselective hydrohalogenation of 1,3-butadiynamide.
Hydrogen halides are generated in situ from halotrimethylsilane (Br, I) and water─or chlorotrimethylsilane and saturated ammonium chloride. Under these conditions, the perfect syn-addition of H-X (X = Cl, Br) across the ynamide triple bond occurs. The hydrobromination of non-symmetrical 1,3-butadiynamides 3f,j ensues only on the triple bond linked to the nitrogen atom, i.e., the more electron-rich carbon-carbon triple bond, affording the 1-en-3-ynamides 19a–b (Scheme 8, (2)). Notably, the hydrobromination of 1,4-diphenyl-butadiyne does not occur under similar reaction conditions. Germylzincation of 1,3-butadiynamide 20 was achieved with excellent regio- and stereoselectivity using a combination of hydrogermanes and ZnEt2 (Scheme 9) [50].
Scheme 9.

Radical germylzincation towards stereodefined ynenamides.
Addition of germanium did not occur in the absence of Et2Zn. The proposed mechanism involves the addition of a germanium-centered radical across the triple bond on β-position to the N-atom in agreement with the underlying polarization of the 1,3-butadiynamide. The resulting α-heteroatom-substituted vinyl radical undergoes ethylzinc group transfer to give the β-zincated vinylgermane 21 along with an ethyl radical keeping the radical chain propagation. The (E)-form of the vinylgermane radical is favored. Retention of the double bond geometry of the vinylzinc intermediate 21 is observed upon hydrolysis or Cu(I)-mediated C-C bond formation with 1-bromophenylacetylene. The resulting stereodefined tri- and tetrasubstituted ynenamides 22a–b and 23 can be further functionalized through displacement of germanium. Vinylgermanes gain increasing attention as alternatives to vinylsilanes and vinylstannanes for the preparation of stereodefined alkenes because of their low toxicity, increased stability towards protonolysis and facile transformation into vinyl halides.
Gold-catalyzed double hydroaminations of symmetrical substituted 1,3-butadiynamides 2a,c,f with anilines readily proceed at room temperature and deliver the corresponding 2,5-diamido-N-arylpyrroles 24a–d (Scheme 10) [51].
Scheme 10.

Gold-catalyzed synthesis of 2,5-diamidopyrroles.
Moreover, the 1,2,5-trisubstituted pyrrole 25 is accessible by using phenylhydrazine as the hydroamination reagent. In this case, thermal activation is required, as well as the use of the more reactive Echavarren’s gold catalyst derived from the bulky and electron-rich di-t-butylphosphinobiphenyl ligand (Scheme 11) [51].
Scheme 11.

Gold-catalyzed synthesis of a 1,2,5-trisubstituted pyrrole with phenylhydrazine.
Copper catalysis in the presence of a zinc additive is also effective in the pyrrole series and proceeds with excellent functional group tolerance [52]. In comparison to the gold-catalyzed protocol, the scope of 2,5-diamidopyrroles available from 1,3-butadiynamides is considerably broadened as primary aliphatic amines, and sterically hindered anilines are now suitable hydroamination reagents (Scheme 12).
Scheme 12.

Copper-catalyzed synthesis of 2,5-diamidopyrroles.
Interestingly, the copper-catalyzed reaction of 1,3-butadiynamide 26 with 2-amino (benzo)thiazoles as nucleophiles does not deliver pyrrole derivatives. Now, fused diazepines 28a–f are formed via a formal [4+3] annulation sequence (Scheme 13) [52].
Scheme 13.

Copper-catalyzed synthesis of diazepines derived from 1,3-diynamides.
Symmetrical substituted 1,3-butadiynamides also serve as substrates in gold catalyzed hydration leading to the 2,5-diamidofurans 29a–c (Scheme 14, (1)) [51]. Interestingly, the dimethanesulfonyl analogue of 29b—the 2,5-diamidofuran 30—was obtained in a better yield using a copper(II) catalyst in the presence of a zinc additive (Scheme 14, (2)) [52].
Scheme 14.

Gold- or copper-catalyzed synthesis of 2,5-diamidofurans.
The metal-catalyst-free synthesis of 2,5-diamido-thiophenes 31a–d using 1,3-butadiynamides 2a–d and Na2S•9H2O as the sulfur-providing source proceeds under very mild conditions (Scheme 15, Conditions A) [26]. Both symmetrical as well as the non-symmetrical 1,3-diynamides undergo a formal 1,4-functionalization of the parent 1,3-diyne unit. In the case of diynamide 2a (R1 = Ph), whose electrophilicity is enhanced by conjugation of the ynamide unit with the N-phenyl group, these reaction conditions were not suitable. This is probably because of side reactions resulting from the addition of ethanol across the triple bond. Switching to acetonitrile as the solvent was the key for success (Conditions B). Now, the corresponding thiophene 31a is obtained in 62% yield. Importantly, the method is likewise effective with unsymmetrical 1,3-butadiynamides 3a–c,e–h giving a straightforward entry to variously functionalized 2-amidothiophenes 32a–g (Scheme 15).
Scheme 15.

Metal-catalyst-free synthesis of 2-amido- and 2,5-diamidothiophenes.
This new approach to 2-amido- or 2,5-diamidothiophenes is extendable to the synthesis of terthiophenes (Scheme 16). The required tetrayne 33 bearing two linked 1,3-diynamide units is accessible via a double Cadiot–Chodkiewicz coupling between a terminal ynamide and bis(bromoalkynyl)thiophene. Application of the developed reaction conditions to substrate 33 delivers the diamido-capped terthiophene 34 having a string of N,S heteroatoms embedded in a highly π-conjugated molecular scaffold. Electron-rich terthiophenes are interesting as active materials in organic electronics such as organic transistors or organic photovoltaic cells.
Scheme 16.

Synthesis of a terthiophene.
4.2. Cycloaddition Reactions
Intramolecular [4+2] cycloadditions of 1,3-butadiynamides 35a–d tethered to an enyne moiety furnish functionalized 7-alkynyl indolines 37a–d (Scheme 17) [53]. Yields improve in the presence of BHT, which not only suppresses the polymerization of enyne substrates, but also facilitates the isomerization of the cyclic allene intermediate 36 into indoline 37 via proton/hydrogen atom transfer. As alkyl-substituted ynamides with only one C–C triple bond give low yields in related [4+2] cycloadditions, the hydrogenation of the triple bond of indolines 37a–d is an alternative and offers a valuable entry to the parent alkyl-substituted indolines. In these Didehydro–Diels–Alder (DHDA) reactions the 1,3-butadiynamide serves as the formal dienophile. Notably, this has to be distinguished from its use in Hexa-Dehydro-Diels–Alder (HDDA) reactions that will be discussed in Chapter 6, where the 1,3-butadiynamide unit functions as the formal 4π cycloaddition partner.
Scheme 17.

Intramolecular [4+2] cycloaddition with 1,3-butadiynamides to give 7-alkynyl indolines via DHDA reaction.
Azide-alkyne [3+2] cycloadditions of 1,3-butadiynamide 3c using rhodium catalysis afford 4-alkynyl 5-amino-triazole 38 [54], whereas the copper-catalyzed addition of azides to terminal 1,3-butadiynamides provide alternative ynamide-derived azide-alkyne click-products 39 (Scheme 18) [35]. Click reactions with 1,3-diynamides are exclusively chemo- and regioselective with stoichiometric amounts of azides. The exclusion of air and moisture is unnecessary in the case of the rhodium catalysis, whereas high yields in copper-mediated azide-alkyne [3+2] cycloadditions are obtained only under strict anhydrous conditions.
Scheme 18.

Intermolecular azide-alkyne [3+2] cycloaddition with 1,3-butadiynamides to give 4-alkynyl triazoles.
The gold-catalyzed cycloaddition of 1,3-butadiynamide 3k with aminide 40 as N-acyl nitrene equivalent gives 5-alkynyloxazole 41 as a single regioisomer in 83% yield (Scheme 19) [55]. This formal [3+2] cycloaddition is based on the use of the robust and air-stable dichloro(pyridine-2-carboxylato)gold(III) complex as a pre-catalyst.
Scheme 19.

Intermolecular [3+2] cycloaddition of aminide 40 to 1,3-butadiynamide 3k to give 5-alkynyloxazole 41.
4.3. Oxidation Reactions
The catalytic oxidation of aryl 1,3-butadiynamides 42 with 8-methylquinoline oxide (43) using a cationic gold catalyst in the presence of a silver salt delivers the 1,4-oxidation products 46 (Scheme 20) [56].
Scheme 20.

Au(I)-catalyzed 1,4-oxidation of 1,3-butadiynamides.
In agreement with other gold-catalyzed oxidations of ynamides with pyridine N-oxides, a striking chemoselectivity for C(1) oxidations is observed leading to the α-carbonyl gold carbene intermediate 44, which, in the case of the ethynylogous ynamide, undergoes a 1,3-carbene migration affording intermediate 45 [57]. The 1,3-migration tautomer 45 is then further oxidized to give the double oxidation products 46. Finally, this oxidative process results in a 1,4-functionalization of the 1,3-diynamide moiety.
Interestingly, a different chemical outcome is observed with 5-hydroxy-1,3-butadiynamides 47a–b, which refrain from C(1) oxidation. Here, an atypical C(3) oxidation and a further gold-mediated oxidative cyclization lead to either furan-3-ones 48a–e or to pyran-4-ones 49a–e with AuCl3 and CyJohnPhosAuCl/AgSbF6, respectively (Scheme 21) [56]. The formation of pyran-4-ones 49 from the less basic N-phenyl N-toluenesulfonylamide-derived substrates 47 (R1 = Ph) is more efficient with 8-iso-propylquinoline oxide, which is illustrated in the synthesis of product 49b with 78 vs. 33% yield. Importantly no exchange between the two cyclic ketones occurs in the presence of the gold catalysts.
Scheme 21.

Au(I)- and Au(III)-mediated oxidative cyclization of 5-hydroxy-1,3-butadiynamides.
DFT calculations support a mechanism for the chemoselective conversion of 47a (R1 = Ph, R2 = H) into cyclic ketones 48b or 49b (Scheme 22).
Scheme 22.

Proposed mechanism for the Au-catalyzed oxidative formation of cyclic ketones.
The gold π-complexed alkynes 50 and 51 in Scheme 22 are interconvertible. The 1,4-functionalization through the oxidation at C(3) relies on an energetically favored complex 51 in comparison to 50 because the energy barrier of the oxidation of 51 with 8-methylquinoline oxide (43) is calculated as lower. The resulting α-oxo gold carbene 52 undergoes 1,2-hydride migration leading to intermediate 53, which evolves to the 3-oxo-5-enol intermediate 54 by releasing a gold species. Complexation of the triple bond of 54 with less alkynophilic (more Lewis acidic) Au(III)Cl3 induces an intramolecular carbonyl addition to the alkyne moiety via a 5-exo-dig cyclization. The latter affords intermediate 56, whereas the formation of a keteniminium species 57 in the case of the cationic gold catalyst facilitates 6-endo-dig cyclization to give intermediate 58. A mechanism for the transformation of the 1,3-diynamide 47b bearing an aryl-substituted hydroxy moiety was unfortunately not provided by the authors. The structure of the corresponding products 48c–e and 49c–e, however, suggests that a less often observed 1,2-aryl migration takes place [58]. This might also explain the net result with substrate 47 bearing an alkyl-substituted hydroxy moiety. The migratory aptitude of an alkyl group to an electron deficient center typically follows the order H > Ph > alkyl.
5. Metal-Catalyzed Cascade-Type Cyclization and Annulation Reactions
5.1. Intramolecular Processes
The activation of 1,3-butadiynamides with transition metal π-acids to form keteniminium ions provide superb opportunities of cascade-type cyclization and annulation reactions. For intramolecular processes, only gold catalysts have been used so far [59]. The involved gold catalysts often vary according to the 1,3-butadiynamides or to the reaction conditions applied.
1,3-Butadiynamides 59 that are linked via the ynamide nitrogen by a two carbon tether to electron-rich benzenes or electron excess heteroaromatics readily undergo a gold-catalyzed cascade reaction with IPrAuNTf2 (IPr = 1,3-bis(diisopropylphenyl)imidazole-2-ylidene), leading to sulfone-containing pyrrolo[2,1-a]tetrahydroisoquinolines 60a–g (Scheme 23) [60]. The cationic nature of the gold catalyst is crucial. The counter anion NTf2− acts as a proton transfer shuttle and facilitates the overall process, which notably involves a formal 1,4-sulfonyl migration. No reaction takes place in the absence of an electron-donating group on the phenyl ring (see 60h). This stands for the electrophilic aromatic substitution step within the reaction cascade. Further functional groups susceptible to react with the gold catalyst, such as alkyne, hydroxy or acetate groups, are tolerated (see 60e–g). Remarkably, other structural motifs are also accessible through this process, such as the pyrrolo-azepine 61 or the heterocyclic systems 62 and 63.
Scheme 23.

Gold-catalyzed synthesis of sulfone-containing pyrrolo[2,1-a]tetrahydroisoquinolines.
The main intermediates involved in this cascade reaction find support from DFT calculations (Scheme 24). The gold keteniminium species 64 resulting from a regioselective attack of the electrophilic gold catalyst to the 1,3-diynamide moiety undergoes an intramolecular arylation (Vilsmeier–Haack-type reaction) followed by re-aromatization through a 1,3-H shift mediated by the counter anion NTf2− to deliver intermediate 66. The isomerization of cis-66 to trans-67 required for the second annulation process is the proposed rate-determining step. This second cyclization takes place via a concerted C-N bond formation and includes a 1,2-methanesulfonyl-(Ms) migration to give intermediate 68. A second 1,2-Ms shift followed by a 1,2-H shift, which is greatly facilitated by the counter anion NTf2−, delivers intermediate 70, which finally furnishes product 60a after demetallation.
Scheme 24.

Proposed mechanism for the gold-catalyzed transformation of 1,3-butadiynamides into sulfone-containing pyrrolo[2,1-a]tetrahydroisoquinolines.
A related gold(I)-catalyzed para-toluenesulfonic acid (PTSA) promoted cycloisomerization of 1,3-diynamides 71 gives access to α,β-unsaturated ketones 72 together with minor amounts of the 1,3-diynamide hydration product 73 (Scheme 25) [61]. The studied transformation was limited to 1,3-diynamides terminated with a phenyl group, and the other substitution pattern was incompatible mismatched with the desired reactivity.
Scheme 25.

Gold(I)-catalyzed para-toluenesulfonic acid (PTSA) promoted the cycloisomerization of 1,3-diynamides to α,β-unsaturated ketones.
C-terminal-functionalized 1,3-butadiynamides 74 linked to a terminal alkyne and an allylic ether were used in a reaction cascade involving a dual gold-catalyzed process. The reaction cascade includes an intramolecular carboalkoxylation and a subsequent charge-accelerated [3,3] sigmatropic rearrangement to deliver carbazoles 75 as the major products (Scheme 26) [62]. Compound 76, resulting from a [1,3] sigmatropic rearrangement, was formed as the minor side product in most cases. However, reversal of regioselectivity took place when the latter process was electronically favored (see 76h).
Scheme 26.

Gold-catalyzed formal HDDA/carboalkoxylation reaction cascade.
Triynes bearing an internal instead of a terminal alkyne moiety are unsuitable for this reaction cascade in agreement with the proposed mechanism that is being initiated by a σ,π dual activation of triyne 74a to give intermediate 77 (Scheme 27). The latter undergoes a 5-exo-dig cyclization to give the gold vinylidene carbenoid 78, which then cyclizes to furnish the ortho-Au phenyl cation 79. The gold-complexed aryne 79 is trapped by the ether group leading to the oxonium species 80. A charge-accelerated [3,3] sigmatropic rearrangement of 80 gives intermediate 81 followed by re-aromatization delivering 82 along the release of the gold ion. The final step of the proposed mechanism is the protodeauration of 82 to furnish product 75a by releasing the active gold catalyst.
Scheme 27.

Proposed mechanism for the Au-catalyzed carboalkoxylation reaction cascade.
5.2. Intermolecular Processes
As highlighted in chapter 4, the metal-catalyzed double hydroaminations of symmetrical 1,3-butadiynamides with anilines gives 2,5-diamido-N-arylpyrroles. Surprisingly, in related transformations, ortho-substituted anilines as hydroamination reagent trigger a cationic driven reaction cascade after the first hydroamination step. For example, silver(I)-catalyzed transformations of 1,3-butadiynamides 83a–f with ortho-substituted anilines 84 bearing electron-donating substituents give 2-amidoquinolines 85a–h (Scheme 28) [63]. The combination of steric and electronic factors is crucial to gain preference for the formation of quinoline over pyrrole-derived products. One purpose of the electron-donating group in ortho position of the aniline is to facilitate the sequential electrophilic aromatic substitution step that rationalizes product formation. Symmetrical substituted 1,3-butadiynamides are also suitable substrates as shown in the example of bis-amido quinoline 85g (96% yield).
Scheme 28.

Ag(I)-catalyzed synthesis of 2-amidoquinolines.
The proposed mechanism is depicted in Scheme 29. Silver-catalyzed intermolecular hydroamination of 1,3-butadiynamide 83 with aniline 84 delivers the enyne intermediate 86, which, after proton transfer and protodeargentation, leads to 87. Silver complexation of the remaining triple bond in 87 facilitates intramolecular hydroarylation via an electrophilic aromatic substitution process to give 89. Subsequent proton transfer and re-aromatization results in the final quinoline 85.
Scheme 29.

Proposed mechanism for the Ag(I)-catalyzed synthesis of 2-amidoquinolines.
The synthesis of 2-aminopyrrolo[1,2-b]pyridazines 92a–g from non-symmetrical 1,3-butadiynamides 83a–f and 1-aminopyroles 91 via a related strategy is also reported (Scheme 30) [64]. The overall reaction cascade proceeds through Au(I)/Ag(I)-mediated C-N/C-C bond formations to end up with readily substituted 2-aminopyrrolo[1,2-b]pyridazines 92.
Scheme 30.

Au(I)-catalyzed synthesis of 2-aminopyrrolo[1,2-b]pyridazines.
The reaction of symmetrical substituted 1,3-butadiynamides 93a–c with ortho-cyano anilines 94 provides aryl-fused 1H-pyrrolo[3,2-c]quinolines 95a–g within a single step via a dual catalyst process (Scheme 31) [65]. Heteroaryl-fused 1H-pyrrolo[3,2-c]quinolines 96a–b are also accessible using 2-amino-3-cyano thiophenes as nucleophiles. Both catalysts—copper(II) acetate and zinc(II) triflate—are necessary for the cascade reaction to be effective. Finally, the overall sequence realizes the formation of one C-C and two new C-N bonds.
Scheme 31.

Cu(II)/Zn(II)-catalyzed synthesis of 1H-pyrrolo[3,2-c]quinolines.
Activation of 1,3-butadiynamides via metal–π coordination of a π-acidic copper(II)-species to form the metal keteniminium 97 is the initiating step of the proposed mechanism (Scheme 32). This facilitates the intermolecular hydroamination to give enamine 98 followed by activation of the cyano group in 98 through coordination with the Zn catalyst. The resulting Zn-complexed 99 can undergo a 6-exo-dig cyclization via intramolecular nucleophilic attack of the enamine moiety producing 100. Activation of the remaining carbon–carbon triple bond by Cu(II) facilitates a 5-endo-dig cyclization providing 101, which upon protodemetallation leads to the 1H-pyrrolo [3,2-c]quinoline product 95. In the case of non-symmetrical 1,3-butadiynamides 102 (R = Ph), tautomerization of intermediate 100 leads to the monocyclization product 103, which is a major side product in the reaction. Quinoline 103 is unable to provide 104 through a second annulation sequence, as shown in a separate control experiment. Therefore, an equimolar mixture of products 103 and 104 resulting from both pathways is observed starting from 1,3-butadiynamide 102 even after prolongation of the reaction time. Notably, diphenyl-1,3-diacetylene did not undergo any reaction under these reaction conditions. This one more time underlines the differences in reactivity between 1,3-diynamides and other 1,3-butadiynes.
Scheme 32.

Proposed mechanism of Cu(II)/Zn(II)-catalyzed synthesis of 1H-pyrrolo[3,2-c]quinolines.
The outcome of gold-catalyzed reactions of 1,3-butadiynamides 102 with anthranils 105 is substrate-dependent, preferentially affording the formal [5+2] annulation products 106 in the case of 1,3-butadiynamides bearing an electron-deficient N-aryl group, while in contrast, [3+2] annulation products 107 are preferentially formed with more electron-rich N-alkyl 1,3-butadiynamides (Scheme 33) [66].
Scheme 33.

Gold-catalyzed reaction between 1,3-butadiynamides and anthranils.
DFT calculations rationalize the observed chemoselectivity. Gold activation of the 1,3-butadiynamide followed by N-attack of anthranils leads to intermediate 109, which evolves into the energetically more favorable α-imino gold carbene 110 (Scheme 34). The latter undergoes carbonyl addition to give 111 bearing a seven-membered cycle (path a). Although intermediate 111 is higher in energy than intermediate 112, resulting from carbene arylation (path b), the free energy barrier for the formation of 111 is smaller than the one of 112. Moreover, the conversion of 111 into quinoline oxide 106 is highly exothermic and expected to be almost barrierless. The preference for path a relies on the presence of the alkynyl substituent, which brings favorable steric and electronic effects to facilitate the formation of seven-membered ring intermediate 111. These results are specific to 1,3-butadiynamides [67].
Scheme 34.

Proposed mechanism for Au(I)-catalyzed reaction of 1,3-butadiynamides with anthranil.
Gold-catalyzed oxidative cascade reactions of (het)aryl-tethered 1,3-butadiynamides, using pyridine N-oxide as the oxidant, encompass efficient protocols to access complex polycyclic heteroaromatics within a single step. Here, the active gold species is an α-carbonyl gold carbene, whose obviously faster reaction with the proximal electron-rich aromatic group overrides the otherwise typical 1,4-dicarbonyl formation (Section 4.3, Scheme 20).
The reaction of 3-methoxyphenyl-tethered 1,3-butadiynamides 113a–b with pyridine N-oxide (114), using catalytic amounts of IPrAuCl in the presence of NaBArF4, leads to furo [2,3-c]isoquinolines 115a–h (Scheme 35) [68]. The nature of the substituent at the terminal 1,3-diynamide end—alkyl or phenyl—for the outcome of the reaction cascade is negligible, but the presence of the 3-methoxy group on the tethered aryl ring is mandatory for the reaction cascade to occur. In the absence of the 3-methoxy group, in the case of substrates N-benzyl 1,3-butadiynamide 120 (Ar = Ph, Scheme 36), the double oxidation product 122 is the only product formed under the reaction conditions (Path b, Scheme 36).
Scheme 35.

Au(I)-catalyzed synthesis of furo[2,3-c]isoquinolines.
Scheme 36.

Proposed mechanism for the synthesis of furo[2,3-c]isoquinolines.
According to the proposed mechanism, the gold-activated 1,3-butadiynamide 113 is oxidized by pyridine N-oxide (114) to give α-carbonyl gold carbenoid 116, which is in situ trapped by the aryl group through CH insertion delivering intermediate 117 (path a, Scheme 36). Carbonyl addition on the activated second triple bond of 117 leads to the fused tricyclic intermediate 118, which undergoes protodeauration to 119. The transformation of intermediate 119 into the final polycyclic heteroaromatic product 115 via release of sulfonic acid is highly favorable and might be the driving force of the total transformation according to the authors. The involvement of 119a (R2 = Ph) was evidenced by conducting the reaction at room temperature that provided 119a (25% isolated yield) along with unreacted 1,3-butadiynamide 113a. Full conversion of 119a into product 115a was achieved upon heating at 80 °C.
Accordingly, as extension, the assembly of fused tetracyclic heteroaromatics 125a–g was achieved via the same strategy using N-methyl indolyl-tethered 1,3-butadiynamides 123 as the substrates and 2-chloropyridine N-oxide (124) as the oxidant (Scheme 37).
Scheme 37.

Gold-catalyzed synthesis of 6H-furo[3′,2′:5,6]pyrido[3,4-b]indoles.
Strikingly, the outcome of the gold-catalyzed oxidative cascade cyclization of methoxyphenyl-tethered 1,3-butadiynamides is tunable by modifying the position of the methoxy group [69]. Indeed, the gold-catalyzed reaction of 2-methoxyphenyl-tethered 1,3-butadiynamides 126 with pyridine N-oxide (114) selectively delivers the polycyclic compounds 127a–i with a barbalan-type carbon skeleton along with the byproduct 128, resulting from hydrolysis and saponification of 126 (Scheme 38).
Scheme 38.

Gold-catalyzed synthesis of polycyclic product 127.
Arene CH insertion from in situ generated α-carbonyl gold carbenoids is not involved here in comparison to the case of the parent α-carbonyl gold carbenoid 116 depicted in Scheme 36. The preferential mechanistic path now is the intramolecular cyclopropanation of the arene unit by the carbenoid 129 to give norcaradiene 130 (Scheme 39). [3,3] Sigmatropic enyne cycloisomerization between the vinyl methoxy ether and the gold-activated alkynyl moiety leads to the formation of oxocarbenium ion 131, which after hydrolysis delivers the polycycle 127.
Scheme 39.

Proposed mechanism for the synthesis of 127.
A different set of polycycles now having an eight-membered ring moiety is available through a related gold-catalyzed dearomatization/cycloisomerization process by placing an additional methoxy group and varying the substitution pattern of dimethoxyphenyl-tethered 1,3-butadiynamides substrates. For example, 2,4-dimethoxyphenyl-tethered 1,3-butadiynamides 132 are selectively converted into the polycyclic products 133a–f upon reaction with pyridine N-oxide (114) using Cy3AuCl/AgF as the catalyst (Scheme 40). In this case, the 2-methoxy group remains intact, whereas the additional 4-methoxy group is involved in the formal [3,3] sigmatropic enyne rearrangement.
Scheme 40.

Au(I)-catalyzed reaction of 2,4-dimethoxyphenyl-tethered 1,3-butadiynamide 132 with pyridine N-oxide (114).
Using 2,5-dimethoxyphenyl-tethered 1,3-butadiynamides 136 as substrates makes the two fused N-heterocyclic structures 137a–d and 138a–d available, depending on whether the reaction is carried out in the presence of water or under anhydrous conditions, respectively (Scheme 41).
Scheme 41.

Au(I)-catalyzed reaction of 2,5-dimethoxyphenyl-tethered 1,3-butadiynamides 136 with pyridine N-oxide (114).
The proposed mechanism involves the generation of a reactive α-carbonyl gold carbene species 139, which undergoes a formal [2+1] cycloaddition with the adjacent arene moiety to give norcaradiene intermediate 140 (Scheme 42). The methoxy-substituted cyclopropane unit facilitates norcaradiene to cycloheptatriene ring expansion, leading to 141. Subsequent intramolecular enyne cycloisomerization delivers intermediate 142, whose hydrolysis furnishes the tricyclic products 137. Under anhydrous conditions, intermediate 142 converts into product 143 via deprotonation and protodeauration. Acidic treatment of 143 induces the cleavage of one carbocycle, delivering the bicyclic compounds 138. Importantly, such a pathway is supported by the isolation of compound 143a (R2 = Ph) and its conversion into the corresponding product 138a by treatment with PTSA.
Scheme 42.

Proposed mechanism for the Au(I)-catalyzed reaction of 2,5-dimethoxyphenyl-tethered 1,3-butadiynamides 136 with pyridine N-oxide (114).
Whereas the majority of the so far reviewed transformations of 1,3-butadiynamides rely on activation of one or two of its carbon–carbon triple bonds by either Brønsted or Lewis acids, or by transition metal catalysts acting both as π-acid and Lewis acid, or Au catalysts transforming 1,3-diynamides into metal carbenoid intermediates, other principles of ynamide activation might result from transforming 1,3-butadiynamides into [4]cumulenimines or related highly unsaturated π-conjugated molecular scaffolds.
Recently, divergent palladium-catalyzed reaction cascades for the selective synthesis of either 2-amino-3-alkynyl-indoles 145a–d or 2-amino-4-alkenylquinolines 146a–f from 1,3-butadiynamides 144 and primary or secondary amines were established (Scheme 43) [70]. Starting from identical or similar substrates, the outcome of the reaction giving either indole or quinoline motifs is switchable, respectively, by the absence of presence of TBAF.
Scheme 43.

Divergent Pd-catalyzed reaction cascades towards indole and quinoline motifs.
Under similar reaction conditions than those applied for the synthesis of 2-aminoindoles through Pd-catalyzed heteroannulation reaction from N-alkynyl-2-haloanilides [71], the 1,3-butadiynamides 144 behave like internal ynamides. The second triple bond remains unaffected through the transformation, whose key intermediate is the σ,π-chelated palladium species 147 (Scheme 44, path a). The latter results from the initial oxidative addition of the Pd(0) catalyst to the iodophenyl moiety of 1,3-butadiynamides 144. Metal–π complexation of the ynamide triple bond in 147 facilitates intermolecular amine addition delivering the 3-alkynyl indoles 145 selectively. Strikingly, in the presence of TBAF, both triple bonds are engaged to convert 1,3-butadiynamides 144 in 2-aminoquinolines 146a–f in one pot (Scheme 44, path b). The combination TBAF/KOH is believed to induce cleavage of the tosyl group either as the first step or after the oxidative addition of the palladium catalyst to the iodophenyl moiety. Now, the key intermediate is the palladium-chelated [4]cumulenimine 148. The intermolecular amine addition to 148 selectively occurs at the α-carbon delivering the allene-derived intermediate 149. Subsequent intramolecular Heck-type reaction furnishes the annulated π-allyl palladium species 150, which undergoes β-hydrogen elimination to produce 4-alkenyl quinolines 146.
Scheme 44.

Proposed mechanism for the Pd-catalyzed divergent synthesis of either 2-amino-3-alkynyl-indoles (path a) or 2-amino-4-alkenylquinolines via [4]cumulenimine intermediate (path b).
The [4]cumulenimine structure in 148 is unprecedented. However, the deuterium labeling experiment shows the selective deuterium incorporation into the C3 and C11 positions of the quinoline products 146 in agreement with the formation of the transient species 148.
6. HDDA Cascade Reactions
The hexa-dehydro-Diels–Alder (HDDA) reaction of 1,3-diynamides tethered to 1,3-butadiynes delivers highly reactive arynes (dehydrobenzenes) after a formal intramolecular [4π + 2π] cycloaddition process [17]. They are subsequently trapped by in situ intra- or inter-molecular additions to the dehydrobenzene carbon–carbon triple bond, leading to a variety of new heterocyclic structures and useful products. The HDDA reaction itself was discovered in 1997 [72,73] and further developed. However, since 2012, it has received considerable attention, and the underlying process was coined with the term HDDA [18]. The field of HDDA reactions is rapidly expanding, including substrate variations and development of new aryne-trapping modes. The latter takes vast advantage of the fact that typical reagents necessary for generating arynes are absent and therefore cannot interfere [19,20,21]. Studies have shown that reagent-free HDDA reactions proceed through a stepwise mechanism via diradical intermediates. Significant acceleration of the process is observed when the formal 2π cycloaddition partner in the underlying [4π + 2π] cycloaddition process bears an alkynyl substituent (1,3-butadiynyl unit) [33,74]. This is why tetraynes rather than triynes are frequently used in HDDA cascade reactions.
1,3-Butadiynamides tethered to monoalkynes or 1,3-butadiyne moieties are privileged substrates for HDDA reactions because aryne formation proceeds chemo- and regioselectively due to the intrinsic polarization of the 1,3-diynamide unit. Notably, intramolecular HDDA reactions with 1,3-diynamides belong to transformations relying on a 1,4-functionalization of 1,3-diynamides, although the underlying process is step-wise. The follow-up reaction of those 1,3-diynamide-originated arynes continues being highly regioselective, i.e., for the addition of nucleophiles to the aryne. For example, the thermal reaction of tetrayne 152 with triethylamine or trifluoroethanol regioselectively furnishes indolines 153 and 154, respectively (Scheme 45, (1)) [75]. A novel entry to functionalized carbazoles 157a–d from triyne substrates 155 is accessible through perfectly regioselective nucleophilic addition to HDDA-generated carbazolynes 156 (Scheme 45, (2)) [76].
Scheme 45.

Regioselective “trapping reactions” of 1,3-butadiynamide-derived arynes.
The discriminating reactivity of HDDA-generated arynes from 1,3-butadiynamides was further highlighted by chemo- and regioselective transformations using structurally complex multifunctionalized “aryne-trapping agents” taken from the nature pool. For example, the reaction of tetrayne 158 with the cinchona alkaloid quinidine, which displays several potential reacting sites and/or reacting modes, delivers the single indoline product 159 (Scheme 46) [77].
Scheme 46.

Reaction of a HDDA-generated aryne with quinidine.
Experimental results of intramolecular HDDA reactions with 1,3-diynamide were rationalized by DFT calculations (Scheme 47) [78]. Amongst possible diradical intermediates formed in the principal first step of the HDDA reaction of tetrayne 158, the aza-cycloheptynes 163 and 164 can be ruled out, because they are higher in energy by 35.9 and 41.7 kcal/mol, respectively, compared to the pyrrolidine species 160. This largely originates from the twofold propynyl stabilization inherent in biradical 160 and the high triple bond strain energy in arynes 163 and 164. The formation of intermediate 160 is the rate-determining step. Steric factors govern the selective formation of aryne 161 in the second step. The activation barrier to aryne 161 and 162 is 4.3 and 6.6 kcal/mol, respectively, and relates to differences in product stability. Aryne 162 is less stable than 161 by 3.4 kcal/mol because of the steric hindrance between the methanesulfonyl group and the propynyl moiety.
Scheme 47.

Mechanistic pathway of the formation of a single HDDA-generated aryne from 1,3-butadiynamide-derived substrates.
Computational studies, moreover, explain why nucleophilic additions to indoline aryne 161 take place selectively on the C-6 rather than the C-7 position (Scheme 47) [79]. Transition state distortion energies determine regioselectivity. In other words, the aryne 161 displays unsymmetrical bending distortion, and the nucleophilic addition occurs at its flatter and more electropositive end.
The silver-catalyzed HDDA reaction is an interesting alternative to the sole thermal version, eventually affording different reaction outcomes [80]. Ag(I) complexation of the aryne allows for its stabilization while retaining high reactivity. The reactive intermediate resulting from the interaction of aryne with silver salts is described as the silver-bound aryl cations 165, 166 or by its mesomeric 1,2-carbene-silver carbenoid 167 (Scheme 48).
Scheme 48.

Ag(I)-complexed aryne species in silver-catalyzed HDDA reactions.
6.1. Sole Thermally Induced HDDA Cascade Reactions
Aryne trapping via C-C bond formation: The HDDA-generated benzyne 161 reacts selectively with phenols and delivers 2-hydroxybiaryl compounds 169a–f, and not the expected diaryl ethers, which are archetypal products of arynes generated by elimination reactions (Scheme 49, (1)) [81]. Indeed, sole thermal HDDA reactions proceed under neutral conditions, without the presence of any base or metal reagent, in contrast to classical methods of benzyne generation. The biaryl junction occurs at the ortho-position to the hydroxyl group via a “concerted phenol-ene-type” reaction. On the other hand, in the presence of cesium carbonate, the thermal reaction of tetrayne 158 with 4-methoxyphenol gives diaryl ether 170 as a single product (Scheme 49, (2)) [81].
Scheme 49.

Biaryl vs. diaryl ether synthesis through aryne reactions with phenols.
Inter- and intramolecular Alder-ene reactions with HDDA-generated arynes lead to a variety of differently substituted indolines. The reaction between 1,3-butadiynamides 171a–c with methyl methacrylate regioselectively delivers the ortho-isomers of indolines ortho-172a–c (Scheme 50) [82].
Scheme 50.

Intermolecular Alder-ene reactions with 1,3-butadiynamide-derived arynes.
However, the steric bulk of the R2 substituent has an influence on the Alder-ene reaction outcome. The conversion of 1,3-butadiynamide 171c substituted with a bulky silyl group (R2 = SiEt3) preferentially gives indoline meta-173 with 2-methyl-hepten-3-one.
Intramolecular HDDA reactions followed by intramolecular Alder-ene sequences were realized with 1,3-diynamides 174a–d to deliver the linear annulated indolines 176a–d (Scheme 51, (1)) [83]. The chemoselective formation of aryne 175 makes the Alder-ene reaction the most favorable pathway amongst all other possible aryne reacting modes. Extending the tether from three to four atoms delivers eight-membered ring products like compound 176d. Tetrayne 177 bearing a shorter two-atom tether with a methyl-substituted alkene also undergoes an Alder-ene reaction to deliver tricyclic compound 178 (Scheme 51, (2)) [84]. Isoprenyl-tethered tetrayne 179a displaying an even shorter one-atom link fails to produce the corresponding Alder-ene product 181a (R1 = 1-hexynyl) even under more drastic conditions [85]. However, increase of steric pressure with the rather bulky isopropanol group in triyne 179b (R1 = C(Me)2OH) facilitates the formation of the corresponding benzocyclobutene 181b (Scheme 51, (3)).
Scheme 51.

Alder-ene reactions of 1,3-butadiynamide-(179a–b)-derived arynes 180a–b.
Thermal HDDA reactions of allene-tethered 1,3-butadiynamides 182 deliver the corresponding aryne intermediates 183, whose reactivity and selectivity are divergent. The outcome of the intramolecular reaction of the allene with the aryne moiety depends on the substitution pattern of the allene and on the length of the tether (Scheme 52) [86]. The Alder-ene reaction involving an allylic C-H bond is favored with trisubstituted allenes (path a) leading to the eight-membered ring containing product 184. In contrast, the allenic C-H bond is preferentially involved with mono- and 1,3-disubstituted allenes affording the seven-membered Alder-ene product 185 (path b). Finally, [2+2] cycloaddition is the preferred path of intramolecular aryne reactions with a 1,1-disubstituted terminal allene furnishing the tetracyclic compound 186 (path c).
Scheme 52.

Intramolecular Alder-ene reactions or [2+2] cycloadditions with ynamide-derived tetraynes tethered with an allene.
The sole thermal reaction of N-arenesulfonyl 1,3-butadiynamides 187, spacing a terminal alkene moiety by a two-atom tether, leads to the pentacyclic structures 189 or 190 via an unprecedented aryne-mediated dearomatization of the phenyl connected to the sulfonyl moiety (Scheme 53) [84]. This transformation reveals the 1,2-dicarbene character of HDDA-generated arynes (intermediate 188). The efficiency of the reaction increases with cation-stabilizing substituents on the alkene (R2) and electron-withdrawing substituents on the arenesulfonyl group (R3). Interestingly, compounds 189a–c, bearing a haloalkene moiety (R2 = halogen), do not undergo subsequent acetic acid elimination to give the corresponding aromatization products 190. It is also worth noting that a ketone-containing triyne is a suitable substrate delivering the corresponding pentacyclic product 191 at a slightly higher reaction temperature of 120 °C.
Scheme 53.

Synthesis of pentacyclic products via HDDA-aryne-mediated arene dearomatization reaction.
A plausible mechanism, underlined by DFT calculations carried out on the simplified aryne structure 192, is given in Scheme 54. Alkene cyclopropanation is feasible due to the 1,2-dicarbene character of the HDDA-generated aryne and gives carbene 193. The latter undergoes nucleophilic addition onto the electron-deficient phenylsulfonyl moiety delivering 1,3-zwittterion 194. Subsequent ring expansion of the cyclopropane generates the 1,6-zwitterion 195, which leads to the final product 196 via intramolecular proton transfer.
Scheme 54.

Main intermediates of the proposed mechanistic pathway.
Acenes with indolylnaphthalene or indolylanthracene units are accessible through iterative intramolecular HDDA reaction cascades starting from 1,3-butadiynamide 197 (Scheme 55) [87]. A first intramolecular HDDA cycloaddition gives aryne 198 consecutively undergoing a second intramolecular HDDA reaction to provide the new aryne 199 as the reactive intermediate. The latter is trapped either by [4+2] cycloaddition with α-pyrone to give indolylanthracene 200 or by dichlorination with dilithium tetrachlorocuprate, leading to indolylnaphthalene 201, respectively, as the sole products.
Scheme 55.

Domino HDDA reactions towards polyacenes.
Aryne trapping via C-N bond formation: In situ trapping of HDDA-generated aryne 202 with simple tertiary amines such as triethylamine leads to the zwitterionic species 203, which delivers the same product 204 as being provided from 152 with diethylamine. Disproportionation of the zwitterion 203 by release of ethylene delivers the final product 204 (Scheme 56, (1)) [75]. This olefin elimination sequence was confirmed in the case of tertiary amines bearing activated β-protons such as β-aminoester 205 (Scheme 56, (2)) [88].
Scheme 56.

Tertiary amine addition onto HDDA-generated aryne.
However, in protic solvents, the preferential pathway does not involve intra- but intermolecular protonation of the zwitterionic species (209 → 210). Now, a quaternary ammonium center is formed following ring opening by a nucleophile in the case of cyclic tertiary amines (210 → 211) [88]. This reaction sequence also delivers poly-heterocyclic structures 211a–d based on three-component reactions (TCR) with HDDA-generated arynes 209 (Scheme 57) [31]. The addition of the amine onto benzyne 209 must be faster than that of the protic nucleophile. In some cases, such as acetic acid, the expected TCR product 211b (54%) was formed along with the direct benzyne–acetate addition product (27%). Thus, a two-step sequence based on the addition of triflic acid to generate ammonium triflates and their subsequent nucleophilic ring opening by nucleophilic displacement of the triflate in the second step broadens the scope of suitable nucleophiles.
Scheme 57.

Three-component reactions of HDDA-generated benzynes with (bi)cyclic tertiary amines and protic nucleophiles.
The TCR strategy is applicable to six-membered N-heteroaromatics as the nitrogen nucleophile. For instance, the reaction of 1,3-butadiynamide 158 with quinoline in chloroform provides the addition product 213 via betaine 212 (Scheme 58, (1)) [89]. Catching the aryne generated from 158 with triflic acid and N-heteroaromatics like quinoline or quinazolines delivers isolable N-arylinium triflates 214a–b, which undergo nucleophilic addition of Grignard reagents in a second reaction to give the heterocyclic products 215a–b (Scheme 58, (2)).
Scheme 58.

Reaction of HDDA-generated arynes with six-membered N-heteroaromatics.
The reaction of HDDA-generated benzyne starting from 1,3-butadiynamide 158 and diaziridines 216a–b delivers N-arylhydrazones 217a–b (Scheme 59, (1)) [90]. The more nucleophilic but also more hindered N-benzyl nitrogen atom in 216a–b adds to the aryne moiety. This reasons the formation of meta-isomer of 217a–b along with the expected major ortho-isomer.
Scheme 59.

Trapping of HDDA-generated benzynes with (1) diaziridines and (2) arylhydrazines.
Arylhydrazines are ambident nucleophiles, but in the case of para-nitro-phenylhydrazine 219, only the β-nitrogen atom (NH2) engages in the trapping of arynes thermally generated from tetraynes 158 (EWG = Ms) or 218 (EWG = Ts) (Scheme 59, (2)) [91]. The reaction is highly regioselective and subsequent oxidation of the resulting 1,2-diarylhydrazines 220a–b with MnO2 delivers azoarene products 221a–b.
The reaction of HDDA-generated arynes with C,N-diarylimine 222 proceeds efficiently, whereas this reaction is known to be poor yielding with o-benzynes generated by classical methods, i.e., by ortho-elimination of arene compounds (Scheme 60) [92]. Aromatization of the resulting dihydroacridine 223 by oxidation with MnO2 gives acridine 224. The conversion of 158 to 223 probably involves a formal [2+2] cycloaddition of an imine to a highly reactive aryne species. Initial imine addition on benzyne 161 provides betaine 225, which cyclizes to benzazetidine intermediate 226 to provide azo-quinomethide 227 after electrocyclic ring opening. Finally, a 6π electrocyclization followed by a proton shift gives dihydroacridine 223.
Scheme 60.

Trapping of HDDA-generated benzynes with a C,N-diarylimine.
Aryne trapping via C-O bond formation: Intramolecular HDDA reactions generated with 1,3-butadiynamide 228, linked via two carbons to a silyl ether unit, result in fully substituted benzene derivatives via formal aryne insertion into the O-silicon bond. For example, furanyl-annulated indoline 229 and carbazole 230 are products from aryne reactions involving C-O bond formation (Scheme 61) [18,76].
Scheme 61.

Intramolecular reaction of HDDA-generated benzynes from 1,3-butadiynamides tethered with a silyl ether.
Intermolecular trapping of HDDA-generated benzynes from tetrayne 158 with hydroxy-containing cyclic ethers such as glycidol 231a is also possible (Scheme 62, (1)) [93]. The reaction preferentially proceeds via the addition of the cyclic ether oxygen to the aryne. The resulting betaine 232 is protonated followed by ring opening of the cyclic oxonium ion delivering aryl ether 233a. Addition of the alcohol hydroxy of 231a to the aryne becomes a competitive pathway when a large excess of glycidol (100 equiv) is used. In the case of trisubstituted epoxide 231b, another concurrent pathway based on C–C-bond cleavage and aldehyde liberation is observed leading to aryl enol derivative 234 (Scheme 62, (2)). The use of the cyclopropanol derivative 236 as the nucleophile readily delivered the cyclobutanone compound 238 via a cationic-driven cyclopropanol to the cyclobutanone ring-enlargement reaction (Scheme 62, (3)).
Scheme 62.

Diversity of HDDA–aryne reaction modes with glycidol derivatives via Pinacol-like rearrangements or oxirane fragmentation.
HDDA reaction-generated arynes trapped by 3,3-disubstituted enals 240 offer a new entry to benzopyran motifs, in particular to the pyranocarbazole skeleton, as found in the products 241a–b (Scheme 63) [76]. Betaine 242 is the primary addition product and undergoes a formal [2+2] cycloaddition to the benzoxetene 243. Subsequent 4π-electrocyclic ring opening gives (Z)-dienone 244, which finally undergoes 6π-electrocyclic ring closure to restore the aromaticity and gives the pyran moiety. The use of exocyclic conjugated enals delivered a variety of fused spirocyclic benzopyran structures like 241c [94].
Scheme 63.

Catching HDDA-generated arynes with conjugated enals towards benzopyran structures.
With di(4-methoxy)phenyl sulfoxide (246) as an aryne-trapping agent, dimeric dibenzofuran S-shaped helicene 247 is obtained as major product along with carbazoles 248 and 249 starting from triyne 245 (Scheme 64) [95]. Remarkably, five new fused rings assemble within a single step under sole thermal activation.
Scheme 64.

Synthesis of dimeric dibenzofuran helicenes.
Mechanistically, the reaction of HDDA-generated benzyne 250 with sulfoxide 246 first leads to adduct 251 (Scheme 65). The latter interacts with a second benzyne molecule 250 to give tetracarbo-ligated σ-sulfurane 252. The hypervalent S(IV) species 252 is the key intermediate of this novel process. Reductive elimination of diarylsulfide 253 delivers the helicene product 247. The 1:1 adduct carbazole 248 is the SNAr reaction product of 251 via the Meisenheimer intermediate 254 (Scheme 65).
Scheme 65.

Mechanistic rationale for the formation of dibenzohelicene 247.
Aryne trapping via C-S bond formation: HDDA reactions of tetrayne 218 with its 1,3-butadiynamide moiety in the presence of either thiirane (255a) or tetrahydrothiophene 255b both furnish vinyl sulfide 258 (Scheme 66, (1)) [96]. The initial formation of a betaine 256 (o-sulfonium/arylcarbanion) is followed by intramolecular proton transfer leading to a more stable S-aryl sulfur ylide 257, which undergoes ring cleavage. Moreover, in the presence of a protic nucleophile, a three-component reaction takes place (Scheme 66, (2)). This becomes possible when the sulfide reacts faster with the aryne than the protic nucleophile. Intermolecular protonation of the resulting aryl carbanion leads to sulfonium 259. Subsequent ring opening by nucleophile addition affords the final products 260a–c. It is worthy of note that thiirane (255a) is not a suitable component for such a process because fragmentation to the vinyl sulfide is faster than nucleophile addition.
Scheme 66.

Reaction of HDDA-generated arynes with cyclic sulfides.
The HDDA reaction of tetraynes 158 (EWG = Ms) or 218 (EWG = Ts) with aryl thioamides 261 affords the dihydrobenzothiazine-derived molecules 262a–c in a regio- and diastereoselective fashion (Scheme 67) [97]. Notably, these are the first examples of the use of thioamides as aryne-trapping agents. The proposed mechanism involves the formation of benzothietene 263 and its ring opening to give intermediate 264 or its resonance form o-thiolatoaryliminium 265. Intramolecular 1,3-hydrogen atom migration delivers the iminium zwitterion 267, whose cyclization leads to products 262a–c. The analogous product 268 was also obtained using a thiourea as reaction partner.
Scheme 67.

Reaction of HDDA-generated arynes with aromatic thioamides.
Aryne trapping via C-B bond formation: The hydroboration of the aryne that was generated by an HDDA reaction with tetrayne 269, with the N-heterocyclic carbene borane (NHC-borane) 270, provides the highly functionalized arylborane compound 271 as a single isomer (Scheme 68) [98]. The feasibility of the reaction relies on the fact that NHC-boranes are deactivated and do not hydroborate the starting tetrayne unlike most other borane reagents.
Scheme 68.

Trapping of HDDA-generated arynes with NHC-boranes.
6.2. Silver-Catalyzed HDDA Reactions
Ag(I)-bound aryne trapping via C-C bond formation: Alkane C-H bonds can be activated by 1,3-butadiyne-HDDA-reaction generated arynes in the presence of silver(I) salts (Scheme 69) [99]. Importantly, under Ag(I)-free reaction conditions, the alkane C-H insertion fails to appear. According to mechanistic studies, the C(sp3)-H bond-breaking and C(sp2)-H bond-forming processes are concerted and induced via the silver-carbenoid intermediate 273.
Scheme 69.

Reaction of HDDA-generated aryne via silver-catalyzed alkane C-H insertion.
The fact that Ag(I) salts drive the character of the aryne reactivity from an alkyne towards a carbene character was nicely shown with the silyl-substituted substrates 275 bearing a primary C-H bond (Scheme 70, (1)) [100]. In the presence of a secondary or tertiary C-H bond on the β-carbon of the silyl group, hydride transfer occurs instead of C-H insertion (Scheme 70, (2)). This result is plausible considering a favorable hyperconjugation of the β-silicon stabilized carbocation 280, whose reaction with water delivers the formal aryne hydrogenation products 281a–b.
Scheme 70.

Intramolecular trapping of HDDA-generated aryne from silyl-substituted substrates.
Silver-catalyzed reactions of non-activated benzenes 283 with 1,3-butadiynamide-282–HDDA reaction-generated arynes lead to the hydroarylation products 284a–c (Scheme 71) [101]. Diels–Alder reaction products of arynes with arenes, as usually found with “free arynes”, stay out and are not formed. The chemical outcome of this transformation is rationalized by an electrophilic aromatic substitution through the formation of a Wheland-type intermediate 286 (arenium ion) and subsequent water-catalyzed proton transfer.
Scheme 71.

Intermolecular hydroarylation of silver-complexed HDDA-generated arynes.
Ag(I)-bound aryne trapping via C-N bond formation: Nitriles are too weak nucleophiles to react with arynes. However, in the presence of a silver catalyst, the trapping of HDDA-generated aryne is taking place with nitriles readily via the formation of nitrilium ion intermediates 291a–b. The latter can react with water or with acetic acid under anhydrous conditions to give indolinyl amide 292 or imide 293, respectively, starting from tetrayne 288 (Scheme 72, (1)) [102].
Scheme 72.

Reaction of silver-complexed HDDA-generated arynes with nitriles.
Under anhydrous conditions and in the absence of carboxylic acids, tetrayne 288 converts into quinazoline 296 by incorporation of two benzonitrile molecules (289c) (Scheme 72, (2)) [103]. Now, the nitrilium ion 291c interacts with a second nitrile molecule to give the resonance-stabilized complex 294. Transformation of 294 into bis-nitrile adduct 295 and the subsequent ring closure finally delivers quinazoline 296.
Ag(I)-bound aryne trapping via C-O bond formation: Water, unlike alcohols or carboxylic acids, is a less suitable aryne-trapping agent to deliver subsequent phenols. This is probably because of the immiscibility of water in organic solvents where transient arynes are generated. Silver trifluoroacetate (AgO2CCF3) turned out to be a suitable water surrogate to obtain phenol-related compounds [75,104]. Indeed, the reaction of 1,3-butadiynes 152 or 297 with AgO2CCF3 leads via transient HDDA reaction-generated arynes 298 to trifluoroacetoxy organosilver arenes 299, whose hydrolysis on silica gel furnishes the phenolic compounds 300a–b (Scheme 73).
Scheme 73.

Synthesis of phenolic compounds from HDDA-generated arynes.
Ag(I)-bound aryne trapping with fluorine-containing reagents: Fluorination, trifluoromethylation and trifluoromethylthiolation of HDDA-generated arynes from 1,3-butadiynes 301 were successfully achieved by using AgBF4, AgCF3 (in situ generated from AgF and TMSCF3) and AgSCF3 in stoichiometric quantities, respectively (Scheme 74) [105]. The trifluoromethylation reaction is not proceeding with terminal (R = H) or tertiary alkyl-substituted 1,3-butadiynamides as starting materials (see 303b, 303d). A catalytic protocol for aryne fluorination appeared. It relies on the use of AgBF4 (10 mol%) as the catalyst and the BF4•pyridinium salt (1.5 equiv) as the fluoride source. Importantly, fluorination fails in the absence of silver salts.
Scheme 74.

Silver-mediated fluorination, trifluoromethylation and trifluoromethylthiolation of HDDA-generated arynes.
6.3. HDDA Reactions Catalyzed by Other Metals Than Silver
The hydrohalogenative aromatization of ynamide-derived tetraynes 305 readily proceeds in halogenated hydrocarbons CH2X2 (X = Cl, Br, I) as the solvent in the presence of Grubbs-type ruthenium alkylidene complex 306 and delivers novel halogenated indoline derivatives 308a–e (Scheme 75) [106]. The proposed mechanism involves the activation of CH2 × 2 by the ruthenium catalyst for the HX transfer to ruthenium-complexed aryne intermediate 307.
Scheme 75.

Ruthenium-catalyzed hydrohalogenation of HDDA-generated arynes.
The addition of 1-bromoalkynes 309 or terminal alkynes 311 to 1,3-butadiyne-218-HDDA-reaction generated aryne proceeds efficiently under copper catalysis (Scheme 76) [107]. Interestingly, the insertion of the alkynyl moiety into the aryne carbon–carbon triple bond to give either indolines 310a–e or 312a–e, respectively, takes place at complementary positions.
Scheme 76.

Copper-catalyzed bromo- and hydroalkynylation of HDDA-generated arynes.
Two different catalytic cycles A and B are proposed to rationalize the outcome of these reactions (Scheme 77). In the case of the bromoalkynylation, CuBr adds to in situ generated aryne 313 (from 1,3-diynamide 218) to give aryl copper (I) species 314. Oxidative addition of this Cu(I) species to 1-bromoalkyne 309 gives the aryl copper(III) intermediate 315. Subsequent reductive elimination produces the 4,7-diethynyldihydroindole 310 and regenerates the copper(I) catalyst. In the case of the catalytic hydroalkynylation reaction (catalytic cycle B), aryne 313 undergoes carbocupration with in situ generated copper acetylide 316 from terminal alkyne 311, providing aryl copper species 317. Proton exchange with terminal alkyne 311 delivers compound 312 and closes the catalytic cycle.
Scheme 77.

Proposed mechanism of copper-catalyzed bromoalkynylation and hydroalkynylation of HDDA-generated arynes.
The catalytic reaction of tetrayne 218 with homopropargyl alcohol (318) selectively gives the hydroalkynylation product 319, whereas compound 320 resulting from the addition of the alcohol moiety on the transient aryne 313 is the major compound in the absence of copper catalyst (Scheme 78).
Scheme 78.

Different outcomes of the reaction of HDDA-generated aryne with homopropargyl alcohol with and without copper catalyst.
7. Conclusions and Perspectives
The present review covers the synthesis, molecular properties and use of 1,3-butadiynamides in heterocyclic chemistry. Based on this comprehensive summary and its compiled reactions, which not only focused on applications in organic synthesis, but also on mechanistic aspects, it becomes obvious that 1,3-butadiynamides—the ethynylogous variant of ynamides—are more than just simple alkynes: they should be considered as a new class of compounds showing its own specific reactivity.
Their potential as building blocks for the construction of complex molecular scaffolds has emerged only recently, although the synthesis of symmetrical and unsymmetrical 1,3-butadiynamides was described more than 15 years ago. Terminal ynamides are useful precursors of 1,3-butadiynamides for their use in the Glaser–Hay coupling reaction or the Cadiot–Chodkiewicz cross-coupling reaction with 1-bromoalkynes. The latter emerged to a broadly used and indispensable synthetic method to access various highly functionalized non-symmetrical 1,3-butadiynamides. Alternative protocols based on the direct N-buta-1,3-diynylation of amides appeared just recently and will find further attention.
The extended conjugation and polarized character of 1,3-butadiynamides facilitate their use in addition and cycloaddition reactions with predictable regioselectivity. Many of them involve gold catalysis along with the successful use of other metal salts (Pd, Ag, Cu/Zn). Moreover, 1,3-butadiynamides are easily derivatized by introducing judicious functional groups tethered to the N-atom and/or to the C-terminal of the 1,3-diynyl moiety. Such diversely functionalized 1,3-butadiynamides serve as highly useful molecular scaffolds in the development of new reaction cascades.
Intensive studies have been dedicated to thermal or metal-catalyzed reactions with 1,3-butadiynamides tethered to a monoalkyne or 1,3-butadiyne moiety. These tetra- or triynes having a 1,3-butadiynamide unit are privileged substrates for the HDDA reaction because they lead to the in situ formation of a single aryne isomer, and its intermolecular trapping takes place regioselectively. Reagent-free, highly chemo- and regioselective aryne formation with regioselective follow-up reactions by inter- or intramolecular aryne trapping not only enhanced the chemistry of 1,3-butadiynamides, but also boosted the understanding of different reaction channels available for in situ generated aryne species. HDDA reactions with 1,3-butadiynamide units can be performed solely thermally or in the presence of sub-stoichiometric amounts of Ag(I)-salts. The latter often changes the mode of reactivity towards a more carbenoid character of the aryne intermediate and opens new alternative reaction channels.
Most described 1,3-butadiynamides are derived from sulfonamides. It is conceivable that the synthesis of carbamate- or oxazolidinone-related 1,3-butadiynamides will be further explored and that new reaction sequences will be discovered, as the strength of the nitrogen bound EWG group will alter the 1,3-butadiynamide reactivity.
The more classical activation of 1,3-butadiynamides relies on the in situ generation of a keteniminium species to accelerate follow-up transformations. Recently, a new reaction mode—the in situ liberation of [4]cumulenenimines from 1,3-butadiynamides—was revealed. This finding will probably initiate the development of novel reaction cascades and broaden the array of available structurally complex molecules.
Finally, the examples of optically active 1,3-butadiynamides are scarce, and the possibility of chirality transfer during reaction cascades has not been investigated yet. Asymmetric or enantioselective synthesis relying on the transformation of 1,3-butadiynamides will certainly be a future topic to gain access to the chiral world of heterocycles. Surely, the chemistry of 1,3-butadiynamides is still at its infancy and more new reactions and more sophisticated applications, especially in the field of heterocyclic chemistry, will appear in near future.
Acknowledgments
The continuous support of our work by the CNRS, the Conseil Régional de Normandie, the European FEDER funding, and the Agence Nationale de la Recherche (ANR-22-CE07-0010) is gratefully acknowledged.
Author Contributions
All authors contributed equally to this article. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This work was funded by the Agence Nationale de la Recherche (ANR-22-CE07-0010).
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Witulski B., Stengel T. N-Functionalized 1-Alkynylamides: New Building Blocks for Transition Metal Mediated Inter- and Intramolecular [2+2+1] Cycloadditions. Angew. Chem. Int. Ed. 1998;37:489–492. doi: 10.1002/(SICI)1521-3773(19980302)37:4<489::AID-ANIE489>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 2.Iftikhar R., Mazhar A., Iqbal M.S., Khan F.Z., Askary S.H., Sibtain H. Ring Forming Transformation of Ynamides via Cycloaddition. RSC. Adv. 2023;13:10715–10756. doi: 10.1039/D3RA00139C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu Y.-C., Zhao Y., Wan B., Chen Q.-A. Reactivity of Ynamides in Catalytic Intermolecular Annulations. Chem. Soc. Rev. 2021;50:2582–2625. doi: 10.1039/D0CS00283F. [DOI] [PubMed] [Google Scholar]
- 4.Shandilya S., Gogoi M.P., Dutta S., Sahoo A.K. Gold-Catalyzed Transformation of Ynamides. Chem. Rec. 2021;21:4123–4149. doi: 10.1002/tcr.202100159. [DOI] [PubMed] [Google Scholar]
- 5.Campeau D., Rayo D.F.L., Mansour A., Muratov K., Gagosz F. Gold-Catalyzed Reactions of Specially Activated Alkynes, Allenes and Alkenes. Chem. Rev. 2021;121:8756–8867. doi: 10.1021/acs.chemrev.0c00788. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y.-B., Qian P.-C., Ye L.-W. Brønsted Acid-Mediated Reactions of Ynamides. Chem. Soc. Rev. 2020;49:8897–8909. doi: 10.1039/D0CS00474J. [DOI] [PubMed] [Google Scholar]
- 7.Lynch C.C., Sripada A., Wolf C. Asymmetric Synthesis with Ynamides: Unique Reaction Control, Chemical Diversity and Applications. Chem. Soc. Rev. 2020;49:8543–8583. doi: 10.1039/D0CS00769B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan T.-D., Wang Z.-S., Qian P.-C., Ye L.-W. Radical Reactions of Ynamides. Small Methods. 2021;5:2000673. doi: 10.1002/smtd.202000673. [DOI] [PubMed] [Google Scholar]
- 9.Mahe C., Cariou K. Ynamides in Free Radical Reactions. Adv. Synth. Catal. 2020;362:4820–4832. doi: 10.1002/adsc.202000849. [DOI] [Google Scholar]
- 10.Hong F.-L., Ye L.-W. Transition Metal-Catalyzed Tandem Reactions of Ynamides for Divergent N-Heterocycle Synthesis. Acc. Chem. Res. 2020;53:2003–2019. doi: 10.1021/acs.accounts.0c00417. [DOI] [PubMed] [Google Scholar]
- 11.Zhou B., Tan T.-D., Zhu X.-Q., Shang M., Ye L.-W. Reversal of Regioselectivity in Ynamide Chemistry. ACS Catal. 2019;9:6393–6406. doi: 10.1021/acscatal.9b01851. [DOI] [Google Scholar]
- 12.Prabagar B., Ghosh N., Sahoo A.K. Cyclization and Cycloisomerization of π-Tethered Ynamides: An Expedient Synthetic Method to Construct Carbo- and Heterocycles. Synlett. 2017;28:2539–2555. [Google Scholar]
- 13.Cook A.M., Wolf C. Terminal Ynamides: Synthesis, Coupling Reactions, and Additions to Common Electrophiles. Tetrahedron Lett. 2015;56:2377–2392. doi: 10.1016/j.tetlet.2015.03.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X.-N., Yeom H.-S., Fang L.-C., He S., Ma Z.-X., Kedrowski B.L., Hsung R.P. Ynamides in Ring Forming Transformations. Acc. Chem. Res. 2014;47:560–578. doi: 10.1021/ar400193g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evano G., Coste A., Jouvin K. Ynamides: Versatile Tools in Organic Synthesis. Angew. Chem. Int. Ed. 2010;49:2840–2859. doi: 10.1002/anie.200905817. [DOI] [PubMed] [Google Scholar]
- 16.DeKorver K.A., Li H., Lohse A.G., Hayashi R., Lu Z., Zhang Y., Hsung R.P. Ynamides: A Modern Functional Group for the New Millennium. Chem. Rev. 2010;110:5064–5106. doi: 10.1021/cr100003s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fluegel L.L., Hoye T.R. Hexadehydro-Diels–Alder Reaction: Benzyne Generation via Cycloisomerization of Tethered Triynes. Chem. Rev. 2021;121:2413–2444. doi: 10.1021/acs.chemrev.0c00825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoye T.R., Baire B., Niu D., Willoughby P.H., Woods B.P. The hexadehydro-Diels–Alder reaction. Nature. 2012;490:208–212. doi: 10.1038/nature11518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holden (née Hall) C., Greaney M.F. The Hexadehydro-Diels–Alder Reaction: A New Chapter in Aryne Chemistry. Angew. Chem. Int. Ed. 2014;53:5746–5749. doi: 10.1002/anie.201402405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diamond O.J., Marder T.B. Methodology and Applications of the Hexadehydro-Diels-Alder (HDDA) Reaction. Org. Chem. Front. 2017;4:891–910. doi: 10.1039/C7QO00071E. [DOI] [Google Scholar]
- 21.Yu H., Xu F. Advances in the Synthesis of Nitrogen-Containing Heterocyclic Compounds by in situ Benzyne Cycloaddition. RSC Adv. 2023;13:8238–8253. doi: 10.1039/D3RA00400G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez D., Castedo L., Saa C. Homocoupling of 1-Alkynyl Tosylamides. Synlett. 2004;2004:377–379. doi: 10.1002/chin.200424095. [DOI] [Google Scholar]
- 23.Doan T.-H., Talbi I., Lohier J.-F., Touil S., Alayrac C., Witulski B. Synthesis, Crystal Structure, Optical, Electrochemical and Thermal Properties of the Ynamide: Bis-(N-4-methylbenzenesulfonyl, N-n-butyl)-1,3-butadiyne-1,4-diamide. J. Mol. Struct. 2016;1116:127–134. doi: 10.1016/j.molstruc.2016.03.026. [DOI] [Google Scholar]
- 24.Takai R., Shimbo D., Tada N., Itoh A. Ligand-Enabled Copper-Catalyzed N-Alkynylation of Sulfonamide with Alkynyl Benziodoxolone: Synthesis of Amino Acid-Derived Ynamide. J. Org. Chem. 2021;86:4699–4713. doi: 10.1021/acs.joc.1c00101. [DOI] [PubMed] [Google Scholar]
- 25.Witulski B., Schweikert T., Schollmeyer D., Nemkovich N.A. Synthesis and Molecular Properties of Donor-π-Spacer-Acceptor Ynamides with up to Four Conjugated Alkyne Units. Chem. Commun. 2010;46:2953–2955. doi: 10.1039/b919275a. [DOI] [PubMed] [Google Scholar]
- 26.Talbi I., Alayrac C., Lohier J.-F., Touil S., Witulski B. Application of Ynamides in the Synthesis of 2-(Tosylamido)- and 2,5-Bis(tosylamido)thiophenes. Org. Lett. 2016;18:2656–2659. doi: 10.1021/acs.orglett.6b01101. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y.-P., Danheiser R.L. Synthesis of 2-Iodoynamides and Regioselective [2+2] Cycloadditions with Ketene. Tetrahedron Lett. 2011;52:2111–2114. doi: 10.1016/j.tetlet.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tracey M.R., Zhang Y., Frederick M.O., Mulder J.A., Hsung R.P. Sonogashira Cross-Couplings of Ynamides. Syntheses of Urethane- and Sulfonamide-Terminated Conjugated Phenylacetylenic Systems. Org. Lett. 2004;6:2209–2212. doi: 10.1021/ol0493251. [DOI] [PubMed] [Google Scholar]
- 29.Dunetz J.R., Danheiser R.L. Copper-Mediated N-Alkynylation of Carbamates, Ureas, and Sulfonamides. A General Method for the Synthesis of Ynamides. Org. Lett. 2003;5:4011–4014. doi: 10.1021/ol035647d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kohnen A.L., Dunetz J.R., Danheiser R.L. Synthesis of Ynamides by N-Alkynylation of Amine Derivatives. Preparation of N-Allyl-N-(Methoxycarbonyl)-1,3-Decadiynylamine. Org. Synth. 2007;84:88–101. [PMC free article] [PubMed] [Google Scholar]
- 31.Ross S.P., Hoye T.R. Multiheterocyclic Motifs via Three-Component Reactions of Benzynes, Cyclic Amines, and Protic Nucleophiles. Org. Lett. 2018;20:100–103. doi: 10.1021/acs.orglett.7b03458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y., Hsung R.P., Tracey M.R., Kurtz K.C.M., Vera E.L. Copper Sulfate-Pentahydrate-1,10-Phenanthroline Catalyzed Amidations of Alkynyl Bromides. Synthesis of Heteroaromatic Amine Substituted Ynamides. Org. Lett. 2004;6:1151–1154. doi: 10.1021/ol049827e. [DOI] [PubMed] [Google Scholar]
- 33.Wang T., Niu D., Hoye T.R. The Hexadehydro-Diels–Alder Cycloisomerization Reaction Proceeds by a Stepwise Mechanism. J. Am. Chem. Soc. 2016;138:7832–7835. doi: 10.1021/jacs.6b03786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mansfield S.J., Smith R.C., Yong J.R.J., Garry O.L., Anderson E.A. A General Copper-Catalyzed Synthesis of Ynamides from 1,2-Dichloroenamides. Org. Lett. 2019;21:2918–2922. doi: 10.1021/acs.orglett.9b00971. [DOI] [PubMed] [Google Scholar]
- 35.Kawakarni R., Usui S., Tada N., Itoh A. Late-Stage Diversification Strategy for Synthesizing Ynamides through Copper-Catalyzed Diynylation and Azide-Alkyne Cycloaddition. Chem. Commun. 2023;59:450–453. doi: 10.1039/d2cc05575a. [DOI] [PubMed] [Google Scholar]
- 36.Gohrai S., Lee D. Selectivity for Alkynyl or Allenyl Imidamides and Imidates in Copper-Catalyzed Reactions of Terminal 1,3-Diynes and Azides. Org. Lett. 2021;23:697–701. doi: 10.1021/acs.orglett.0c03861. [DOI] [PubMed] [Google Scholar]
- 37.Garner M.H., Hoffmann R., Rettrup S., Solomon G.C. Coarctate and Möbius: The Helical Orbitals of Allene and Other Cumulenes. ACS Cent. Sci. 2018;4:688–700. doi: 10.1021/acscentsci.8b00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garner M.H., Jensen A., Hyllested L.O.H., Solomon G.C. Helical Orbitals and Circular Currents in Linear Carbon Wires. Chem. Sci. 2019;10:4598–4608. doi: 10.1039/C8SC05464A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garner M.H., Corminboeuf C. Helical Electronic Transitions of Spiroconjugated Molecules. Chem. Commun. 2021;57:6408–6411. doi: 10.1039/D1CC01904J. [DOI] [PubMed] [Google Scholar]
- 40.Ohashi S., Inagaki S. Orbital Phase Control of Conformations of Alkyne Derivatives. Tetrahedron. 2001;57:5361–5367. doi: 10.1016/S0040-4020(01)00452-5. [DOI] [Google Scholar]
- 41.Hendon C.H., Tiana D., Murray A.T., Carbery D.R., Walsh A. Helical Frontier Orbitals of Conjugated Linear Molecules. Chem. Sci. 2013;4:4278–4284. doi: 10.1039/c3sc52061g. [DOI] [Google Scholar]
- 42.Balakrishnan A.R., Suresh R., Vijayakumar S. Amine Terminated Polyynes as Candidates for Molecular Wire Applications: A DFT Study. Phys. E Low-Dimens. Syst. Nanostruct. 2022;137:115045. doi: 10.1016/j.physe.2021.115045. [DOI] [Google Scholar]
- 43.Bro-Jorgensen W., Garner M.H., Solomon G.C. Quantification of the Helicality of Helical Molecular Orbitals. J. Phys. Chem. A. 2021;125:8107–8115. doi: 10.1021/acs.jpca.1c05799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petrov A.R., Daniliuc C.G., Jones P.G., Tamm M. A Novel Synthetic Approach to Diaminoacetylenes: Structural Characterization and Reactivity of Aromatic an Aliphatic Ynediamines. Chem. Eur. J. 2010;16:11804–11808. doi: 10.1002/chem.201002211. [DOI] [PubMed] [Google Scholar]
- 45.Tokutome Y., Okuno T. Preparations, Crystal Polymorphs and DFT Calculations of N1,N1,N4,N4-Tetraphenylbuta-1,3-diyne-1,4-diamine. J. Mol. Struct. 2013;1047:136–142. doi: 10.1016/j.molstruc.2013.05.013. [DOI] [Google Scholar]
- 46.Mayerle J.J., Flandera M.A. Bis(1-carbazolyl)butadiyne. Acta Cryst. 1978;B34:1374–1376. doi: 10.1107/S0567740878005646. [DOI] [Google Scholar]
- 47.Matsuda H., Nakanishi H., Hosomi T., Kato M. Synthesis and Solid-State Polymerization of a New Diacetylene: 1-(N-Carbazolyl)penta-1,3-diyn-5-ol. Macromolecules. 1988;21:1238–1240. doi: 10.1021/ma00183a010. [DOI] [Google Scholar]
- 48.Tabata H., Kuwamoto K., Okuno T. Conformational Polymorphs and Solid-State Polymerization of 9-(1,3-Butadiynyl)carbazole Derivatives. J. Mol. Struct. 2016;1106:452–459. doi: 10.1016/j.molstruc.2015.11.022. [DOI] [Google Scholar]
- 49.Ide M., Ohashi K., Mihara S., Iwasaha T. Regio- and Stereoselective hydrohalogenation of Ynamide Components in 1,3-Butadiynes with in situ Generated HX. Tetrahedron Lett. 2014;55:2130–2133. doi: 10.1016/j.tetlet.2014.02.028. [DOI] [Google Scholar]
- 50.De la Vega-Hernandez K., Romain E., Coffinet A., Bijouard K., Gontard G., Chemla F., Ferreira F., Jackowski O., Perez-Luna A. Radical Germylzincation of α-Heteroatom-Substituted Alkynes. J. Am. Chem. Soc. 2018;140:17632–17642. doi: 10.1021/jacs.8b09851. [DOI] [PubMed] [Google Scholar]
- 51.Kramer S., Madsen J.L.H., Rottländer M., Skrydstrup T. Access to 2,5-Diamidopyrroles and 2,5-Diamidofurans by Au(I)-Catalyzed Double Hydroamination or Hydration of 1,3-Diynes. Org. Lett. 2010;12:2758–2761. doi: 10.1021/ol1008685. [DOI] [PubMed] [Google Scholar]
- 52.Choudhary S., Gayyur, Ghosh N. Cu(II)-Catalyzed [4+1] and [4+3] Annulation Reactions: A Modular Approach to N-Aryl/Alkyl Substituted 2,5-Diamidopyrroles and Diazepines. Org. Biomol. Chem. 2022;20:7017–7021. doi: 10.1039/D2OB01458K. [DOI] [PubMed] [Google Scholar]
- 53.Dunetz J.R., Danheiser R.L. Synthesis of Highly Substituted Indolines and Indoles via Intramolecular [4+2] Cycloaddition of Ynamides and Conjugated Enynes. J. Am. Chem. Soc. 2005;127:5776–5777. doi: 10.1021/ja051180l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liao Y., Lu Q., Chen G., Yu Y., Li C., Huang X. Rhodium-Catalyzed Azide-Alkyne Cycloaddition of Internal Ynamides: Regioselective Assembly of 5-Amino-Triazoles under Mild Conditions. ACS Catal. 2017;7:7529–7534. doi: 10.1021/acscatal.7b02558. [DOI] [Google Scholar]
- 55.Davies P.W., Cremonesi A., Dumitrescu L. Intermolecular and Selective Synthesis of 2,4,5-Trisubstituted Oxazoles by a Gold-Catalyzed Formal [3+2] Cycloaddition. Angew. Chem. Int. Ed. 2011;50:8931–8935. doi: 10.1002/anie.201103563. [DOI] [PubMed] [Google Scholar]
- 56.Skaria M., Hsu Y.-C., Jiang Y.-T., Lu M.-Y., Kuo T.-C., Cheng M.-J., Liu R.-S. Gold-Catalyzed Oxidations of 1,3-Diynamides with C(1) versus C(3) Regioselectivity: Catalyst-Dependent Oxidative Cyclizations in the C(3) Oxidation. Org. Lett. 2020;22:4478–4482. doi: 10.1021/acs.orglett.0c01480. [DOI] [PubMed] [Google Scholar]
- 57.Lee D., Kim M. Advances in the Metallotropic [1,3]-Shift of Alkynyl Carbenoids. Org. Biomol. Chem. 2007;5:3418–3427. doi: 10.1039/b710379d. [DOI] [PubMed] [Google Scholar]
- 58.Yanai H., Kawazoe T., Ishii N., Witulski B., Matsumoto T. Regioselective Synthesis of 4-Aryl-1,3-dihydroxy-2-naphthoates through 1,2-Aryl-Migrative Ring Rearrangement Reaction and their Photoluminescence Properties. Chem. Eur. J. 2021;27:11442–11449. doi: 10.1002/chem.202101459. [DOI] [PubMed] [Google Scholar]
- 59.Kumar M., Kaliya K., Maurya S.K. Recent Progress in the Homogeneous Gold-Catalysed Cycloisomerisation Reactions. Org. Biomol. Chem. 2023;21:3276–3295. doi: 10.1039/D2OB02015G. [DOI] [PubMed] [Google Scholar]
- 60.Liu J., Chakraborty P., Zhang H., Zhong L., Wang Z.-X., Huang X. Gold-Catalyzed Atom-Economic Synthesis of Sulfone-Containing Pyrrolo [2,1-a]isoquinolines from Diynamides: Evidence for Consecutive Sulfonyl migration. ACS Catal. 2019;9:2610–2617. doi: 10.1021/acscatal.8b04934. [DOI] [Google Scholar]
- 61.Choudhary S., Gayyur, Ghosh N. Gold(I)-Catalyzed and PTSA-Promoted Cycloisomerization of Ynamides to Access Pyrrole Substituted α,β-Unsaturated Ketones. Eur. J. Org. Chem. 2023;26:e202201223. doi: 10.1002/ejoc.202201223. [DOI] [Google Scholar]
- 62.Wang H.F., Guo L.N., Fan Z.B., Tang T.H., Zi W. Gold-Catalyzed Formal Hexadehydro-Diels–Alder/Carboalkoxylation Reaction Cascades. Org. Lett. 2021;23:2676–2681. doi: 10.1021/acs.orglett.1c00581. [DOI] [PubMed] [Google Scholar]
- 63.Liu N., Sun H., Wang J., Zhang Z., Wang T. Ag(I)-Catalyzed Synthesis of 2-Aminoquinolines from 1-Aminobutadiynes and Anilines. Adv. Synth. Catal. 2021;363:5443–5447. doi: 10.1002/adsc.202100952. [DOI] [Google Scholar]
- 64.Wang J., Du J., Wang T. Synthesis of 2-Aminopyrrolo[1,2-b]pyridazines via Gold(I)-Catalyzed Chemoselective Hydroamination/Hydroarylation Cascade of 1,3-Diynamides with 1-Aminopyrroles. Adv. Synth. Catal. 2023;365:1088–1092. doi: 10.1002/adsc.202300075. [DOI] [Google Scholar]
- 65.Gayyur, Choudhary S., Kant R., Ghosh N. Synergetic Copper/Zinc Catalysis: Synthesis of Aryl/Heteroaryl-Fused 1H-Pyrrolo[3,2-c]pyridines. Chem. Commun. 2022;58:1974–1977. doi: 10.1039/D1CC05514C. [DOI] [PubMed] [Google Scholar]
- 66.Pandit Y.B., Jiang Y.-T., Jian J.-J., Chen T.-C., Kuo T.-C., Cheng M.-J., Liu R.-S. Gold-Catalyzed [5+2]-Annulations of 1,3-Diyn-1-amides with Anthranils Bearing no C(6)-Substituents. Org. Chem. Front. 2021;8:2563–2568. doi: 10.1039/D1QO00026H. [DOI] [Google Scholar]
- 67.Tian X., Song L., Farshadfar K., Rudolph M., Rominger F., Oeser T., Ariafard A., Hashmi A.S.K. Acyl Migration versus Epoxidation in Gold Catalysis: Facile, Switchable, and Atom-Economic Synthesis of Acylindoles and Quinoline Derivatives. Angew. Chem. Int. Ed. 2020;59:471–478. doi: 10.1002/anie.201912334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu J., Zhu L., Wan W., Huang X. Gold-Catalyzed Cascade Cyclization of 1,3-Diynamides: Polycyclic N-Heterocycle Synthesis via Construction of a Furopyridinyl Core. Org. Lett. 2020;22:3279–3285. doi: 10.1021/acs.orglett.0c01086. [DOI] [PubMed] [Google Scholar]
- 69.Xia J., Liu J., Yu Y., Zhang J., Huang X. Divergent Access to Polycyclic N-Heterocyclic Compounds through Büchner-Type Dearomatisation Enabled Cycloisomerization of Diynamides under Gold Catalysis. Org. Lett. 2022;24:4298–4303. doi: 10.1021/acs.orglett.2c01807. [DOI] [PubMed] [Google Scholar]
- 70.Lenko I., Mamontov A., Alayrac C., Legay R., Witulski B. Media-Driven Pd-Catalyzed Reaction Cascades with 1,3-Diynamides Leading Selectively to Either Indoles or Quinolines. Angew. Chem. Int. Ed. 2021;60:22729–22734. doi: 10.1002/anie.202110221. [DOI] [PubMed] [Google Scholar]
- 71.Witulski B., Alayrac C., Tevzadze-Saeftel L. Palladium-Catalyzed Synthesis of 2-Aminoindoles by a Heteroannulation Reaction. Angew. Chem. Int. Ed. 2003;42:4257–4260. doi: 10.1002/anie.200351977. [DOI] [PubMed] [Google Scholar]
- 72.Bradley A.Z., Johnson R.P. Thermolysis of 1,3,8-Nonatriyne: Evidence for Intramolecular [2+4] Cycloaromatization to a Benzyne Intermediate. J. Am. Chem. Soc. 1997;119:9917–9918. doi: 10.1021/ja972141f. [DOI] [Google Scholar]
- 73.Miyawaki K., Suzuki R., Kawano T., Ueda I. Cycloaromatization of a Non-Conjugated Polyenyne System: Synthesis of 5H-Benzo[d]fluoreno[3,2-b]pyrans via Diradicals Generated from 1-[2-{4-(2-Alkoxymethylphenyl)butan-1,3-diynyl}]phenylpentan-2,4-diyn-1-ols and Trapping Evidence for the 1,2-Didehydrobenzene Diradical. Tetrahedron Lett. 1997;38:3943–3946. [Google Scholar]
- 74.Liang Y., Hong X., Yu P., Houk K.N. Why Alkynyl Substituents Dramatically Accelerate Hexadehydro-Diels–Alder (HDDA) Reactions: Stepwise Mechanisms of HDDA Cycloadditions. Org. Lett. 2014;16:5702–5705. doi: 10.1021/ol502780w. [DOI] [PubMed] [Google Scholar]
- 75.Karmakar R., Yun S.Y., Wang K.-P., Lee D. Regioselectivity in the Nucleophile Trapping of Arynes: The Electronic and Steric Effects of Nucleophiles and Substituents. Org. Lett. 2014;16:6–9. doi: 10.1021/ol403237z. [DOI] [PubMed] [Google Scholar]
- 76.Wang T., Hoye T.R. Hexadehydro-Diels–Alder (HDDA)-Enabled Carbazolyne Chemistry: Single Step, de Novo Construction of the Pyranocarbazole Core of Alkaloids of the Murraya koenigii (Curry Tree) Family. J. Am. Chem. Soc. 2016;138:13870–13873. doi: 10.1021/jacs.6b09628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ross S.P., Hoye T.R. Reactions of Hexadehydro-Diels–Alder Benzynes with Structurally Complex Multifunctional Natural Products. Nat. Chem. 2017;9:523–530. doi: 10.1038/nchem.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen M., He C.Q., Houk K.N. Mechanism and Regioselectivity of an Unsymmetrical Hexadehydro-Diels–Alder (HDDA) Reaction. J. Org. Chem. 2019;84:1959–1963. doi: 10.1021/acs.joc.8b02865. [DOI] [PubMed] [Google Scholar]
- 79.Cheong P.H.-Y., Paton R.S., Bronner S.M., Im G.-Y.J., Garg N.K., Houk K.N. Indolyne and Aryne Distortions and Nucleophilic Regioselectivities. J. Am. Chem. Soc. 2010;132:1267–1269. doi: 10.1021/ja9098643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Karmakar R., Lee D. Reactions of Arynes Promoted by Silver Ions. Chem. Soc. Rev. 2016;45:4459–4470. doi: 10.1039/C5CS00835B. [DOI] [PubMed] [Google Scholar]
- 81.Zhang J., Niu D., Brinker V.A., Hoye T.R. The Phenol–Ene Reaction: Biaryl Synthesis via Trapping Reactions between HDDA-Generated Benzynes and Phenolics. Org. Lett. 2016;18:5596–5599. doi: 10.1021/acs.orglett.6b02830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gupta S., Xie P., Xia Y., Lee D. Reactivity and Selectivity in the Intermolecular Alder–Ene Reactions of Arynes with Functionalized Alkenes. Org. Lett. 2017;19:5162–5165. doi: 10.1021/acs.orglett.7b02438. [DOI] [PubMed] [Google Scholar]
- 83.Karmakar R., Mamidipalli P., Yun S.Y., Lee D. Alder-Ene Reactions of Arynes. Org. Lett. 2013;15:1938–1941. doi: 10.1021/ol4005905. [DOI] [PubMed] [Google Scholar]
- 84.Karmakar R., Le A., Xie P., Xia Y., Lee D. Reactivity of Arynes for Arene Dearomatization. Org. Lett. 2018;20:4168–4172. doi: 10.1021/acs.orglett.8b01466. [DOI] [PubMed] [Google Scholar]
- 85.Gupta S., Lin Y., Xia Y., Wink D.J., Lee D. Alder-Ene Reactions Driven by High Steric Strain and Bond Angle Distortion to Form Benzocyclobutenes. Chem. Sci. 2019;10:2212–2217. doi: 10.1039/C8SC04277B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Le A., Lee D. Selectivity between an Alder-ene Reaction and a [2+2] Cycloaddition in the Intramolecular Reactions of Allene-Tethered Arynes. Org. Chem. Front. 2021;8:3390–3397. doi: 10.1039/D1QO00459J. [DOI] [Google Scholar]
- 87.Xiao X., Hoye T.R. The Domino Hexadehydro-Diels–Alder Reaction Transforms Polyynes to Benzynes to Naphthynes to Anthracynes to Tetracynes (and Beyond?) Nat. Chem. 2018;10:838–844. doi: 10.1038/s41557-018-0075-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ross S.P., Baire B., Hoye T.R. Mechanistic Duality in Tertiary Amine Additions to Thermally Generated Hexadehydro-Diels–Alder Benzynes. Org. Lett. 2017;19:5705–5708. doi: 10.1021/acs.orglett.7b02888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Arora S., Zhang J., Pogula V., Hoye T.R. Reactions of Thermally Generated Benzynes with Six-Membered N-Heteroaromatics: Pathway and Product Diversity. Chem. Sci. 2019;10:9069–9076. doi: 10.1039/C9SC03479J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Arora S., Palani V., Hoye T.R. Reactions of Diaziridines with Benzynes Give N-Arylhydrazones. Org. Lett. 2018;20:8082–8085. doi: 10.1021/acs.orglett.8b03439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sneddon D.S., Hoye T.R. Arylhydrazine Trapping of Benzynes: Mechanistic Insights and a Route to Azoarenes. Org. Lett. 2021;23:3432–3436. doi: 10.1021/acs.orglett.1c00897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Arora S., Sneddon D.S., Hoye T.R. Reactions of HDDA Benzynes with C,N-Diarylimines (ArCH = NAr’) Eur. J. Org. Chem. 2020;2020:2379–2383. doi: 10.1002/ejoc.201901855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang J., Hoye T.R. Divergent Reactivity during the Trapping of Benzynes by Glycidol Analogs: Ring Cleavage via Pinacol-Like Rearrangements vs. Oxirane Fragmentations. Org. Lett. 2019;21:2615–2619. doi: 10.1021/acs.orglett.9b00595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang T., Oswood C.J., Hoye T.R. Trapping of Hexadehydro-Diels–Alder Benzynes with Exocyclic, Conjugated Enals as a Route to Fused Spirocyclic Benzopyran Motifs. Synlett. 2017;28:2933–2935. doi: 10.1055/s-0036-1589091. [DOI] [Google Scholar]
- 95.Ritts C.B., Hoye T.R. Sulfurane [S(IV)]-Mediated Fusion of Benzynes Leads to Helical Dibenzofurans. J. Am. Chem. Soc. 2021;143:13501–13506. doi: 10.1021/jacs.1c07187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen J., Palani V., Hoye T.R. Reactions of HDDA-Derived Benzynes with Sulfides: Mechanism, Modes, and Three-Component Reactions. J. Am. Chem. Soc. 2016;138:4318–4321. doi: 10.1021/jacs.6b01025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Palani V., Chen J., Hoye T.R. Reactions of Hexadehydro-Diels–Alder (HDDA)-Derived Benzynes with Thioamides: Synthesis of Dihydrobenzothiazino-Heterocyclics. Org. Lett. 2016;18:6312–6315. doi: 10.1021/acs.orglett.6b03199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Watanabe T., Curran D.P., Taniguchi T. Hydroboration of Arynes Formed by Hexadehydro Diels–Alder Cyclizations with N-Heterocyclic Carbene Boranes. Org. Lett. 2015;17:3450–3453. doi: 10.1021/acs.orglett.5b01480. [DOI] [PubMed] [Google Scholar]
- 99.Yun S.Y., Wang K.P., Lee N.-K., Mamidipalli P., Lee D. Alkane C–H Insertion by Aryne Intermediates with a Silver Catalyst. J. Am. Chem. Soc. 2013;135:4668–4671. doi: 10.1021/ja400477r. [DOI] [PubMed] [Google Scholar]
- 100.Mamidipalli P., Yun S.Y., Wang K.-P., Zhou T., Xia Y., Lee D. Formal Hydrogenation of Arynes with Silyl Cβ–H Bonds as an Active Hydride Source. Chem. Sci. 2014;5:2362–2367. doi: 10.1039/C3SC53478B. [DOI] [Google Scholar]
- 101.Lee N.-K., Yun S.Y., Mamidipalli P., Salzman R.M., Lee D., Zhou T., Xia Y. Hydroarylation of Arynes Catalyzed by Silver for Biaryl Synthesis. J. Am. Chem. Soc. 2014;136:4363–4368. doi: 10.1021/ja500292x. [DOI] [PubMed] [Google Scholar]
- 102.Ghorai S., Lee D. Aryne Formation via the Hexadehydro Diels–Alder Reaction and their Ritter-Type Transformations Catalyzed by a Cationic Silver Complex. Tetrahedron. 2017;73:4062–4069. doi: 10.1016/j.tet.2017.02.008. [DOI] [Google Scholar]
- 103.Ghorai S., Lin Y., Xia Y., Wink D.J., Lee D. Silver-Catalyzed Annulation of Arynes with Nitriles for Synthesis of Structurally Diverse Quinazolines. Org. Lett. 2020;22:626–630. doi: 10.1021/acs.orglett.9b04395. [DOI] [PubMed] [Google Scholar]
- 104.Karmakar R., Ghorai S., Xia Y., Lee D. Synthesis of Phenolic Compounds by Trapping Arynes with a Hydroxy Surrogate. Molecules. 2015;20:15862–15880. doi: 10.3390/molecules200915862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang K.-P., Yun S.Y., Mamidipalli P., Lee D. Silver-Mediated Fluorination, Trifluoromethylation, and Trifluoromethylthiolation of Arynes. Chem. Sci. 2013;4:3205–3211. doi: 10.1039/c3sc50992c. [DOI] [Google Scholar]
- 106.Karmakar R., Wang K.P., Yun S.Y., Mamidipalli P., Lee D. Hydrohalogenative Aromatization of Multiynes Promoted by Ruthenium Alkylidene Complexes. Org. Biomol. Chem. 2016;14:4782–4788. doi: 10.1039/C6OB00524A. [DOI] [PubMed] [Google Scholar]
- 107.Xiao X., Wang T., Xu F., Hoye T.R. CuI-Mediated Bromoalkynylation and Hydroalkynylation Reactions of Unsymmetrical Benzynes: Complementary Modes of Addition. Angew. Chem. Int. Ed. 2018;57:16564–16568. doi: 10.1002/anie.201811783. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.