


Abstract
Deregulated expression of the proto-oncogene c-myc in Burkitt lymphoma (BL) cells carrying a t(2;8) translocation is mediated by a synergistic interaction of the translocated immunoglobulin (Ig) κ gene intron (κEi) and 3′ (κE3′) enhancers and characterized by a strong activation of the promoter P1. We have investigated the functional role of distinct κ enhancer sequence motifs in P1 activation on both minichromosomes and reporter gene constructs. Stable and transient transfections of BL cells revealed critical roles of the κEi and κE3′ elements κB and PU, respectively. Joint mutation of κB and PU completely abolished P1 activity, implying that an interaction of κB- and PU-binding factors is essential for the enhancer synergism. Mutation of the E box 1 and E box 2 motifs markedly decreased P1 activity in transient but not in stable transfection experiments. Co-expression of the NF-κB subunit p65(RelA) and Sp1, an essential factor for P1 transcription, in Drosophila melanogaster SL2 cells synergistically enhanced promoter activity. Our results support a model which proposes cross-talk between promoter and enhancer binding factors as the basic mechanism for κ enhancer-mediated c-myc activation in BL cells.
INTRODUCTION
Chromosomal translocations by which the proto-oncogene c-myc on region 8q24 is juxtaposed to one of the immunoglobulin (Ig) gene loci on chromosomes 14q32 (IgH), 2p11 (Igκ) or 22q11 (Igλ) are consistently found in Burkitt lymphoma (BL) cells. Up-regulation of c-myc expression as a result of translocation to the Ig loci is seen as a causal event in the development of BL cells. As a general consequence, the normal c-myc allele is transcriptionally silent or expressed at low rate while the translocated c-myc gene is strongly expressed, characteristically by enhanced transcription from promoter P1 (1–4). The reconstruction of the variant t(2;8) translocation on an Epstein–Barr virus (EBV)-based minichromosome encompassing the c-myc gene, a LY91-derived t(2;8) breakpoint and elements of the Igκ locus allowed study of the influence of the latter on overall c-myc activation, differential promoter usage and other BL cell-specific features. Stable transfection of BL Raji cells with these episomal constructs demonstrated the preferential activation of promoter P1 by a cooperative action of the κ intron enhancer (κEi), the κ 3′ enhancer (κE3′) and the κ matrix attachment region (κMAR) (5–7). The c-myc gene on the episomally replicating minichromosome showed a chromatin structure which is indistinguishable from that of the gene embedded in its normal chromosomal surrounding (7,8). Activation by the κ elements was independent of their positioning either adjacent to or as far as 30 kb from the c-myc promoters, mirroring the variant chromosomal translocation t(2;8) where the translocation breakpoints relative to c-myc and the Igκ gene were found in a range of 1–350 kb (9,10). The current model for such an activation over long distances assumes physical links between the κ enhancers and the c-myc promoters by an interaction of multiprotein complexes assembled on both enhancer and promoter regions, thereby looping out intermediate DNA segments (2,9,11,12). This interaction is presumably based on sequence-specific binding of members of the multiprotein complexes to enhancer and promoter elements and could be either direct or indirect due to the interposition of mediator proteins. Within the P1 core promoter, the TATA box and the promoter-proximal of two adjacent Sp1 sites were defined as essential and sufficient for Igκ enhancer-activated P1 expression (12).
Developmental stage-specific transcription of the κ light chain gene is controlled by the lymphoid-specific κEi and κE3′ enhancers, both being inactive at the proB and preB cell stages and active at the mature B cell and plasma cell stages (13,14). Both enhancers contain several binding motifs for general and tissue-specific nuclear factors. The developmental stage-specific activity of κEi is mainly dependent on the nuclear factor NF-κB whose binding to κEi is inducible in preB and other cell types and constitutive in mature B cells and plasma cells (15,16). Deletion or mutation of the κB site abolished both the constitutive activity and inducibility of κEi, identifying it as a crucial enhancer element (17,18). Moreover, a tandem dimer of two κB sites was shown to be as active as the whole κEi and conferred tissue specificity and inducibility (16). Situated downstream of the κB site are three consensus E box motifs termed E1, E2 and E3 (reviewed in 14). Mutations of E1, E2 and E3 in transfection experiments decreased enhancer activities to 20, 10 and 30% of wild-type level, respectively, but did not affect the inducibility of κEi (18). While no stringent orientational and spacing requirements for the κB and E2 sequence motifs were observed, their location within the core enhancer fragment seems to be crucial for enhancer activity (19). Mutation of the κB motif or E box 2, but not of the E box 3, severely impaired the synergy of κEi and κE3′ (20). Binding of distinct nuclear protein factors was demonstrated for the murine E2 [basic helix–loop–helix (bHLH) transcription factors E12, E47, ITF1 and ITF2] and E3 sites (USF, TFE3 and TFEB), while no protein–DNA interaction was detectable for E1 (reviewed in 13,14).
The finding that NF-κB-deficient cell lines were capable of transcribing Ig genes even in the absence of κEi led to identification of the κE3′ enhancer (21–24). Functional analysis of κE3′ revealed a B cell-specific enhancer able to activate transcription 2- to 5-fold stronger than κEi (7,25). Several positive regulatory elements in the core region of human κE3′ were characterized, including the consensus binding site for the B cell- and macrophage-specific transcription factor Spi-1 (the human homolog of PU.1), an E box motif for factors of the bHLH type and a cAMP-responsive element (CRE; 24–26). In the murine κE3′ enhancer, PU.1 was found to recruit an additional protein, NF-EM5 (also known as Pip or IRF4), to bind just downstream of the PU site (27).
Here we have defined the role of the κB, E1, E2 and PU sites in κ enhancer-mediated activation of the c-myc P1 promoter. Mutation analysis of the κB and PU sites in the context of stable as well as transient transfections demonstrated their critical importance for the synergism of κEi and κE3′ in P1 activation. Our data propose functional interactions of members of the NF-κB/Rel family with factors of the Spi-1/PU family, on the one hand, and with factors binding to the P1 promoter, on the other, as crucial parts of the basic mechanism of c-myc P1 activation through the Igκ elements.
MATERIALS AND METHODS
DNA constructs
Constructs pRF115-3, pKH199-7, pRF211-1 and pKH80-4 were described previously (6,7). Mutated constructs were created with the Muta-Gene Phagemid mutagenesis kit according to the instructions of the manufacturer (Bio-Rad, USA) using the following oligonucleotides: κBmut, 5′-GCCAGGTGGCCTC-TTGGAAACTAGTCTCTGGGGGATTCCACCCGTTGGG-3′; PUmut, 5′-GTGCTCAAGGTTCTGTTTTCAGACTAGTA-AAGGGTCTTCTCCTTGACC-3′; E1mut, 5′-CTGACCCTC-AGCAACTGCCAGAATTCCTCTTGGAAATCCCCCTCT-GG-3′; E2mut, 5′-GAGGTCAACTGTAATCTTGGCAGAA-TTCCTAAGAGAAGTGGCTAGCTTC-3′. Mutagenesis of the κB site in plasmid pKH73-4 (7) with oligonucleotide κBmut resulted in plasmid pKH460-6. Digestion with BamHI and SacI, modification of the restriction sites using XbaI oligonucleotide linkers and insertion of the mutated κEi-containing fragment into the SpeI site of plasmid pKH74-2 (7) gave rise to plasmid pKH487-6. Mutagenesis of the κE3′ PU site in pKH80-4 with oligonucleotide PUmut resulted in plasmid pKH459-2. For plasmid pKH484-15, the mutagenized κE3′ fragment of pKH459-2 was isolated by digestion with SacII and inserted into the SacII site of pKH460-6. Plasmids pKH487-6, pKH459-2 and pKH484-15 were digested with BamHI and SacI followed by modification of the restriction sites with BamHI oligonucleotide linkers and digestion with BamHI. Exchange of the wild-type enhancer-containing BamHI fragment of pKH199-7 for the obtained mutant enhancer-containing fragments resulted in plasmids pKH497-8, pKH486-6 and pKH496-4, respectively. Mutagenesis of plasmid pKH80-4 with oligonucleotides E1mut or E2mut gave rise to plasmids pKH102-0 and pKH107-0 mutated in the E1 and E2 boxes, respectively. These plasmids were digested with Ecl136II and the restriction sites modified with BamHI oligonucleotide linkers followed by digestion with BamHI. Exchange of the wild-type enhancer-containing BamHI fragment of pKH199-7 for the obtained mutant enhancer-containing fragments resulted in plasmids pCG615-6 and pCG616-6, respectively. Construct pCG57 was described before (12). Plasmid pCMV-lacZ was obtained from Stratagene (USA). Plasmids pPacSp1 and pPac0 were kindly provided by K. J. Goodrich and D. Tautz, respectively. Plasmids pPacSp3 and pRSV-p65 were kindly provided and described by G. Hagen (28) and R. Schmid (29), respectively. Plasmid DNA used for transient transfection of Raji cells was purified using an endotoxin-free plasmid purification kit (Qiagen, Germany) in order to avoid induction of NF-κB by co-transfected endotoxin.
Cell lines, Southern blot analysis, tissue culture and transfection experiments
For generation of stable cell lines, BL Raji cells (30) were grown, transfected and selected as described (7,31). The copy numbers of the constructs in the transfectants and the presence of the introduced mutations were determined by Southern blot analysis. Total cellular DNA of representative cell lines of each transfected construct was digested with BamHI, SpeI (specific for the κB and PU mutations; see Fig. 1) or EcoRI (specific for the E1 and E2 box mutations; see Fig. 1). The blots were probed either with the HindIII/ClaI-digested vector pHEBOPL (5,32) encompassing EBV oriP or with the BamHI–SacI fragment of KH80-4 encompassing the κ enhancers. Copy numbers in the range 10–60 per cell were calculated relative to pKH199-7 by use of hybridization signals of EBV sequences present in Raji cells.
Figure 1.

In the case of the t(2;8) translocation, the Igκ region is localized 3′ of c-myc. Depicted are the following elements in the κ locus: J gene segments (J), the matrix attachment region (MAR), the κ intron and 3′ enhancers (κEi and κE3′), the Cκ gene (Cκ). The wild-type (wt) sequences of the binding motifs κB, E1, E2, E3 and PU and the base substitutions (mut) introduced into κB, E1, E2 and PU by site-directed mutagenesis changing the motifs to restriction enzyme recognition sites are shown as enlargements beneath.
Luciferase reporter gene assays were done as described by de Wet et al. with slight modifications (33,34). Raji cells were transfected with a mixture of 10 µg of a reporter gene plasmid and 2 µg of plasmid pCMV-lacZ in a total volume of 20 µl. Protein extracts were prepared after 48 h growth. Aliquots of 10 µl of extracts were used for measurement of luciferase and β-galactosidase activities using the Luciferase (Promega, USA) and β-Galactosidase (Tropix, USA) assay systems according to the manufacturers’ instructions. The obtained relative luciferase units were normalized to the respective β-galactosidase units. Luciferase and β-galactosidase activities were determined as the means of three to four independent experiments. Each sample was measured in duplicate.
Drosophila melanogaster SL2 cells (35), derived from embryonic stem cells, were kindly provided by R. Rivera-Pomar and cultured at a density of 3–8 × 106 cells/ml in Schneiders medium (Gibco, USA) supplemented with 10% FCS, glutamine and antibiotics at 26°C. Cells were seeded on the day of transfection into 6-well plates at a density of ~5 × 105 cells/2 ml complete medium and were left undisturbed for 1 h to adhere. Alternatively, cells were seeded 1 day prior to transfection to reach 50–80% confluency after 18–24 h. For each transfection, 2 µg of a c-myc reporter gene construct were mixed with 4 µg of expression plasmids pPacSp1, pPacSp3 and/or pRSV-p65 and made up to 10 µg total DNA with pPac0 (vector only). Transfection was performed by calcium phosphate-mediated DNA precipitation as described (36). Cells were harvested 48 h post-transfection and assayed for luciferase activity as described (12). Reporter gene activities were normalized to the total protein content of the cell extracts determined with a protein quantification kit (Bio-Rad, USA).
RNA analysis
Total cellular RNA of two to four representative cell lines containing the DNA constructs pRF115-3, pKH199-7, pKH497-8, pKH486-6 and pKH496-4 was prepared by extraction with guanidinium thiocyanate followed by centrifugation in cesium chloride (37). Total cellular RNA of cells transfected with plasmids pCG615-6 and pCG616-6 was prepared using the Qiagen RNeasy kit according to the manufacturer’s recommendations (Qiagen, Germany). All RNA samples were quantitated by spectrophotometric analysis and staining of formaldehyde/agarose gels with ethidium bromide. Nuclease S1 analysis with a probe specific for the first exon of c-myc was carried out as described previously (38). Signals were captured and quantitated using a Fuji BAS1000 phosphorimager system (Fuji, Japan). The analysis of cell lines containing the DNA constructs pRF115-3, pKH497-8, pKH486-6 and pKH496-4 on the one hand and of those transfected with plasmids pCG615-6 and pCG616-6 on the other was performed in two separate experiments. The determined P1 and P2 values were subsequently normalized to a unique scale by the values obtained for the pKH199-7-containing cell lines analyzed in both experiments.
Western blot analysis
Cellular extracts for western blot analysis were prepared in lysis buffer (10 mM Tris–HCl pH 7.5, 1 mM EDTA, 1% Triton, 10 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride) and sonicated. Proteins were separated by SDS–PAGE and blotted onto nitrocellulose membrane. Equal loading of the gels was verified by staining with Ponceau red (Sigma, USA). Immunoreactive proteins were detected using polyclonal rabbit antisera specific for p65(RelA), cRel, RelB and p52 and polyclonal goat antiserum specific for p50 (Santa Cruz Biotechnologies, USA) and subsequent incubation with peroxidase-coupled secondary antibodies (Santa Cruz Biotechnologies, USA; Sigma, USA) followed by Enhanced Chemiluminiscence (ECL; Amersham, USA).
RESULTS
Mutations of κB, PU, E box 1 and E box 2 and their impact on enhancer-mediated P1 transcription in the context of minichromosomes mimicking a t(2;8) translocation
The Igκ sequences necessary and sufficient for c-myc activation had previously been narrowed down to a 0.7 kb fragment encompassing κEi, a 1.2 kb fragment encompassing κE3′ and a 0.7 kb fragment containing κMAR. These elements had been inserted into the episomally replicating plasmid pKH199-7 which contains the c-myc gene (nucleotide sequences –2332 to +5754 relative to the P1 transcription start site) (7). To analyze the role of the κB, PU, E1 and E2 sequence motifs in c-myc activation, the respective DNA sequences in pKH199-7 were mutated by site-directed mutagenesis (see Materials and Methods and Figs 1 and 2A). Plasmid pKH199-7, its mutated derivatives and a construct without the κ elements (pRF115-3) were each transfected into Raji cells and several stable transfectants were obtained for each construct after selection with hygromycin B.
Figure 2.

c-myc promoter usage in Raji transfectants. (A) Schematic representation of the episomally replicating constructs used for stable transfection of Raji cells. To analyze the role of the κB and PU sites and of the E1 and E2 boxes, a series of pKH199-7 derived plasmids was constructed carrying mutations in the respective DNA sequences. The mutated enhancer elements are depicted as smaller circles and ellipses compared to the wild-type elements. (B) Nuclease S1 analysis of total cellular RNA of the respective transfectants and untransfected Raji cell lines. The result of one independent cell line for each construct is shown (the direction of the gel run is marked by an arrow). Transcripts were analyzed using a uniformly labeled, single-stranded DNA probe spanning the c-myc promoter region and first exon shown in (C). Signals corresponding to the endogenous translocated allele are marked P1t and P2t, signals from the transfected constructs P1 and P2. The sizes of the expected fragments (in bp) are given in (C). The signals of the P1 and P2 transcripts captured by a phosphorimager were quantified and standardized to the P1t value of each RNA and to the copy number of the respective plasmid. Analysis of the cell lines was performed in two separate experiments (depicted by a dotted horizontal line, B). The resultant values were normalized to a unique scale and plotted as means ± standard deviation of two to four individual RNA determinations in a histogram shown in (D). RU, relative scan units.
Southern blot analysis confirmed the integrity of the transfected plasmids and the presence of the mutations that were introduced (data not shown; see Materials and Methods and Fig. 1). Northern blot analysis of total RNA of the transfectants revealed c-myc mRNA of the correct size with a level of expression considerably above that of untransfected cells (data not shown). The transfectants were analyzed by nuclease S1 mapping using a probe specific for c-myc exon 1 (Fig. 2C). The short deletion in this exon of the t(8;14) translocation chromosome of Raji cells allows differentiation between transcripts derived from the P1 and P2 promoters of the endogenous translocated allele (P1t and P2t) and those derived from the transfected constructs (P1 and P2). The results of one independent cell line for each construct are shown in Figure 2B. The signals were scanned densitometrically and the individual P1 and P2 values normalized relative to the respective P1t value and to the copy number of the transfected plasmid (see Materials and Methods) and plotted in a histogram (Fig. 2D). Transfectants of pKH199-7 revealed a strong induction of P1-derived transcripts with a P1/P2 ratio of about one, similar to what has been described for a construct encompassing the complete c-myc gene under the control of the Igκ enhancers (7). Mutation of the κB motif resulted in a strong decrease in P1 expression to 17% compared to pKH199-7, while mutation of the PU site was less dramatic, reducing P1 activity to 64%. Joint mutation of both sites resulted in a dramatic reduction in P1 transcription down to basal transcription levels. In contrast, mutation of the E1 and E2 boxes did not decrease but rather activated P1 transcription. Of note, a decrease in P1 activity by single mutations was accompanied by an increase in transcription from the second promoter P2. Combined mutation of κB and PU, however, also strongly reduced P2 activation ~10-fold compared to pKH199-7.
Different roles of PU, E1 and E2 for P1 activation depending on the context of transfection
The effect of mutations in the κ enhancer elements was also examined in transient reporter gene experiments, which allowed the separate analysis of enhancer-mediated transcription of the two c-myc promoters P1 and P2 (12,39). We first tested the enhancer mutants in constructs containing the P1 promoter (nucleotides –1058 to +66 relative to P1, corresponding to the KpnI–XhoI fragment), the luciferase gene (LUC) and both Igκ enhancers inserted 3′ of LUC. Earlier experiments showed that upstream (positions –2332 to –1058 relative to P1) or downstream (positions +66 to +5754) c-myc sequences have no influence on κ enhancer-mediated P1 activation in transient transfection assays (12,39). Notably, all LUC fusion constructs tested lacked the κMAR element, as it acted as a repressor of the κ enhancers in transient transfection experiments (7).
The constructs depicted in Figure 3 (left) were transiently transfected into Raji cells and the relative luciferase activities of the respective constructs were determined after 48 h (Fig. 3, right). Mutation of the κB site led to a strong decrease in luciferase activity to 7% compared to pKH80-4, similar to the impact of the κB mutation in stable transfected constructs (Fig. 2). In contrast to the stable situation, mutation of the PU site reduced luciferase activity to 8% of wild-type level, an effect equivalent to destruction of the κB site. Combined mutation of both sites completely abolished the enhancement. Luciferase activity was reduced to a level similar to the basal activity obtained with a construct without the κ enhancers (pRF211-1), corresponding to the reduction in P1 enhancement caused by the joint mutations in the stable transfection. Mutation of the E1 and E2 boxes reduced luciferase activity to 12 and 17% of wild-type level, respectively, in contrast to the lack of reduction found in the stable situation.
Figure 3.

Role of Igκ enhancer elements for P1 activation in transient luciferase reporter gene assays. The left hand part shows a schematic representation of a series of pKH80-4-derived plasmids carrying mutations in the respective DNA sequences. The mutated enhancer elements are depicted as smaller circles and ellipses compared to the wild-type elements. All constructs contain the luciferase gene (LUC) under control of the c-myc P1 promoter. Luciferase activities measured after transfection of the respective constructs into Raji cells are shown to the right. Three independent transfection experiments with each of the constructs were performed. The mean values ± standard deviation are presented as percent luciferase activities relative to construct pKH80-4, which is set to 100.
We also determined the effects of the enhancer mutations on promoter P2 by insertion of the c-myc fragment corresponding to nucleotide positions +66 to +513 (relative to P1) instead of the P1 promoter fragment in the constructs depicted in Figure 3. The effects of the κB, E1 and E2 mutations on P2 activity mirrored those determined in the stable situation (Fig. 2 and data not shown). The PU mutation, however, which left P2 transcription unchanged in the stable context, reduced the enhancer effect on P2 ~7-fold in the transient transfection.
Interaction of NF-κB and κEi is essential for P1 activation
Mutation of the κB site had the most dramatic effect on P1 activation, independent of the context of transfection. Binding to κB motifs and transactivation of distinct promoters can be achieved by complexes formed by various members of the Rel family (40,41). To determine the cellular composition of possible c-myc-activating NF-κB complexes, we analyzed expression of Rel family factors in the BL cell lines Raji [t(8;14), EBV+], LY91 [t(2;8), EBV+] and DG75 [t(8;14), EBV–] by western blot analysis. Factors RelA (p65), RelB, c-Rel and p52 were detectable in roughly the same amounts in the tested BL cell lines in whole cell extracts (Fig. 4a) as well as in phorbol ester-induced nuclear extracts (data not shown). Factor p50 was not detectable in LY91 cells, implying that c-myc-activating NF-κB complexes in t(2;8) BL cells are formed by homo- or heterodimers of other Rel family members. In contrast, in a non-BL cell line (HeLa), c-Rel was undetectable and RelB and p52 were only weakly detectable while p50 seemed to be present in high amounts.
Figure 4.
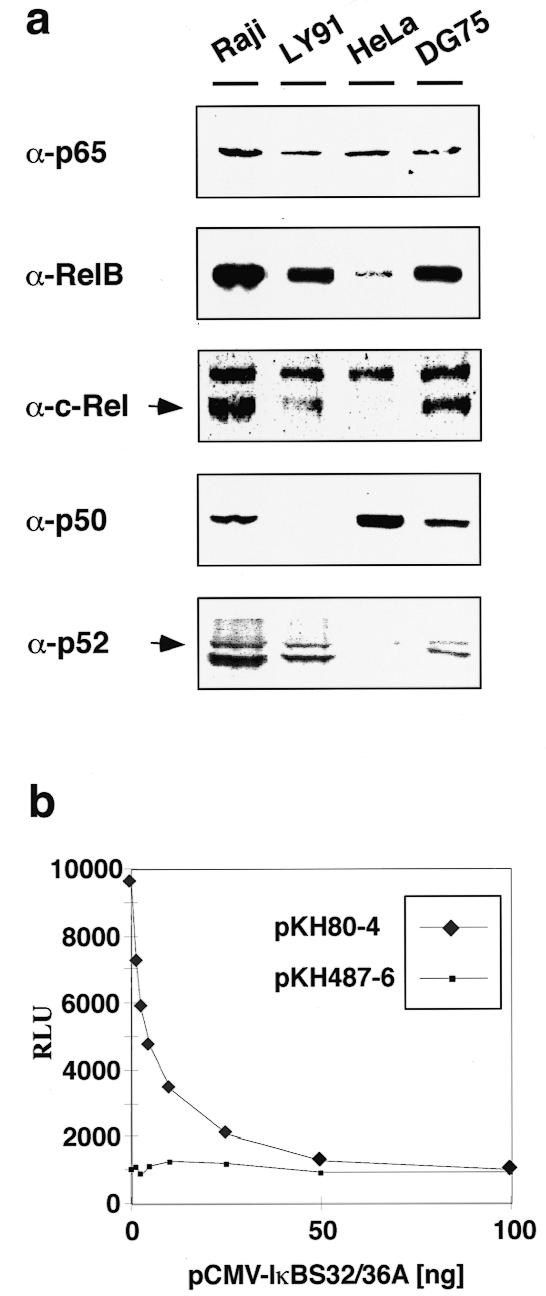
(a) Western blot analysis of the Rel components of the BL cell lines Raji, LY91 and DG75 and of the fibroblast cell line HeLa. (b) Co-expression of the IκB-α mutant IκBS32/36A. Raji cells were transfected with 10 µg of either plasmid pKH80-4 or pKH487-6 (for constructs see Fig. 3) and increasing amounts of plasmid pCMV-IκBS32/36A. Luciferase expression is presented as means of two independent transfection experiments for each pCMV-IκBS32/36A titration point as relative luciferase units (RLU).
To further address the functional role of NF-κB in Igκ enhancer-mediated P1 activation, we performed co-transfection experiments in Raji cells using plasmid pCMV-IκBS32/36A which expresses IκBS32/36A, a dominant negative mutant of the NF-κB inhibitor IκB-α (42). IκBS32/36A was found to potently suppress activation of the entire NF-κB family (43), thus most likely inhibiting all possible c-myc-activating NF-κB complexes formed in Raji cells (see Fig. 4a). Increasing amounts of transfected DNA of pCMV-IκBS32/36A led to a concomitant decrease in the activity of pKH80-4 to a final level concurrent with the activity of pKH487-6, which harbors the κB site mutation (Fig. 4b, constructs depicted in Fig. 3). The activity of pKH487-6 remained unchanged at any amount of co-transfected pCMV-IκBS32/36A. As a control, co-transfection of increasing amounts of a CMV-lacZ expression plasmid had no effect on either pKH80-4 or pKH487 6, further confirming the specificity of the inhibitory effect of IκBS32/36A (data not shown). Thus, the observed decline in P1 activity caused by deprivation of nuclear NF-κB due to its interception in the cytoplasm by IκBS32/36A demonstrated that interaction of NF-κB with the κB site of κEi is essential for κ enhancer-mediated P1 activation.
Synergistic activation of c-myc P1 by p65(RelA) and Sp1
The model for c-myc activation by the κ enhancer elements assumes protein–protein interactions of enhancer-binding proteins with either factors of the transcription initiation complex, activator proteins, or both. We have shown previously that binding of transcription factor Sp1 to the P1 core promoter as well as the P1 TATA box are required for Igκ enhancer-activated P1 transcription (12). Synergistic transcriptional induction of the HIV-1 LTR by Sp1 and the p65(RelA) NF-κB subunit and a direct protein–protein interaction of a NF-κB complex (p65/p52) and Sp1 have been demonstrated in vitro (44). For the analysis of putative interactions of NF-κB with Sp1 as a prerequisite for P1 activation in vivo, co-transfection experiments were performed in D.melanogaster SL2 cells. Due to the lack of homologous mammalian transcription factors [e.g. Sp1-related factors (45) and Rel family factors other than a p50(NFKB1)-related protein (44)], SL2 cells serve as an ideal heterologous cell system for such a task. Constructs with and without the Igκ enhancers (pKH80-4 and pRF211-1, respectively) as well as derivatives of pKH80-4 containing mutations either in the promoter-proximal Sp site (pCG57) or in the κB site of κEi (pKH487-6) were transfected into SL2 cells. Each construct displayed equal luciferase activities at very low levels, indicating the lack of endogenous factors promoting faithful basal transcription and enhancement (Fig. 5). These levels remained unchanged by co-transfection of pCMV-p65. Supplying transcription factor Sp1 by co-transfection of pPacSp1 led to a marked increase in P1-driven luciferase activity dependent on the integrity of the Sp1 binding site. Simultaneous co-expression of Sp1 and p65(RelA) in cells containing construct pKH80-4 further increased its activity 2-fold compared to co-expression of Sp1 only. In contrast, no further elevation of activity levels was observed in cells carrying constructs with mutations in the Sp or κB sites, or those lacking the κ elements. This demonstrated that correct protein–DNA interaction is a prerequisite for the cooperative activation of P1 expression by Sp1 and p65(RelA). Co-expression of transcription factor Sp3, a member of the Sp family which also binds to the P1 promoter in vitro (12), had no effect on P1 activation when expressed separately or simultaneously with p65 (data not shown). This finding further supports the specificity of the functional interaction of p65(RelA) and Sp1 in SL2 cells leading to enhanced P1 activity.
Figure 5.

Activation of c-myc P1 by Sp1 and NF-κB (p65) in D.melanogaster SL2 cells. (Upper) Reporter gene constructs used for transfection of SL2 cells. All plasmids contain the luciferase gene (LUC) under control of the c-myc P1 promoter encompassing a CT element, two Sp binding sites (open boxes), the TATA box (open oval circle) and, with the exception of pRF211-1, the Igκ enhancers (closed oval circles). The constructs carry either wild-type sequences or mutations (depicted by crosses) in the promoter-proximal Sp binding site or in the κB site. (Lower) Luciferase activities presented as relative luciferase units (RLU) determined after co-transfection of SL2 cells with reporter gene constructs and Sp1 and/or p65 expression plasmids.
DISCUSSION
In this paper we have addressed the individual importance of the κB, E1 and E2 consensus sequences of κEi and the PU site of κE3′ for BL-specific activation of c-myc promoter P1. Mutant enhancer-mediated P1 expression was analyzed in Raji cells in the context of either stably replicating minichromosomes comprising c-myc and the Igκ elements MAR, κEi and κE3′ or in transient transfection experiments utilizing luciferase reporter gene constructs encompassing the P1 promoter, κEi and κE3′. Alteration of the κB motif by base exchange led to a 6-fold decrease in P1-driven c-myc transcription, as determined by nuclease S1 analysis of stable transfectants, which suggests a major role of the κB site in c-myc expression displayed in BL cells. This role was confirmed in P1-dependent reporter gene assays where luciferase activity of κB mutant constructs was reduced to nearly basal levels. An equivalent effect of a κB site mutation was found for murine κEi-mediated activation of a c-fos reporter gene construct in mouse myeloma cells (18). The fact that NF-κB is indeed the limiting factor in κEi-mediated P1 activation was shown through expression of the NF-κB inhibitor IκBS32/36A. Deprivation of nuclear NF-κB due to its irreversible cytoplasmic trapping by this IκB-α mutant decreased enhancer-mediated P1 luciferase activity to the same extent as mutation of the κB site. Thus, three independent sets of experiments indicated that NF-κB is a crucial factor for κ enhancer-activated P1 transcription, and that NF-κB activates P1 via the κB motif. The observed strong reduction in enhancement as a consequence of the prevented correct interaction between NF-κB and κEi suggests a lack of functional substitution of NF-κB-mediated P1 activation by other κEi- and κE3′-binding protein factors. This result is in line with earlier findings showing that activation of c-myc requires the interaction of κEi and κE3′, while κE3′ by itself was not sufficient (6).
Individual mutation of the E1 and E2 box motifs showed no decremental effects on κ enhancer-mediated P1 c-myc transcription in the context of Raji cells stably transfected with EBV-derived minichromosomes. In the context of transient transfection studies, however, the E1 and E2 box mutations caused an ~8- and 6-fold reduction in enhancement, respectively, relative to enhancement by the wild-type enhancer. Comparable effects of E1 and E2 box mutations on c-fos promoter activation (~5- and 10-fold reductions in enhancement, respectively) were found for murine κEi (18). Thus, the effect of mutation of E1 and E2 on enhancement of P1 expression varies dependent on the context of the DNA template. Transiently transfected DNA differs from cellular minichromosomal DNA in its chromatin structure (46,47), which might confer an abnormal accessibility to transcription factors on the DNA (48). Accordingly, the P1 promoter could be abnormally accessible to E1- and E2-interacting factors in transient transfection which might explain the excessive impact of the E box mutations on P1 activation that was not found in the context of stably transfected constructs. Differential activation of chromosomal and injected DNA templates was also shown recently by Alberts et al. (49), who demonstrated that activation of a chromosomally located reporter gene required two kinds of signals, while presence of only one of these signals was sufficient for activation of the same transfected or microinjected template. However, signaling induced by binding of factors to the E1 and E2 boxes seems to be negligible for P1 activation in the chromosomal context but crucial in the transient transfection context. This circumstance could be the consequence of a functional redundancy displayed by the κ E boxes or by other κEi factor-binding elements with respect to the κ E boxes that is revealed only in a chromosomal environment. A partial redundancy of factor binding sites was found in the Igµ heavy chain gene intronic enhancer where mutation of single elements did not significantly reduce enhancer activity (13,50).
It is assumed that the Igκ enhancers activate c-myc by physical linkage of enhancer elements to the c-myc gene via protein factors (2,9,11). Our experiments showed that NF-κB is a major factor in c-myc P1 activation, suggesting an interaction of NF-κB complexes with factors binding in the vicinity of P1. The crucial P1 promoter binding factor for enhancer-mediated P1 transcription and therefore the prime candidate for an interaction with NF-κB is the ubiquitous transcription factor Sp1 (12). Simultaneous co-expression of Sp1 and p65(RelA) with a P1–luciferase–κ enhancer reporter gene plasmid in Drosophila SL2 cells led to a 2-fold activation of P1 luciferase expression compared to co-expression of Sp1 alone. In contrast, co-expression of p65(RelA) alone did not activate P1 luciferase expression. This shows a functional cooperation of p65(RelA) and Sp1 in enhanced P1 activation. The observed cooperativity of Sp1 and p65(RelA) was dependent on the presence of the Igκ enhancers as well as the P1-proximal Sp site, since missense of these elements or mutations in the Sp or κB sites abolished cooperativity, and, in the case of the Sp site mutation, even total activation. Although a protein–protein interaction of p65(RelA) with TATA box-binding protein (TBP) and other factors of the basal transcription complex has been shown before (51), these potential interactions by themselves do not seem to result in P1 activation. However, Sp1-mediated transcription is assumed to be accomplished by a direct interaction of Sp1 with at least two components of the TFIID complex, TBP and TBP-associated factor TAFII110 (52–54). Moreover, previous results indicated that a TATA box is required for Igκ enhancer-activated P1 transcription (12). Therefore, P1 activation owing to co-expression of p65(RelA) and Sp1 could be the result of formation of a multiprotein complex consisting of p65(RelA), Sp1 and other transcription factors, including members of the basal transcription machinery. In addition, co-activators like CREB (CRE-binding protein)-binding protein, CBP or the related p300 protein could be involved in Sp1/NF-κB-mediated P1 activation. Members of this protein family are presumed to serve as a bridge between sequence-specific transcription factors and components of the basal transcription machinery (55), and were also found in Drosophila (56). In such a scenario, other κEi- and κE3′-interacting protein factors like E box-binding proteins, PU.1 (Spi-1)/NF-EM5 or those binding to the CRE site could contribute to such a higher order complex and thereby cooperatively activate P1.
Destruction of the κE3′ PU site reduced enhancer-mediated P1 transcription in stable transfectants moderately, by one third. Similar to the situation described for the κ E boxes, the effect of PU motif mutation was much stronger in the context of transient transfection experiments, reducing P1 luciferase activity to 8% compared to the wild-type level. As for the κEi E box mutations, this diversity in the impact of mutations in dependence on the chromosomal context could be explained in two ways: either by a large degree of functional redundancy displayed by κE3′-binding factors which is discernible only in a chromosomal chromatin environment or by an abnormal accessibility of P1 to PU-interacting factors in transiently transfected DNA, which admits a more crucial role for the PU site than in the context of stably transfected constructs. Reduction of enhancement by only one third in stable transfectants implies that the strong activation capacity of κE3′ depends only in part on correct binding of both PU.1 (Spi-1) and the adjacent binding protein factor NF-EM5, whose binding requires the presence of DNA-bound PU.1 (27,57). Several other protein–DNA interaction sites, including a CRE site, an E box situated next to PU and an upstream GC sequence motif, have been shown to contribute to κE3′ function to various degrees by transient expression assays (24–26,58,59). A recent mutational analysis, also in the context of transient transfections, demonstrated the necessity but also sufficiency of each of the motifs PU/NF-EM5, the E box and the 5′-region of CRE for the synergy of κE3′ and κEi (60). The apparently minor contribution of the PU site to κE3′ function found in the minichromosomal context of our study might in contrast hint at an overall higher degree of functional redundancy displayed by κE3′-binding factors compared to κEi, where the remaining binding elements were not able to compensate for loss of NF-κB binding. However, simultaneous mutation of κB and PU resulted in total elimination of κ enhancer-mediated P1 activation, showing that the combined absence of PU.1 (Spi-1)/NF-EM5 and NF-κB binding cannot at all be counterbalanced by binding of other protein factors to κE3′ or κEi. Thus, the conclusions of Liu and co-workers (60) regarding the PU motif might be extended to the situation of stable transfections if the κB site of κEi is also mutated. The strong synergism of κE3′ and κEi for enhancement of P1 transcription observed in stably replicating episomes as well as in transient transfection studies might therefore be promoted in large part by enhancer binding of factors NF-κB and PU.1/NF-EM5 and their subsequent involvement in formation of a transactivating multiprotein complex (6,7). The role of PU.1 in formation of such a multiprotein complex could lie in its ability to change DNA topology in a characteristic way (61) rather than in direct transactivation, since PU.1-mediated transactivation was shown to be independent of its transactivation domain (62). In vitro formation of such a complex comprising the PU.1, NF-EM5, E box-binding and CRE-binding (ATF/CREM, c-Fos and c-Jun) factors was reported recently (62). In addition, architectural proteins like LEF-1 or YY1 (NF-E1), which also bind to distinct sites in κE3′ (63,64), could assist in formation of such a multiprotein complex connecting κE3′ and κEi.
In minichromosomal environments, the κB, PU and E1 box mutations caused a slight and the E2 box mutation a strong increase in P2 activation levels, suggesting no or even negative effects of the respective binding proteins on initiation of P2 transcription. Moreover, the individual κB or PU mutations apparently do not impair the cooperativity of κEi and κE3′ regarding the P2 activation described previously (6,7). However, simultaneous mutation of κB and PU caused a strong reduction in c-myc transcription from promoter P2, implying that correct DNA interaction of at least one of the factors NF-κB or PU.1 (Spi-1) is required for P2 activation and enhancer cooperativity.
Our data present two lines of indication for formation of an enhancer- and promoter-based multiprotein complex bridging the distances between the DNA sequences encompassing κEi, κE3′ and the c-myc promoters, leading to enhanced c-myc expression. The observed functional synergisms of κB and PU motif-binding factors on the one hand and of κB-binding p65(RelA) and P1-binding Sp1 on the other imply protein–protein interactions of NF-κB components, PU.1 (Spi-1)/NF-EM5 and Sp1 in the context of a higher order protein complex. In this model, κ E box-binding proteins and other κ enhancer-interacting factors might serve as peripheral stabilizing factors rather than as essential components of an activating multiprotein complex.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Barbara Christoph and Barbara Baier for perfect technical help and E. Schneider for critical comments on the manuscript. We are grateful to K. J. Goodrich, G. Hagen, R. Schmid and D. Tautz for providing plasmids and to R. Rivera-Pomar for providing Drosophila SL2 cells. C.G. is a fellow of Die Studienstiftung des deutschen Volkes. This work was supported by a grant of Deutsche Forschungsgemeinschaft (Po325/1-3).
REFERENCES
- 1.Taub R., Moulding,C., Battey,J., Murphy,W., Vasicek,T., Lenoir,G.M. and Leder,P. (1984) Cell, 36, 339–348. [DOI] [PubMed] [Google Scholar]
- 2.Bornkamm G.W., Polack,A. and Eick,D. (1988) In Klein,G. (ed.), Cellular Oncogene Activation. Marcel Decker, New York, NY, pp. 223–273.
- 3.Spencer C.A., LeStrange,R.C., Novak,U., Hayward,W.S. and Groudine,M. (1990) Genes Dev., 4, 75–88. [DOI] [PubMed] [Google Scholar]
- 4.Spencer C.A. and Groudine,M. (1991) Adv. Cancer Res., 56, 1–48. [DOI] [PubMed] [Google Scholar]
- 5.Polack A., Strobl,L., Feederle,R., Schweizer,M., Koch,E., Eick,D., Wiegand,H. and Bornkamm,G.W. (1991) Oncogene, 6, 2033–2040. [PubMed] [Google Scholar]
- 6.Polack A., Feederle,R., Klobeck,G. and Hortnagel,K. (1993) EMBO J., 12, 3913–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hortnagel K., Mautner,J., Strobl,L.J., Wolf,D.A., Christoph,B., Geltinger,C. and Polack,A. (1995) Oncogene, 10, 1393–1401. [PubMed] [Google Scholar]
- 8.Siebenlist U., Hennighausen,L., Battey,J. and Leder,P. (1984) Cell, 37, 381–391. [DOI] [PubMed] [Google Scholar]
- 9.Magrath I. (1990) Adv. Cancer Res., 55, 134–270. [DOI] [PubMed] [Google Scholar]
- 10.Mautner J., Behrends,U., Hortnagel,K., Brielmeier,M., Hammerschmidt,W., Strobl,L., Bornkamm,G.W. and Polack,A. (1996) Oncogene, 12, 1299–1307. [PubMed] [Google Scholar]
- 11.Hayday A.C., Gillies,S.D., Saito,H., Wood,C., Wiman,K., Hayward,W.S. and Tonegawa,S. (1984) Nature, 307, 334–340. [DOI] [PubMed] [Google Scholar]
- 12.Geltinger C., Hortnagel,K. and Polack,A. (1996) Gene Expr., 6, 113–127. [PMC free article] [PubMed] [Google Scholar]
- 13.Libermann T.A. and Baltimore,D. (1991) In Cohen,P. and Foulkes,J.G. (eds), The Hormonal Control Regulation of Gene Transcription. Elsevier Science (Biomedical Division), Amsterdam, The Netherlands, pp. 399–421.
- 14.Staudt L.M. and Lenardo,M.J. (1991) Annu. Rev. Immunol., 9, 373–398. [DOI] [PubMed] [Google Scholar]
- 15.Sen R. and Baltimore,D. (1986) Cell, 46, 705–716. [DOI] [PubMed] [Google Scholar]
- 16.Pierce J.W., Lenardo,M. and Baltimore,D. (1988) Proc. Natl Acad. Sci. USA, 85, 1482–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Queen C. and Stafford,J. (1984) Mol. Cell. Biol., 4, 1042–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lenardo M., Pierce,J.W. and Baltimore,D. (1987) Science, 236, 1573–1577. [DOI] [PubMed] [Google Scholar]
- 19.Schanke J.T. and Van Ness,B.G. (1994) J. Immunol., 153, 4565–4572. [PubMed] [Google Scholar]
- 20.Fulton R. and Van Ness,B. (1994) Nucleic Acids Res., 22, 4216–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Atchison M.L. and Perry,R.P. (1987) Cell, 48, 121–128. [DOI] [PubMed] [Google Scholar]
- 22.Meyer K.B. and Neuberger,M.S. (1989) EMBO J., 8, 1959–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blasquez V.C., Xu,M., Moses,S.C. and Garrard,W.T. (1989) J. Biol. Chem., 264, 21183–21189. [PubMed] [Google Scholar]
- 24.Pongubala J.M. and Atchison,M.L. (1991) Mol. Cell. Biol., 11, 1040–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Judde J.G. and Max,E.E. (1992) Mol. Cell. Biol., 12, 5206–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pongubala J.M. and Atchison,M.L. (1995) J. Biol. Chem., 270, 10304–10313. [DOI] [PubMed] [Google Scholar]
- 27.Pongubala J.M., Nagulapalli,S., Klemsz,M.J., McKercher,S.R., Maki,R.A. and Atchison,M.L. (1992) Mol. Cell. Biol., 12, 368–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hagen G., Muller,S., Beato,M. and Suske,G. (1994) EMBO J., 13, 3843–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmid R.M., Perkins,N.D., Duckett,C.S. Andrews,P.C. and Nabel,G.J. (1991) Nature, 352, 733–736. [DOI] [PubMed] [Google Scholar]
- 30.Pulvertaft R.I.V. (1965) J. Clin. Pathol., 18, 261–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toneguzzo F., Hayday,A.C. and Keating,A. (1983) Nature, 302, 575–581.6300689 [Google Scholar]
- 32.Sugden B., Marsh,K. and Yates,J. (1985) Mol. Cell. Biol., 5, 410–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Wet J.R., Wood,K.V., DeLuca,M., Helinski,D.R. and Subramani,S. (1987) Mol. Cell. Biol., 7, 725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zimber-Strobl U., Kremmer,E., Grässer,F., Marschall,G., Laux,G. and Bornkamm,G.W. (1993) EMBO J., 12, 167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schneider I. (1972) J. Embryol. Exp. Morph., 27, 353–365. [PubMed] [Google Scholar]
- 36.DiNocera P.P. and Dawid,I.B. (1983) Proc. Natl Acad. Sci. USA, 80, 7095–7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 38.Eick D. and Bornkamm,G.W. (1989) EMBO J., 8, 1965–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geltinger C. (1997) Dissertationsverlag NG Kopierladen GmbH, München, Germany.
- 40.Siebenlist U., Franzoso,G. and Brown,K. (1994) Annu. Rev. Cell Biol., 10, 405–455. [DOI] [PubMed] [Google Scholar]
- 41.Baeuerle P.A. and Henkel,T. (1994) Annu. Rev. Immunol., 12, 141–179. [DOI] [PubMed] [Google Scholar]
- 42.Traenckner E.B.-M., Pahl,H.L., Henkel,T., Schmidt,K.N., Wilk,S. and Baeuerle,P.A. (1995) EMBO J., 14, 2876–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baichwal V.R. and Baeuerle,P.A. (1997) Curr. Biol., 7, R94–96. [DOI] [PubMed] [Google Scholar]
- 44.Perkins N.D., Edwards,N.L., Duckett,C.S., Agranoff,A.B., Schmid,R.M. and Nabel,G.J. (1993) EMBO J., 12, 3551–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Courey J.A. and Tjian,R. (1988) Cell, 55, 887–898. [DOI] [PubMed] [Google Scholar]
- 46.Jeong S.W. and Stein,A. (1994) J. Biol. Chem., 269, 2197–2205. [PubMed] [Google Scholar]
- 47.Jeong S. and Stein,A. (1994) Nucleic Acids Res., 22, 370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Archer T.K., Lefebvre,P., Wolford,R.G. and Hager,G.L. (1992) Science, 255, 1573–1576. [DOI] [PubMed] [Google Scholar]
- 49.Alberts A.S., Geneste,O. and Treisman,R. (1998) Cell, 92, 475–487. [DOI] [PubMed] [Google Scholar]
- 50.Dang W., Nikolajczyk,B.S. and Sen,R. (1998) Mol. Cell. Biol., 18, 6870–6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmitz M.L., Stelzer,G., Altmann,H., Meisterernst,M. and Baeuerle,P.A. (1995) J. Biol. Chem., 270, 7219–7226. [DOI] [PubMed] [Google Scholar]
- 52.Hoey T., Weinzierl,R.O.J., Gill,G., Chen,J.-L., Dynlacht,B.D. and Tjian,R. (1993) Cell, 72, 247–260. [DOI] [PubMed] [Google Scholar]
- 53.Emili A., Greenblatt,J. and Ingles,C.J. (1994) Mol. Cell. Biol., 14, 1582–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gill G., Pascal,E., Tseng,Z.H. and Tjian,R. (1994) Proc. Natl Acad. Sci. USA, 91, 192–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Janknecht R. and Hunter,T. (1996) Nature, 383, 22–23. [DOI] [PubMed] [Google Scholar]
- 56.Eckner R. (1996) Biol. Chem., 377, 685–688. [PubMed] [Google Scholar]
- 57.Eisenbeis C.F., Singh,H. and Storb,U. (1995) Genes Dev., 9, 1377–1387. [DOI] [PubMed] [Google Scholar]
- 58.Meyer K.B., Sharpe,M.J., Surani,M.A. and Neuberger,M.S. (1990) Nucleic Acids Res., 18, 5609–5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Costa M.W. and Atchison,M.L. (1996) Biochemistry, 35, 8662–8669. [DOI] [PubMed] [Google Scholar]
- 60.Liu X., Prabhu,A. and Van Ness,B. (1999) J. Biol. Chem., 274, 3285–3293. [DOI] [PubMed] [Google Scholar]
- 61.Nikolajczyk B.S., Nelsen,B. and Sen,R. (1996) Mol. Cell. Biol., 16, 4544–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pongubala J.M. and Atchison,M.L. (1997) Proc. Natl Acad. Sci. USA, 94, 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Park K. and Atchison,M.L. (1991) Proc. Natl Acad. Sci. USA, 88, 9804–9808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meyer K.B. and Ireland,J. (1994) Nucleic Acids Res., 22, 1576–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]