


Abstract
A new deprotection procedure enables a medium scale preparation of phosphodiester and phosphorothioate oligonucleotides substituted with a protected thiol function at their 5′-ends and an amino group at their 3′-ends in good yield (up to 72 OD units/µmol for a 19mer phosphorothioate). Syntheses of 3′-amino-substituted oligonucleotides were carried out on a modified support. A linker containing the thioacetyl moiety was manually coupled in two steps by first adding its phosphoramidite derivative in the presence of tetrazole followed by either oxidation or sulfurization to afford the bis-derivatized oligonucleotide bound to the support. Deprotection was achieved by treating the fully protected oligonucleotide with a mixture of 2,2′-dithiodipyridine and concentrated aqueous ammonia in the presence of phenol and methanol. This procedure enables (i) cleavage of the oligonucleotide from the support, releasing the oligonucleotide with a free amino group at its 3′-end, (ii) deprotection of the phosphate groups and the amino functions of the nucleic bases, as well as (iii) transformation of the 5′-terminal S-acetyl function into a dithiopyridyl group. The bis-derivatized phosphorothioate oligomer was further substituted through a two-step procedure: first, the 3′-amino group was reacted with fluorescein isothiocyanate to yield a fluoresceinylated oligonucleotide; the 5′-dithiopyridyl group was then quantitatively reduced to give a free thiol group which was then substituted by reaction with an Nα-bromoacetyl derivative of a signal peptide containing a KDEL sequence to afford a fluoresceinylated peptide–oligonucleotide conjugate.
INTRODUCTION
Ever since the pioneering work of Zamecnik and co-workers (1), major advances have been made in the development of oligonucleotides as potential regulators of gene expression (for reviews see 2–4). However, the use of oligodeoxyribonucleotides as therapeutic agents is still in its infancy due to several problems. To be active, oligodeoxyribonucleotides must efficiently cross the cell membrane, be resistant to serum and cellular nucleases and specifically bind their targets on mRNA or DNA with high affinity. Besides the attempts developed to improve their resistance to cellular nucleases and their binding properties (4,5), many investigations have been devoted to increase the amounts of oligonucleotides able to reach their targets inside cells. Among reported strategies, peptides have been used either in association as complexes (6–8) or through a covalent linkage (9–15). The covalent conjugation to peptides was carried out in aqueous solution, through the 5′- or the 3′-ends of the oligonucleotides via specific reactions between both moieties (7,9,15–18), although other coupling strategies including on-line synthesis of peptido-oligonucleotides (19,20) or synthesis by a fragment coupling approach have been proposed (21). In order to investigate the role of peptides covalently linked to the oligonucleotides the use of nuclease-resistant oligonucleotides as well as the possibility of detecting them inside cells are required. Because phosphorothioate oligodeoxyribonucleotides are rather nuclease resistant and because their inhibitory activity includes a mechanism involving the action of RNase H, they were selected for the preparation of bisfunctionalized derivatives. Heterobifunctional oligodeoxyribonucleotides have already been described in the phosphodiester series (22,23). Among them, those involving functional groups at the oligonucleotide ends are best suited because their substitution usually does not disturb the binding properties of the oligonucleotides. In most cases, substitution of the bases decreases hybridization efficiency of the modified oligonucleotides either by steric hindrance related to the presence of the substituent group or by altering the properties of one atom involved in base pairing. In the same way, substitutions of an internucleotide phosphate induce chirality of the phosphorus atom leading to two oligonucleotide diastereoisomers possessing different hybridization capabilities. We chose to prepare phosphorothioate oligodeoxyribonucleotides containing a primary amino group via a linker at their 3′-ends and a thiol-protected function at their 5′-ends in order to couple a fluorescent probe through a disubstituted thiourea linkage and a peptide via a thioether bond, which is stable inside cells. The preparation of oligonucleotides containing a primary amino group via a linker at their 3′-ends can be achieved using a modified support (24–26). The synthesis of oligodeoxyribonucleotides containing a thiol masked function at their 5′-ends has already been described for the phosphodiester series by several groups (27–30). However, scaling up synthesis using these protocols very often leads to poor yields. We report here a new deprotection procedure enabling medium scale preparation of these heterobifunctional oligodeoxyribonucleotides. This method, first worked out for the phosphodiester series, was then successfully used for the medium scale preparation of phosphorothioate oligodeoxyribonucleotides, which to our knowledge has not yet been reported. Among the oligonucleotides synthesized, a 21mer phosphorothioate complementary to a region of the HIV-1 envB mRNA was selectively coupled via its 3′-end to a fluorescein derivative and via its 5′-end to a signal peptide containing the KDEL motif [involved in the recycling of proteins from the cis-golgi and ERGIC compartments to the endoplasmic reticulum (ER); 31] previously coupled to a heterobifunctional phosphodiester oligonucleotide (10).
MATERIALS AND METHODS
General methods
All chemicals were used as obtained unless otherwise stated. All solvents were dried and distilled as described (32). Analyses and purifications by ion exchange chromatography were carried out on a Pharmacia FPLC with a DEAE column (8 µm, 100 × 10 mm; Waters) with a linear gradient of NaCl in 25 mM Tris–HCl buffer (pH 7) containing 10% CH3CN. Reversed phase chromatography analyses was performed on a 600E instrument (system controller) equipped with a photodiode array detector (Waters 990) using a LiChrospher 100RP18 (5 µm) column (125 × 4 mm) from Merck with a linear gradient of CH3CN in 0.1 M aqueous triethylammonium acetate (pH 7) at a flow rate of 1 ml/min or a Nova Pak (C18) column (150 × 3.9 mm). Reversed phase purification was performed on a 600E instrument (system controller) using a Radial Pak System containing a PrePack cartridge (100 × 25 mm) filled with C18 (10 µm) from Millipore with a linear gradient (12.5–35% CH3CN) in 0.1 M aqueous triethylammonium acetate (pH 7) at a flow rate of 6 ml/min. Oligonucleotide syntheses were performed on either a Pharmacia Gene Assembler or an Expedite Nucleic Acid Synthesis system 8909 from Perseptive Biosystems. Peptide synthesis was performed on an Applied Biosystem 433A Peptide Synthesizer. UV/visible spectra were recorded on a Uvikon 860 spectrophotometer. Absorption coefficients of oligonucleotides were calculated as described previously (33). Fluorescein isothiocyanate (FITC) was purchased from Molecular Probes (Eugene, OR) and a size exclusion gel (BioGel-P2) from Bio-Rad (Hercules, CA).
Preparation of the modified support 1. This preparation was achieved following a previously published procedure (26) using aminopropyl CPG 500 instead of long chain alkylamine CPG. The modified support 1 was obtained with a loading of 42 µmol/g.
Oligonucleotide chain assembly. Oligonucleotide chain assembly was carried out following phosphoramidite chemistry (34) on modified support 1. Syntheses were performed on either a Pharmacia Gene Assembler on a 13 µmol scale using 9 equiv. of commercial phosphoramidites per cycle with a cycle time of 10 min and a coupling time of 1.5 min, or an Expedite Nucleic Acid Synthesizer 8909 on a 15 µmol scale. The oxidation step was carried out with iodine in a THF/pyridine/water solution (obtained from the manufacturer) and the sulfurization step was performed using 3H-1,2-benzodithiole-3-one 1,1-dioxide (Beaucage reagent, 50 mM in acetonitrile) (35). After addition of the last nucleotide an additional detritylation step was performed on the synthesizer to give oligonucleotide 2 (Scheme 1).


Scheme 1. Synthesis of the heterobifunctional oligonucleotide 5.
Coupling of the phosphoramidite derivative of the S-acetylated linker 3 on the 5′-terminal hydroxyl of oligodeoxyribonucleotide 2 bound to the support. To a small vial, stoppered with a septum, was added oligodeoxyribonucleotide 2 (13 or 15 µmol) bound to the support. A needle was placed in the septum and the vial was left in a dessicator under vacuum for at least 4 h. The dessicator was then filled with nitrogen before its opening. A mixture of phosphoramidite 3 (30 equiv., 0.1 M solution in acetonitrile) and tetrazole (100 equiv., 0.45 M solution in acetonitrile) was added. This mixture was shaken gently by hand from time to time for 30 min, the supernatant was removed with a syringe and the oxidation step was performed upon adding a 2 ml iodine solution (the solution used in the synthesizer). The solution was removed after 1 min and the support was extensively washed with acetonitrile. Alternatively, the sulfurization step was performed using a 2 ml Beaucage reagent solution (the solution used in the synthesizer). The support was then dried for 10 min at 30°C using a rotavapor.
Deprotection of the oligonucleotide derivatized by the S-acetylated linker 4 and transformation of the released thiol function into a pyridyl disulfide group. The oligodeoxyribonucleotide 4 bound to the support was left in the vial and 2,2′-dithiodipyridine (140 equiv.), phenol (10 equiv./phosphate), methanol (3 ml) and concentrated (28%) aqueous ammonia (5 ml) were added successively. The mixture was left at room temperature for 60 h under gentle stirring. The mixture was centrifuged and the supernatant was recovered. The support was washed twice with a water:methanol (2:1 v /v) mixture and once with water. Ammonia and methanol were removed by evaporation under reduced pressure. The other reagents, including 2,2′-dithiodipyridine, phenol and pyridyl-2-thione, were extracted with ethylacetate; after complete removal of ethylacetate by evaporation using a rotavapor, the aqueous solution was filtered through a 0.45 µM disposable filter.
Purification and analysis of oligodeoxyribonucleotides 5 substituted with a protected thiol group at their 5′-ends and an amino function at their 3′-ends. Phosphodiester oligonucleotides 5 were purified by ion exchange chromatography on a DEAE column (8 µm, 100 × 10 mm; Waters) eluted with a linear gradient of 0–1.5 M NaCl in 25 mM Tris–HCl buffer (pH 7) containing 10% acetonitrile. Acetonitrile was removed by evaporation; the solution was desalted by chromatography using a (10 × 100 mm) column packed with LiChroprep RP-18 (40–63 µm) (Merck). The purified modified phosphodiester oligonucleotides 5 were analyzed by reversed phase chromatography on a C18 column. Phosphorothioate oligonucleotides 5 were purified by reversed phase chromatography using a Radial Pak System containing a PrePack cartridge (100 × 25 mm) filled with C18 (10 µm) (Millipore) with a linear gradient of 12.5–35% acetonitrile in a 0.1 M triethylammonium acetate buffer (pH 7) solution at a flow rate of 6 ml/min and then analyzed by reversed phase chromatography on a C18 column. The presence of the 2-pyridyl disulfide group was confirmed on an aliquot according to Carlsson et al. (36).
Preparation of the bis-derivatized oligodeoxyribonucleotide phosphorothioate 9
Preparation of the fluorescein–oligophosphorothioate conjugate 6. The procedure used was adapted from a previously published method (37). An oligonucleotide 5e with a primary amino group at its 3′-end (8 OD units) was dissolved in 50 µl 2 M sodium acetate, 0.2 M carbonate/bicarbonate buffer, pH 9.3. FITC (isomer I, 25 equiv.) was added to the oligonucleotide solution and the mixture was gently stirred at 37°C; more FITC (25 equiv.) was added 5 h later. After overnight incubation, the fluoresceinylated oligonucleotide was precipitated by adding 2.5 vol. cold (–20°C) absolute ethanol. The free fluorescein remaining was removed by size exclusion chromatography at 4°C on a BioGel-P2 column (2 × 25 cm) eluted with 5% n-butanol in water. The first fluorescent peak was recovered and lyophilized and then analyzed by reversed phase chromatography and UV/visible spectroscopy (Fig. 1Ib and IIb) as well as by PAGE (Fig. 2). Polyacrylamide gel analysis (Fig. 2, lane 2) showed the presence of two bands (b and e) identified as the fluoresceinylated oligonucleotide 6 and a product which could correspond to the dimer of oligonucleotide 7 (see below). Polyacrylamide gel analysis of the starting bis-derivatized oligonucleotide 5e (Fig. 2, lane 1) also displayed two bands. The main one (a) corresponds to oligonucleotide 5e and the second (d), with a lower mobility, could be the dimer of oligonucleotide 5e obtained after loss of the 2′-thiopyridyl group of 5e. Reversed phase analysis indicated the presence of a more retarded peak (Rt = 26.1 min; Rt5e = 24.1 min; Fig. 1Ia) with a shoulder containing fluorescein (Fig. 1Ib and IIb). The shoulder corresponds to the dimer of oligonucleotide 7. Starting from 8 OD units of 5e, 7.8 OD units of crude oligonucleotide 6 were obtained.
Figure 1.

(I) Reversed phase monitoring of the coupling reaction between oligonucleotide 5e and both FITC and the peptide. (a) Oligonucleotide 5e; (b) crude fluorescein–oligonucleotide conjugate 6; (c) bis-derivatized oligonucleotide 9. Chromatographic analyses were performed on a Nova-Pack reversed phase (C18) column (150 × 3.9 mm) using a linear gradient from 5 to 40% acetonitrile in 0.1 M TEAA, pH 7.0, over 30 min with a flow rate of 1 ml/min. Detection: λ=260 and 495 nm. (II) UV/visible spectra of compounds 5e (a), 6 (b) and 9 (c) recorded between λ=220 and 600 nm.
Figure 2.
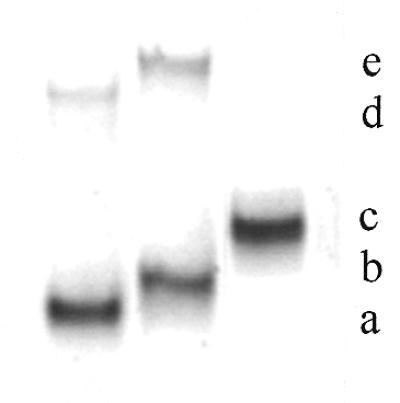
Polyacrylamide (15%) gel electrophoresis of oligonucleotide conjugates stained with methylene blue. Lane 1, heterobifunctionalized phosphorothioate oligonucleotide 5e; lane 2, crude fluorescein–oligonucleotide conjugate 6; lane 3, purified bis-derivatized peptide–fluoresceinyl oligonucleotide 9. (a) Heterobifunctionalized phosphorothioate oligonucleotide 5e; (b) crude fluorescein–oligonucleotide conjugate 6; (c) fluoresceinyl oligonucleotide–peptide conjugate 9; (d) dimerized product of (a); (e) dimerized product of (b).
Synthesis of the Nα-bromoacetyl peptide (containing a KDEL signal sequence) 8. The peptide YGEEDTSEKDEL (corresponding to the C-terminal sequence of the ER-resident luminal protein GRP78) (31) was prepared by solid phase synthesis on an Applied Biosystems 433A peptide synthesizer, monitored by conductimetry, using a 9-fluorenylmethoxycarbonyl strategy starting from a p-hydroxymethyl phenoxymethyl resin. At the end of the synthesis, bromoacetic anhydride was added to form the Nα-bromoacetyl-protected peptide still bound to the resin (10). The peptide was deprotected and released from the resin by treatment with 95% TFA (v/v) in water for 4 h in the presence of phenol (5% w/v) used as a scavenger. The peptide was precipitated in dry diethylether. Purity was assessed by analytical HPLC on a 150 × 3.9 mm reversed phase C18 column (Nova-Pack C18, 4 µm; Waters) using a linear gradient of 5–80% acetonitrile in water containing 0.1% TFA (Rt8 = 17.8 min). The peptide was characterized by matrix-assisted laser desorption ionization time-of-flight mass spectrometry with a Finnigan MAT Laser-mat 2000 instrument (San Jose, CA) (calculated mass 1533.5 Da; measured mass 1535.4 ± 2 Da).
Deblocking of the thiol function of oligonucleotide 6. The fluoresceinylated oligonucleotide 6 (7.8 OD units) was dissolved in 2 M ammonium acetate, 0.05 M sodium phosphate buffer, pH 7.0, (200 µl) and degassed under vacuum by successive freezing and thawing of the solution under vacuum. Then, a water solution of tris(2-carboxyethyl)phosphine (TCEP, 1.2 equiv. dissolved in 2 µl) (10,38) was added under a nitrogen atmosphere. The mixture was left at room temperature for 30 min. The presence of the thiol-containing oligonucleotide 7 was checked by reversed phase analysis on a Nova-Pack C18 column using the conditions described in Figure 1. Rt6 = 26.1 min, Rt7 = 25.1 min (data not shown).
Coupling of peptide 8 to give the bis-derivatized oligonucleotide 9. Two equivalents (0.14 mg in 14 µl of 2 M ammonium acetate, 0.05 M sodium phosphate buffer, pH 7.0) of the Nα-bromoacetyl peptide 8 were added directly into the solution of the thiol-containing oligonucleotide 7 (7.8 OD). After a 3 h reaction, polyacrylamide gel electrophoresis analysis showed the presence of a main product (yield ≈ 70%) with a lower mobility than that of the thiol-containing oligonucleotide 7 as well as two side products identified as oligonucleotide 7 and its corresponding dimer (data not shown). The absence of other bands indicated that coupling of more than one peptide per oligonucleotide did not occur. The reaction mixture was separated by PAGE using the conditions reported for the analysis and the bis-derivatized oligonucleotide 9 was recovered by soaking the gel in 1 ml of a 50 mM solution of ammonium phosphate, pH 7.0, for 2 h at 4°C followed by precipitation upon adding 2.5 vol. of absolute ethanol. Finally, after solubilization of the precipitate in distilled water and desalting by chromatography on a BioGel-P2 column (250 × 20 mm) eluted in water, 3.6 OD units of purified oligonucleotide 9 were obtained (Fig. 2, lane 3, band c). The UV/visible absorbance ratio (A490/A260) confirms the presence of one molecule of fluorescein per oligonucleotide (Fig. 1IIc). The bis-derivatized oligonucleotide 9 was characterized by mass spectrometry. The negative ion electrospray mass spectra were obtained using a Platform Quadrupole Mass Spectrometer (VG Biotech, Fisons Instruments, Altrincham, UK) equipped with an electrospray atmospheric pressure ionization source. Calculated mass 8789.5 Da; found mass 8785 ± 2 Da.
RESULTS AND DISCUSSION
Synthesis of 5′,3′-bis-substituted oligonucleotides 5
Oligonucleotide chain assembly. This synthesis was performed as described in Scheme 1 on a 13 or 15 µmol scale on the modified tritylated support 1 (42 µmol/g) obtained according to a published procedure (26). The oligonucleotide chain assembly was performed via phosphoramidite chemistry (34). The oxidation step was carried out using either iodine or 3H-1,2-benzodithiol-3-one 1,1-dioxide (35), giving the oligonucleotide phosphodiester or phosphorothioate, respectively. After addition of the last nucleotide, an additional detritylation step was performed to deblock the 5′-terminal hydroxyl function. The acetylthiolated linker (30) was then coupled via its phosphoramidite derivative 3 to the 5′-end of the protected oligonucleotide still bound to the support (protected at both the base and the phosphate levels) to afford the oligonucleotide 4.
Deprotection, purification and characterization. The deprotection step was first performed using concentrated ammonia in the presence of 2,2′-dithiodipyridine. This treatment allows cleavage of the oligonucleotide from the support, releasing the primary aliphatic amino function at the 3′-end of the oligonucleotide, as well as complete removal of the protecting groups from the bases, the phosphates and the thiol function at the 5′-end. The thiol function then reacts with 2,2′-dithiodipyridine to afford the thiol-protected oligonucleotide 5. After deprotection of the oligonucleotide chain and transformation of the thioester at the 5′-end of the oligonucleotide into a dithiopyridyl group, oligonucleotide analyses and purifications were performed by chromatography. In both the phosphodiester and the phosphorothioate series, coupling of the thiol-containing linker could be ascertained by comparing the reversed phase analyses of the crude products obtained after deprotection of oligonucleotide 2 containing a hydroxyl group at its 5′-end and of those released upon treatment of oligonucleotide 4 with concentrated aqueous ammonia and 2,2′ dithiodipyridine (140 equiv.), respectively. In the latter case, oligonucleotide 5 containing the dithiopyridyl group eluted later than the oligonucleotide with a 5′ free hydroxyl group (obtained after deprotection of oligonucleotide 2). After purification on a reversed phase column, the presence of the dithiopyridyl group at the 5′-end of the oligonucleotide could be shown by reductive cleavage of the disulfide bridge with either dithiothreitol or tris-(2-carboxyethyl)phosphine, releasing free pyridine-2-thione detected at λ = 343 nm (36). When the synthesis was performed on a small scale (µmol), in most cases oligonucleotide 5 was obtained in good yield. However, when the synthesis was performed on a larger scale the yield based on compound 5 was usually low. This was not because coupling of the thiol-containing linker failed (only a small amount of 5′-hydroxyl-containing oligonucleotide was recovered), but rather due to the formation of a major product whose retention time was between that of the oligonucleotide with the hydroxyl group at the 5′-end and that of the expected product 5 containing the dithiopyridyl group. The product which remained unchanged after prolonged treatment with dithiothreitol was not the oligonucleotide dimer obtainable by mild oxidation of the oligonucleotide containing the thiol. We suspected rather that it was a reaction product [ODN-linker-SCH2CH2CN] between the electrophilic acrylonitrile [CH2=CH2CN] released from the phosphates during the deprotection step and the nucleophilic thiol [ODN-linker-SH] function as previously reported by Kuijpers and van Boeckel (30). This was then confirmed by electrospray mass analysis performed on the side product obtained during synthesis of the bis-derivatized oligonucleotide 5a (calculated mass 7850.9; measured mass 7851 ± 2 Da). In our hands, the use of a large excess of 2,2′-dithiodipyridine as described by these authors did not fully prevent formation of the cyanoethyl adduct. In order to prevent formation of this unwanted product, we chose to add to the deprotection mixture a large excess (10 equiv. per released acrylonitrile molecule) of phenol which, in the presence of concentrated ammonia, was in the phenoxide form and could trap acrylonitrile. Using this new deprotection procedure, we observed that the unwanted product mentioned above was absent and that the expected modified oligonucleotide, in both the phosphodiester and the phosphorothioate series, was obtained in good yield (Table 1).
Table 1. Structures of the bis-derivatized oligonucleotides 5.

R1 = Py-SS(CH2CH2O)3-P(O,S)-
R2 = -P(O,S)(CH2)6NH2
Bis-derivatization of the oligonucleotide 5e by a fluorescein residue and a peptide
This was achieved following the general procedure depicted in Scheme 2. The oligonucleotide 5e was allowed to react with FITC (50 equiv.) overnight at pH 9.3 in order to form a disubstituted thiourea involving the primary amino function of the oligonucleotide. The excess FITC was eliminated in the supernatant upon precipitation of the oligonucleotide derivative with ethanol and by a gel filtration step. Both PAGE and reversed phase chromatography analyses indicated that the expected fluoresceinylated oligonucleotide 6 was sufficiently pure to be used for coupling with a peptide. The absorbance ratio (A490/A260) of the fluorescein–oligonucleotide conjugate 6 (Fig. 1IIb) confirms incorporation of one fluorescein residue per oligonucleotide. Polyacrylamide (15%) gel electrophoresis analysis of oligonucleotides 5e and 6 (Fig. 2) shows, by visualizing fluorescein upon illumination at 260 nm and by methylene blue staining of the oligonucleotide, a different electrophoretic mobility for the oligonucleotide before and after labeling with fluorescein. Following the optimized protocol, we were able to keep the thiopyridyl group at the 5′-end of the oligonucleotide. Indeed, after treatment of the derivative 6 with TCEP (a selective reducing agent for disulfide bridges) a single new peak was obtained indicating that >90% of the fluoresceinylated oligonucleotide 7 was in the thiol form (data not shown). The coupling reaction of this fluorescent oligonucleotide 7 with Nα-bromoacetyl peptide 8, performed at pH 7, gave the oligonucleotide phosphorothioate 9 covalently linked to the fluorescein via its 3′-end and to the peptide via a thioether linkage at its 5′-end. After a 3 h reaction, reversed phase chromatography and PAGE analyses confirmed the presence of a new product (yield ≈ 70%) with a different electrophoretic mobility on PAGE analysis as well as with a retention time by HPLC chromatography different from those of the fluoresceinylated oligonucleotide 6 and the thiol-free oligonucleotide derivative 7; in addition, a trace amount of a fluoresceinylated compound with a mobility corresponding to a dimeric oligonucleotide 7 was detected by electrophoresis. Electrospray mass spectrometry confirmed the calculated molecular weight of the conjugate (calculated mass 8789.5 Da; found mass 8785 ± 2 Da). These results confirm that only one peptide was linked to the oligonucleotide, despite the fact that bromoacetamido derivatives were reported to be sufficiently reactive to substitute internucleotide phosphorothioates leading to phosphothiolotriester linkages (39). These results may be due to (i) the higher reactivity of the thiol group relative to that of the internucleotide phosphorothioate, (ii) steric hindrance due to a particular conformation of the peptide and/or (iii) the coupling conditions, including the use of a high salt concentration and a short reaction time.
Scheme 2. Synthesis of the bis-derivatized oligonucleotide 9.
CONCLUSIONS
We report here the medium scale synthesis of oligonucleotides bearing a protected thiol group at their 5′-ends and a primary amino function at their 3′-ends in both the phosphodiester and the phosphorothioate series. The use of a new deprotection procedure, involving the presence of phenol which prevents addition of the released acrylonitrile to the thiol group (an important side reaction), allows the preparation of bis-substituted oligonucleotides in good yield. This procedure is reliable and several heterobifunctional phosphorothioate oligonucleotides (from 19mers to 25mers) have been obtained in yields ranging from 32 to 72 OD units/µmol. Furthermore, we show that the bis-derivatization of a phosphorothioate oligonucleotide led to a well-defined compound in two steps, successively coupling of the 3′-terminal amino group to FITC and of the 5′-terminal thiol function to a bromoacetyl peptide. Using the reported coupling conditions, formation of phosphothiolotriester bonds between the bromoacetylated peptide and the internucleotide phosphorothioate was not detected, showing that the expected thioether linkage was specifically formed.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr D. Dupret (Appligene-Oncor, Strasbourg, France; present address: Proteus, Nîmes, France) for the gift of compound 3, A. Deroussent (Institut Gustave Roussy, Villejuif, France) for running the electrospray mass spectroscopy and Dr E. Bonfils for helpful discussions. This work was supported by grants from the Agence Nationale de Recherches sur le SIDA (ANRS) and ARC (6103). Y.A. is an engineer at the Institut National de la Santé et de la Recherche Médicale. S.B., now Assistant Professor at the University of Orléans, received a post-doctoral fellowship from the ANRS. L.M. received a fellowship from the MENESR. A.C.R. is Research Director at the Institut National de la Santé et de la Recherche Médicale. M.M. is Professor of Biochemistry at the University of Orléans.
REFERENCES
- 1.Stephenson M.L. and Zamecnik,P.C. (1978) Proc. Natl Acad. Sci. USA, 75, 285–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neidle S. (1997) Anticancer Drug Des., 12, 433–442. [PubMed] [Google Scholar]
- 3.Giovannangeli C. and Hélène,C. (1997) Antisense Nucleic Acid Drug Dev., 7, 413–421. [DOI] [PubMed] [Google Scholar]
- 4.Soyfer V.N. and Potoman,V.N. (1996) In Garber,R.C. (ed.), Triple-Helical Nucleic Acids. Springer Verlag, New York, NY.
- 5.Freier S.M. and Altmann,K.-H. (1997) Nucleic Acids Res., 25, 4429–4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wyman T.B., Nicol,F., Zelphati,O., Scario,P.V., Plank,C. and Szoka,F.C. (1997) Biochemistry, 36, 3008–3017. [DOI] [PubMed] [Google Scholar]
- 7.Wadhwa M.S., Collard,W.T., Adami,R.C., McKenzie,D.L. and Rice,K.G. (1997) Bioconjugate Chem., 8, 81–88. [DOI] [PubMed] [Google Scholar]
- 8.Morris M.C., Vidal,P., Chaloin,L., Heitz,F. and Divita,G. (1997) Nucleic Acids Res., 25, 2730–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eritja R., Pons,A., Escarceller,M., Giralt,E. and Albericio,F. (1991) Tetrahedron Lett., 47, 4113–4120. [Google Scholar]
- 10.Arar K., Monsigny,M. and Mayer,R. (1993) Tetrahedron Lett., 34, 8087–8090. [Google Scholar]
- 11.Bongartz J.P., Aubertin,A.M., Milhaud,P.G. and Lebleu,B. (1994) Nucleic Acids Res., 22, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arar K., Aubertin,A.-M., Roche,A.-C., Monsigny,M. and Mayer,R. (1995) Bioconjugate Chem., 6, 573–577. [DOI] [PubMed] [Google Scholar]
- 13.Soukchareun S., Tregear,G.W. and Haralambidis,J. (1995) Bioconjugate Chem., 6, 43–53. [DOI] [PubMed] [Google Scholar]
- 14.Pichon C., Arar,K., Stewart,A.J., DucDodon,M., Gazzolo,L., Courtoy,P.J., Mayer,R., Monsigny,M. and Roche,A.-C. (1996) Mol. Pharmacol., 51, 431–438. [PubMed] [Google Scholar]
- 15.Prochiantz A. (1996) Curr. Opin. Neurobiol., 6, 629–634. [DOI] [PubMed] [Google Scholar]
- 16.Vives E. and Lebleu,B. (1997) Tetrahedron Lett., 38, 1183–1186. [Google Scholar]
- 17.Tung C.-H., Wang,J., Leibowitz,M.J. and Stein,S. (1995) Bioconjugate Chem., 6, 292–295. [DOI] [PubMed] [Google Scholar]
- 18.Jensen O.N., Kulkarni,S., Aldrich,J.V. and Barofsky,D.F. (1996) Nucleic Acids Res., 24, 3866–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haralambidis J., Duncan,L., Angus,K. and Tregear,G.W. (1987) Tetrahedron Lett., 28, 5199–5202. [Google Scholar]
- 20.Truffert J.-C., Asseline,U., Brack,A. and Thuong,N.T. (1996) Tetrahedron, 52, 3005–3016. [Google Scholar]
- 21.Peyrottes S., Mestre,B., Burlina,F. and Gait,M.J. (1998) Tetrahedron, 54, 12513–12522. [Google Scholar]
- 22.Agrawal S. and Zamecnik,P.C. (1990) Nucleic Acids Res., 18, 5419–5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asseline U., Bonfils,E., Kurfürst,R., Chassignol,M., Roig,V. and Thuong,N.T. (1992) Tetrahedron, 48, 1233–1254. [Google Scholar]
- 24.Nelson P.S., Frye,R.A. and Liu,E. (1989) Nucleic Acids Res., 17, 7187–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Asseline U. and Thuong,N.T. (1990) Tetrahedron Lett., 31, 81–84. [Google Scholar]
- 26.Petrie C., Reed,M., Adams,A. and Meyer,R. (1992) Bioconjugate Chem., 3, 85–87. [DOI] [PubMed] [Google Scholar]
- 27.Connolly B.A. (1985) Nucleic Acids Res., 13, 4485–4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ansorge W., Sproat,B., Stegemann,J., Schwager,C. and Zenke,M. (1987) Nucleic Acids Res., 15, 4593–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gaur R.K., Sharma,P., Kumar,P. and Gupta,K.C. (1989) Nucleic Acids Res., 17, 4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuijpers W. and van Boeckel,C.A. (1993) Tetrahedron, 49, 10931–10944. [Google Scholar]
- 31.Sorger P.K. and Pelham,H.R.B. (1987) J. Mol. Biol., 194, 341–344. [DOI] [PubMed] [Google Scholar]
- 32.Atkinson T. and Smith,M. (1984) In Gait,M.J. (ed.), Oligonucleotides Synthesis: A Practical Approach. IRL Press, Oxford, UK, pp. 35–81.
- 33.Puglisi J.D. and Tinocco,I. (1989) Methods Enzymol., 180, 304–325. [DOI] [PubMed] [Google Scholar]
- 34.Beaucage S. and Caruthers,M. (1981) Tetrahedron Lett., 20, 94–102. [Google Scholar]
- 35.Iyer R., Egan,W., Regan,J. and Beaucage,S. (1990) J. Am. Chem. Soc., 112, 1253–1254. [Google Scholar]
- 36.Carlsson J., Drevin,H. and Axen,R. (1987) Biochem. J., 173, 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith L.M., Fung,S., Hunkapiller,M.W., Hunkapiller,T. and Hood,L.E. (1985) Nucleic Acids Res., 13, 2399–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burns J.A., Bulter,J.C., Moran,J. and Whitesides,G.M. (1991) J. Org. Chem., 56, 2648–2650. [Google Scholar]
- 39.Fidanza J.A., Ozaki,H. and McLaughlin,L. (1994) In Agrawal,S. (ed.), Protocols for Oligonucleotide Conjugates. Synthesis and Analytical Techniques. Humana Press, Totowa, NJ, pp. 121–143.