


Abstract
Drug-like molecules that bind RNA with sequence selectivity would provide valuable tools to elucidate gene expression pathways and new avenues to the treatment of degenerative and chronic conditions. Efforts at discovering such agents have been hampered, until recently, by the limited knowledge of RNA recognition principles. Several recent structures of aminoglycoside–RNA complexes have begun to reveal the structural basis for RNA–drug recognition. However, the absence of suitable chemical scaffolds known to bind the RNA major groove, where specificity could be provided by the diversity of functional groups exposed on the RNA bases, has represented a major obstacle. Here we report an investigation of the structural basis for recognition of an RNA stem–loop by neomycin, a naturally occurring aminoglycoside antibiotic. We found that neomycin binds the RNA stem–loop that regulates alternative splicing of exon 10 within the gene coding for human tau protein. Mutations within this splicing regulatory element destabilise the RNA structure and cause frontotemporal dementia and Parkinsonism linked to chromosome 17 (FTDP-17), an autosomal dominant condition leading to neurodegeneration and death. The three-dimensional structure of the RNA–neomycin complex shows interaction of the drug in the major groove of the short RNA duplex, where familial mutations cluster. Analysis of the structure shows how aminoglycosides and related drugs bind to the RNA major groove, adding to our understanding of the principles of drug–RNA recognition.
INTRODUCTION
The essential role of RNA in many biological processes and in the progression of disease makes the discovery of small RNA-binding molecules an emerging priority in drug discovery. Most interest has so far been directed to the area of infectious diseases, due to the existence of natural RNA-binding antibiotics as well as favourable patterns of drug resistance. However, appealing opportunities for new RNA-binding agents exist in the treatment of chronic conditions as well. Until recently, the discovery of chemically accessible RNA-binding drugs has been hampered by poor understanding of the principles of RNA recognition (1). However, several recent structures of aminoglycoside–RNA complexes have begun to reveal the structural basis for RNA–drug recognition (2–5). A majority of studies on RNA-binding therapeutic candidates have focused on the direct read-out of the sequence of single-stranded RNA targets, for example by antisense methods (6). However, natural RNAs fold into complex structures, very often containing double helical regions capped by hairpin or internal loops and bulges. In analogy with the elegant work on sequence-specific minor groove recognition of double-stranded DNA by polyamine analogues (7,8), the availability of small molecular weight compounds that bind to double-stranded RNA with at least some sequence selectivity would be of great value. Here we report a structural investigation of the complex between neomycin and an RNA regulatory element implicated in neurodegeneration showing how aminoglycoside antibiotics bind the major groove of double-stranded RNA.
The present work was motivated by our interest in the structure and function of the RNA regulatory element found in the gene coding for human tau protein. Intronic mutations in this gene have been mapped to a stem–loop structure that regulates alternative splicing of tau exon 10 (Fig. 1). These mutations cause an autosomal dominant inherited condition, frontotemporal dementia and Parkinsonism linked to chromosome 17 (FTDP-17), which invariably leads to neurodegeneration and death through the formation of filamentous tau protein deposits similar to those found in Alzheimer’s disease and other neurodegenerative conditions (9–16). Intronic mutations have been identified in different families at positions –1, +3, +13, +14 and +16 (with the first nucleotide of the splice donor site of the intron following exon 10 taken as +1) (9,10). We have previously reported the three-dimensional structure of the tau exon 10 splicing regulatory element RNA determined using NMR spectroscopy and have shown that the RNA forms a stable stem–loop structure in vitro (17). We have also shown that the intronic FTDP-17 mutations disrupt the stem–loop structure and reduce its thermodynamic stability. The thermodynamic stability of the stem–loop structure was strongly correlated with the efficiency of splice site utilisation, suggesting that in familial FTDP-17 the efficiency with which exon 10 is spliced into tau mRNA is controlled by the stability of the RNA stem–loop. A corollary to this mechanism is that stabilisation of the stem–loop structure would reverse this effect and could correct the pathological imbalance in the production of tau protein isoforms containing either three or four microtubule-binding repeats. A very attractive way to achieve such an effect is through small molecular weight RNA-binding compounds that selectively bind the RNA stem–loop to modulate splice site utilisation. Such drugs would provide a test of the hypothesis that intronic mutations and the resulting unbalanced expression of tau isoforms causes dementia. Furthermore, they would provide invaluable tools for testing the role of expression of tau protein in sporadic conditions such as Pick’s disease, progressive supranuclear palsy and cortical degeneration (18,19).
Figure 1.

Sequence and secondary structures of wild-type and mutant human tau exon 10 splicing regulatory elements. Exonic sequences are boxed and shown in capital and intronic sequences are in small letters, with the FTDP-17 mutations circled. Base paired nucleotides are connected by a dash.
Here we report an investigation of the interaction of the aminoglycoside antibiotic neomycin B with wild-type and mutant FTDP-17 stem–loops. We found that neomycin binds the RNA stem–loops with micromolar affinity and stabilises their structures. The three-dimensional structure of the complex between the splicing RNA regulatory element and neomycin presented here provides new insight into the structural basis of RNA–drug recognition.
MATERIALS AND METHODS
RNA synthesis
RNA oligonucleotides were prepared by in vitro transcription using T7 RNA polymerase and synthetic DNA templates of the appropriate sequence. RNA was purified by polyacrylamide gel electrophoresis, followed by electroelution, extensive dialysis and size exclusion chromatography (17,20). Isotopically labelled samples (100% 13C-15N) were prepared in the same manner, using suitable isotopically labelled nucleotide triphosphate precursors. These were prepared from isotopically labelled NMPs (CIL, Boston) after rephosphorylation and purification by filtration through 0.2 µm filters, followed by ethanol precipitation. Neomycin B from Sigma was used without further purification. The sequence and secondary structure of the RNAs used in this study are shown in Figure 1.
Samples for UV melting experiments were dialysed against 150 mM NaCl, 0.1 mM EDTA in 10 mM sodium phosphate buffer, pH 6.7. Samples for NMR were dialysed against low- salt buffer consisting of 10 mM sodium phosphate buffer, pH 6.7, so as to reduce the likelihood of RNA dimerisation. At the high concentrations required for biophysical studies, RNA has the potential to form dimeric structures. The presence of the stem–loop structure was unambigously established from the very favourable linewidth and relaxation properties of the NMR resonances and from diffusion measurements that directly monitor the molecular mass of the sample (21). The RNA concentration for the UV melting experiments was 1–2 µM, and 0.8–1.5 mM for NMR experiments.
UV melting
UV melting experiments were recorded on a CARY UV/VIS spectrophotometer equipped with a temperature-controlled heating unit. The heating rate was 0.5°C/min and the data could generally be reproduced within experimental error after one heating/cooling cycle. Reproducibility of the melting temperatures was ~2°C in independent experiments.
NMR spectroscopy
NMR spectra were acquired on Bruker AMX-500, DMX-600 or DRX-800 spectrometers equipped with triple resonance gradient probes. Data were processed using Felix 2.30 (MSI). Acquisition and processing parameters were as reported in other publications from our laboratory (22,23).
Two-dimensional spectra in H2O were recorded at 4°C and lower pH (5.0–5.5) to reduce the rate of exchange with solvent of imino and amino resonances and to facilitate assignments. Base imino (G and U) and amino (C, A and G) resonances were unambiguously separated in 1H-15N HSQC spectra. Assignments were complete for exchangeable NH and NH2 protons from the base-paired regions, but could not be obtained for NH and NH2 protons from the apical loop region of the structure because their fast exchange with solvent prevented the observation of NOE interactions. NMR spectra recorded in D2O were acquired at 27°C. A 2QF-COSY spectrum was recorded with reduced spectral width (2500 Hz) to facilitate the quantitative analysis of scalar correlations involving sugar resonances. 3D HCCH-COSY and HCCH-TOCSY experiments were used for sugar resonance assignments. Sugar and base resonances were correlated using long range 1H-15N HSQC spectra. Several 2D 1H-31P and 3D triple resonance 1H-13C-31P experiments were recorded to assign the phosphorous spectrum. Assignments of non-exchangeable resonances were >95% complete.
Constraints for structure determination
Numbers and categories of experimental constraints are shown in Table 1. Distance constraints between non-exchangeable protons were obtained from 2D NOESY build-ups recorded at 100 and 200 ms mixing times. In addition, two 3D NOESY-HMQC spectra at 100 and 200 ms mixing time were recorded in succession at 27°C. Finally, a 3D HMQC-NOESY-HMQC spectrum was recorded at 150 ms mixing time. Cross-peaks corresponding to covalently constrained distances were used as reference in NOESY experiments recorded at 100 ms. Cross-peaks with volumes equal to or larger than H5–H6 peaks at 100 ms mixing time were attributed upper limits of 2.5 Å (‘strong’ peaks). Cross-peaks with volumes larger than or equal to intranucleotide H1′–H2′ cross-peaks were attributed upper limits of 3 Å (medium peaks). Cross-peaks with volumes larger than or equal to H1′–H3′ or intranucleotide H1′–H6 cross-peaks were attributed 4 Å upper limits (weak). Cross-peaks weaker than H1′–H3′ peaks, and those that could not be volume integrated reliably due to partial spectral overlap, were attributed upper limits of 5 Å. NOE interactions observed only at long mixing time were attributed upper limits of 6 Å. Interproton distances derived from NOE cross-peaks involving exchangeable resonances were generally given a single generous upper limit of 6 Å. However, cross-peaks involving base-paired guanosine NH and cytosine NH2, as well as uracil NH and adenine H2, resonances were attributed 3 Å upper limits.
Table 1. Experimental constraints and structure statistics.
| Experimental constraints | |
| RNA–RNA distance constraints | 564 |
| Neomycin | 47 |
| Intermolecular | 16 |
| Hydrogen bonding and planarity constraints | 90 |
| Dihedral constraints | 124 |
| Total experimental constraints | 841 |
| Structure analysisa | |
| Largest NOE violation in converged structure | 0.25 Å |
| Angle violations in converged structures (>5°) | 0 |
| RMS deviation from average structure (all atoms)b | |
| Full structure | 2.89 ± 0.55 Å |
| Double helical stem (G-1-A+5; U+12-C+17) | 0.77 ± 0.18 Å |
| Neomycin and drug binding site (U0-A+4; U+11-A+15) | 1.57 ± 0.71 Å |
| Neomycin | 1.55 ± 0.82 Å |
aAverage deviations from ideal covalent geometry are as reported from other publications from our laboratory (22).
bBased on 19 converged structures out of 43, as per Figure 2b. Thirteen structures within this ensemble belong to the same conformational family, while six differ from the remainder more significantly. Within this ensemble of 13 structures, RMS deviation for the neomycin binding site is 0.97 ± 0.24 Å and for neomycin is 1.03 ± 0.50 Å; other statistics do not differ significantly between the two sets.
Hydrogen bonding constraints were introduced when a slow rate of exchange with solvent and a large downfield shift of NH and NH2 resonances were observed, together with characteristic patterns of NOE interactions expected for Watson–Crick pairs. Two distance constraints were used for each hydrogen bond, one between heavy atoms (3 ± 0.15 Å) and one between the hydrogen atom and the acceptor (1.9 ± 0.15 Å). Very weak planarity restraints (8% of the standard X-PLOR value of 300) were also introduced to generate planar base pairs. Dihedral angle restraints were also introduced based on the analysis of 2D and 3D correlated spectra as described (22). Typical uncertainties on the dihedral constraints were ±30° (β, γ, δ and ɛ) and ±120° (α and ζ).
Structure determination and analysis
Structures of the RNA–neomycin complex were calculated starting from random coordinates, without making any assumption on the structure of the complex or any manual or automatic docking step. When we tried to automatically dock neomycin against the predetermined RNA structures, we obtained very poor convergence and unsatisfactory agreement with the experimental data (Fig. 2). The method used in the present study is less likely to bias the structure, as compared to docking methods, and at the same time provides larger numbers of structures in good agreement with the experimental data. Intramolecular RNA–RNA distance constraints were supplemented with 16 NOE-based intermolecular (RNA–neomycin) distance constraints and 47 intramolecular (neomycin–neomycin) distance constraints. Structure calculations by restrained molecular dynamics were performed using an X-PLOR-based simulated annealing protocol (22). Dihedral energy terms designed to reproduce ideally staggered rotamers were never introduced to avoid any bias. Electrostatic interactions were not introduced in the structure calculation. Charges were not assigned to neomycin in the structure calculation, nor in the calculation of the electrostatic surface shown in Figure 4, because the protonation state of amines in neomycin is not known in its RNA-bound state, and partial charges would therefore be unreliable. Pseudo-energies of NOE violations and total energies (not shown) are very low, indicating good and similar convergence for the 19 best structures; no violation >0.25 Å was found for any of these structures. RMS deviations from the average structure were calculated using Clusterpose (24).
Figure 2.
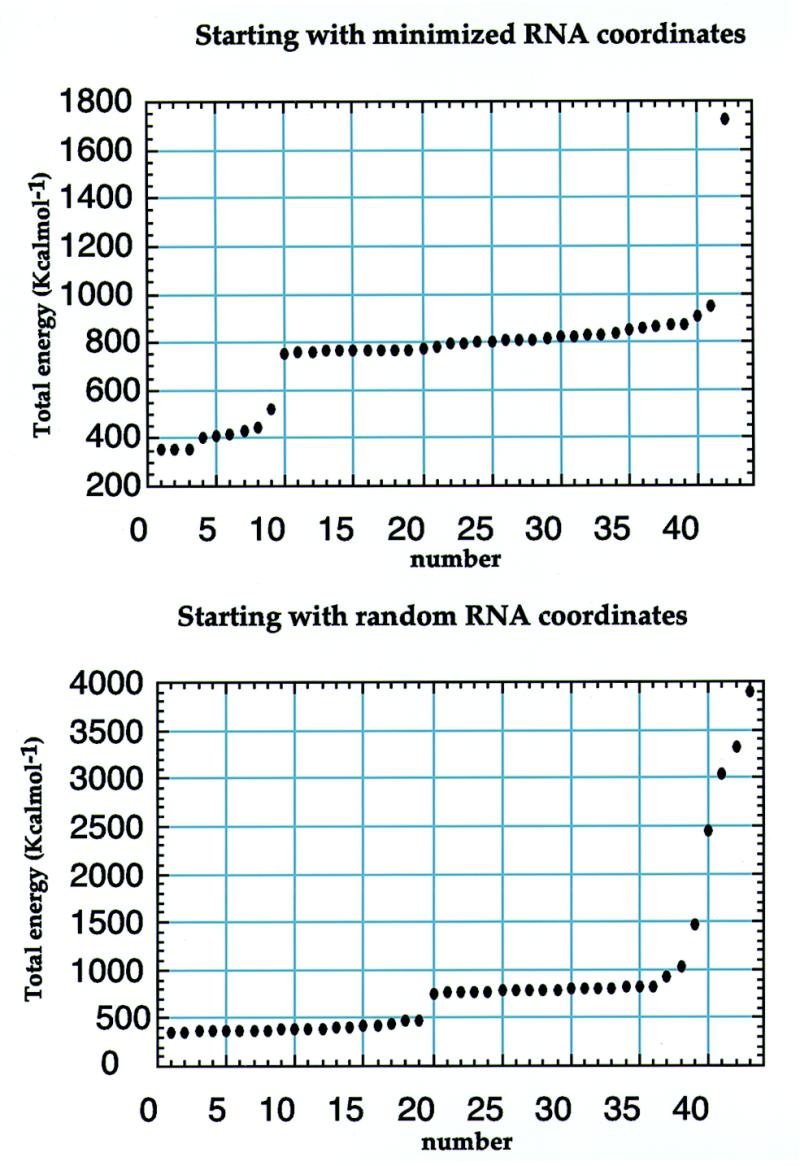
Total energy of structures calculated using either predetermined RNA structures (top) or completely random RNA coordinates (bottom) as starting coordinates. The docking procedure based on a predetermined RNA structure compromises the convergence of the computational protocol.
Figure 4.

Surface representation with electrostatic potential map of the human tau exon 10 splicing regulatory element (left) and of its complex with neomycin B (right). Red indicates negative electrostatic potential, while blue indicates positive potential. This view into the major groove highlights the region of deep negative electrostatic potential where neomycin (green) binds.
RESULTS
FTDP-17 is caused in some families by mutations clustered within the RNA stem–loop that regulates utilisation of the splice site following exon 10 of the human tau gene (Fig. 1). These mutations destabilise the stem–loop structure and increase splicing of exon 10, leading to an unbalanced production of tau protein isoforms containing either three or four microtubule-binding repeats. In the current model, the onset of neurodegeneration is controlled by the stability of the RNA regulatory element. We wanted to test whether small molecular weight compounds would bind the stem–loop and modulate its thermodynamic stability. FTPD-17 intronic mutations belong to a growing list of neurodegenerative and chronic conditions associated with mutations in non-coding regions of genes. Small molecular weight compounds that selectively bind RNA regulatory elements and modulate their activity would provide invaluable tools for dissecting the mechanisms by which pathological conditions arise and could, in due course, lead to new therapeutic avenues.
We recently reported the structure of tau exon 10 splicing regulatory element RNA (17). As shown in Figure 3 (left), the upper part of the structure forms a stable stem–loop with a double helix of 6 bp capped by an apical loop containing six unpaired nucleotides. The loop does not form a unique structure, but adopts multiple conformations in rapid exchange; the potential G·U base pair capping the double helical stem does not form. The conformation of the double helical 6 bp stem is typical of the A-form family of RNA structures. FTDP-17 mutations disrupt this regular structure by introducing wobble pairs or mismatches that reduce its thermodynamic stability. The surface representation of the tau exon 10 splicing regulatory element RNA shows that the bulged adenine at position –2, separating the two double helical regions, and the apical loop increase major groove accessibility, compared to perfectly double-stranded RNA helices. The RNA major groove presents numerous hydrogen bond donors and acceptors and a deep minimum of negative electrostatic potential (Fig. 4, left). Increased accessibility at the junction between the stem and the apical loop, as well as near the unpaired adenosine, could allow small molecules to bind the RNA major groove.
Figure 3.

Three-dimensional structure of wild-type splicing regulatory element RNA (left) and of its complex with neomycin (right). Nucleotides mutated in FTDP-17 are in yellow.
Stabilisation of the structure of the tau exon 10 splicing regulatory element by neomycin B
We performed UV melting experiments on the wild-type and several mutant RNAs to measure the thermodynamic stability of the RNA in the presence of neomycin B. Representative UV melting spectra for mutant +3 in the absence and presence of 1 µM neomycin B are shown in Figure 5. Unfolding of RNA secondary structure leads to an increase in UV absorption at 260 nm (25). The melting temperature (Tm), defined as the mid-point of the transition from fully structured to fully unstructured RNA, was estimated from the maximum in the derivative of the UV melting profile. Tm values in the absence and presence of neomycin are reported in Figure 1. Increasing the neomycin concentration to 2 µM did not have any significant effect on thermodynamic stability. Taken together, these results demonstrate that neomycin binds to the RNA regulatory element and that the neomycin–RNA complex is significantly more stable for both wild-type and mutant RNA sequences. These results also demonstrate that neomycin binds to each RNA with micromolar affinity and stabilises each of the RNAs investigated, the effect being similar for all RNAs (ΔTm = 8–12°C). The presence of divalent metal ions does not significantly reduce the large increase in melting temperature observed when neomycin is added to RNA. The difference in Tm of the RNA–neomycin complexes observed in the presence and absence of 10 mM MgCl2 is only 2°C. As a result of the presence of neomycin, the stability of mutant tau splicing regulatory element RNAs is increased to a level comparable to that of the wild-type RNA. However, the +3 and +14 mutants remain significantly less stable than the wild-type RNA even in the presence of neomycin B.
Figure 5.

UV melting profiles of the +3 mutant (Fig. 1) recorded in the absence (broken line) or presence of 2 µM neomycin.
Interaction of neomycin B with the tau exon 10 splicing regulatory element RNA
Having established that neomycin binds to the tau exon 10 splicing regulatory element RNA, we used NMR spectroscopy to map the site of interaction. The binding site of neomycin onto the RNA regulatory element was mapped by chemical shift perturbation analysis of a complex containing isotopically labelled RNA and unlabelled neomycin, after spectral assignments for the RNA in the complex were obtained as described in Materials and Methods. Chemical shift changes in the RNA were of surprisingly modest magnitude. They clustered in two regions of the RNA, the lower stem, including the A–2 bulge, and the upper part of the stem–loop, including the first few unpaired nucleotides in the apical loop (G+5, C+9 and U+10). The clearest changes manifested as broadening in the spectrum within the lower part of the structure and as chemical shift changes, never larger than 0.1 p.p.m. in the 1H dimension or 0.5 p.p.m. in either 15N or 13C, in the upper part of the stem–loop. The small changes in chemical shift in the upper part of the structure demonstrate that the RNA structure was not changed when the drug was bound. This conclusion was confirmed by a thorough investigation of the pattern of NOE interactions in 2D and 3D spectra recorded using labelled RNA. The neomycin spectrum was assigned using TOCSY and NOESY spectra. Assignments for the paromomycin–ribosomal decoding site complex kindly made available to us (S.Yoshikawa and J.Puglisi, personal communication) made it easier to assign the neomycin spectrum. NOESY spectra recorded using unlabelled components, as well as 3D and 2D heteronuclear filtered spectra using isotopically labelled RNA in complex with unlabelled neomycin, were used to observe intermolecular NOE interactions between neomycin B and the RNA.
Numerous intermolecular NOE interactions could be observed and 16 could be unambiguously assigned to specific RNA and neomycin resonances (Fig. 6). The observation of intermolecular NOE interactions between rings I, II and IV of neomycin and the RNA bases indicated that neomycin interacts with the major groove edge of the RNA bases in the upper part of the stem–loop. We could not observe any intermolecular NOE interaction involving the lower part of the structure, almost certainly because binding occurs with intermediate exchange kinetics and because this broadens the NMR spectra. In the absence of intermolecular NOE interactions, it is impossible to determine how neomycin interacts with this region of the RNA, and we therefore focused our investigation within the apical portion of the stem–loop. The unambiguous assignment of intermolecular RNA–neomycin NOE interactions made it possible to determine the structure of the complex with neomycin bound to the upper part of the RNA stem–loop. These intermolecular NOE interactions were used in the standard structure determination protocol used by our group (22). From a technical point of view, it is worth remarking that satisfactory convergence of the computed structures was only obtained when the structures were calculated starting from completely random initial RNA and drug coordinates. When we tried to determine the structure of the complex by docking the drug against a preformed RNA structure, convergence rates were very low and only a few structures had satisfactory agreement with the data (Fig. 2). Even if the computational protocol is identical, starting with a preformed RNA structure in what amounts to an automatic docking procedure prevents thorough conformational sampling. Statistics of data collection and of the final structure are shown in Table 1. The superposition shown in Figure 7B highlights the good definition of the structure of the complex at and near the neomycin binding site. The apparent loss of definition at the base of the double helical stem is caused by the well-known absence of long range distance constraints in RNA (23); the apical loop remains flexible. A view of a converged structure highlighting the four sites where intronic mutations occur is shown in Figure 3 (right). Close-up views of the neomycin–RNA interaction are shown in Figure 7.
Figure 6.
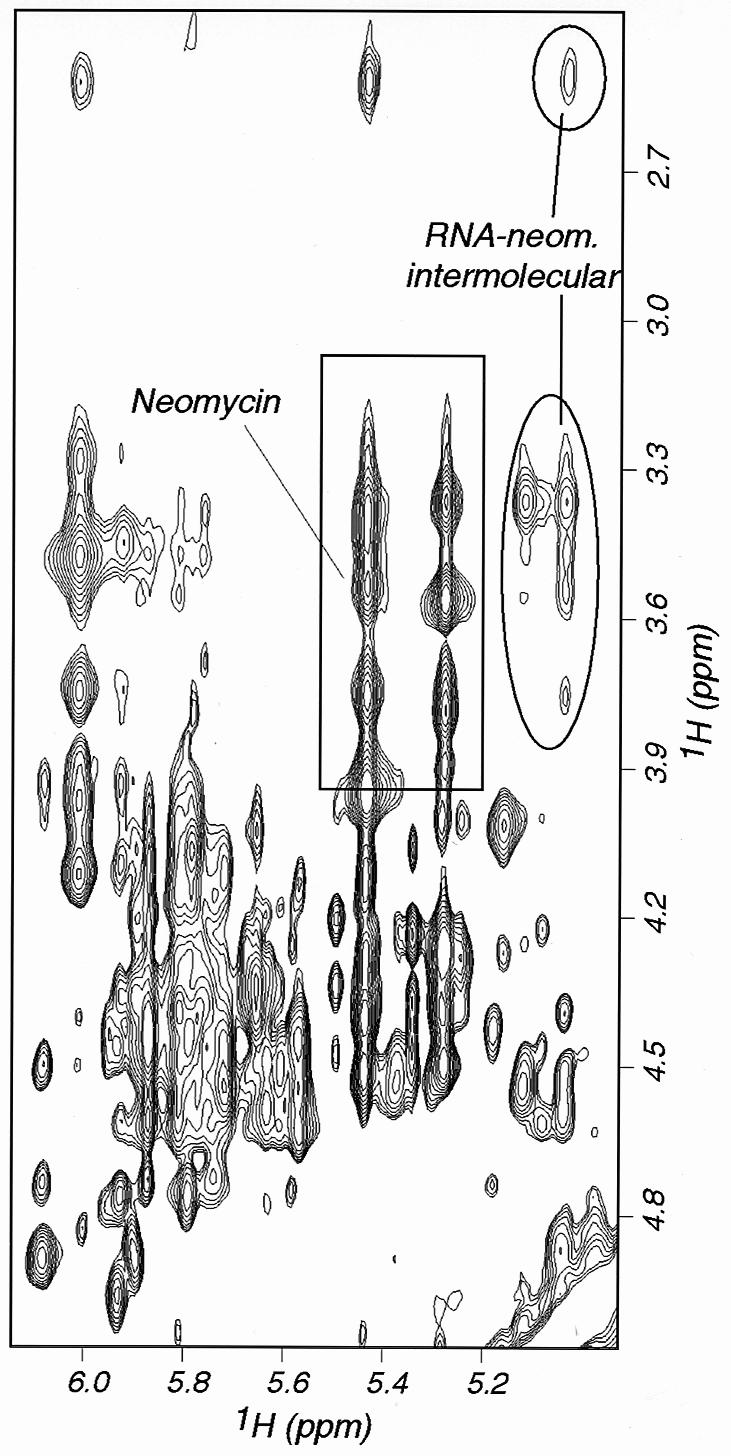
Two-dimensional NOESY experiment recorded on the complex between neomycin and wild-type tau exon 10 splicing regulatory element RNA. Intramolecular neomycin cross-peaks are boxed; intermolecular NOE interactions between neomycin and RNA are circled.
Figure 7.
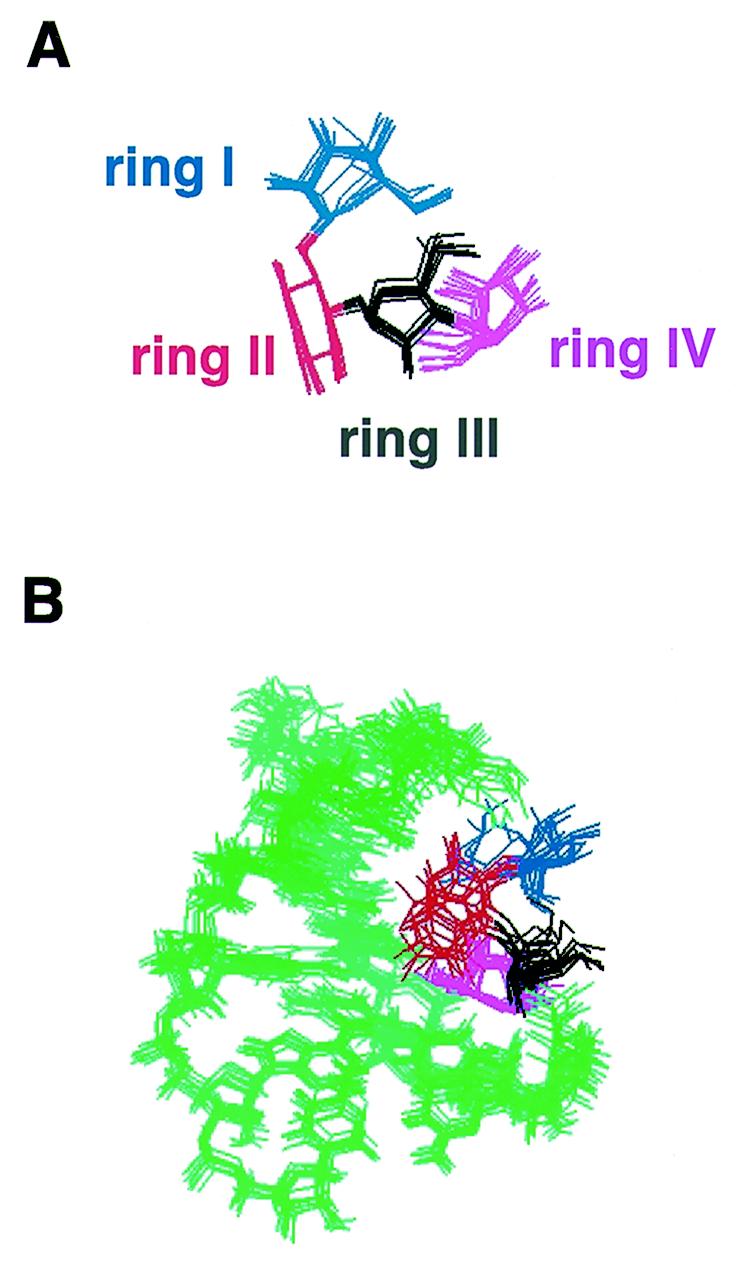
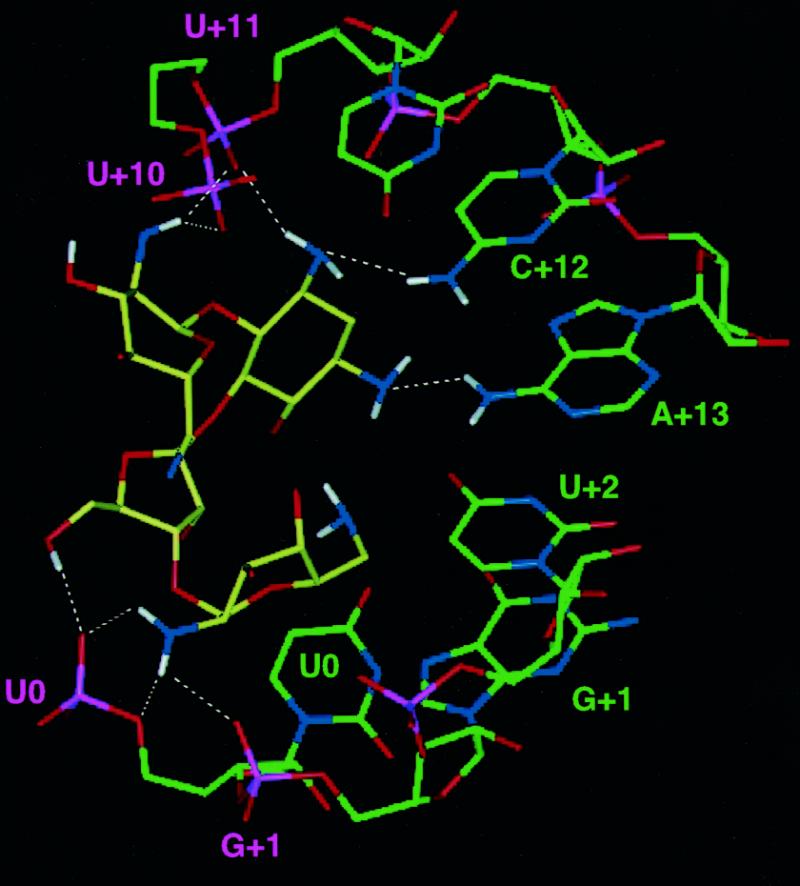
(A) Superposition of neomycin highlighting the good definition of its structure in the complex. (B) Superposition of 10 converged structures viewed down the major groove of the RNA. The orientation of rings I and II relative to the RNA is not as well defined as for the other two rings. (C) Close-up view of the RNA–neomycin interaction from a low energy structure. Intermolecular electrostatic interactions and hydrogen bonds are indicated by dashed lines. Bases have been labelled with different colours according to whether interactions with neomycin involved base (green) or phosphate (purple) groups.
Structure of the complex between neomycin B and tau exon 10 splicing regulatory element RNA
Neomycin binds in the major groove of the RNA stem–loop, as is also observed in other complexes between aminoglycoside antibiotics and RNA (Fig. 3, right). Since the major groove in A-form RNA is very narrow, neomycin binds close to the top of the double helical tract, where the groove is made accessible by an interruption in the progression of the double helix. The length of the stem–loop, 6 bp, is sufficient to provide a binding pocket for neomycin. The intermolecular interaction appears to be largely of electrostatic origin, the positively charged neomycin binding a region of deep negative electrostatic potential (Fig. 4). Favourable electrostatic interactions are supplemented by extensive van der Waals contacts, favoured by the shape complementarity between neomycin itself and the RNA major groove. The antibiotic adopts a well-defined L-shaped conformation (Fig. 7A and B), rings II–IV forming a nearly co-linear array that lines the floor of the major groove covering approximately 5 bp (U0–A+15 and A+4–U+11). These base pairs coincide with the sites where intermolecular NOE interactions were observed. The alignment of rings II–IV in the major groove of the RNA double helix is remarkably similar to that observed for the complex between paromomycin and a ribosomal A-site oligonucleotide mimic (2), as well as an RNA aptamer (5), despite the very different conformations of these three RNAs (Fig. 8). Furthermore, this arrangement is also closely reminiscent of the model of the complex between lividomycin and an A-form double helix obtained by automatic docking and molecular dynamics (26).
Figure 8.

Surface representation of the three aminoglycoside–RNA complexes. The neomycin rings I–IV are coloured blue, red, yellow and purple, respectively. In the aptamer structure (3), ring I is almost completely buried and cannot be seen from this visual angle.
The best defined portion of the present structure is composed of rings II–IV, with ring I being less well defined (Fig. 7A and B). This is because the intermolecular NOE interactions on which the structure is based are largely provided by ring IV. Although we could not observe intermolecular NOEs involving ring III, ring–ring interactions with ring II and between rings I and II, together with steric constraints, position this portion of the drug precisely. The structures of aminoglycosides with specific targets [aptamer (5) or the ribosomal A-site (2)] are more precisely defined than the present structure. This is due in part to a larger number of intermolecular NOE interactions (41 in the aptamer complex versus 16 in the present study) but also to differences in the procedures used in the selection of converged structures. A much smaller portion of all calculated structures was chosen for analysis in the aptamer study (3), leading to an artificial impression of increased precision. Despite these technical differences, the lower number of intermolecular NOE cross-peaks reflects a real distinction between the systems. The RNA makes a less intimate interaction with neomycin in the present system (Fig. 8) and a larger portion of the drug surface is buried in the two complexes of RNA with aminoglycosides characterised so far, when compared with the present structure. The total buried surface area is 810 ± 35 Å2 in the present structure, 1070 Å2 in the average structure of the A-site complex (2) and 1060 Å2 in the lowest energy structure reported for the aptamer complex (5). Another important difference concerns ring III and ring IV, which form the majority of intermolecular interactions observed here and create the bulk of the interaction surface. This is in contrast to what was observed in the A-site complex, where rings III and IV contributed little to RNA binding. In all structures, ring I is positioned at ~90° with respect to rings II–IV. However, in the A-site structure, ring I lies in a tight pocket created by a bulged nucleotide and an unusual, non-planar A–A pair. It is this binding pocket for ring I created by the RNA structure that confers specificity in recognition of the A-site oligonucleotide. In contrast, the interaction of ring I with the splicing regulatory element RNA is not extensive, with ring I facing the outside of the major groove and pointing towards the solvent (Fig. 7B).
Specific intermolecular interactions observed in the present structure involve all four neomycin rings (Fig. 7C). Since the protonation state of neomycin is not known in the RNA-bound state, electrostatic interactions were not included in the calculation and hydrogen bonds and electrostatic interactions are likely to be underestimated in the present structure. Consequently, the observation of numerous interactions of this kind (Fig. 7C) is a strong result of the present study. The N2 amine hydrogens from ring I are in close proximity (<2.5 Å) to phosphate oxygen of nucleotides +10 and +11, but there are no RNA base functional groups within 5 Å of ring I. Ring II is in van der Waals contact with the C+12 base. The N3 amine hydrogen from ring II is in hydrogen bonding contact with the U+11 phosphate backbone, while the N3 nitrogen is hydrogen bonded to the exocyclic amino group of C+12, providing a bifurcated interaction from the N3 substituent with both backbone oxygen and base groups. The N1 nitrogen of ring II is similarly in contact with the exocyclic amino group of A+13. Rings III and IV are close to nucleotides at positions +0, +1 and +2 on the opposite RNA strand. The hydroxyl group at position 5 of ring III forms a hydrogen bond to the U0 phosphate, while the other hydroxyl at position 2 is also in electrostatic contact with the same phosphate group. Ring III is also in van der Waals contact with U0. Ring IV fits into the major groove more intimately than any of the other rings and is in van der Waals interaction with the bases of U0, G+1 and U+2. The hydroxyl at position 4 is in electrostatic contact with the U+2 base carbonyl, while the hydroxyl group at position 3 is in electrostatic contact with the phosphate of G+1. Finally, the amine at position 2 of ring IV is in electrostatic contact with the phosphates of U0 and hydrogen bonds to the phosphate of G+1.
DISCUSSION
The development of small molecular weight compounds that recognise specific RNA structures would provide the basis to discover new chemotherapeutic compounds to treat infectious and chronic conditions. Aminoglycoside antibiotics have received considerable attention, both in an effort to develop suitable mimics with more favourable synthetic and pharmacological properties (27) and as paradigms to investigate the structural and thermodynamic principles underlying RNA–drug recognition (28,29). In addition to the landmark structure of the complex between paromomycin and an oligonucleotide mimic of the decoding site of the ribosome (2), two other structures of complexes between aminoglycoside and RNA aptamers selected to bind to them have revealed principles of RNA–aminoglycoside recognition (4,5). In each case, the drugs are buried in a deep cleft defined by the RNA structure, which folds upon binding of the drug and, in the aptamer case, wraps up the drug, trapping it between the floor of the RNA major groove and an exposed purine base. A unique structure of the RNA binding sites, created by folding upon drug binding, provides specificity in recognition. However, naturally occurring RNA-binding targets derived from eukaryotic or viral mRNAs do not resemble the aptamer structures. RNA regulatory elements from viral or eukaryotic mRNAs that constitute potential pharmacological targets resemble instead the present stem–loop structure, containing one or more double helical regions interrupted by internal loops, hairpin loops or bulges that provide binding sites for regulatory proteins (30). Although the major groove of extended RNA double helices is generally not accessible, dislocations at bulge or hairpin loops open up the major groove and provide potential binding sites (30). In this context, it would be very valuable to have available small molecules that bind double-stranded RNAs with sequence selectivity. Binding of neomycin to the RNA double helical region was reported for the aptamer complexes investigated by Patel and co-workers (5). However, these authors focused their investigation on the interaction with the single-stranded region of the RNA. The present work describes the structure of the complex between the aminoglycoside antibiotic neomycin B and an RNA regulatory element derived from the tau exon 10 splicing regulatory element, and provides structural insight into recognition of RNA double helices by RNA-binding drugs.
Neomycin interacts with the edges of the bases in the major groove of the RNA stem–loop. The major groove presents a region of deep electrostatic potential (Fig. 4). Electrostatic energy is likely to be the driving force in guiding binding, but shape complementarity is also likely to play a major role. It is remarkable that the orientation of neomycin B in the floor of the major groove is very similar between the present complex and the aptamer and ribosomal decoding site complexes. This observation suggests very strongly that the conformational preferences of rings II–IV of aminoglycoside antibiotics closely match the progression and steric properties of A-form RNA. This shape complementarity was scored for in the computational search for new RNA-binding drugs using DOCK (26). A database search for compounds that recognise the shape of the major groove of A-form RNA and that do not recognise the minor groove of B-form DNA identified aminoglycosides among potential major groove binders. The present structure provides a stereochemical rationale for this result and validates the computational analysis. These considerations suggest that the ability of aminoglycosides to target a variety of partially double-stranded RNA structures, for example the HIV RRE and several ribozymes, could be due to these same conformational preferences. They also suggest that synthetically accessible structural mimics of aminoglycoside antibiotics could provide general major groove RNA binders. The binding mode illustrated here is based on recognition of general conformational features rather than a specific sequence, and therefore could provide the basis for the design of generic RNA major groove binding molecules, where the well-known diversity of functional groups could be exploited to provide specificity in recognition.
The most important difference between the present structure and previous studies of aminoglycoside–RNA complexes is represented by the extent to which the drug is buried in the RNA structure. Ring I makes the least intimate interactions with the RNA and is facing the solvent, whereas rings I and II provide the majority of intermolecular contacts in the decoding site and aptamer complexes. As binding becomes tighter and more specific from the present structure to the decoding site and the aptamer complex, a larger portion of the drug surface becomes buried (Fig. 8). This corresponds to a progressively more complex architecture of the RNA-binding pocket. In both cases of specific recognition, selectivity is built into the RNA structure itself and is achieved through adaptive binding, i.e. by modulation of the target structure during recognition. In the present structure, instead, the limited length of the RNA helix, 6 bp, provides natural boundaries and binding occurs by rigid docking to a pre-formed RNA structure.
The present investigation was motivated by the discovery that intronic mutations at the exon 10–5′ intron boundary of the tau gene are sufficient to produce a dementia disease (9,11), suggesting that correcting the effects of these mutations could slow down disease progression. Intronic FTDP-17 mutations reduce the thermodynamic stability of the stem–loop structure investigated here and alter utilisation of the splice site following exon 10 of the tau gene (17). RNA-binding compounds that recognise the tau exon 10 regulatory element and down-regulate splice site utilisation could restore the physiological balance between tau isoforms and therefore provide investigative tools to dissect how unbalanced production of tau isoforms leads to neurodegeneration. Our results show that neomycin binds to the regulatory element and produces a marked increase in the thermodynamic stability of both wild-type and mutant regulatory elements. Exogenous regulation of alternative splicing of exon 10 of the tau gene through RNA-binding drugs may also provide investigative tools to study diseases other than FTDP-17. Most late onset neurodegenerative conditions are characterised by the presence of an intracellular filamentous pathology, which is believed to cause neurodegeneration (31). Tau protein, α-synuclein and lengthy glutamine repeats account for the filamentous assemblies in the majority of these diseases. Assembly into filaments has been shown to be a nucleation-dependent process which is highly concentration dependent (32–35). It is therefore likely that a reduction in the levels of these proteins could be of therapeutic benefit. RNA-binding compounds would be of great value as tools to understand the post-transcriptional regulation of the proteins that assemble into filaments in nerve cells. This understanding may lead to the demonstration that small molecules that regulate expression of these proteins are effective therapeutic and neuroprotective agents. Neomycin and related aminoglycoside antibiotics bind a wide range of RNA structures and sequences and clearly do not display the selectivity that would be necessary for therapeutic or investigative applications. However, the present results provide a starting point for the rational design of new chemical entities and combinatorial libraries from which the desired selectivity could be built.
Acknowledgments
ACKNOWLEDGEMENTS
It is a pleasure to thank Mr P. Cole for sample preparation, Dr A. Ramos for advice on structure calculation, Dr Paul Barker for help in collecting UV melting data and Prof. I. Tinoco Jr (University of California) for advice on RNA thermodynamics and UV melting experiments. Prof. J. Puglisi and Dr S. Yoshikawa (Stanford University Medical School) provided assignments of neomycin B. L.V. was supported by a studentship from the European Union and by the Medical Research Council.
REFERENCES
- 1.Afshar M., Prescott,C.D. and Varani,G. (1999) Curr. Opin. Biotechnol., 10, 59–63. [DOI] [PubMed] [Google Scholar]
- 2.Fourmy D., Recht,M.I., Blanchard,S.C. and Puglisi,J.D. (1996) Science, 274, 1367–1371. [DOI] [PubMed] [Google Scholar]
- 3.Jiang L., Suri,A.K., Fiala,R. and Patel,D.J. (1997) Chem. Biol., 4, 35–50. [DOI] [PubMed] [Google Scholar]
- 4.Jiang L. and Patel,D.J. (1998) Nature Struct. Biol., 5, 769–774. [DOI] [PubMed] [Google Scholar]
- 5.Jiang L., Majumdar,A., Hu,W., Jaishree,T.J., Xu,W. and Patel,D.J. (1999) Structure, 7, 817–827. [DOI] [PubMed] [Google Scholar]
- 6.Branch A.D. (1998) Trends. Biochem. Sci., 23, 45–50. [DOI] [PubMed] [Google Scholar]
- 7.Gottesfeld J.M., Neely,L., Trauger,J.W., Baird,E.E. and Dervan,P.B. (1997) Nature, 387, 202–205. [DOI] [PubMed] [Google Scholar]
- 8.Trauger J.W., Baird,E.E. and Dervan,P.B. (1996) Nature, 382, 559–561. [DOI] [PubMed] [Google Scholar]
- 9.Hutton M., Lendon,C.L., Rizzu,P., Baker,M., Froelich,S., Hoelden,H., Pickering-Brown,S., Chakraverty,S., Isaacs,A., Grover,A., Hackett,J., Adamson,J.,S.,L., Dickson,D., Davies,P., Petersen,R.C., Stevens,M., de Graaf,E., Wauters,E., van Baren,J., Hillebrand,M., Joosse,M., Kwon,J.M., Nowotny,P., Che,L.K., Norton,J., Morris,J.C., Reed,L.A., Trojanowski,J., Basun,H., Lannfelt,L., Neystat,M., Fahn,S., Dark,F., Tannenberg,T., Dodd,P.R., Hayward,N., Kwok,J.B.J., Schoefiled,P.R., Andreadis,A., Snowden,J., Craufurd,D., Neary,D., Owen,F., Oostra,B.A., Hardy,J., Goate,A., van Swieten,J., Mann,D., Lynch,T. and Heutink,P. (1998) Nature, 393, 702–705. [DOI] [PubMed] [Google Scholar]
- 10.Spillantini M.G., Murrell,J.R., Goedert,M., Farlow,M.R., Klug,A. and Ghetti,B. (1998) Proc. Natl Acad. Sci. USA, 95, 7737–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goedert M., Crowther,R.A. and Spillantini,M.G. (1998) Neuron, 21, 955–958. [DOI] [PubMed] [Google Scholar]
- 12.Hardy J. and Gwinn-Hardy,K. (1998) Science, 282, 1075–1079. [DOI] [PubMed] [Google Scholar]
- 13.Iijima M., Tabira,T., Poorkaj,P., Schellenberg,G.D., Trojanowski,J.Q., Lee,V.M.-Y., Schmidt,M.L., Takahashi,K., Nabika,T., Matsumoto,T., Yamashita,Y., Yoshioka,S. and Ishino,H. (1999) NeuroReport, 10, 497–501. [DOI] [PubMed] [Google Scholar]
- 14.Morris H.R., Perez-Tur,J., Janssen,J.C., Brown,J., Lees,A.J., Wood,N.W., Hardy,J., Hutton,M. and Rossor,M.N. (1999) Ann. Neurol., 45, 270–271. [PubMed] [Google Scholar]
- 15.Goedert M., Spillantini,M.G., Crowther,R.A., Chen,S.G., Parchi,P., Tabaton,M., Lanska,D.J., Markesbery,W.R., Whilemsen,K.C., Dickson,D.W., Petersen,R.B. and Gambetti,P. (1999) Nature Med., 5, 454–457. [DOI] [PubMed] [Google Scholar]
- 16.Hulette C.M., Pericak-Vance,M.A., Roses,A.D., Schmechel,D.E., Yamaoka,L.H., Gaskell,P.C., Welsh-Bohmer,K.A., Crowther,R.A. and Spillantini,M.F. (1999) J. Neuropathol. Exp. Neurol., 58, 859–866. [DOI] [PubMed] [Google Scholar]
- 17.Varani L., Hasegawa,M., Spillantini,M.G., Smith,M.J., Murrell,J.R., Ghetti,B., Klug,A., Goedert,M. and Varani,G. (1999) Proc. Natl Acad. Sci. USA, 96, 8229–8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delacourte A., Sergeant,N., Wattez,A., Gaureau,D. and Robitaille,Y. (1998) Ann. Neurol., 43, 193–204. [DOI] [PubMed] [Google Scholar]
- 19.Sergeant N., Wattez,A. and Delacourte,A. (1999) J. Neurochem., 72, 1243–1249. [DOI] [PubMed] [Google Scholar]
- 20.Price S.R., Oubridge,C., Varani,G. and Nagai,K. (1998) In Smith,C. (ed.), RNA–Protein Interaction: A Practical Approach. Oxford University Press, Oxford, UK.
- 21.Lapham J., Rife,J.P., Moore,P.B. and Crothers,D.M. (1997) J. Biomol. NMR, 10, 255–262. [DOI] [PubMed] [Google Scholar]
- 22.Varani G., Aboul-ela,F. and Allain,F.H.-T. (1996) Prog. NMR Spectrosc., 29, 51–127. [Google Scholar]
- 23.Allain F.H.-T. and Varani,G. (1995) J. Mol. Biol., 250, 333–353. [DOI] [PubMed] [Google Scholar]
- 24.Diamond R. (1995) Acta Crystallogr., D51, 127–135. [DOI] [PubMed] [Google Scholar]
- 25.Puglisi J.D. and Tinoco,I.,Jr (1990) Methods Enzymol., 180, 304–325. [DOI] [PubMed] [Google Scholar]
- 26.Chen Q., Shafer,R.H. and Kuntz,I.D. (1997) Biochemistry, 36, 11402–11407. [DOI] [PubMed] [Google Scholar]
- 27.Park W.K.C., Auer,M., Jaksche,H. and Wong,C.-H. (1996) J. Am. Chem. Soc., 118, 10150–10155. [Google Scholar]
- 28.Recht M.I., Fourmy,D., Blanchard,S.C., Dahlquist,K.D. and Puglisi,J.D. (1996) J. Mol. Biol., 262, 421–436. [DOI] [PubMed] [Google Scholar]
- 29.Recht M.I., Douthwaite,S. and Puglisi,J.D. (1999) EMBO J., 18, 3133–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varani G. and Pardi,A. (1994) In Mattaj,I. and Nagai,K. (eds), RNA–Protein Interactions. Oxford University Press, Oxford, UK.
- 31.Goedert M., Spillantini,M.G. and Davies,S.W. (1998) Curr. Opin. Neurobiol., 8, 619–632. [DOI] [PubMed] [Google Scholar]
- 32.Goedert M., Jakes,R., Spillantini,M.G., Hasegawa,M., Smith,M.J. and Crowther,R.A. (1996) Nature, 383, 550–553. [DOI] [PubMed] [Google Scholar]
- 33.Scherzinger E., Lurz,R., Turmaine,M., Mangiarini,L., Hollenbach,R., Bates,G.P., Davies,S.W., Lehrach,H. and Wanker,E.E. (1997) Cell, 90, 549–558. [DOI] [PubMed] [Google Scholar]
- 34.Friedhoff P., Schneideau,A., Mandelkow,E.M. and Mandelkow,E. (1998) Biochemistry, 37, 10223–10230. [DOI] [PubMed] [Google Scholar]
- 35.Crowther R.A., Jakes,R., Spillantini,M.G. and Goedert,M. (1998) FEBS Lett., 436, 309–312. [DOI] [PubMed] [Google Scholar]