


Abstract
Naturally occurring hammerhead ribozymes are produced by rolling circle replication followed by self-cleavage. This results in monomer-length catalytic RNAs which have self-complementary sequences that can occupy their trans-binding domains and potentially block their ability to cleave other RNA strands. Here we show, using small self-processed ribozymes, that this self-binding does not necessarily inhibit trans-cleavage and can result in greatly elevated discrimination against mismatches. We utilized a designed 63 nt circular DNA to encode the synthesis of a self-processed ribozyme, MDR63. Rolling circle transcription followed by self-processing produced the desired 63 nt ribozyme, which potentially can bind mdr-1 RNA with 9+9 nt of complementarity or bind itself with 4+5 nt of self-complementarity by folding back its ends to form hairpins. Kinetics of trans-cleavage of short complementary and mismatched RNAs were measured under multiple turnover conditions, in comparison to a standard 40 nt ribozyme (MDR40) that lacks the self-complementary ends. The results show that MDR63 cleaves an mdr-1 RNA target with a kcat/Km almost the same as MDR40, but with discrimination against mismatches up to 20 times greater. Based on folding predictions, a second self-processed ribozyme (UG63) having a single point mutation was synthesized; this displays even higher specificity (up to 100-fold) against mismatches. The results suggest that self-binding ends may be generally useful for increasing sequence specificity of ribozymes.
INTRODUCTION
The hammerhead ribozyme was originally identified as the catalytic RNA motif responsible for self-processing of plant infecting viroid RNAs (1–4). The replication pathway for these infectious RNAs involves rolling circle replication to produce a multimeric copy and requires a self-cleavage step to produce monomer-length viroid RNAs in amplified quantities. Plant viroids are typically 200–400 nt in length, but the catalytic function can be made considerably smaller. Minimal hammerhead RNA motifs have been identified that can cleave other RNAs with multiple turnovers (5–7) and such agents are under investigation as therapeutic agents for specific gene inhibition (8–10). It has been observed, however, that the specificity of RNA cleavage by hammerhead ribozymes is low, especially if the ribozyme/target RNA recognition helices are long (11). When these helices are too short, however, cleavage site specificity is likely to be adversely affected in the presence of complex nucleic acid sequences. Because cleavage of mismatched targets is undesirable in therapeutic and diagnostic strategies, there would be considerable value in finding ways to increase sequence specificity of ribozyme cleavage.
We have previously shown that small synthetic circular single-stranded DNAs can behave as efficient templates for RNA polymerases, despite their lack of promoter sequences (12). The resulting rolling circle transcription produces long multimeric RNAs in a mechanism that mimics the first step of viroid RNA replication. Further, when ribozyme RNAs and their cleavage substrates are encoded in such circular vectors, the repeating RNAs undergo self-processing, yielding monomer-length ribozyme RNAs as the chief products and in amplified amounts. This biomimetic strategy has been successfully used in the synthesis of hammerhead, hairpin and hepatitis delta ribozyme motifs (13–15).
As with viroid monomer RNAs, however, the monomer ribozymes produced from such a self-processing mechanism necessarily contain not only the minimal catalytic RNA, but also self-complementary RNAs which are the remnants of self-cleavage. These sequences are located at the ends of the minimal ribozyme and have the potential to fold back and occupy the domains of the ribozyme that are necessary for binding to another RNA before cleaving it. Thus there is the possibility that this self-binding might interfere with trans-cleaving ability. For this reason, internal self-complementarity might well be expected to strongly affect the trans-cleaving properties of ribozymes. Importantly, although all hammerhead ribozymes in their natural form do possess such complementarity, the effect on trans-cleaving efficiency and specificity has apparently not been investigated.
Here we show that self-complementary ends resulting from self-processing do not necessarily inhibit trans-cleaving activity of a hammerhead ribozyme. Moreover, we find that sequence specificity is markedly increased by this potential structure. The results suggest that rolling circle transcription coupled with self-processing may be a generally useful strategy for synthesis of ribozymes that have enhanced properties for RNA cleavage.
MATERIALS AND METHODS
Preparation of oligonucleotides and circular DNAs
DNA and RNA oligonucleotides were synthesized on solid supports using the phosphoramidite method on an Applied Biosystems model 392 DNA/RNA synthesizer. Oligodeoxyribonucleotides were deprotected by treatment with concentrated 25% ammonia at 55°C for 8 h (16). Synthesized RNAs were removed from the solid support and base blocking groups were removed by treatment with concentrated 25% ammonia in ethanol (3:1 v/v) at 55°C for 8 h. After drying in vacuum, the 2′-silyl protecting groups were removed by resuspending the pellet in 50 equiv. of 1.0 M tetrabutylammonium fluoride per equivalent of silyl and the mixtures incubated overnight at room temperature. The oligoribonucleotides were then passed through a C-18 Sep-Pak cartridge column for desalting. After deblocking, DNA and RNA oligonucleotides were purified by electrophoresis on polyacrylamide denaturing gels. After elution from the gels, the oligonucleotides were desalted again with C18 Sep-Pak cartridges. Construction of circular DNAs was done by sequential enzymatic ligations. Ligations of 5′-phosphorylated 35 and 28 nt oligonucleotides were performed sequentially using T4 DNA ligase and 24 nt splint oligonucleotides as described previously (13,14,17). Sequences were: MDR63, 5′-pGAC TGA GGA GTT CGT CTG TCT TTC AGT TTC GTC CT-3′ and 5′-pCAC GGA CTC ATC AGA ATG GCA ACA CAT T-3′; UG63, 5′-pGAC TGA GGA GTT CGT CTG TCT TTC AGT TTC GTC CT-3′ and 5′-pCAC GGA CTC ATC AGA ATG GCA ACC CAT T-3′. Single-strand concentrations of purified DNA and RNA oligonucleotides were determined by measuring the absorbance at 260 nm at high temperature. Single-strand extinction coefficients were calculated from mononucleotide and dinucleotide data with a nearest neighbor approximation (18). The MDR40 short ribozyme was synthesized by T7 RNA polymerase transcription of a synthesized DNA template and purified by 15% denaturing gel electrophoresis (19). Sequences of RNA targets of varying length were 5′-UCA GUA AAU GG-3′, 5′-UUU CAG UCA AUG GCA-3′, 5′-UGU UUC AGU CAA UGG CAA C-3′ and 5′-CUG UGU UUC AGU CAA UGG CAA CAC A-3′.
Transcription reactions
Conditions for an internally labeled rolling circle transcription reaction were: 1 µM circular DNA, 2 U Escherichia coli RNA polymerase (RNAP) holoenzyme or 25 U T7 RNAP, 0.5 mM ATP, CTP and GTP, 60 µM GTP, 0.30 µCi [α-32P]UTP in 25 mM Tris–HCl (pH 8.1) buffer containing 20 mM NaCl, 15 mM MgCl2, 0.4 mM spermine–HCl, 100 µg/ml acetylated bovine serum albumin, 10 mM DTT and 12.5 U/ml RNase inhibitor, in a total reaction volume of 15 µl. Unlabeled MDR63 and UG63 were prepared from a rolling circle transcription reaction as indicated above using E.coli RNAP with all four rNTPs at 0.5 mM. Reactions were incubated at 37°C for 12 h and the reaction was stopped by the addition of 1 vol of stop solution (30 mM EDTA, 8 M urea). Gel analysis was on a 10% polyacrylamide denaturing gel run at 4°C.
Sequencing of monomer ribozymes
5′-End-labeling with T4 polynucleotide kinase was done for monomer RNAs following standard procedures. RNase T1 cleavage and alkaline hydrolysis was performed on the 32P-labeled monomers after they were ethanol precipitated and redissolved in water. Alkaline hydrolysis was carried out in 50 mM sodium bicarbonate (pH 9.0) buffer containing 1 mM EDTA for 10 min at 90°C. RNase T1 cleavage conditions were as follows: 32P-labeled RNA and 0.064 U/µl RNase T1 (US Biochemical) in 20 mM sodium citrate (pH 3.5) buffer containing 6 M urea and 1 mM EDTA, reacted at 50°C for 5 min. All reactions were stopped by rapid cooling on dry ice prior to immediate analysis on a 15% polyacrylamide denaturing gel.
Cleavage reactions
Multiple turnover experiments were performed with RNA substrate in at least 10-fold excess over the ribozyme (20). The 32P-labeled RNA substrate and 50 nM ribozyme were separately heated to 90°C for 1 min, cooled slowly and incubated at 37°C for 20 min. All cleavage reactions under the multiple turnover conditions were initiated by mixing the substrate and the ribozyme. In the case of single turnover conditions, each ribozyme and substrate was heat treated in reaction buffer separately and then allowed to reach reaction temperature. Reactions were initiated by combining various concentrations (50 nM–1.5 µM) of ribozyme and a 5 nM 32P-labeled RNA substrate. A different reaction protocol was also used in which ribozyme (50 nM–1.5 µM) and 32P-labeled RNA substrate (5 nM) were heat treated together in 25 mM Tris–HCl (pH 8.1) buffer containing 20 mM NaCl and 0.4 mM spermine. After equilibration to 37°C, the reaction was initiatied by the addition of MgCl2 to a final concentration of 15 mM. The reactions were terminated by adding an equal volume of 100 mM Na2EDTA, 9 M urea, 0.02% bromophenol blue and 0.02% xylene cyanol. The labeled product and substrate were separated by electrophoresis on 20% polyacrylamide denaturing gels. The RNA cleavage yields were determined by quantitation of radioactivity in the bands of labeled products and substrate with a radioanalytical scanner (Molecular Dynamics Storm 860). Initial rates corresponding to the first 20% of reaction were used to obtain rate constants and the kcat and Km values were calculated from non-linear least square fits (KaleidaGraph; Ablebeck Software). Rate constants for reaction under single turnover conditions were determined from the slopes of semi-logarithmic plots of the 32P-labeled RNA substrate concentration, normalized to the final extent of cleavage, versus time.
RESULTS
Design of circular vector and in vitro transcription
In the case of some kinetic investigations of the mechanism of the hammerhead ribozyme, short recognition stems (e.g. 5–6 nt) are used because the relatively low binding affinity leads to high activity (21). In the case of self-processing ribozymes, however, a short recognition stem would not be sufficient for high trans-cleavage activity (shown below), since the ribozyme must have complementarity (at least 4 or 5 nt) with itself to make the intramolecular hammerhead ribozyme structure. Thus, there must be enough binding energy between the substrate and the recognition domains of the ribozyme to overcome this self-binding. For that reason, 9 nt of recognition was designed as the stem length on each side of the cleavage domain. For a minimal hammerhead RNA designed to bind 9+9 nt of an RNA target, a total size of 39–40 nt is necessary. To generate a hammerhead RNA with this activity by self-processing, one must also include ~16–28 nt of sequence that can be cleaved by this ribozyme. We previously showed that an 83 nt DNA circle could be transcribed to yield self-processing hammerhead RNAs (13,22); in the present case we shortened the length by 20 nt, giving a 63 nt construct, MDR63 (Fig. 1). Because of concerns that too much self-complementarity might greatly limit trans-cleaving activity, we designed MDR63 to have 9+9 nt of complementarity for mdr-1 RNA (23), but to have less complementarity (4+5 nt) for itself. RNA folding analysis (24–27) predicts free energies (37°C) of –0.2 and –3.2 kcal/mol for the two self-complementary hairpins in MDR63 (Fig. 1A) and –11.3 and –10.3 kcal/mol for the helices formed by binding a perfectly complementary target (as in Fig. 2). Also synthesized for comparison was the minimal hammerhead MDR40, which is identical to MDR63 but lacks the self-complementary ends (Fig. 1C).
Figure 1.

(A) Sequences and predicted secondary structures of the 63 nt circular DNA encoding MDR63 and the product, the self-processed MDR63 hammerhead ribozyme. (B) Sequence of the self-processed UG63 ribozyme. The single base changed from MDR63 is marked by an asterisk. (C) Sequence of the minimal MDR40 ribozyme, which is the same as the above two ribozymes but without the self-complementary ends.
Figure 2.

Illustration of the MDR63 ribozyme–substrate complex and sequence of the four mismatched substrates studied. Mismatched bases are marked by an asterisk. Self-complementary bases are underlined; any self-binding that involves these must be displaced by an incoming substrate RNA.
Studies were then carried out to determine whether the circular DNA encoding MDR63 was transcribed in vitro by E.coli or T7 RNA polymerase (RNAP). Figure 3A shows the products after 12 h of transcription at 37°C. Both enzymes produced virtually identical products, which appear as several bands on the polyacrylamide gel. These were presumed to be monomer, dimer and higher order multimers, similar to previous findings with transcription of circular ribozyme DNAs (13–15). RNase T1 sequencing confirmed that the fastest migrating band was the expected MDR63 linear monomer RNA (Fig. 3B). This ribozyme was then prepared in larger amounts in unlabeled form for kinetics studies.
Figure 3.
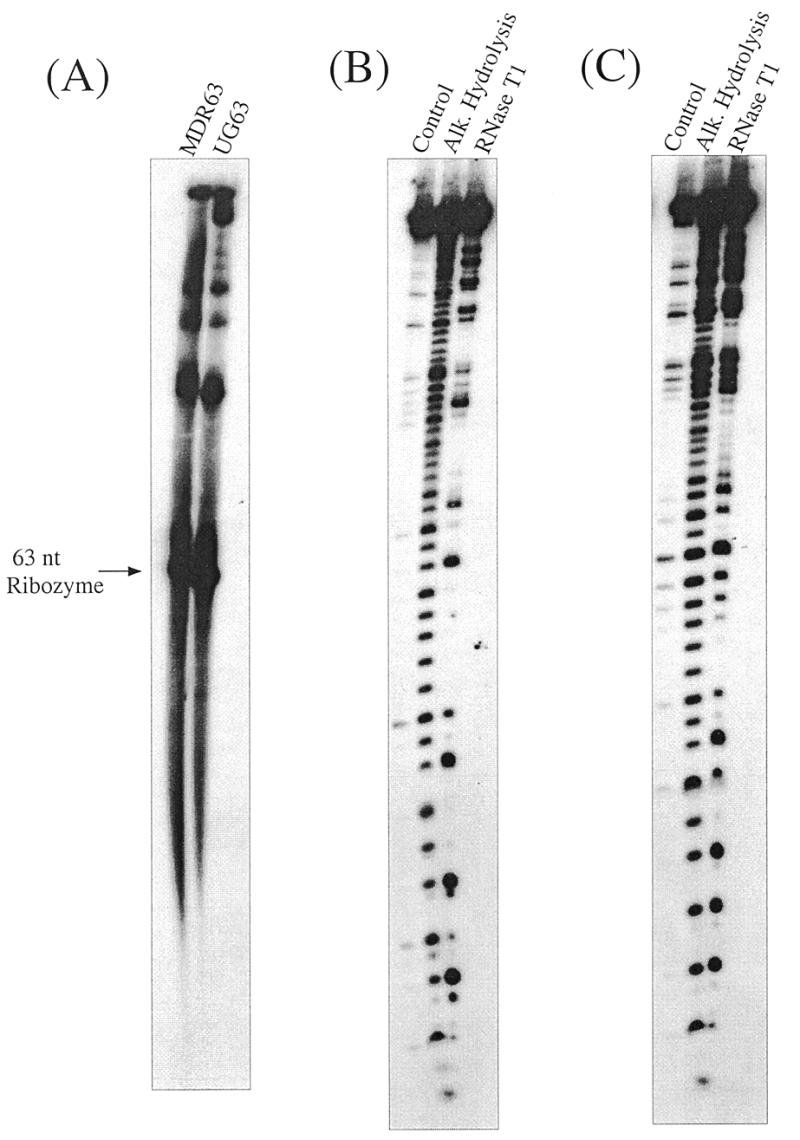
Transcription and characterization of self-processed ribozymes. (A) Autoradiogram of denaturing 10% polyacrylamide gel showing in vitro transcription of the 63 nt circular DNAs encoding MDR63 and UG63 by E.coli RNAP (after 12 h) and showing the monomer bands that arise after self-processing. (B) Autoradiogram of denaturing 15% polyacrylamide gel showing RNase T1 sequencing of the monomeric MDR63 hammerhead ribozyme. (C) Sequencing of the monomeric UG63 hammerhead ribozyme.
Basic trans-cleavage properties of MDR63
Kinetics studies were initially carried out under multiple turnover conditions to find the trans-binding helix length that is optimum for highest cleavage efficiency with this self-complementary ribozyme. The effect of length was investigated with four target RNAs of increasing size: 11, 15, 19 and 25 nt (see Materials and Methods for sequences). These have 5+5, 7+7, 9+9 and 12+12 nt of complementarity for the ribozyme, respectively. Kinetics studies were carried out under multiple turnover conditions in a buffer containing 15 mM MgCl2, 20 mM NaCl and 0.4 mM spermine at 37°C, conditions similar to those used for transcription of the ribozyme itself. This allowed us to examine cleavage under the conditions where self-cleavage was known to occur successfully.
Observed second order rate constants (kcat/Km values) were 8.0 × 103, 6.0 × 104, 2.6 × 105 and 1.7 × 104 M–1 min–1 with the 11mer, 15mer, 19mer and 25mer substrates, respectively. Thus, the experiments showed that 9+9 nt of complementarity gave the highest cleavage efficiency under these conditions.
We also measured the kinetics under single turnover conditions. There are two protocols commonly used to initiate cleavage under single turnover conditions: one is initiation by combining ribozyme–substrate complex and Mg2+. Another is initiation by combining the ribozyme–Mg2+ and the substrate–Mg2+ complexes. When the ribozyme–Mg2+ complex or the substrate–Mg2+ complex has unfavorable secondary structure, both saturated cleavage rate constants (kobs) are the same (28). However, if there is a trap step or inhibition step that depends on secondary structure in the reaction mechanism, both saturated cleavage rate constants would not be the same. The stable ribozyme–Mg2+ complex or substrate–Mg2+ complex causes the saturated kobs to decrease because of a small fraction of active complex (28,29). When we carried out the two experiments, the saturated kobs (19mer substrate) by combining ribozyme–substrate complex with Mg2+ was 1.45 min–1 (data not shown). On the other hand, the saturated kobs by combining the ribozyme–Mg2+ complex and the substrate–Mg2+ complex was 0.17 min–1, suggesting that there is a trap step or inhibition step based on competition against binding of target in the reaction mechanism. The 0.17 min–1 value is in agreement with the kcat value, 0.19 min–1, measured for MDR63 under multiple turnover conditions. The kcat value is also smaller than the predicted rate of product dissociation of 0.54 and 2.76 min–1 for both the products (30). These data show that the kcat value for MDR63 under multiple turnover conditions is unlikely to be the rate of product dissociation because one only has to consider the steps up to the chemical step in single turnover conditions. Thus, the kinetic parameters with MDR63 under multiple turnover conditions contain the competition against binding of targets. Further, the catalytic efficiency of the minimal MDR40 hammerhead was found to be 3.6 × 105 M–1 min–1 under multiple turnover conditions. Thus, with the 19mer substrate RNA both the short ribozyme and the self-complementary, self-processed one have very similar activity under multiple turnover conditions. Therefore, we conclude that any self-binding that might be occurring with MDR63 does not adversely affect its trans-cleaving ability.
Specificity of MDR63 against mismatched substrates
To investigate sequence specificity for RNA cleavage by the MDR63 ribozyme, we prepared four mismatched 19 nt target RNAs (mis1, mis2, mis3 and mis4; Fig. 2). The mismatches were located within or near self-binding domains, two on each side of the catalytic domain. They were placed away from the cleavage site, however, because substitution near this site affects not only the substrate binding affinity but also the catalytic rate of cleavage (31). Again for comparison we studied the shortened MDR40 ribozyme.
Kinetics studies were then carried out under multiple turnover conditions with these two ribozymes and the complementary and mismatched 19mer target RNAs. Plots of the initial rates as a function of concentration showed hyperbolic shapes in all cases, suggesting that all ribozyme–substrate combinations behaved with Michaelis–Menten kinetics (Fig. 4). The kinetics data are given in Table 1. Specificity was defined as the ratio kcat/Km for mismatched and complementary targets. For the standard MDR40 ribozyme, specificities were 0.33, 0.39, 0.53 and 0.42 for mis1–mis4, respectively. Thus, these mismatched RNAs are cleaved with efficiencies one-third to one-half that of the complementary RNA by this standard ribozyme. Interestingly, the MDR63 ribozyme gave quite different results. Specificities were 0.02, 0.02, 0.36 and 0.21 for the same mismatches. Thus, the larger self-complementary ribozyme displays mismatch discrimination that is up to 20-fold greater than that of the ribozyme lacking this self-binding potential.
Figure 4.

Examples of plots of initial rate of ribozyme cleavage at 37°C versus substrate concentration, under multiple turnover conditions. (A) Plots for the cleavage of wild-type substrates by MDR63 (open circle) and MDR40 (closed circle). (B) Plots for the cleavage of mis1 mismatched substrate by MDR 63 (open circle) and MDR 40 (closed circle). Curves were obtained with non-linear least square fits to Michaelis–Menten kinetics.
Table 1. Kinetic parameters for RNA cleavage by the ribozymesa.
| Ribozyme | Substrate | kcat (min–1) | Km (µM) | kcat/Km × 10–5 (M–1 min–1) | Specificityb | MDR63/MDR40c | UG63/MDR40d |
|---|---|---|---|---|---|---|---|
| MDR40 | Wild-type | 0.32 | 0.88 | 3.6 | 1 | ||
| MDR40 | mis1 | 0.32 | 2.6 | 1.2 | 0.33 | ||
| MDR40 | mis2 | 0.33 | 2.3 | 1.4 | 0.39 | ||
| MDR40 | mis3 | 0.91 | 4.8 | 1.9 | 0.53 | ||
| MDR40 | mis4 | 0.76 | 4.9 | 1.5 | 0.42 | ||
| MDR63 | Wild-type | 0.19 | 0.74 | 2.6 | 1 | 0.74 | |
| MDR63 | mis1 | 0.05 | 8.2 | 0.06 | 0.02 | 0.05 | |
| MDR63 | mis2 | 0.04 | 8.2 | 0.05 | 0.02 | 0.03 | |
| MDR63 | mis3 | 0.54 | 5.7 | 0.95 | 0.36 | 0.50 | |
| MDR63 | mis4 | 0.20 | 3.7 | 0.54 | 0.21 | 0.36 | |
| UG63 | Wild-type | 0.54 | 1.5 | 3.6 | 1 | 1 | |
| UG63 | mis1 | 0.06 | 23.7 | 0.02 | 0.006 | 0.02 | |
| UG63 | mis2 | 0.05 | 14.0 | 0.03 | 0.009 | 0.02 | |
| UG63 | mis3 | 0.31 | 11.9 | 0.27 | 0.075 | 0.14 | |
| UG63 | mis4 | 0.07 | 11.0 | 0.06 | 0.017 | 0.04 |
aAll experiments were done in 50 mM Tris–HCl (pH 8.1) with 15 mM MgCl2, 20 mM NaCl and 0.4 mM spermine at 37°C.
bSpecificity = (kcat/Km with mismatched substrate)/(kcat/Km with wild-type substrate).
cMDR63/MDR40 = (kcat/Km with MDR63)/(kcat/Km with MDR40).
dUG63/MDR40 = (kcat/Km with UG63)/(kcat/Km with MDR40).
A single base change yields higher specificity
Examination of the specificity data with the MDR63 ribozyme shows that the two mismatches on one side of the ribozyme are much better discriminated than those on the other side (Table 1). Interestingly, the higher specificity occurred on the side predicted to have the stronger self-complementary hairpin sequence with 5 bp of possible duplex. We postulated that having stronger self-complementarity on the other side might possibly increase the sequence specificity in that trans-binding domain. To test this we designed a new self-processed ribozyme (UG63, Fig. 1B) having 1 bp of additional self-complementarity on the 5′-side of the ribozyme. This requires only a single U→G base change in the putative self-binding domain. Importantly, this mutation is located at a site where normal binding of substrate does not occur. Thus, if it affected cleavage it would be indirectly rather than by direct interaction with the target. RNA folding algorithms predict that the UG63 ribozyme can form a stronger hairpin, with a free energy of –3.5 kcal/mol, 3.1 kcal/mol more stable than the corresponding hairpin in MDR63. If self-binding is important in competing against mismatched target binding, then one would predict that higher sequence specificity against mis3 and mis4 might result.
As was done previously, a circular DNA encoding this second ribozyme was constructed. It was successfully transcribed in vitro by E.coli RNAP and gave products that appeared the same as those with the MDR63 circular vector (Fig. 3A). The fastest traveling band was isolated and was confirmed to be the UG63 mutant by RNase T1 sequencing (Fig. 3C).
Kinetics studies were carried out under conditions of multiple turnover for this second ribozyme using the same complementary and mismatched 19mer RNAs studied previously. The data are given in Table 1. The results show that the specificity of the UG63 ribozyme is considerably higher with the mis3 and mis4 mismatched RNAs, as compared to the previous MDR63 ribozyme. It cleaves them only 8 and 1.7% as efficiently as the complementary RNA, giving a 7- and 25-fold advantage over the standard 40mer ribozyme at these positions. Interestingly, it appears that specificity may also be slightly higher at the mis1 and mis2 mismatch positions; here the UG63 self-processed ribozyme has a 50- to 100-fold advantage over the short ribozyme. The high specificities for mis1 and mis2 are seen to depend mainly on Km. Because of the extra base pair, the UG63 self-processed ribozyme has a greater energy penalty than MDR63 in binding to the substrate. Consistent with this, the Km value for wild-type substrate with UG63 is larger than that for MDR63. The higher specificities for mis1 and mis2 appear to be due to this energy penalty. The results suggest that competition between self-binding and target binding plays an important role in the sequence specificity of these self-processed ribozymes.
DISCUSSION
Previous studies of hammerhead ribozyme discrimination against mismatched targets have revealed levels of specificity similar to those of the present control MDR40 ribozyme. For example, when substrate RNAs were designed to have 8+7, 8+6 and 8+5 nt of complementarity for a hammerhead ribozyme, the specificities were 0.66, 0.66 and 0.5 for singly mismatched substrates, respectively (11). Mismatch discrimination is expected to be low when chemical cleavage is rate limiting, since most mismatches do not affect the active site geometry. If, on the other hand, product release is rate limiting, mismatched targets can be cleaved even more rapidly than complementary ones under multiple turnover conditions. Thus, minimal hammerhead ribozymes generally display quite low levels of sequence discrimination and the present self-binding strategy offers a substantial degree of improvement, with specificities of 0.075–0.006 for singly mismatched substrates with the UG63 ribozyme. This specificity appears to be due both to increases in Km with a mismatch (by factors of 7- to 16-fold) and to decreases in kcat with a mismatch (by 2- to 9-fold). It appears that both effects can be explained by self-binding of the RNA ends in UG63 relative to the MDR40 control (see discussion below).
Interestingly, this apparent self-binding does not inhibit the rate of substrate cleavage under multiple turnover conditions. Our data show that MDR63 and UG63 cleave a 19 nt wild-type substrate with a kcat/Km almost the same as the control MDR40. RNA folding algorithms predict a free energy (37°C) of –11.7 kcal/mol for the MDR63 folded structure with hairpins as in Figure 1 and –12.7 kcal/mol for UG63. On the other hand, the free energy for the complex between MDR63 or UG63 and the 19 nt target RNA is calculated to be –23.4 kcal/mol, using a catalytic core value of +3.3 kcal/mol (32). Thus, from a thermodynamic standpoint trans-cleavage is reasonable, since trans-binding is much more favorable than self-binding. From a kinetic standpoint, the kcat values suggest that at 37°C the self-binding hairpins are melted at least as rapidly as the slowest step in the turnover cleavage mechanism. If one assumes a rate of 3.4 × 104 s–1 for self-annealing of one hairpin in MDR63 (33), then the rate constant for opening of this hairpin is expected to be 136 s–1. This suggests that there are multiple chances for the target RNA to bind before the productive complex is formed. With a considerably stronger hairpin, however, one expects that the rate of cleavage would be slowed. Calculations suggest that binding domain hairpins longer than 9 bp in length (in this sequence context) might be kinetically inhibitory.
It is worth noting that the activity of MDR40 is low (by a factor of ~30) relative to a ‘well-behaved’ hammerhead ribozyme (HH16) studied by Hertel et al. (28). It has been observed previously that lower activities than predicted are seen in many ribozymes and they cannot always be simply explained (30). For example, when substrate RNAs were designed to have 8+8, 8+5 and 8+3 nt of complementarity for a previously studied hammerhead ribozyme, the kcat/Km values were 1.2 × 107, 0.5 × 107 and 1.4 × 107 M–1 min–1, respectively (11). Further, when two hammerhead ribozymes were designed to have the same binding energies (ΔG°37 = –17.7 and –17.6 kcal/mol) against the substrate RNAs, the kcat/Km values were 8.0 × 106 and 3.6 × 105 M–1 min–1, respectively (11,21). This difference in kcat/Km values is similar in magnitude to that observed for MDR40 here. Such unpredicted differences depend on kinetically or thermodynamically inactive conformations in the reaction mechanism (11). For example, if the substrate binds in an alternative unreactive complex with the ribozyme, known as non-productive binding (34), the observed kcat values decrease so that kcat/Km values also decrease. Since our 19 nt substrate has a UUUC sequence complementary to GAAA within the ribozyme core, it is possible that a non-productive complex lowers the overall activity for this sequence. Although the activity with MDR40 appears to be low, MDR63 and UG63 also have the same UUUC sequence. Therefore, the conditions with MDR40, MDR63 and UG63 are equal and MDR40 is expected to behave as a valid control.
MDR63 and UG63 display much greater abilities to distinguish mismatches than the standard MDR40 ribozyme and other hammerhead ribozymes in general (11). We observe that these high specificities depend both on increases in Km and decreases in kcat for mismatched targets. It is unclear whether the Km is the same as the binding constant (Kd) of the ribozyme to the substrate because the Kd value was not determined by a separate method. From the standpoint of thermodynamic RNA folding predictions, however, the Km effects can be understood by analysis of free energies of self-binding versus target binding. In the case of MDR40, the free energies for mis1 and mis2 target binding are calculated to be –17.5 and –17.8 kcal/mol (as compared to –21.7 for the complementary target). In the case of MDR63, the calculated free energy (–3.4 kcal/mol) for hairpin loop structures at self-complementary ends must be subtracted from these values and so the net free energy of binding is predicted to be –14.6 and –14.9 kcal/mol for mis1 and mis2, respectively (as compared to –18.8 kcal/mol for the complementary target). In fact, both experimentally determined Km values of 8.2 µM for mis1 and mis2 with MDR63 are ~3.5-fold larger than those with MDR40. Similar effects are seen at the 5′-side, with the mis3 and mis4 targets. The free energy of target binding by MDR40 for these two is predicted to be –18.0 and –15.0 kcal/mol. The calculated net free energy for MDR63 is –15.1 and –12.1 kcal/mol. For UG63 it is –11.8 and –8.8 kcal/mol. As predicted by the free energy of target binding, the experimentally observed Km value of 11.9 µM for mis3 with UG63 is the largest of the three ribozymes. The Km value of 11.0 µM for mis4 with MDR63 is also the largest. Thus, the difference in Km observed for these three ribozymes correlates with the calculated relative binding free energies with the RNA targets, suggesting that competition between self-binding and mismatched targets contributes to this sequence discrimination.
The observed high specificity of the self-processed ribozymes is not due to Km effects alone. Unlike the short MDR40 ribozyme and most hammerhead ribozymes in general (11), the MDR63 and UG63 ribozymes display significantly decreased kcat values with mismatched target RNAs. For the superior UG63 ribozyme, kcat drops by as much as 180-fold with a mismatched target, while the MDR40 ribozyme shows either no effect or an increase in kcat. If kcat reflects only the chemical cleavage step, the decrease in kcat might be due to alteration of the geometry near the cleavage site, although in the present case the positions of the mismatches were designed to have no effect on the catalytic rate of cleavage (31). In some cases the kcat value reflects a rate limiting product release step, however, that seems an unlikely explanation in the present case, since the products are very similar for both short and self-processed ribozymes. Thus it appears that the results can be best explained by the fraying model for hammerhead ribozyme catalysis (35). In this model (Fig. 5) there are two non-productive ribozyme–target complexes that can be formed when only one side of the ribozyme binds the target (termed ‘open states’). These non-productive complexes compete with productive complex formation (the ‘closed state’) and the observed kcat value depends on the internal equilibrium constant for these complexes multiplied by the intrinsic kcat value. Although the intrinsic kcat is not affected by mismatches, the internal equilibrium between open and closed states certainly would be. In the case of the self-processed ribozymes, the internal equilibrium with mismatched targets would lie much more toward the side of the open complexes because of the competition with self-binding domains. Thus under the reaction conditions there is a much smaller fraction of active complex formed with mismatched targets than with complementary ones and this is reflected in the observed kcat values.
Figure 5.

Schematic diagram of the fraying model, illustrating how self-complementarity in the ribozyme can increase specificity by competition between self-binding and binding of mismatched substrates. Self-binding is expected to increase the Km with mismatched substrates by shifting the initial equilibrium away from the active complex. This is also expected to affect the observed rate of the chemical cleavage step (kcat·[active complex]), again by lowering the concentration of the active complex, especially in the case of mismatched substrates.
Herschlag has carried out an analysis of specificity in ribozyme systems, leading to the prediction of strategies for increasing specificity between correct and incorrect substrates (36). One of these was the choice of A+U-rich targets, which permits substrate dissociation to be faster than chemical cleavage. A second was the use of high ionic strengths, which would increase the rate of equilibration between substrate and ribozyme. The third suggestion was to make use of unfavorable interactions between substrate and ribozyme, although a specific mechanism for carrying this out was not suggested. The present self-processed ribozymes also appear to utilize this third mechanism for increasing sequence specificity, decreasing substrate binding affinity by competition with self-structure. This strategy for hammerhead ribozymes has apparently not been tested previously. Interestingly, it was reported recently that the energetic penalty for conformational rearrangement with a group II intron ribozyme led to a reduction in the substrate binding energy so that the sequence specificity was enhanced (37,38). A related observation was made by Uhlenbeck, who observed that competition by secondary structure in a target RNA led to significant increases in sequence specificity of cleavage (33). The present study demonstrates how this concept can be successfully applied to ribozyme design, as opposed to target selection.
Although it was not carried out in a catalytic ribozyme system, another related observation merits mention in this context. Roberts and Crothers studied the specificity of binding of duplex DNA by a triplex-forming oligonucleotide and found that specificity could be increased by designing a self-binding domain (termed a ‘stringency clamp’) into the oligonucleotide (39). This was important in demonstrating the value of competition between self-binding and target binding in a hybridization experiment.
Acknowledgments
ACKNOWLEDGEMENTS
We thank the DOD Breast Cancer Research Program, administered by the US Army and the National Institutes of Health (GM46625) for support of this work.
REFERENCES
- 1.Buzayan J.M., Gerlach,W.L. and Bruening,G. (1986) Nature, 323, 349–353. [Google Scholar]
- 2.Hutchins C.J., Rathjen,P.D., Forster,A.C. and Symons,R.H. (1986) Nucleic Acids Res., 14, 3627–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prody G.A., Bakos,J.T., Buzayan,J.M., Schneider,I.R. and Bruening,G. (1986) Science, 231, 1577–1580. [DOI] [PubMed] [Google Scholar]
- 4.Forster A.C. and Symons,R.H (1987) Cell, 49, 211–220. [DOI] [PubMed] [Google Scholar]
- 5.Uhlenbeck O.C. (1987) Nature, 328, 596–600. [DOI] [PubMed] [Google Scholar]
- 6.Haseloff J. and Gerlach,W.L. (1988) Nature, 334, 585–591. [DOI] [PubMed] [Google Scholar]
- 7.Zhou D.-M. and Taira,K. (1998) Chem. Rev., 98, 991–1026. [DOI] [PubMed] [Google Scholar]
- 8.Eckstein F. and Lilley,D.M.J. (eds) (1996) RNA Catalysis: Nucleic Acids and Molecular Biology. Springer Verlag, Berlin, Germany, Vol. 10.
- 9.Chen H., Ferbeyre,G. and Cedergren,R. (1997) Nature Biotechnol., 15, 273–277. [DOI] [PubMed] [Google Scholar]
- 10.Kuwabara T., Warashina,M., Nakayama,A., Ohkawa,J. and Taira,K. (1999) Proc. Natl Acad. Sci. USA, 96, 1886–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hertel K.J., Herschlag,D. and Uhlenbeck,O.C. (1996) EMBO J., 15, 3751–3757. [PMC free article] [PubMed] [Google Scholar]
- 12.Daubendiek S.L., Ryan,K. and Kool,E.T. (1995) J. Am. Chem. Soc., 117, 7818–7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daubendiek S.L. and Kool,E.T. (1997) Nature Biotechnol., 15, 273–277. [DOI] [PubMed] [Google Scholar]
- 14.Diegelman A.M. and Kool,E.T. (1998) Nucleic Acids Res., 26, 3235–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diegelman A.M. and Kool,E.T. (1999) Chem. Biol., 9, 569–576. [DOI] [PubMed] [Google Scholar]
- 16.Usman N., Ogilvie,K.K., Jiang,M.-V. and Cedergren,R. (1987) J. Am. Chem. Soc., 109, 7845–7854. [Google Scholar]
- 17.Diegelman A.M. and Kool,E.T. (1999) In Glick,G.D. (ed.), Current Protocols in Nucleic Acid Chemistry. John Wiley & Sons, New York, NY, in press.
- 18.Richards E.G. (1975) In Fasman,G.D. (ed.), Handbook of Biochemistry and Molecular Biology: Nucleic Acids, 3rd Edn. CRC Press, Cleveland, OH, Vol. I, p. 197.
- 19.Milligan J.F., Groebe,D.R., Witherell,G.W. and Uhlenbeck,O.C. (1987) Nucleic Acids Res., 15, 8783–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hendry P., McCall,M.J. and Lockett,T.J. (1997) In Turner,P.C. (ed.), Methods in Molecular Biology. Humana Press, Totowa, NJ, Vol. 74.
- 21.Fedor M.J. and Uhlenbeck,O.C. (1992) Biochemistry, 31, 12042–12054. [DOI] [PubMed] [Google Scholar]
- 22.Kool E.T. (1998) Acc. Chem. Res., 31, 502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi H., Dorai,T., Holland,J.F. and Ohnuma,T. (1994) Cancer Res., 54, 1271–1275. [PubMed] [Google Scholar]
- 24.Turner D.H., Sugimoto,N. and Freier,S.M. (1988) Annu. Rev. Biophys. Biophys. Chem., 17, 167–192. [DOI] [PubMed] [Google Scholar]
- 25.Serra M.J. and Turner,D.H. (1995) Methods Enzymol., 259, 242–261. [DOI] [PubMed] [Google Scholar]
- 26.Xia T., SantaLucia,J.,Jr, Burkard,M.E., Kierzek,R., Schroeder,S.J., Jiao,X., Cox,C. and Turner,D.H. (1998) Biochemistry, 37, 14719–14735. [DOI] [PubMed] [Google Scholar]
- 27.Mathews D.H., Sabina,J., Zuker,M. and Turner,D.H. (1999) J. Mol. Biol., 288, 911–940. [DOI] [PubMed] [Google Scholar]
- 28.Hertel K.J., Herschlag,D. and Uhlenbeck,O.C. (1994) Biochemistry, 33, 3374–3385. [DOI] [PubMed] [Google Scholar]
- 29.Walstrum S.A. and Uhlenbeck,O.C. (1990) Biochemistry, 29, 10573–10576. [DOI] [PubMed] [Google Scholar]
- 30.Stage-Zimmermann K.T. and Uhlenbeck,O.C. (1998) RNA, 4, 875–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Werner M. and Uhlenbeck,O.C. (1995) Nucleic Acids Res., 23, 2092–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hertel K.J., Stage-Zimmermann,T.K., Ammons,G. and Uhlenbeck,O.C. (1998) Biochemistry, 37, 16983–16988. [DOI] [PubMed] [Google Scholar]
- 33.Pörschke D. (1974) Biophys. Chem., 1, 381–384. [DOI] [PubMed] [Google Scholar]
- 34.Fersht A. (1985) Enzyme Structure and Mechanism, 2nd Edn. W.H. Freeman and Co., New York, NY.
- 35.Hertel K.J., Peracchi,A., Uhlenbeck,O.C. and Herschlag,D. (1997) Proc. Natl Acad. Sci. USA, 94, 8497–8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herschlag D. (1991) Proc. Natl Acad. Sci. USA, 88, 6921–6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qin P.Z. and Pyle,A.M. (1999) J. Mol. Biol., 291, 15–27. [DOI] [PubMed] [Google Scholar]
- 38.Xiang Q., Qin,P.Z., Michels,W.J., Freeland,K. and Pyle,A.M. (1998) Biochemistry, 37, 3839–3849. [DOI] [PubMed] [Google Scholar]
- 39.Roberts R.W. and Crothers,D.M. (1991) Proc. Natl Acad. Sci. USA, 88, 9397–9401. [DOI] [PMC free article] [PubMed] [Google Scholar]