


Abstract
Endonucleolytic processing of precursor tRNAs (ptRNAs) by RNase P yields 3′-OH and 5′-phosphate termini, and at least two metal ions are thought to be essential for catalysis. To determine if the hydrolysis reaction catalyzed by bacterial RNase P (RNAs) involves stabilization of the 3′-oxyanion leaving group by direct coordination to one of the catalytic metal ions, ptRNA substrates with single 3′-S-phosphorothiolate linkages at the RNase P cleavage site were synthesized. With a 3′-S-phosphorothiolate-modified ptRNA carrying a 7 nt 5′-flank, a complete shift of the cleavage site to the next unmodified phosphodiester in the 5′-direction was observed. Cleavage at the modified linkage was not restored in the presence of thiophilic metal ions, such as Mn2+ or Cd2+. To suppress aberrant cleavage, we also constructed a 3′-S-phosphorothiolate-modified ptRNA with a 1 nt 5′-flank. No detectable cleavage of this substrate was seen in reactions catalyzed by RNase P RNAs from Escherichia coli and Bacillus subtilis, independent of the presence of thiophilic metal ions. Ground state binding of modified ptRNAs was not impaired, suggesting that the 3′-S-phosphorothiolate modification specifically prevents formation of the transition state, possibly by excluding catalytic metal ions from the active site.
INTRODUCTION
Ribonuclease P is a ubiquitous metalloenzyme that cleaves tRNA precursor transcripts to generate the mature 5′-ends of tRNAs. Processing of precursor tRNAs (ptRNAs) by RNase P is an essentially irreversible reaction generating 3′-OH and 5′-phosphate termini. A solvent hydroxide or activated water molecule is thought to act as the nucleophile in an SN2 in-line displacement mechanism (1,2). Two (3) or three (2) metal ions were proposed to mediate catalysis by bacterial RNase P RNA. The two metal ion mechanism (3), orginally proposed by Steitz and Steitz (4) for RNA-catalyzed hydrolysis and phosphoryl transfer reactions, involves direct metal ion coordination to the 3′-oxyanion leaving group in the transition state (Fig. 1). Substitution of an oxygen leaving group by sulfur provides a means to test this possibility, because different metal ions differ in their ability to coordinate oxygen versus sulfur. For example, Mg2+ strongly resists coordination to sulfur, preferring oxygen by ~104, whereas softer cations such as Mn2+, Zn2+ and Cd2+ will readily coordinate sulfur. Thus a switch in metal ion specificity upon sulfur substitution from Mg2+ to a more thiophilic metal ion provides strong evidence that a metal ion stabilizes the leaving group by direct coordination in the transition state. This has been documented for the first and second steps of the self-splicing reactions catalyzed by the Tetrahymena group I intron (5,6) and by the ai5γ group II intron from Saccharomyces cerevisiae (7) and for the first step of pre-mRNA splicing catalyzed by the spliceosome (8). In these reactions, sulfur substitution of the leaving group has a large deleterious effect on the reaction rate in the presence of Mg2+, but a substantial restoration of activity in the presence of thiophilic metal ions was observed, indicating that direct metal ion coordination to the leaving group occurs during catalysis.
Figure 1.

Possible mechanism for ptRNA cleavage by RNase P RNA (adapted from 3). The model includes main features of the general two metal ion mechanism proposed for protein- and RNA-catalyzed hydrolytic and phosphoryl transfer reactions (4).
Here we have synthesized ptRNAs with a single 3′-S-phosphorothiolate internucleotide linkage to investigate the proposed direct metal ion coordination to the 3′-oxyanion leaving group (Fig. 1). Modified ptRNAs were analyzed as substrates for bacterial RNase P (RNAs) under single turnover conditions (E >> S) to ensure that the rate of the chemical step was being monitored (3).
MATERIALS AND METHODS
Synthetic oligoribonucleotides Ino (5′-CCCUUUIGCGGGA-3′), Ino-S [5′-CCCUUU(IS)GCGGGA-3′, where IS is 3′-thioinosine], Cyt (5′-CGCGGGAGUAGCUCAGUC-3′) and Cyt-S [5′-(CS)GC-GGGAGUAGCUCAGUC-3′, where CS is 3′-thiocytidine] were synthesized at the 1 µmol scale on a Millipore solid phase RNA/DNA synthesizer. Coupling of unmodified nucleoside phosphoramidites (Glen Research) followed standard procedures (9); synthesis and coupling of the protected cytidine 3′-S-phosphorothioamidite were as described by Sun et al. (10). Both oligonucleotides were deprotected following standard techniques (9), purified by anion exchange HPLC, further purified and desalted by reverse phase HPLC, dried in vacuo and resuspended in water.
Assembly of ptRNAGly variants
Assembly and ligation of ptRNAGly variants was performed according to the strategies illustrated in Figure 2, essentially as described (3).
Figure 2.

Precursor ptRNAGly variants carrying a single 3′-S-phosphorothiolate linkage at the RNase P cleavage site. Highlighted nucleotides mark the sites of modification. Unmodified ptRNAIno and modified ptRNAIno-S (a), which carry a 7 nt 5′-flank and inosine or 3′-thioinosine at position –1, respectively, were obtained by ligation of three RNA fragments (see Materials and Methods and Results), with the sites of ligation indicated by boxes (A6/G7 and C17/G18). Unmodified and modified constructs of the type shown in (b) carried a 1 nt 5′-flank and were assembled from two RNA fragments (ligation between C17 and G18); natural precursor tRNAGly contains a cytidine at position –1 (17). The canonical RNase P cleavage site is located between nt –1 and +1. Unmodified ptRNAGly, identical to ptRNAIno except for a C residue at position –1, is termed ‘wild-type’ ptRNAGly in Table 1.
Preparation of RNase P components
RNAs were obtained by in vitro run-off transcription using T7 RNA polymerase as described previously (11–13). RNase P RNA from Bacillus subtilis was transcribed from plasmid pDW66 (14) linearized with DraI. RNase P RNA from Escherichia coli was transcribed from PCR templates (12) or from a pSP64 derivative encoding E.coli wild-type RNase P RNA (15), linearized with BamHI. RNase P RNA from Thermus thermophilus was transcribed from plasmid pT7M1HB8 linearized with NarI (16). The 3′-portion (starting at G+18) of T.thermophilus tRNAGly was prepared by T7 transcription in the presence of 10 mM GMP, 2.5 mM GTP essentially as described (12) using a PCR template amplified from plasmid pTT675 (17). After T7 transcription, RNA preparations were digested with DNase I and then extracted with phenol:chloroform (1:1). RNAs were purified on 5–10% polyacrylamide–8 M urea gels, visualized by UV shadowing, excised from the gel and eluted overnight at 4°C in 200 mM Tris–HCl pH 7.0, 1 mM EDTA. RNAs were recovered by ethanol precipitation in the presence of 75 mM NaOAc (pH 7.0), dissolved in water and concentrations were determined by UV spectroscopy (1 OD260 = 37 µg/ml). The E.coli RNase P protein was prepared as described (18).
5′-End-labeling of RNAs
5′-End-labeling using [γ-32P]ATP and T4 polynucleotide kinase was performed as described (19).
Kinetics
Single turnover experiments were performed as described (20) with trace amounts (<1 nM) of 5′-32P-end-labeled ptRNA and 5 µM RNase P RNA in 50 mM MES, pH 6.0 or 7.0 (at 37°C), and either 1.0 M NH4OAc, 15 mM divalent cations (E.coli RNase P RNA) or 1.0 M NH4OAc, 100 mM divalent cations (B.subtilis RNase P RNA), unless stated otherwise. RNAs were separated on 25% polyacrylamide–8 M urea sequencing gels (in the presence of 10 mM DTT for electrophoresis of 3′-S-phosphorothiolate-modified RNAs) and quantified as described (20).
Determination of apparent equilibrium dissociation constants (Kd app)
For measurement of apparent dissociation constants by the spin column assay (21), increasing amounts of RNase P RNA and trace amounts of radiolabeled ptRNA (<1 nM) were pre-incubated as described (20) in 50 mM MES, pH 6.0 (at 37°C), 0.8 or 1.0 M NH4OAc, 0.05% Nonidet-P40, 0.1% SDS and 0.05 or 0.1 M Ca(OAc)2. About 5–10% of the radioactivity was measured in the eluate in the absence of the ribozyme and ~65–75% eluted at high concentrations of RNase P RNA (end-point). The fraction of ptRNA in the complex was calculated as [c.p.m.eluate/(c.p.m.eluate + c.p.m.column)][E] > 0 – [c.p.m.eluate/(c.p.m.eluate + c.p.m.column)][E] = 0 ([E] = RNase P RNA concentration) and Kd app values were determined by non-linear regression analysis as described (20).
Nuclease P1, RNase T1 and snake venom phosphodiesterase I (SVPD) hydrolysis reactions
For limited digestion by nuclease P1, ~105 Cerenkov c.p.m. of the RNA substrate and 2.5 µg of carrier RNA were preincubated in buffer P1 (40 mM NH4OAc, pH 5.3, 0.4 mM ZnSO4) for 5 min at 70°C, followed by addition of 0.001–0.01 U nuclease P1 (Boehringer Mannheim) to a final reaction volume of 10 µl and further incubation for 1 min at 70°C. For limited digestion by RNase T1, ~105 Cerenkov c.p.m. of the RNA probe and 2.5 µg of carrier RNA were incubated for 10 min at 55°C with 0.2 U RNase T1 (Pharmacia) in buffer T1 (20 mM sodium citrate, 7 M urea, 1 mM EDTA, pH 5) in a 10 µl final volume. For limited digestion by SVPD from Crotalus adamanteus (Boehringer Mannheim), ~105 Cerenkov c.p.m. of the RNA probe and 2.5 µg of carrier RNA were incubated for 5 min at 37°C in 10 mM MgCl2, 10 mM DTT, 50 mM HEPES pH 7.5 with 0.1 or 0.2 µg SVPD in a total volume of 5 µl, unless stated otherwise. Enzymatic reactions were stopped by addition of equal volumes of gel loading buffer [2.3 M urea, 66% formamide, 10–20 mM DTT, 0.05% (w/v) each BPB and XCB in 1× TBE].
Chemical cleavage reactions
For cleavage of 3′-S-phosphorothiolate linkages by I2, ~2 × 104 Cerenkov c.p.m. of the RNA probe and 2.5 µg of carrier RNA were lyophilized and dissolved in 9 µl 10 mM HEPES (pH 7.5), followed by addition of 1 µl I2/EtOH (1 mg/ml I2 in 20% EtOH) and incubation for 10 min at 37°C. For cleavage of 3′-S-phosphorothiolate linkages by AgNO3, ~2 × 104 Cerenkov c.p.m. of the RNA probe and 2.5 µg of carrier RNA were lyophilized and dissolved in 4 µl H2O (or buffer, if indicated), followed by addition of 1 µl 250 mM aqueous AgNO3 solution and incubation for 30 min at 30°C. The primary reaction products are the 5′-phosphate group and the silver salt of the 3′-thiolate (22). Reaction mixtures were then diluted to 50 µl with H2O and adjusted to 5 mM DTT, and insoluble DTT–silver precipitates were removed by centrifugation (22) for 5 min in a table centrifuge at 13 000 r.p.m. (~11 000 g). RNAs in the supernatant were concentrated by ethanol precipitation and redissolved in gel loading buffer (see above).
RESULTS
Assembly of ptRNAs
Precursor tRNAs carrying a single 3′-S-phosphorothiolate modification at the RNase P cleavage site (Fig. 2) were synthesized by combining chemical and enzymatic RNA synthesis techniques. For construction of ptRNAIno and ptRNAIno-S (Fig. 2, constructs a), RNA oligonucleotides 13 nt in length, either unmodified (Ino, Fig. 3) or carrying a single 3′-S-phosphorothiolate modification (Ino-S, Fig. 3), were synthesized by phosphoramidite chemistry (see Materials and Methods). The modified or unmodified 13mer, a second 11 nt RNA oligonucleotide (obtained by either chemical or enzymatic synthesis) and a transcript representing the 3′-terminal portion of the tRNAGly were annealed to a bridging DNA oligonucleotide (complementary to nt +37 to –3 of the ptRNA) for ligation by T4 DNA ligase (3; Fig. 2). For chemical synthesis reasons, the resulting ptRNA carried an inosine at position –1. The ptRNACyt and ptRNACyt-S (Fig. 2, constructs b) carrying a single C at the 5′-terminus (as encoded in the T.thermophilus tRNAGly gene; 17), were constructed by ligating a modified 18mer (Cyt-S; see Materials and Methods) or unmodified 18mer (Cyt) to the aforementioned 3′-portion of the tRNAGly.
Figure 3.
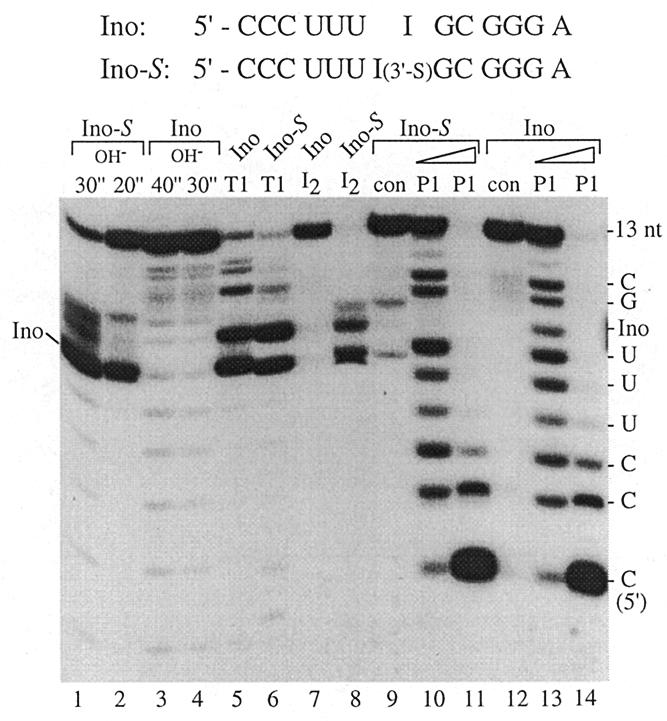
Characterization of 5′-end-labeled 13mers: Ino, unmodified 13mer; Ino-S, modified 13mer. Lanes 1–4, limited alkaline hydrolysis (incubation period in s); lanes 5 and 6, limited digestion by nuclease T1; lanes 7 and 8, iodine hydrolysis; lanes 10, 11, 13 and 14, limited digestion (1 min at 70°C) with nuclease P1 (lanes 10 and 13, 0.001 U; lanes 11 and 14, 0.01 U); lanes 9 and 12, control lanes in the absence of nuclease P1. Assignment of nucleotides to P1 hydrolysis bands is shown in the right margin; products corresponding to alkaline hydrolysis and RNase T1 cleavage at the 3′-side of the inosine residue at position 7 are indicated on the left. Note that alkaline hydrolysis and RNase T1 cleavage generates 3′-phosphate termini, whereas nuclease P1 cleavage products carry 3′-OH termini. Hydrolysis products were analyzed on 25% polyacrylamide–8 M urea sequencing gels in the presence of 10 mM DTT (for further details see Materials and Methods).
Characterization of the 3′-S-phosphorothiolate linkage
Initially, the modified and unmodified 13mer RNA oligonucleotides, carrying an inosine at position 7, were characterized using chemical and enzymatic probes. Figure 3 demonstrates that the thiolate linkage is more labile to alkaline hydrolysis than normal phosphodiester bonds (lanes 1 and 2 versus 3 and 4), is susceptible to RNase T1 (lanes 5 and 6) and iodine hydrolysis (lane 7 versus 8) and relatively resistant to digestion with nuclease P1 (lane 10 versus 13). These observations are similar to those obtained for a dinucleotide containing the same modified linkage (23) and confirm that the modified 13mer RNA oligonucleotide contained a single 3′-S-phosphorothiolate linkage connecting nt 7 and 8. Multiple iodine cleavage products (lane 8) may represent different oxidation states of the terminal sulfur (23).
Cleavage site selection
We analyzed the 5′-cleavage products generated by E.coli RNase P RNA-catalyzed processing of ptRNAIno and ptRNAIno-S. Representative results are illustrated in Figure 4. Processing of the unmodified ptRNAIno by E.coli RNase P RNA in the presence of Mg2+ (Fig. 4b, lane 14) or Mn2+ (Fig. 4c, lane 1 or 6) resulted predominantly in the expected 7 nt 5′-cleavage product. A low extent of aberrant cleavage occurred at position –2/–1 (~5% in the presence of 15 mM Mg2+ or Mn2+), yielding a 6 nt 5′-cleavage product, which can be attributed to formation of an I–1:U+73 base pair. It has been shown that the stability of the –1/+73 base pair is correlated with the propensity of E.coli RNase P RNA to cleave at the aberrant –2/–1 site (24,25). Processing of the 3′-S-phosphorothiolate-modified ptRNAIno-S under various conditions, some of which are shown in Figure 4 (a, lanes 4–7; b, lanes 11–13, 15 and 16) resulted exclusively in a 5′-cleavage product that migrated at the position of a 6 nt RNA fragment, suggesting aberrant cleavage between positions –2 and –1 (Fig. 2). This conclusion is supported by the absence of changes in gel mobility when the thiol-modifying reagent ICH2C(O)NH2 was present during RNase P processing (Fig. 4a, lane 4, and b, lane 11), when processing was performed under reducing conditions (10 mM DTT; Fig. 4a, lane 5, and b, lane 12) or when the acetate salts of the processing buffer were replaced by chloride salts (Fig. 4a, lane 6, and b, lane 13). The E.coli RNase P holoenzyme also produced the shorter 5′-cleavage product (Fig. 4b, lane 15). Finally, neither the presence of more thiophilic divalent metal ions (Mn2+, Cd2+, Zn2+ or Co2+) nor the use of other RNase P RNAs (e.g. from B.subtilis or T.thermophilus) changed the observed cleavage site selection for ptRNAIno-S (data not shown).
Figure 4.

Analysis of 5′-cleavage products derived from RNase P (RNA)-catalyzed cleavage of 5′-end-labeled control ptRNAIno and ptRNAIno-S. (a) Lanes 1 and 2, limited digestion of 13mer Ino-S with 0.2 (lane 1) or 0.1 µg/5 µl (lane 2) SVPD; lane 3, AgNO3 cleavage of 13mer Ino-S under RNase P RNA processing conditions [1 M NH4OAc, 15 mM Mg(OAc)2, pH 6.0]; lane 7, 5′-cleavage products derived from processing of ptRNAIno-S (<1 nM) by E.coli RNase P RNA (P RNA, 5 µM) under conditions of 1 M NH4OAc, 15 mM Mn(OAc)2, pH 6.0; lanes 4 and 5, as lane 7, but in the presence of 40 mM ICH2C(O)NH2 (lane 4) or 10 mM DTT (lane 5); lane 6, as lane 7, but using chloride instead of acetate salts; lane 8, [5′-32P]CMP obtained by RNase P RNA cleavage as in lane 7 and additional treatment of the 5′-cleavage product with 0.3 mU SVPD. (b) Lanes 9 and 10, limited digestion of 13mer Ino (lane 9) and 13mer Ino-S (lane 10) with 0.005 U of nuclease P1. Lanes 11–13, identical to lanes 4–6 of (a); lanes 14 and 16, processing of ptRNAIno (lane 14) or ptRNAIno-S (lane 16) by E.coli RNase P RNA in the presence of 1 M NH4OAc, 15 mM Mg(OAc)2, pH 6.0; lane 15, processing of ptRNAIno-S in the presence of 1 µM E.coli RNase P RNA and 2 µM E.coli RNase P protein C5 under conditions of 10 mM Mg(OAc)2, 0.1 M NH4OAc, 50 mM MES–acetate, pH 7.0; lane 17, 3′-S-phosphorothiolate-specific cleavage of ptRNAIno-S by AgNO3 in RNase P RNA cleavage buffer [1 M NH4OAc, 15 mM Mg(OAc)2, pH 6.0]. (c) Derivatization of free thiol groups by ICH2C(O)NH2. Lanes 1 and 2, 5′-cleavage products derived from processing of <1 nM ptRNAIno (lane 1) or ptRNAIno-S (lane 2) by E.coli RNase P RNA (5 µM) under conditions of 1 M NH4OAc, 15 mM Mn(OAc)2, pH 6.0; lane 3, AgNO3 hydrolysis of 13mer Ino-S in H2O; lanes 4 and 5, as lane 3, but additional incubation in 80 mM HEPES pH 7.5 (lane 4) or 80 mM HEPES pH 7.5, plus 50 mM ICH2C(O)NH2 (lane 5). Lanes 6–9, RNase P RNA processing of ptRNAIno (lanes 6 and 7) or ptRNAIno-S (lanes 8 and 9) as in lanes 1 and 2, respectively, followed by removal of salts by ethanol precipitation and washing of the precipitate with 70% EtOH, resolving of the pellet in H2O (lanes 6 and 8) or additional treatment with 40 mM ICH2C(O)NH2 in 50 mM HEPES pH 7.5 (lanes 7 and 9). Hydrolysis products were analyzed on 25% polyacrylamide–8 M urea sequencing gels in the presence of 10 mM DTT (for further details see Materials and Methods).
To further substantiate our conclusion that the 3′-S-phosphorothiolate modification prevented RNase P-catalyzed processing at the canonical cleavage site, we performed a limited digestion of the 13mer Ino-S (used for the assembly of ptRNAIno-S; Fig. 2) by SVPD, which, like RNase P, generates 5′-phosphate termini and efficiently hydrolyzes 3′-S-phosphorothiolate linkages (22). As shown in Figure 4a (lanes 1 and 2), the 7 nt fragment carrying a 3′-thiol group showed no significant deviation in gel mobility under the electrophoresis conditions applied, although the sulfur is expected to be deprotonated and therefore to carry an extra negative charge compared with a 3′-OH group (10,23). 3′-S-phosphorothiolate-specific hydrolysis of 13mer Ino-S or ptRNAIno-S by AgNO3, which also yields 3′-SH termini (23), resulted in two main cleavage products under RNase P processing conditions that showed reduced gel mobility compared with the RNase P RNA cleavage product of ptRNAIno-S (Fig. 4a, lane 3, and b, lane 17). It is unclear why more than one band results from AgNO3-catalyzed hydrolysis; this may reflect different oxidation states of the terminal sulfur, similar to the multiple oxidation states observed after iodine cleavage of 3′-S-phosphorothiolate linkages (22). We next analyzed the susceptibility of RNase P RNA- and AgNO3-generated hydrolysis products to derivatization by iodoacetamide [ICH2C(O)NH2; Fig. 4c]. As demonstrated in lane 5, we were able to derivatize AgNO3 hydrolysis products, resulting in a single product band that migrated at the same position as the unmodified 7 nt 5′-flank generated by RNase P cleavage of unmodified ptRNAIno (lane 1). This is consistent with the observation that an unmodified 8 nt RNA oligonucleotide showed nearly the same gel mobility under similar electrophoresis conditions as an 8 nt RNA fragment carrying a 3′-terminal sulfur derivatized by iodoacetamide (7). Unlike the AgNO3 hydrolysis products, the 5′-cleavage product derived from RNase P RNA processing of ptRNAIno-S remained unaffected by incubation with ICH2C(O)NH2 (Fig. 4c, lane 9), migrating faster than ICH2C(O)NH2-derivatized AgNO3 hydrolysis products (lane 5).
In summary, the data presented in Figures 3 and 4 indicate that the 13mer Ino-S and ptRNAIno-S carried a single 3′-S-phosphorothiolate modification at the RNase P cleavage site and that this modification prevents cleavage by bacterial RNase P (RNA), resulting in a complete shift of the cleavage site to the next unmodified phosphodiester in the 5′-direction under the various conditions tested.
Single turnover kinetics
Single turnover experiments with ptRNAIno-S and the control substrate ptRNAIno were performed with trace amounts (<1 nM) of 5′-32P-end-labeled ptRNAs and 5 µM E.coli RNase P RNA in the presence of 1 M NH4OAc at pH 6.0. Metal ion-dependent cleavage rates (kobs) are summarized in Table 1. The rates for cleavage of the unmodified ptRNAIno were in the range 0.2–0.5 min–1 in the presence of Mg2+ and/or Mn2+, ~3- to 7-fold slower than the rate observed for the normal ptRNAGly carrying the same 7 nt 5′-flank but a C at position –1 (3). This suggests that a purine at position –1 somewhat reduces cleavage efficiency of ptRNAGly by E.coli RNase P RNA. Processing of ptRNAIno-S at the aberrant cleavage site (position –2/–1) was less efficient, but occurred at a 10-fold higher rate in the presence of Mn2+ compared with Mg2+. The dependence of cleavage efficiency and cleavage site selection by E.coli RNase P RNA on metal ion identity has previously been described for other ptRNA substrates (24).
Table 1. Cleavage of ptRNAIno, ptRNAIno-S and ‘wild-type’ ptRNAGly by E.coli RNase P RNAa.
| ptRNA species | Metal ions | kobs (min–1) |
|---|---|---|
| ptRNAIno | 15 mM Mg2+ | 0.46 ± 0.1 (0.023)b |
| 12.5 mM Mg2+, 2.5 mMMn2+ | 0.21 ± 0.02 | |
| 15 mM Mn2+ | 0.23 ± 0.07 (0.012)b | |
| ptRNAIno-S (cleavage at nt –2/–1) | 15 mM Mg2+ | 0.015 ± 0.01 |
| 15 mM Mg2+, 2.5 mM Mn2+ | 0.04 | |
| 15 mM Mn2+ | 0.16 ± 0.01 | |
| ptRNAGly (cleavage at nt –1/+1)c | 15 mM Mg2+ | 3.3 ± 1.0 |
| 12.5 mM Mg2+, 2.5 mMMn2+ | 1.5 ± 0.4 | |
| 15 mM Mn2+ | 0.75 ± 0.25 |
akobs values for ptRNAIno and ptRNAIno-S represent mean values of two independent experiments and errors indicate deviations between individual experiments.
bCleavage at position –2/–1 in parentheses estimated as follows (33):
kobs = kobs(–1/+1) + kobs(–2/–1) 1
and
kobs(–2/–1) = [c.p.m. P(–2/–1)t / {c.p.m. P(–2/–1)t + c.p.m. P(–1/+1)t}] × kobs 2
with P = product, t = time, (–2/–1) = aberrant cleavage site and (–1/+1) = normal cleavage site, and based on ~5% cleavage at position –2/–1 in the presence of 15 mM Mg2+ or Mn2+. Assuming that kobs ≈ kobs(–1/+1) and kobs = 0.23 min–1 at 15 mM Mn2+, it follows from equation 2 that kobs(–2/–1) = 0.05 × 0.23 min–1 = 0.012 min–1.
cTaken from Warnecke et al. (3).
Construction and analysis of modified ptRNA with a 1 nt 5′-flank
Our results obtained with ptRNAIno-S did not exclude the possibility that slow cleavage at the modified canonical site was masked by relatively fast cleavage at position –2/–1. Use of a ptRNA with a 1 nt 5′-flank would exclude a shift of the cleavage site to the –2/–1 position and may permit analysis of slow processing at the modified site. Towards this goal, we first investigated processing of unmodified ptRNACyt by E.coli RNase P RNA to make sure that the 1 nt flank is removed with reasonable efficiency and that no cleavage occurs at position +1/+2. Processing exclusively resulted in formation of the 1 nt 5′-cleavage product, as confirmed by co-electrophoresis with the radiolabeled dinucleotide pCpG (data not shown). The single turnover rate of cleavage was determined to be ~0.2 min–1 (<1 nM ptRNACyt, 4 µM E.coli RNase P RNA, 1 M NH4OAc and 15 mM Mg2+, pH 6.0). This rate is 2.3-fold lower than that measured for ptRNAIno under very similar conditions (Table 1). It seems that the favorable effect of a C at position –1 (see above) in ptRNACyt is counteracted by the presence of a 1 nt 5′-flank compared with a 7 nt 5′-flank in ptRNAIno. Kinetic experiments with B.subtilis RNase P indeed provided evidence that substrate affinity and the cleavage rate constant may decrease for a ptRNA with a 1 nt 5′-flank relative to substrates with longer 5′-precursor segments (26). We then constructed ptRNACyt-S, carrying the same 1 nt 5′-flank and a single 3′-S-phosphorothiolate linkage at the RNase P cleavage site (Fig. 2b). The presence of the modified linkage was verified by iodine hydrolysis, AgNO3 hydrolysis and derivatization of the AgNO3 hydrolysis product by ICH2C(O)NH2 (data not shown). We then analyzed processing of ptRNACyt and ptRNACyt-S by E.coli RNase P RNA under standard single turnover conditions in the presence of either 15 mM Mg2+ or Mn2+. No significant processing within 3 h occurred in the case of ptRNACyt-S under either condition (Fig. 5a, lanes 4–6 and 10–12), as inferred from the fact that we were unable to observe any loss of 5′-end-label from the substrate over this period. In contrast, processing of ptRNACyt was almost complete after 10 min (lanes 1–3 and 7–9).
Figure 5.

Processing of <1 nM 5′-end-labeled ptRNACyt (U) or ptRNACyt-S (S) in the presence of 5 µM (a and b) E.coli or (c) B.subtilis RNase P RNA at 37°C. Aliquots were withdrawn at indicated time points and analyzed on 25% polyacrylamide–8 M urea sequencing gels in the presence of 10 mM DTT. (a) Reaction conditions: 1 M NH4OAc, pH 6.0, and either 15 mM Mg(OAc)2 or 15 mM Mn(OAc)2. (b) Reaction conditions: 1 M NH4OAc, pH 6.0, and either 12.5 mM Mg(OAc)2/2.5 mM Cd(OAc)2 or 15 mM Cd(OAc)2. [5′-32P]pCpG (outer lane on the right) served as a size standard. Two imaging plates were required for analysis of this gel, resulting in a small gap within lane 4. (c) Reaction conditions: 1 M NH4OAc, 40 mM ICH2C(O)NH2, pH 7.0, and either 100 mM Mg(OAc)2, 100 mM Mn(OAc)2, 80 mM Mg(OAc)2/20 mM Cd(OAc)2, 80 mM Mg(OAc)2/20 mM Zn(OAc)2 or 80 mM Mg(OAc)2/20 mM Co(OAc)2. Lane Ag/ICH2C(O)NH2, incubation of ptRNACyt-S in the presence of 1 M NH4OAc, 15 mM Mg(OAc)2, pH 7.0, 50 mM AgNO3 and 40 mM ICH2C(O)NH2 for 30 min at 30°C. The indicated iodoacetamide derivative (pCSH deriv.) has a lower gel mobility than pCOH, consistent with previous observations (23). Lane Con, untreated ptRNACyt-S. Note that products in lane Ag/ICH2C(O)NH2 merged into the outer lane (lane Con). Two imaging plates were required for analysis of this gel, resulting in a small gap within lane 12.
We have previously shown that cleavage of a ptRNA with a single Rp-phosphorothioate modification by E.coli RNase P RNA is most efficiently restored in the presence of Cd2+ (3). However, the presence of Cd2+ did not restore processing of ptRNACyt-S to any significant extent (Fig. 5b). The 5′-end-label remained entirely on the substrate or, in some experiments and after prolonged incubation periods, formation of aberrant high molecular weight products due to cleavage somewhere in the 3′-portion of the mature tRNA moiety was observed (Fig. 5b; see the 21 h time point in the simultaneous presence of Mg2+ and Cd2+). However, we were unable to detect accumulation of a low molecular weight cleavage product that could have corresponded to the pCSH mononucleotide in any of our experiments. This holds true for a variety of additional reaction conditions tested, such as pH 7.0 instead of 6.0 and elevated metal ion concentrations (100 mM Mg2+, 100 mM Mn2+, 80 mM Mg2+ + 20 mM Cd2+, Zn2+ or Co2+). In contrast, unmodified ptRNACyt was efficiently converted to mature tRNA under all of these conditions (data not shown).
We also tested processing of ptRNACyt and ptRNACyt-S by B.subtilis RNase P RNA in the presence of different metal ions and metal ion combinations (Fig. 5c). Since processing reactions were performed in the presence of ICH2C(O)NH2, which did not interfere with RNase P activity (Fig. 4a and b, lanes 4 and 11, respectively), we expected a 5′-cleavage product of ptRNACyt-S to co-migrate with the iodoacetamide derivative of pCSH in lane Ag/ICH2C(O)NH2. However, neither significant accumulation of such a cleavage product nor of any other product suggestive of RNase P RNA processing was observed.
Binding of ptRNACyt and ptRNACyt-S to RNase P RNAs
In a previous study we have observed that an Sp-phosphorothiote modification at the RNase P cleavage site reduced the affinity of ptRNA ground state binding to E.coli RNase P RNA ~30-fold (3). Here we analyzed ground state binding of ptRNACyt and ptRNACyt-S to RNase P RNAs from E.coli and B.subtilis using the gel filtration centrifuge column assay (21). Binding to E.coli RNase P RNA was assayed under conditions of 1 M NH4OAc, 0.05 M Ca(OAc)2, pH 6.0, and binding to B.subtilis RNase P RNA in 0.8 M NH4OAc, 0.1 M Ca(OAc)2, pH 6.0. Ca2+ promotes binding to RNase P RNA with similar efficiency to Mg2+ (2,12), but essentially abolishes cleavage by RNase P RNA at pH 6.0 (2,27). Apparent Kd values for the binding of ptRNACyt and ptRNACyt-S to E.coli and B.subtilis RNase P RNA were in the range 1–10 nM (data not shown), indicating that the 3′-S-phosphorothiolate modification had no significant effect on substrate binding in the presence of Ca2+. Assuming that this also holds for enzyme–substrate ground state binding in the presence of Mg2+ or Mn2+, the defect caused by the sulfur modification seems to be in subsequent catalytic steps.
DISCUSSION
Two ptRNA variants carrying a 3′-S-phosphorothiolate modification at the canonical RNase P cleavage site were subjected to processing by bacterial RNase P (RNAs). In the context of ptRNAIno-S carrying a 7 nt 5′-flank, the sulfur substitution prevented cleavage of the modified phosphodiester by bacterial RNase P activities and directed cleavage to the next unmodified phosphodiester in the 5′-direction. This was observed with different bacterial RNase P RNAs and with the E.coli RNase P holoenzyme and was independent of the absence or presence of thiophilic metal ions. Our experimental results do not reveal the molecular nature of the observed inhibition effects, although metal ion exclusion is one possible explanation (as discussed below). In addition, several chemical and structural differences introduced by the sulfur modification may have contributed to the loss of catalytic function. In general, substitution of sulfur for bridging or non-bridging phosphate oxygens may lead to steric effects due to the larger van der Waals radius of sulfur versus oxygen and/or to somewhat different angles and lengths of P–S and S–C bonds. Also, sulfur has a lower electronegativity and has a lower propensity to donate electron density into the phosphorus d orbitals than oxygen (28). As described above, sulfur has a higher affinity for transition metal ions (such as Mn2+, Zn2+ and Cd2+) compared with alkaline earth ions such as Mg2+ (29–31).
In the hydrolysis reactions catalyzed by E.coli RNase P (RNA) (3,32) as well as B.subtilis RNase P RNA (20), we have now observed the following effects of sulfur substitutions at the scissile phosphodiester. (i) Under rate limiting chemistry, an Rp-phosphorothioate modification is cleaved in the presence of Mg2+ only, although at a ≥1000-fold decreased rate; Mn2+, and particularly Cd2+, largely restore the rate of cleavage, indicating direct metal ion coordination to the (pro)-Rp substituent at the cleavage site (3). An (ii) Sp-phosphorothioate or (iii) 3′-S-phosphorothiolate modification essentially abolishes cleavage at the modified internucleotide linkage, even in the presence of thiophilic metal ions, and directs cleavage to the next unmodified phosphodiester in the 5′-direction (3,20; this study). As mentioned in Results, ptRNAIno was cleaved to ~5% at position –2/–1 in the presence of 15 mM Mg2+ or Mn2+, corresponding to a rate of ~0.02 min–1 at 15 mM Mg2+ and ~0.01 min–1 at 15 mM Mn2+ (Table 1; 33). In comparison, ptRNAIno-S was cleaved at position –2/–1 at a rate of 0.015 min–1 at 15 mM Mg2+ and 0.16 min–1 at 15 mM Mn2+ (Table 1). This suggests that the rate of cleavage at –2/–1 is accelerated at least 10-fold in the presence of the 3′-S-phosphorothiolate modification with Mn2+, but not with Mg2+.
Recently, it has been shown for the crystal structure of an Sp-phosphorothioate-modified substrate bound to the 3′→5′ exonucleolytic active site of the large fragment of DNA polymerase I from E.coli that the bulky sulfur atom displaces essential catalytic metal ions from the active site (34). Analogously, the Sp-phosphorothioate and 3′-S-phosphorothiolate modifications could lead to exclusion of metal ion(s) from the active site of bacterial RNase P (RNA)–substrate complexes, possibly because of the bulkiness of the sulfur atom or because the sulfur substitution prevents correct positioning of the scissile bond in the active site. Recent X-ray analyses of a heptamer deoxynucleotide containing a single 3′-S-phoshorothiolate linkage bound to the 3′→5′ exonucleolytic active site of E.coli DNA polymerase I in the presence of different metal ions (Mg2+, Mn2+ or Zn2+) or combinations thereof have revealed little effect of the sulfur modification on positioning of the DNA in the active site (31). However, the sulfur substitution abolished binding of Mg2+ to metal ion site B (corresponding to metal ion site B in Fig. 1), whereas Mn2+ and Zn2+ were still able to bind this site. Although Mn2+ was able to occupy metal ion site B in the presence of the 3′-S-phoshorothiolate modification, exonuclease activity was ~600-fold reduced for the modified versus the all-oxygen substrate in the presence of Mn2+ (31,35). Thus, in the presence of a sulfur modification, thiophilic metal ions such as Mn2+, though able to accept sulfur as a ligand, could still be positioned in a manner that is sub-optimal for stabilization of the transition state. It should also be noted that the 3′→5′ exonuclease of E.coli DNA polymerase I is much more efficient at cleaving all-oxygen, single-stranded DNA substrates in the presence of Mn2+ than in the presence of Mg2+ (31,35), which is not the case for RNase P cleavage at the canonical site (Table 1; 3). It is conceivable that the aforementioned 600-fold reduction effect in the exonuclease system may be exceeded in other systems, such as the RNase P hydrolysis reaction. This may reduce the cleavage activity below the limit of detection, particularly when the enzyme catalyzes cleavage of the natural all-oxygen substrates less efficiently with Mn2+ than with Mg2+. Thus, failure to observe a rescue of processing of the 3′-S-phosphorothiolate-modified substrate in the presence of thiophilic metal ions does not preclude the possibility that the 3′-bridging oxygen is part of the coordination sphere of catalytic metal ions in RNase P (RNA)–substrate complexes.
Our experimental observations reveal some mechanistic resemblance to the hydrolytic spliced exon reopening (SER) reaction catalyzed by self-splicing group II introns (7,36). The SER reaction is, by the criterion of stereospecificity, analogous to the reverse of step two of group II intron splicing (36). Podar et al. (36) have shown that the SER reaction proceeds in the presence of an Rp-phosphorothioate modification at the exon–exon junction, whereas hydrolysis is essentially abolished in the presence of an Sp-phosphorothioate modification. Very similar to the bacterial RNase P system, Sp-diastereomeric group II intron substrates were cleaved with low efficiency at neighboring unmodified linkages. Likewise, essentially no cleavage at the canonical exon–exon junction was observed in the SER reaction when the 3′-bridging oxygen was replaced with sulfur (7). Again, aberrant cleavage occurred at neighboring unmodified phosphodiester bonds. A difference between the group II intron-catalyzed SER reaction and the reaction catalyzed by bacterial RNase P RNA is the absence of an obvious inhibition effect due to the Rp modification in the SER hydrolysis reaction (36). There is, however, the formal possibility that the SER reaction was not studied under conditions of rate limiting chemistry, which may have masked a potential Rp-phosphorothioate inhibition effect, as recently documented for an Sp-phosphorothioate modification in the first step of group II intron-catalyzed splicing (37). In conclusion, the above-mentioned similarities between the hydrolytic SER reaction catalyzed by group II self-splicing introns and the hydrolysis reaction catalyzed by bacterial RNase P RNA provide evidence that the two reactions are mechanistically related.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Sengen Sun for the synthesis of RNA oligonucleotides and Rita Held for technical assistance. Financial support for these studies from the Deutsche Forschungsgemeinschaft (Ha 1672/7-1/7-2/4-2/4-3) is acknowledged.
REFERENCES
- 1.Guerrier-Takada C., Haydock,K., Allen,L. and Altman,S. (1986) Biochemistry, 25, 1509–1515. [DOI] [PubMed] [Google Scholar]
- 2.Smith D. and Pace,N.R. (1993) Biochemistry, 32, 5273–5281. [DOI] [PubMed] [Google Scholar]
- 3.Warnecke J.M., Fürste,J.P., Hardt,W.-D., Erdmann,V.A. and Hartmann,R.K. (1996) Proc. Natl Acad. Sci. USA, 93, 8924–8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steitz T.A. and Steitz,J.A. (1993) Proc. Natl Acad. Sci. USA, 90, 6498–6502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piccirilli J.A., Vyle,J.S., Caruthers,M.H. and Cech,T.R. (1993) Nature, 361, 85–88. [DOI] [PubMed] [Google Scholar]
- 6.Weinstein L.B., Jones,B.C.N.M., Cosstick,R. and Cech,T.R. (1997) Nature, 388, 805–808. [DOI] [PubMed] [Google Scholar]
- 7.Sontheimer E.J., Gordon,P.M. and Piccirilli,J.A. (1999) Genes Dev., 13, 1729–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sontheimer E.J., Sun,S. and Piccirilli,J.A. (1997) Nature, 388, 801–805. [DOI] [PubMed] [Google Scholar]
- 9.Gait M.J., Pritchard,C. and Slim,G. (1991) In Eckstein,F. (ed.), Oligonucleotides and Analogs: A Practical Approach. Oxford University Press, Oxford, UK, pp. 25–48.
- 10.Sun S., Yoshida,A. and Piccirilli,J.A. (1997) RNA, 3, 1352–1363. [PMC free article] [PubMed] [Google Scholar]
- 11.Schlegl J., Fürste,J.P., Bald,R., Erdmann,V.A. and Hartmann,R.K. (1992) Nucleic Acids Res., 20, 5963–5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hardt W.-D., Schlegl,J., Erdmann,V.A. and Hartmann,R.K. (1993) Nucleic Acids Res., 21, 3521–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hardt W.-D., Erdmann,V.A. and Hartmann,R.K. (1996) RNA, 2, 1189–1198. [PMC free article] [PubMed] [Google Scholar]
- 14.Smith D., Burgin,A.B., Haas,E.S. and Pace,N.R. (1992) J. Biol. Chem., 267, 2429–2436. [PubMed] [Google Scholar]
- 15.Hardt W.-D. and Hartmann,R.K. (1996) J. Mol. Biol., 259, 422–433. [DOI] [PubMed] [Google Scholar]
- 16.Hartmann R.K. and Erdmann,V.A. (1991) Nucleic Acids Res., 19, 5957–5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vogel D.W., Hartmann,R.K., Kröger,B., Ulbrich,N. and Erdmann,V.A. (1987) Biochem. Int., 14, 167–175. [Google Scholar]
- 18.Rivera-León R., Green,C.J. and Vold,B.S. (1995) J. Bacteriol., 177, 2564–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heide C., Pfeiffer,T., Nolan,J.M. and Hartmann,R.K. (1999) RNA, 5, 102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warnecke J.M., Held,R., Busch,S. and Hartmann,R.K. (1999) J. Mol. Biol., 290, 433–445. [DOI] [PubMed] [Google Scholar]
- 21.Beebe J.A. and Fierke,C.A. (1994) Biochemistry, 33, 10294–10304. [DOI] [PubMed] [Google Scholar]
- 22.Cosstick R. and Vyle,J.S. (1990) Nucleic Acids Res., 18, 829–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weinstein L.B., Earnshaw,D.J., Cosstick,R. and Cech,T.R. (1996) J. Am. Chem. Soc., 118, 10341–10350. [Google Scholar]
- 24.Brännvall M. and Kirsebom,L.A. (1999) J. Mol. Biol., 292, 53–63. [DOI] [PubMed] [Google Scholar]
- 25.Krupp G., Kahle,D., Vogt,T. and Char,S. (1991) J. Mol. Biol., 217, 637–648. [DOI] [PubMed] [Google Scholar]
- 26.Crary S.M., Niranjanakumari,S. and Fierke,C.A. (1998) Biochemistry, 37, 9409–9416. [DOI] [PubMed] [Google Scholar]
- 27.Pan T. and Zhong,K. (1994) Biochemistry, 33, 14207–14212. [DOI] [PubMed] [Google Scholar]
- 28.Frey P.A. and Sammons,R.D. (1985) Science, 228, 541–545. [DOI] [PubMed] [Google Scholar]
- 29.Pecoraro V.L., Hermes,J.D. and Cleland,W.W. (1984) Biochemistry, 23, 5262–5271. [DOI] [PubMed] [Google Scholar]
- 30.Sigel R.K.O., Song,B. and Sigel,H. (1997) J. Am. Chem. Soc., 119, 744–755. [Google Scholar]
- 31.Brautigam C.A., Sun,S., Piccirilli,J.A. and Steitz,T.A. (1999) Biochemistry, 38, 696–704. [DOI] [PubMed] [Google Scholar]
- 32.Warnecke J.M., Green,C.J. and Hartmann,R.K. (1997) Nucl. Nucl., 16, 721–725. [Google Scholar]
- 33.Loria A. and Pan,T. (1998) Biochemistry, 37, 10126–10133. [DOI] [PubMed] [Google Scholar]
- 34.Brautigam C.A. and Steitz,T.A. (1998) J. Mol. Biol., 277, 363–377. [DOI] [PubMed] [Google Scholar]
- 35.Curley J.F., Joyce,C.M. and Piccirilli,J.A. (1997) J. Am. Chem. Soc., 119, 12691–12692. [Google Scholar]
- 36.Podar M., Perlman,P.S. and Padgett,R.A. (1995) Mol. Cell. Biol., 15, 4466–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Podar M., Perlman,P.S. and Padgett,R.A. (1998) RNA, 4, 890–900. [DOI] [PMC free article] [PubMed] [Google Scholar]