


Abstract
Telomerase activation is thought to be a critical step in cellular immortalization and carcinogenesis. The human telomerase catalytic subunit (hTERT) is a rate limiting determinant of the enzymatic activity of human telomerase. In the previous study, we identified the proximal 181 bp core promoter responsible for transcriptional activity of the hTERT gene. To identify the regulatory factors of transcription, transient expression assays were performed using hTERT promoter reporter plasmids. Serial deletion assays of the core promoter revealed that the 5′-region containing the E-box, which binds Myc/Max, as well as the 3′-region containing the GC-box, which binds Sp1, are essential for transactivation. The mutations introduced in the E-box or GC-box significantly decreased transcriptional activity of the promoter. Overexpression of Myc/Max or Sp1 led to significant activation of transcription in a cell type-specific manner, while Mad/Max introduction repressed it. However, the effects of Myc/Max on transactivation were marginal when Sp1 sites were mutated. Western blot analysis using various cell lines revealed a positive correlation between c-Myc and Sp1 expression and transcriptional activity of hTERT. Using fibroblast lineages in different stages of transformation, we found that c-Myc and Sp1 were induced to a dramatic extent when cells overcame replicative senescence and obtained immortal characteristics, in association with telomerase activation. These findings suggest that c-Myc and Sp1 cooperatively function as the major determinants of hTERT expression, and that the switching functions of Myc/Max and Mad/Max might also play roles in telomerase regulation.
INTRODUCTION
Telomeres form the ends of eukaryotic chromosomes, composed of tandem arrays of telomeric repeats (1). They are believed to protect against random fusion events and degradation and to stabilize chromosome ends (2). Since DNA polymerase fails to fully synthesize DNA termini, human telomeres in somatic cells undergo progressive shortening with cell division (3). Telomere shortening has been proposed to be a mitotic clock marking progression of a cell toward the end of its replicative lifespan, and it has also been proposed that critically short telomeres trigger cellular senescence and crisis. Synthesis and maintenance of telomeric repeats are mediated by a specialized ribonucleoprotein complex known as telomerase (1). Since activity of telomerase is usually repressed in normal somatic cells, most cells undergo replicative senescence after a definite number of cell divisions. Unlike in normal cells, telomerase is activated in most malignant tumors and tumor-derived cell lines (4,5). Telomeres in tumor cells are relatively short but maintain a certain length, probably as a result of the function of telomerase. These findings suggest that telomerase activation is involved in the attainment of cellular immortality, and that it may therefore be a critical step in carcinogenesis (6,7).
Recently, three major subunits comprising the human telomerase complex have been identified. The RNA component of human telomerase (hTR) provides the template for telomere repeat synthesis (8). Telomerase-associated protein (TP1) has also been cloned as a component of telomerase, the function of which remains unclear (9,10). The most important component responsible for the enzymatic activity of telomerase is human telomerase reverse transcriptase (hTERT) (11,12). Recent studies have found that hTERT is expressed in most malignant tumors but not in normal tissues and that expression of hTERT is closely associated with telomerase activity, while two other factors are constitutively expressed in both tumors and normal tissues (13–17). These findings suggest that hTERT is a rate limiting determinant of enzymatic activity of telomerase.
Analysis of the mechanisms by which hTERT is activated is essential for understanding the molecular basis of telomerase activation and carcinogenesis. Recently, we cloned the 5′-flanking sequence of hTERT. Interestingly, transcriptional activity of hTERT was dependent on the proximal 181 bp region of the promoter, which was essential for transactivation in immortalized and cancer cells (18). This promoter region contains E-boxes and GC-boxes, the consensus binding sequence for Myc and Sp1, respectively. In the present study, we attempted to identify the transcription factors directing hTERT expression and found that Myc and its network proteins as well as Sp1 play crucial roles in the regulation of hTERT transcription during carcinogenesis.
MATERIALS AND METHODS
Cell lines
C33A, ME180, HeLa and SiHa cells were obtained from the American Type Culture Collection, and were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum in the presence of 5% CO2. Normal human primary keratinocytes (NHK) and fibroblasts (NHF) and normal human renal cortical epithelial cells (HRCE) were purchased from Clonetics (San Diego, CA) and were grown according to the supplier’s protocol. Cell lineages of normal skin fibroblasts in different stages of transformation were developed by the introduction of SV40 large T antigen (SV40 LT) (18).
Plasmid construction
The structures of the hTERT promoter–luciferase constructs are shown in Figure 2. Various lengths of DNA fragments upstream of the initiating ATG codon of the hTERT gene were PCR amplified and inserted into luciferase (LUC) reporter vector pGL3-Basic, a promoterless and enhancerless vector (Promega). pGL3-Control (Promega) was used as a positive control plasmid. For the construction of reporter plasmids containing substitution mutations in factor binding sites, site-specific mutagenesis was performed by a PCR-based protocol (19). The c-Myc, Max and Mad1 expression vectors (pEF-Myc, pEF-Max and pEF-Mad) were constructed as described previously (20), with each cDNA driven by the elongation factor (EF) promoter. The Sp1 expression vector (pPacSp1) was provided by Dr R. Tjian (Howard Hughes Medical Institute, University of California, Berkley, CA).
Figure 2.

Deletion and mutational analyses of the hTERT core promoter. Luciferase reporter plasmids with serial deletions of the core promoter or substitution mutations in the factor binding sites were prepared. Crossed-out boxes indicate the mutated sites for binding factors shown above the figure. The mutated sequences are shown in Figure 1. These reporter plasmids were transfected into C33A, ME180 and SiHa cells, and luciferase assays were performed. The luciferase activity of the p181 wild-type plasmid was normalized to 100, and the relative luciferase activity is shown.
Luciferase assay
Transient transfection of luciferase reporter plasmids was performed using LipofectAMINE (Gibco, Gaithersburg, MD) according to the protocols recommended by the manufacturer, or by the calcium phosphate precipitation method (21). Luciferase assays were performed using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI), in which Renilla luciferase plasmids were co-transfected as a control plasmid to standardize the transcription efficiency. All experiments were performed at least three times in each plasmid and represent the relative luciferase activity as an average.
Gel shift assay
Nuclear extracts were prepared from C33A cells as previously described (22). Recombinant c-Myc or Max proteins were expressed as MBP or GST fusion proteins, respectively, in Escherichia coli and were treated with Pre-Scission protease for purification. Twenty micrograms of nuclear extracts or 10–50 ng of recombinant proteins were incubated with 0.5 µg of poly(dC·dG) in the presence or absence of unlabeled competitors on ice for 20 min in a 25 µl reaction volume containing 4% Ficoll 400 (Pharmacia), 25 mM HEPES (pH 7.9), 50 mM KCl, 1 mM EDTA, 1 mM dithiothreitol and 4 mg/ml bovine serum albumin. Following incubation, 5000 c.p.m of 32P-end-labeled oligonucleotide probes (5′-GC-GCTCCCCACGTGGCGGAGGG-3′) containing the E-box in the core promoter at –165 were added and the reactions incubated at 4°C for an additional 20 min. The underlined sequences of the probes were altered to TTT in the mutated oligonucleotides for competition assay. Following electrophoresis on a 4% polyacrylamide gel, the gel was dried and subjected to autoradiography. The Myc/Max consensus and its mutant oligonucleotides (sc-2509 and sc-2510; Santa Cruz) or specific antibodies against c-Myc (N-262; SantaCruz) and Max (C-124; SantaCruz) were added to the binding reactions for competition or supershift assays, respectively.
Western blot analysis
For western blot analysis for c-Myc, Max and Sp1 expression, cell extracts from a variety of cell lines were prepared by the method of Schreiber et al. (23). Twenty micrograms of proteins were electrophoresed on a 10% SDS–polyacrylamide gel and transferred to polyvinylidine difluoride membranes. Filters were incubated with the specific antibodies against c-Myc (N-262), Max (C-124) and Sp1 (PEP2; SantaCruz) followed by reaction with horseradish peroxidase-linked anti-rabbit IgG. Immunoreactive bands were visualized using the ECL detection system (Amersham) as suggested by the manufacturer.
RESULTS
Identification of cis elements in the core promoter of the hTERT gene essential for transcriptional activation
In a previous study, we cloned 3.3 kb of 5′-flanking sequences of the hTERT gene (18). We identified a transcription start site 77 bp upstream of the ATG initiation codon. The first base in the mRNA is labeled +1, while the first base of the promoter is labeled –1. Deletion analyses of the hTERT promoter revealed that the proximal 181 bp region spanning –181 to –1 functions as a core promoter, which is active in immortalized and cancer cells but not in normal cells (18). The core promoter is GC-rich and lacks TATA boxes (Fig. 1). In the previous gel shift assays, we found that Sp1 specifically binds to GC-boxes at five sites (Fig. 1). We also found that E-boxes (CACGTG), which are known to bind factors such as Myc and Max by a basic helix–loop–helix leucine zipper domain (bHLH-Zip), are located at –165 in the core promoter as well as at +44 in the 5′-untranslated region (5′-UTR).
Figure 1.

Sequences of the hTERT core promoter and consensus motifs for factor binding sites. The start site of transcription is shown by an arrow. The –1 indicates the first nucleotide 5′ to the start site of transcription, while 1 indicates the first nucleotide of the mRNA. The published cDNA sequence is shown in italics. The initiating ATG codon is shown in bold. The E-box is boxed and the Sp1 consensus motifs are underlined. The sequences which were mutated in the functional analyses of cis elements are indicated by asterisks, above which substituted sequences are indicated.
To determine the cis elements essential for transcriptional activation of hTERT, luciferase assays were performed with reporter plasmids with serial deletions of the core promoter using a variety of cell types (Fig. 2). An E-box is located at the 5′-end of the core promoter, and deletion of this site (p150) resulted in a 60% reduction in transcriptional activity in C33A and ME180 cells. Abrogation of this E-box by substitution mutations as shown in Figure 1 (Myc-MT1) led to a 70% reduction in transcriptional activity, suggesting that this E-box is essential for transactivation. In contrast, these deletions or mutations had only marginal effects in SiHa cells, suggesting that the role of this E-box depends on cell type. The effects of mutations in another E box at +44 (Myc-MT2) on transcriptional activity were also cell type-specific, and only marginal changes in transcription were observed in C33A and SiHa cells, while 60% reduction was observed in ME180 cells. Further deletions of the core promoter from –150 to –32 led to only a slight reduction in transcriptional activity in C33A and ME180 cells. In all types of cells examined, the most proximal 32 bp region (p32), containing a GC-box which binds Sp1 (18), retained 20–40% of the transcriptional activity of p181, while the 5′-UTR (p+18) by itself appeared to have no significant transcriptional activity. Abrogation of each Sp1 site by substitution mutation (Sp1MT1–Sp1MT5) reduced transcription to various extents, while that of all Sp1 sites (Sp1MT1–5) led to a >90% loss of transcriptional activity. These findings suggest that the 5′-region of the core promoter, containing an E-box, as well as the most proximal 3′-region, containing a GC-box which binds Sp1, are important for transactivation and that multiple Sp1 sites function as potent cis-acting elements.
Myc/Max specifically binds the E-boxes in the hTERT promoter
Initial gel shift analyses in our previous study using the E-box at –165 as a probe revealed binding of an unknown factor, which was competed by Myc/Max consensus oligonucleotides but not by anti-Myc or anti-Max antibodies (18). However, gel shift analyses in the present study using increasing amounts of nuclear extracts from C33A, with some modifications of binding conditions, revealed an additional band with slower mobility on the same probe (Fig. 3A). This band was competed by homologous competitors as well as Myc/Max consensus oligonucleotides, but not by mutated oligonucleotides in which the E-box sequences were altered. The band was also competed by addition of antibodies against c-Myc and Max. These findings suggest that a Myc/Max heterodimer binds to the E-box at –165. Binding of Myc/Max to another E-box in the 5′-UTR was also observed, but with lower binding activity (data not shown). This binding was further confirmed using recombinant proteins. Three major bands (bands A–C) were observed using purified cMyc and Max proteins (Fig. 3B). The fastest migrating band (A) was observed using only Max protein, sometimes coupled with an extra band with similar mobility. The two more slowly migrating bands (B and C) appeared on addition of c-Myc. No band was observed with only c-Myc protein. These three (or four) bands were competed by homologous competitors or Myc/Max consensus oligonucleotides but not by mutated oligonucleotides in which the E-box sequences were altered. Band C was supershifted by addition of anti-c-Myc antibody, while all the bands were supershifted by addition of anti-Max antibody. Based on these results, we conclude that band A was derived from the Max/Max homodimer, while the coupled extra bands may be products of degradation, and band C was derived from the Myc/Max heterodimer. The identity of band B remains unclear, but this band may also be products of degradation of Myc/Max, since recombinant Myc proteins are extremely unstable and easily degraded (24). Taken together, these findings suggest that the Myc/Max heterodimer and Max/Max homodimer can bind E-boxes in the hTERT promoter.
Figure 3.
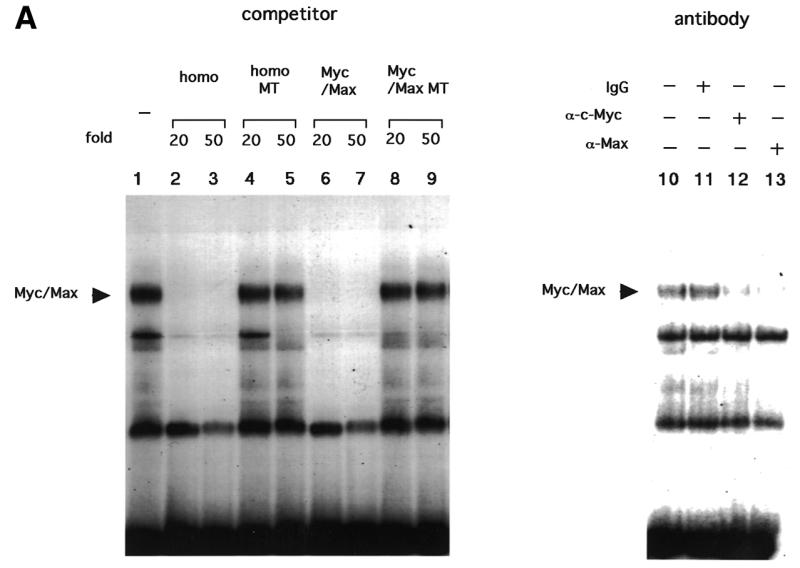

Gel shift analyses representing Myc/Max binding to the E-box in the core promoter. Twenty micrograms of nuclear extracts from C33A cells (A) or various amounts of recombinant proteins (B) were subjected to gel shift assays using the E-box region at –165 as probe (Materials and Methods) in the presence or absence of competitors or antibodies. homo, homologous competitors; homo MT, homologous competitors in which E-box sequences are mutated; Myc/Max, Myc/Max consensus oligonucleotides; Myc/Max MT, Myc/Max consensus oligonucleotides in which E-box sequences are mutated. S indicates the supershifted bands.
Myc/Max and Sp1 activate hTERT transcription while Mad/Max represses it
Myc proteins belong to the bHLH-Zip family, members of which bind to E-boxes and regulate transcription of target genes as obligate heterodimers with a partner protein, Max (25). Max is also a bHLH-Zip family member and can also form heterodimers with proteins of the Mad family (26,27). To examine the roles played by these proteins in transcriptional regulation of hTERT, expression vectors for c-Myc or Mad1 together with those for Max were transfected into various cell types, and luciferase assays were performed using reporter plasmids containing the core promoter (p181). In SiHa cells, Myc/Max introduction significantly activated hTERT transcription. Levels of activation differed depending upon the Myc:Max ratio used (Fig. 4A). An increase in the Myc:Max ratio, keeping the total amounts of expression vectors constant, led to enhanced activation, and ~5-fold activation was observed at a ratio of 10:1. No significant activation was observed with the introduction of only Max, while a 4-fold activation was observed with introduction of only Myc. The transactivation by Myc/Max was eliminated in reporter plasmids containing mutations in both E-boxes (MycMT1+2). In normal human keratinocytes, Myc/Max transactivated hTERT transcription more significantly (Fig. 4B). However, unlike SiHa cells, this transactivation required both Myc and Max, and the highest degree of activation (~7-fold of the control) was observed with a Myc:Max ratio of 1:1. However, Myc/Max activation was less significant in C33A cells (~2-fold activation; data not shown). These findings suggest that Myc/Max transactivates hTERT in a cell type-specific manner. We also examined the effects of Sp1 on transcriptional regulation using NHF and SiHa cells. Overexpression of Sp1 led to significant activation of hTERT transcription in both cells (Fig. 4C). The effects of overexpression were more significant in NHF (4-fold activation) than in SiHa (2-fold activation) cells, possibly due to the difference in levels of constitutive Sp1 expression in the two cell types. These findings suggest that Sp1 also functions as a potent transactivator of hTERT.
Figure 4.





Myc/Max, Sp1 and Mad/Max induction assays. Various amounts of Max expression vectors together with c-Myc expression vectors were co-transfected with wild-type (p181) or mutant reporter plasmid Myc MT1+2 (see Fig. 2) into SiHa cells (A) or normal human foreskin keratinocytes (NHK) (B). Sp1 expression vectors were transfected with p181 or mutant reporter plasmid Sp1MT1–5 into normal human skin fibroblasts (NHF) (C). Mad1 expression vectors together with Max expression vectors were co-transfected with p181 or MycMT1+2 into C33A cells (D). c-Myc and Max expression vectors were also co-transfected with Sp1-MT1-5 into NHK cells (E). As a control, blank vectors in which cDNA sequences for c-Myc, Max, Mad1 or Sp1 were deleted were used. The luciferase activity in control samples was normalized to 1.0. The ratios of Myc/Max and Mad/Max introduced are shown below the figure. Bars indicate SD.
In contrast, introduction of Mad together with Max significantly reduced hTERT transcription (Fig. 4D). In C33A cells, ~60% repression was observed with a 1:1 Mad:Max ratio. Mutations in both E-boxes completely eliminated this repression. Repression was also observed in other types of cells, but at lower levels (data not shown).
We further examined whether Myc/Max can activate hTERT transcription in the absence of Sp1. Myc/Max expression vectors were co-transfected into NHK cells together with reporter plasmids containing mutations in all Sp1 sites (Sp1MT1–5), and luciferase assays were performed. Only marginal activation was observed by Myc/Max (Fig. 4E). These findings suggest that Myc/Max cooperates with Sp1 to activate hTERT transcription.
Correlation of Myc and Sp1 expression levels with transcriptional activity of hTERT
In our previous study, we showed that the transcriptional activity of hTERT varied among cell types (18). Table 1 shows the relative transcriptional activity in each cell type. C33A and ME180 cells had the highest transcriptional activity [60–80% of the positive control (pGL3-Control)], while HeLa cells had a moderate one. SiHa cells exhibited the lowest transcriptional activity, ~5–10% of that of C33A cells. Normal cells (NHK, HRCE, NHF and WI38) had essentially no significant transcriptional activity (<1% of positive control), although epithelial cells such as foreskin keratinocytes (NHK) exhibited detectable activity 10-fold more than that in fibroblast cells (NHF) (evaluated by fold activation compared with the pGL-Basic negative control). Expression of c-Myc was examined in these cell lines by western blot analysis (Fig. 5). The cancer cell lines with strong transcriptional activity (C33A and ME180) expressed the highest levels of c-Myc, while SiHa cells exhibited the weakest transcriptional activity. In normal epithelial cells (NHK and HRCE), Myc expression was modest, while normal fibroblast cells (NHF and WI38) exhibited weak levels of expression. In contrast to Myc, Max was constitutively expressed in both cancer and normal cells, but higher levels of expression were usually observed in cancer cells, especially C33A and SiHa cells. Sp1 expression was also examined. Strong expression of Sp1 was observed in cancer cell lines, while normal cells expressed low or faint levels of Sp1 except for NHK cells, with modest expression. These findings suggest the presence of a close correlation between Myc and Sp1 expression and levels of hTERT transcription.
Table 1. Transcriptional activity of hTERT promoter in various cell lines.

*Luciferase activity of pGL3-Basic was extremely high in C33A cells, resulting in low values of relative transcriptional activity of p181 and p1375 in this cell type.
#ND, not done.
Figure 5.
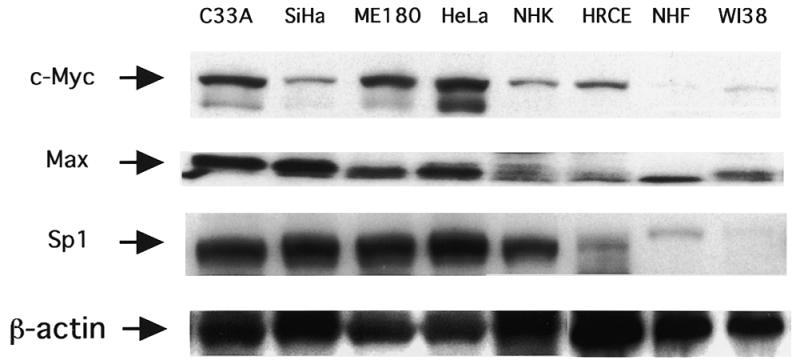
Expression of c-Myc, Max and Sp1 determined by western blot analysis for various cell lines. β-Actin protein was measured to ensure even loading.
We next examined the changes in Myc/Max and Sp1 expression during the process of cellular transformation using human fibroblast (RB) cell lineages. These lineages are at different levels of transformation following introduction of the SV40 LT (18). Normal RB cells and SV40 LT-transfected RBSV cells are mortal, but the latter exhibit an extended lifespan. Neither transcriptional activation of hTERT nor telomerase activation was observed in these cells, as was reported in our previous study (Fig. 6) (18). In contrast, RBI cell clones, which have overcome replicative senescence and obtained immortality, exhibited drastic increases in hTERT transcriptional activity as well as telomerase activity. Western blot analysis revealed weak levels of Myc and Sp1 expression in normal RB and RBSV cells, but significant up-regulation of Myc and Sp1 was obvious in RBI cells (Fig. 6). RBS and RBT clones at the more advanced stages of transformation also exhibited strong Myc and Sp1 expression. In contrast, Max levels were relatively constant throughout the process of transformation. These findings suggest that both Myc and Sp1 expression are up-regulated when normal cells are immortalized and telomerase is activated.
Figure 6.
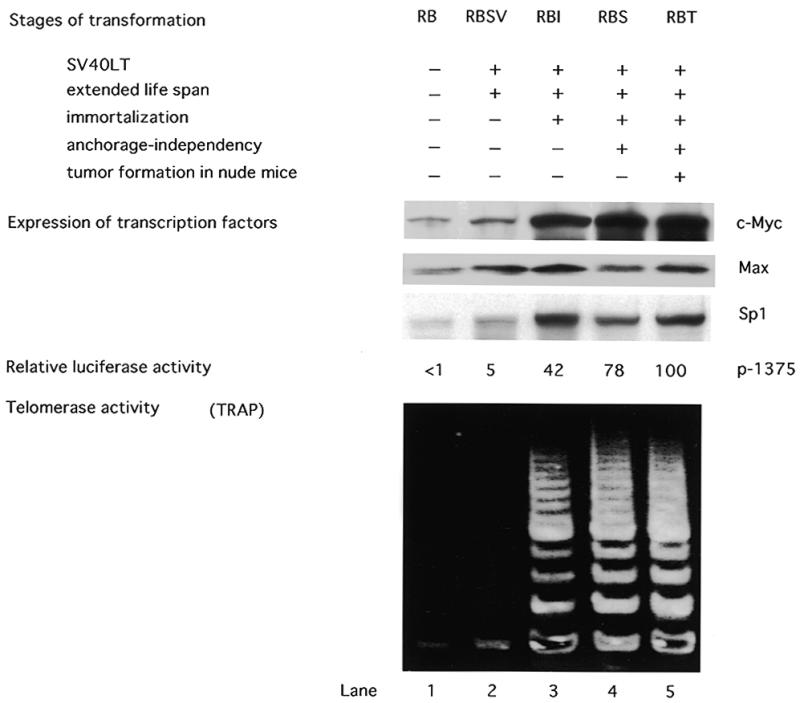
Expression of c-Myc, Max and Sp1 in fibroblast lineages in different stages of transformation. Normal fibroblasts (RB) and transformants obtained by introduction of the SV40 LT were examined for expression of c-Myc, Max and Sp1 as well as transcriptional activity and telomerase activity. Myc, Max and Sp1 expression was examined by western blot analysis. β-Actin protein was measured to ensure even loading. The transcriptional activity in each lineage was examined by luciferase assay using reporter plasmid p1375. The transcriptional activity in RBT clones was normalized to 100, and the relative transcriptional activity is shown. The relative transcriptional activity of p1375 is shown for each lineage. Telomerase activity was examined by the TRAP assay.
DISCUSSION
In the present study, we attempted to identify the cis elements of the hTERT core promoter essential for transactivation, and found that they are concentrated in the 5′- and 3′-regions, which bind Myc/Max and Sp1, respectively. The present study also demonstrated that Myc/Max and Mad/Max regulate hTERT transcription differently in a cell type-specific manner, and that transactivation by Myc/Max is achieved with the cooperative action of Sp1. In the course of this study, two published reports appeared documenting the role of c-Myc in activation of hTERT expression (28,29). We also found that levels of c-Myc and Sp1 expression were correlated with the transcriptional activity of hTERT in various cell types, and expression of c-Myc and Sp1 increased during malignant progression of fibroblast cells, in association with telomerase activity. Taken together, these findings suggest that c-Myc and Sp1 cooperatively function to activate hTERT transcription and up-regulation of these expressions might be critical for telo-merase activation during carcinogenesis.
The cis-acting effect of E-boxes and the Myc or Max requirement for transactivation appear to vary among cell types. Deletion and mutation of E-boxes resulted in significant loss of transcriptional activity in C33A cells, but not in SiHa cells. In an overexpression assay, transactivation was achieved only with Myc in SiHa cells, while both Myc and Max were required for transactivation in normal keratinocytes. In C33A cells, expression of Myc and Max had only marginal effects on transactivation. This diversity in findings may be explained by differences in constitutive expression of Myc/Max among cell types. The present western blot analyses demonstrated strong expression of Myc and Max in C33A cells, but not in normal keratinocytes. In SiHa cells, Myc expression was weak despite strong Max expression. Thus, the cis-acting function of E-boxes may be exaggerated in cells with high levels of Myc/Max, while overexpression of Myc/Max was effective in transactivation only in cells with low levels of their expression. These findings also suggest that high levels of Myc expression are required for transactivation of hTERT.
c-Myc and its network proteins are known to be oncoproteins. How are these proteins involved in telomerase activation during carcinogenesis? Overexpression of Myc is frequently observed in a wide variety of tumor types, and is usually caused by chromosomal translocation involving the c-myc gene as well as by gene amplification (30,31). A study by Hiyama et al. demonstrated a close association between a high level of telomerase activity and amplification of the myc locus in neuroblastomas (32). Thus, up-regulation of Myc due to gene alterations, leading to subsequent activation of hTERT expression, may be one cause of telomerase activation in tumors. A number of in vitro and in vivo studies have demonstrated that Myc proteins exhibit transforming activity in cooperation with other oncoproteins (33). Activation of hTERT may be one of the crucial functions through which Myc exhibits its transforming activity.
Recent studies have demonstrated telomerase activity in some types of normal somatic cells, such as endometrial cells, trophoblasts and hematopoietic progenitors (34–37). This activity is believed to be derived from stem cells, which are highly regenerative and have infinite proliferative capacity. There are several types of evidence that the telomerase activity in these cells is tightly linked to cell proliferation. Human endometrium expresses high levels of telomerase activity only in the proliferative phase in the menstrual cycle, in relation to estrogen levels (34,36). Human chorion expresses telomerase in the early stages of pregnancy, in which trophoblasts exhibit significant proliferation to form the placenta (35). Hematopoietic progenitors also express telomerase activity upon mitogenic stimulation by agents such as phytohemagglutinin and IL-2 (37). The molecular mechanisms of this proliferation-dependent telomerase activation have yet to be determined. Myc proteins play central roles in cell proliferation. The roles of Myc in cell cycle progression have been demonstrated in a large number of studies. One of the major mechanisms through which Myc acts as a cell cycle regulator is activation of G1 cyclin-dependent kinases (CDKs), especially cyclin E/Cdk2 (38). In this activation, Myc activates transcription of a direct target gene, Cdc25A, encoding a phosphatase that removes two inhibitory phosphates from Cdk2, converting the inactive form of cyclin E/Cdk2 to an active form (39). Myc proteins thus play significant roles in cell cycle progression through G1 phase by activating the target gene. In the present study, we have shown that c-Myc also targets the hTERT promoter and activates its transcription. The poliferation-dependent telomerase activation observed in normal cells might be at least in part explained by these binary functions of Myc proteins.
In contrast to Myc, Mad expression is observed in non-proliferating cells and is significantly induced upon cell differentiation (25). It is well known that the Myc/Max complex is replaced by Mad/Max during the process of differentiation and during inhibition of cell growth (33). In the present study, induction of Mad resulted in significant repression of hTERT transcription. These findings suggest that the switching function of Myc/Max and Mad/Max might also modulate telomerase activity associated with cell proliferation.
It has been demonstrated that TATA-less promoters still utilize TATA factors, and several factors have been proposed to help initiation of transcription from TATA-less genes (40). Sp1 has been shown to tether TATA factors and to play significant roles in transcriptional initiation of TATA-less promoters (41). The present study identified a 32 bp minimal promoter region around the transcription start site, which contains one Sp1 site, suggesting that Sp1 may be a compartment of basal transcription factors for hTERT. We also showed that each of the five Sp1 sites cooperatively functioned as a cis-acting element and abrogation of all Sp1 sites resulted in almost complete loss of transcriptional activity. In addition, Sp1 function was required for Myc-mediated activation of hTERT. The findings suggest a central role of Sp1 in activation of hTERT transcription. Although Sp1 is known to be a ubiquitous factor, more than 100-fold differences in its expression level have been found among cell types (42). In the present study, we demonstrated that levels of Sp1 expression were much higher in cancer than normal cells, correlated with telomerase activity. Thus, despite its widespread expression, the level of Sp1 expression may be a critical determinant of telomerase activity not only in cancer but also in normal cells.
In the present study, we demonstrated the roles of Myc and Sp1 in transcriptional regulation of hTERT. However, the hTERT promoter is probably regulated by multiple elements, and the combined action of these will determine the final activity of the promoter. In addition, an increasing number of proteins have been found to interact with c-Myc (33). Analyses of other cis elements essential for promoter regulation as well as of proteins that interact with Myc proteins may provide further insights into molecular mechanisms of telomerase regulation in normal cells and during carcinogenesis.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Robert N. Eisenman (Fred Hutchinson Cancer Research Center) for providing the pBS-Max expression vector and Dr Robert Tjian (Howard Hughes Medical Institute, University of California, Berkley, CA) for providing the SP1 expression vector (pPacSp1). We also thank Drs Laimonis A. Laimins (Department of Microbiology and Immunology, Northwestern University) and Yasuhide Kitagawa (Department of Urology, Kanazawa University) for critically reading the manuscript and giving helpful advice.
REFERENCES
- 1.Greider C.W. (1996) Annu. Rev. Biochem., 65, 337–365. [DOI] [PubMed] [Google Scholar]
- 2.Greider C.W. (1991) Cell, 67, 645–647. [DOI] [PubMed] [Google Scholar]
- 3.Watson J.D. (1972) Nature, 239, 197–201. [DOI] [PubMed] [Google Scholar]
- 4.Kim N.W., Piatyszek,M.A., Prowse,K.R., Harley,C.B., West,M.D., Ho,P.L.C., Oviello,G.M., Wright,W.E., Weinrich,S.L. and Shay,J.W. (1994) Science, 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- 5.Shay J.W. and Bacchetti,S. (1997) Eur. J. Cancer, 33, 787–791. [DOI] [PubMed] [Google Scholar]
- 6.Counter C.M., Avilion,A.A., LeFeuvre,C.E., Stewart,N.G., Greider,C.W. and Harley,C.B. (1992) EMBO J., 11, 1921–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Counter C.M., Hirte,H.W., Bacchetti,S. and Harley,C.B. (1994) Proc. Natl Acad. Sci. USA, 91, 2900–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng J., Funk,W.D., Wang,S.S., Weinrich,A.A.A., Chiu,C.P., Adams,R.R., Chang,E., Allsopp,R.C., Yu,J., Le,S., West,M.D., Harley,C.B. Andrew,W.H., Greider,C.W. and Villeponteau,B. (1995) Science, 269, 1236–1241. [DOI] [PubMed] [Google Scholar]
- 9.Harrington L., McPhail,T., Mar,V., Zhou,W., Oulton,R., Amgen EST Program, Bass,M.B., Arruda I. and Robinson,M.O. (1997) Science, 275, 973–977. [DOI] [PubMed] [Google Scholar]
- 10.Nakayama J., Saito,M., Nakamura,H., Matsuura,A. and Ishikawa,F. (1997) Cell, 88, 875–884. [DOI] [PubMed] [Google Scholar]
- 11.Meyerson M., Counter,C.M., Eaton,E.N., Ellisen,L.W., Steiner,P., Caddle,S.D., Ziaugra,L., Beijersbergen,R.L., Davidoff,M.J., Liu,Q., Bacchetti,S., Haber,D.A. and Weinberg,R.A. (1997) Cell, 90, 785–795. [DOI] [PubMed] [Google Scholar]
- 12.Nakamura T.M., Morin,G.B., Chapman,K.B., Weinrich,S.L., Andrews,W.H., Lingner,J., Harley,C.B. and Cech,T.R. (1997) Science, 277, 955–959. [DOI] [PubMed] [Google Scholar]
- 13.Ito H., Kyo,S., Kanaya,T., Takakura,M., Inoue,M and Namiki,M. (1998) Clin. Cancer Res., 4, 1603–1608. [PubMed] [Google Scholar]
- 14.Kanaya T., Kyo,S., Takakura,M., Ito,H., Namiki,M. and Inoue,M. (1998) Int. J. Cancer, 78, 539–543. [DOI] [PubMed] [Google Scholar]
- 15.Takakura M., Kyo,S., Kanaya,T., Tanaka,M and Inoue,M. (1998) Cancer Res., 58, 1558–1561. [PubMed] [Google Scholar]
- 16.Kyo S., Kanaya,T., Takakura,M., Tanaka,M. and Inoue,M. (1999) Int. J. Cancer, 80, 60–63. [DOI] [PubMed] [Google Scholar]
- 17.Kyo S., Kanaya,T., Takakura,M., Tanaka,M., Yamashita,A., Inoue,H. and Inoue,M. (1999) Int. J. Cancer, 80, 804–809. [DOI] [PubMed] [Google Scholar]
- 18.Takakura M., Kyo,S., Kanaya,T., Hirano,H., Takeda,J., Yutsudo,M. and Inoue,M. (1999) Cancer Res., 59, 551–559. [PubMed] [Google Scholar]
- 19.Higuchi R. (1990) In Innis,M.A., Gelfand,D.H., Sninsky,J.J. and White,T.J. (eds), PCR Protocols. Academic Press, San Diego, CA, pp. 177–183.
- 20.Taira T., Maeda,J., Onishi,T., Kitaura,H., Yoshida,S., Kato,H., Ikeda,M., Tamai,K., Iguchi-Ariga,S.M.M. and Ariga,H. (1998) Genes Cells, 3, 549–565. [DOI] [PubMed] [Google Scholar]
- 21.Graham F.L. and Van der Eb,A.J. (1973) Virology, 52, 456–467. [DOI] [PubMed] [Google Scholar]
- 22.Dignam J.D., Lebobitz,R.M. and Roeder,R.G. (1983) Nucleic Acids Res., 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schreiber E., Matthias,P., Muller,M.M. and Schaffner,W. (1989) Nucleic Acids Res., 17, 6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Date T., Tanihara,K. and Numura,N. (1990) Gene, 90, 141–144. [DOI] [PubMed] [Google Scholar]
- 25.Blackwood E.M. and Eisenman,R.N. (1991) Science, 251, 1211–1217. [DOI] [PubMed] [Google Scholar]
- 26.Ayer D.E., Kretzner,L. and Eisenman,R.N. (1993) Cell, 72, 211–222. [DOI] [PubMed] [Google Scholar]
- 27.Zervos A.S., Gyuris,J. and Brent,R. (1993) Cell, 72, 223–232. [DOI] [PubMed] [Google Scholar]
- 28.Wu K.J., Grandori,C., Amacker,M., Simon-Vermot,N., Polack,A., Lingner,J. and Dalla-Favera,R. (1999) Nature Genet., 21, 220–224. [DOI] [PubMed] [Google Scholar]
- 29.Greenberg R.A., O’Hagan,R.C., Deng,H., Xiao,Q., Hann,S.R., Adams,R.R., Lichtsteiner,S., Chin,L., Morin,G.B. and DePinho,R.A. (1999) Oncogene, 18, 1219–1226. [DOI] [PubMed] [Google Scholar]
- 30.Spencer C.A. and Groudine,M. (1991) Adv. Cancer Res., 56, 1–48. [DOI] [PubMed] [Google Scholar]
- 31.DePinho R.A., Schreiber-Agus,N. and Alt,F.W. (1991) Adv. Cancer Res., 57, 1–46. [DOI] [PubMed] [Google Scholar]
- 32.Hiyama E., Hiyama,K., Yokoyama,T., Matsuura,Y., Piatyszek,M.A. and Shay,J.W. (1995) Nature Med., 1, 249–255. [DOI] [PubMed] [Google Scholar]
- 33.Henriksson M. and Luscher,B. (1996) Adv. Cancer Res., 68, 109–182. [DOI] [PubMed] [Google Scholar]
- 34.Kyo S., Takakura,M., Kohama,T. and Inoue,M. (1997) Cancer Res., 57, 610–614. [PubMed] [Google Scholar]
- 35.Kyo S., Takakura,M., Tanaka,M., Kanaya,T., Sagawa,T., Kohama,T., Ishikawa,H., Nakano,T., Shimoya,K. and Inonue,M. (1997) Biochem. Biophys. Res. Commun., 241, 498–503. [DOI] [PubMed] [Google Scholar]
- 36.Tanaka M., Kyo,S., Takakura,M., Kanaya,T., Sagawa,T., Yamashita,K., Okada,Y., Hiyama,E. and Inoue,M. (1998) Am. J. Pathol., 153, 1985–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hiyama K., Hirai,Y., Kyoizumi,S., Akiyama,M., Hiyama,E., Piatyszek,M.A., Shay,J.W., Ishioka,S. and Yamakido,M. (1995) J. Immunol., 155, 3711–3715. [PubMed] [Google Scholar]
- 38.Bouchard C., Staller,P. and Eilers,M. (1998) Trends Cell Biol., 8, 202–206. [DOI] [PubMed] [Google Scholar]
- 39.Galaktionov K., Chen,X. and Beach,D. (1996) Nature, 382, 511–517. [DOI] [PubMed] [Google Scholar]
- 40.Smale S.T. (1997) Biochim. Biophys. Acta, 1351, 73–88. [DOI] [PubMed] [Google Scholar]
- 41.Pugh B.F. and Tjian,R. (1991) Genes Dev., 5, 1935–1945. [DOI] [PubMed] [Google Scholar]
- 42.Saffer J.D., Jackson,S.P. and Annarella,M.B. (1991) Mol. Cell. Biol., 11, 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]