


Abstract
Type I elements regulate the initiation of DNA replication, elongation of replication forks and transcription of the Tetrahymena thermophila rDNA minichromosome. Previous studies identified a 24 kDa protein, ssA-TIBF, which binds the A-rich strand of type I elements. Here we describe two additional type I element binding activities (native mol. wt ~65 and ~250 kDa) that interact with DNA via previously unidentified 32 and 110 kDa polypeptides. The 65 kDa activity was purified to homogeneity and consists of a homodimer of a 32 kDa polypeptide. In contrast to the other type I element binding factors, the 65 kDa activity partitions preferentially to the nuclear fraction during isolation. Levels of the 65 kDa activity increase dramatically in starved cells, raising the possibility that it might negatively regulate replication or transcription. By comparison, the other two binding activities were elevated slightly during macronuclear development, when the rDNA was undergoing DNA replication. Previous studies indicate that the initiation of rDNA replication is regulated by long range interactions between dispersed type I elements. Competitive DNA binding or cooperative protein–protein interactions between the factors described here may play a regulatory role in replication or expression of the rDNA minichromosome.
INTRODUCTION
DNA–protein interactions regulate a number of important cellular processes. In some instances, unrelated events are controlled by a single multifunctional protein. For example, binding of large T antigen to the SV40 origin region controls both the initiation of replication and transcription from the adjacent viral promoter (reviewed in 1). Similarly, ABF1 regulates replication when bound to its cognate site at the ARS1 origin (2), and transcription when bound to other sites in the Saccharomyces cerevisiae genome (3). In other instances, the binding of more than one protein to a single DNA binding site is central to regulation. This is best illustrated by the dynamic interactions of cro and the lambda repressor that regulate the lytic/lysogenic switch in bacteriophage λ (4). We and others previously identified a cis-acting determinant in the Tetrahymena thermophila rDNA minichromosome that controls: (i) the initiation of DNA replication (5–8), (ii) replication fork movement (9,10); (iii) transcription of the ribosomal RNA genes (11,12; R.E.Pearlman, personal communication). Whether these processes are regulated by distinct DNA binding proteins or a common multifunctional factor is not known.
The regulation of chromosomal DNA replication is poorly understood in eukaryotes. To date, both cis-acting sequences and trans-acting factors have only been identified definitively in S.cerevisiae and Drosophila melanogaster. In S.cerevisiae, the initiation site (origin of replication) and cis-acting regulatory determinants co-localize (2,13). These determinants consist of a core sequence and several flanking auxiliary elements that can differ from one origin to the next (reviewed in 14). For the ARS1 element, these determinants include the essential ARS (autonomously replicating sequence) element, the binding site for the yeast origin recognition complex (ORC) (15), and three closely positioned elements, including a binding site for the transcription factor ABF1. The organization of replication determinants appears to be more complex in other eukaryotes. Although poorly defined in most species, cis-acting regulatory determinants can reside hundreds to thousands of base pairs away from the initiation site (7,16–20). These findings suggest that dispersed determinants are important for the regulation of DNA replication. This is illustrated by a recent study in D.melanogaster which demonstrated that ORC binding to dispersed determinants is required for amplification of the chorion gene loci in terminally differentiated follicle cells (21).
The T.thermophila rDNA minichromosome is a useful eukaryotic model for studying DNA replication and transcription. Genetic and molecular approaches have identified cis-acting elements that regulate both processes (8,22; reviewed in 23). Furthermore, rDNA replication is dynamically controlled. During macronuclear development, the rDNA is amplified from two to 10 000 copies within a single S phase. Subsequent replication in vegetative growing cells is restricted to once (on average) per cell division. Using a classical genetic approach, we and others identified a dispersed, repeated genetic determinant, the type I element, that is required for long-term maintenance of rDNA minichromosomes (5–7,9; D.L.Dobbs and E.H.Blackburn, personal communication). Mutations in or immediately adjacent to this element affect rDNA amplification as well, suggesting that type I elements control a replication-based process (24). Recent studies directly implicate type I elements in the initiation of rDNA replication (8). Four copies of the type I element are present in the 1.9 kb rDNA 5′-non-transcribed spacer (5′-NTS; Fig. 1, types IA–ID). Two copies reside in the tandem duplicated 430 bp segments that encompass the nucleosome-free regions Domain 1 and Domain 2 (D1 and D2) (25). The remaining type I elements map to the rRNA promoter in a region that is similarly devoid of nucleosomes. Two-dimensional gel mapping studies demonstrated that D1 and D2 co-localize with sites for the initiation of DNA replication (26).
Figure 1.

Structural and functional features of the Tetrahymena rDNA minichromosome. (Top) rDNA minichromosomes encode two inverted copies of the rRNA coding region and 5′- and 3′-non-transcribed spacers (NTS), bound by telomeres (thin lines with vertical bars). The 35S rRNA precursor (long arrows) encodes the 17S, 5.8S and 26S rRNAs. (Bottom) Blow-up of the 1.9 kb 5′-NTS region from wild-type C3 rDNA. Terminal arrow, rRNA promoter; horizontal black ovals, positioned nucleosomes (25); black boxes, type I repeats; shaded box, tandemly arrayed type II repeats; open boxes, type III repeats (39); vertical black ovals, replication fork pausing sites (p1–p3) (9). Domains 1 and 2 are 430 bp tandemly duplicated segments that have undergone subsequent sequence divergence. Sequence changes that affect vegetative rDNA replication are depicted for natural (B) and induced (rmm) mutant alleles (reviewed in 23). The type ID +G mutation ablates rRNA transcription (7,12). The sequence of type I elements and flanking DNA are shown at the bottom. Long boxed areas encompass the phylogenetically conserved 33 nt type IA–ID elements. Mutations in the 3′-flanking sequences are indicated for the B (42 bp deletion), rmm8 (G→A transition) and +G (frameshift) alleles. The long arrow above the sequences demarcates the position of the 37mer oligonucleotides (beginning with the sequence GGC) used in DNA binding studies.
In addition to replication initiation, type I elements regulate replication fork movement and transcription. Elongating replication forks pause transiently at three sites in the 5′-NTS at conserved tripartite sequence elements, and mutations in adjacent type I elements diminish fork pausing (Fig. 1, p1–p3; 9,10). Finally, in vivo and in vitro studies demonstrated that the promoter-proximal type I elements are part of the core rRNA gene promoter, providing a strong link to rRNA transcription (11,12, R.E.Pearlman, personal communication).
Previous studies identified a single protein that binds to the type I element in a sequence-specific manner (27–29). This protein, ssA-TIBF (single-strand A type I element binding factor), binds to single-stranded DNA only, interacting with either the A-rich or T-rich strand of the type I element (29). Importantly, it binds differently to wild-type C3 rDNA and a natural variant, B rDNA, which is partially defective for replication initiation and the regulation of replication fork movement (5,9). Here we present evidence for the existence of two additional type I element binding activities. The expression and binding properties of these new activities suggest that they may compete with ssA-TIBF for binding to type I elements in vivo.
MATERIALS AND METHODS
Strains and culture methods
Tetrahymena cultures were grown at 30°C in 2% PPYS (2% proteose peptone, 0.2% yeast extract, 10 µM FeCl3), supplemented with 250 µg/ml penicillin, 100 µg/ml streptomycin and 250 ng/ml amphotericin B (30,31). To study type I element binding activities during the amplification phase of Tetrahymena macronuclear development, strains CU427 and CU428 were grown separately to a density of 2–4 × 105 cells/ml, starved overnight in 10 mM Tris (pH 7.4), and paired in T150 tissue culture flasks at a density of 2 × 105 cells/ml (75 ml/flask). Aliquots of 800 ml of mating cells were harvested 12, 18 and 24 h after pairing. Approximately 100, 4000 and 10 000 copies of the rDNA are present at the respective time points (32).
Extract preparation, protein purification and subcellular fractionation
For protein purification, extracts were prepared from vegetative T.thermophila strain CU428 harvested at a density of 2 × 105 cells/ml. Cell lysates were prepared by two different methods. Vegetative log phase extracts were prepared in HMG buffer [20 mM HEPES, pH 7.1, 1 mM MgCl2, 10% (v/v) glycerol, 0.2 mM PMSF, 2 µg/ml leupeptin, 1 µg/ml pepstatin, 1 mM DTT], followed by ultracentrifugation to generate a whole cell lysate as previously described (28). Alternatively, cytoplasmic and nuclear extracts were prepared from starved cells or mated cells at various times during macronuclear development (33). Samples of 1–2 × 108 cells were harvested by centrifugation (1000 g, 5 min), washed twice with 10 mM Tris (pH 7.5), resuspended in 60 ml of ice-cold TMS buffer (10 mM Tris, pH 7.5, 10 mM MgCl2, 3 mM CaCl2, 0.25 M sucrose, 1 mM DTT, 1 mM PMSF) and lysed by the addition of NP-40 to a final concentration of 0.16%. Following incubation for 30 min, solid sucrose (0.815 g/ml) was added and nuclei were pelleted by centrifugation at 9000 g for 30 min. The (cytoplasmic) supernatant fraction was recentrifuged for 1 h at 100 000 g and the resulting S100 supernatant fraction was collected. The nuclear fraction was resuspended in 1.5 ml of low salt buffer (20 mM HEPES, pH 7.9, 25% glycerol, 1.5 mM MgCl2, 20 mM KCl, 0.2 mM EDTA, 0.2 mM PMSF, 0.2 mM DTT). An equal volume of high salt buffer (low salt buffer adjusted to 1.2 M KCl) was added, incubated for 30 min with gentle rocking, and centrifuged for 1 h at 25 000 g. The nuclear and cytoplasmic supernatant fractions were dialyzed against 20 mM HEPES, pH 7.9, 20% glycerol, 100 mM KCl, 2 mM EDTA, 0.5 mM DTT, 0.2 mM PMSF, and stored at –70°C. Protein concentrations were measured by the Bradford method (34).
Purification of ssA-TIBF and a 32 kDa type I element binding activity was achieved as described (29). All steps were performed at 4°C in buffers containing 1 mM DTT. Briefly, 50–60 ml of HMG S100 extract (prepared from 4 l of log phase cells) were incubated with 0.9% streptomycin sulfate (w/v) to remove nucleic acids and proteins were concentrated by precipitation with ammonium sulfate (0–90% saturation). The pellet was resuspended in and dialyzed against 10 mM potassium phosphate buffer, pH 7.0, loaded onto a Biogel HTP column (Bio-Rad) and eluted with a linear gradient of potassium phosphate. For this and all subsequent steps, type I element binding activity was assessed by electrophoretic mobility shift assays in the absence of UV crosslinking (see below). Peak fractions were dialyzed and applied to a double-stranded DNA cellulose column in TEG buffer (10 mM Tris, pH 8.0, 1 mM EDTA, 20% glycerol, 100 mM NaCl) and eluted with a linear gradient of TEG + sodium chloride. Peak activity fractions were then applied to a single-strand oligonucleotide affinity column [biotinylated 37 nt C3 rDNA type IB element (Fig. 1) in TEG + 1 mM MgCl2] and eluted stepwise with a sodium chloride gradient. Column fractions from the final purification step were analyzed by SDS–PAGE. Glutaraldehyde crosslinking was performed as previously described (29).
Electrophoretic mobility shift assays
For standard binding and oligonucleotide competition studies, sub-saturating amounts of type I element binding activities were incubated with 0.1 pmol of labeled oligonucleotide for 15 min on ice in 12 mM HEPES, pH 7.9, 0.1 mM EDTA, 30 mM KCl, 12.5% glycerol (v/v), 5 mM MgCl2, 1 mM DTT and 5 µg bovine serum albumin. For competition assays, unlabeled oligonucleotides were added to the binding reaction. Where indicated, UV crosslinking was performed by incubating reaction mixtures on ice for 15 min, followed by irradiation for 15 min on ice in a Stratalinker apparatus (Stratagene Inc; distance from lamp 5.5 cm). Samples were electrophoresed at room temperature in 0.6× TGE (50 mM Tris, 220 mM glycine, 6 mM EDTA) + 12.5% glycerol or 1× TBE (90 mM Tris–borate, 2 mM EDTA, pH 8.0) buffer. Electrophoresis was typically carried out for 4 h at 120 V to resolve different DNA–protein complexes. Consequently, unbound oligonucleotides were run off the gel. Dried gels were exposed to X-ray film or quantitated by phosphorimager analysis on a Packard Cyclone apparatus. Oligonucleotides were labeled on their 5′-end with [γ-32P]ATP using T4 polynucleotide kinase or used as cold competitor DNAs. All oligonucleotides were purified by denaturing gel electrophoresis prior to use.
RESULTS
Identification of three distinct type I element binding proteins
As type I elements regulate three distinct chromosomal processes (replication initiation, elongation and transcription), one or more type I element binding proteins may be involved. Previous studies identified a single protein, ssA-TIBF, that binds to type I elements in vitro. We set out to determine whether type I elements interact specifically with additional proteins by incubating S100 cell extracts with a 37mer oligonucleotide corresponding to the A-rich strand of the wild-type C3 type IB element (Fig. 1, C3 type IB). Gel mobility shift analyses detected three DNA–protein complexes with extracts prepared from log phase Tetrahymena cultures (Fig. 2A, –UV). A fourth complex was observed when reaction mixtures were crosslinked with UV light prior to gel electrophoresis (Fig. 2A, +UV). In some experiments the UV-dependent complex resolved into two species.
Figure 2.
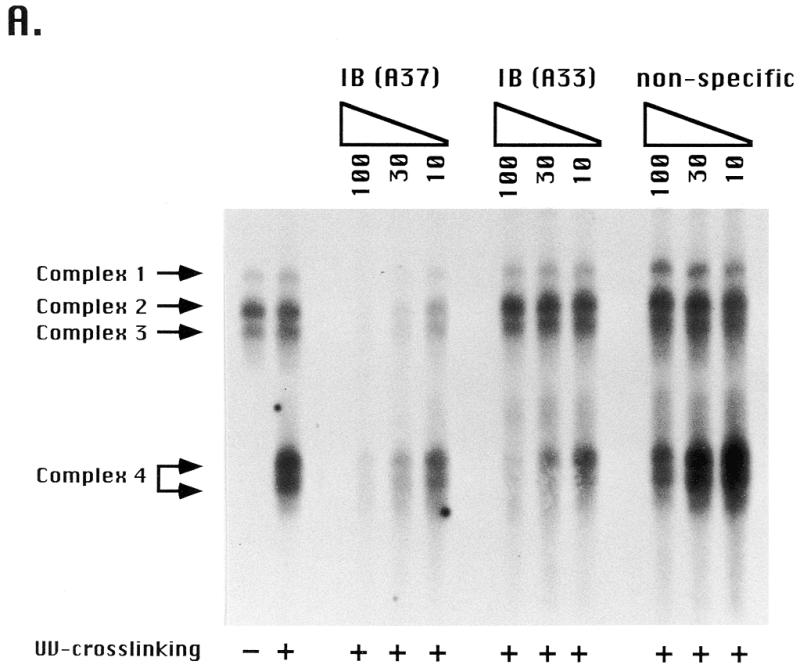
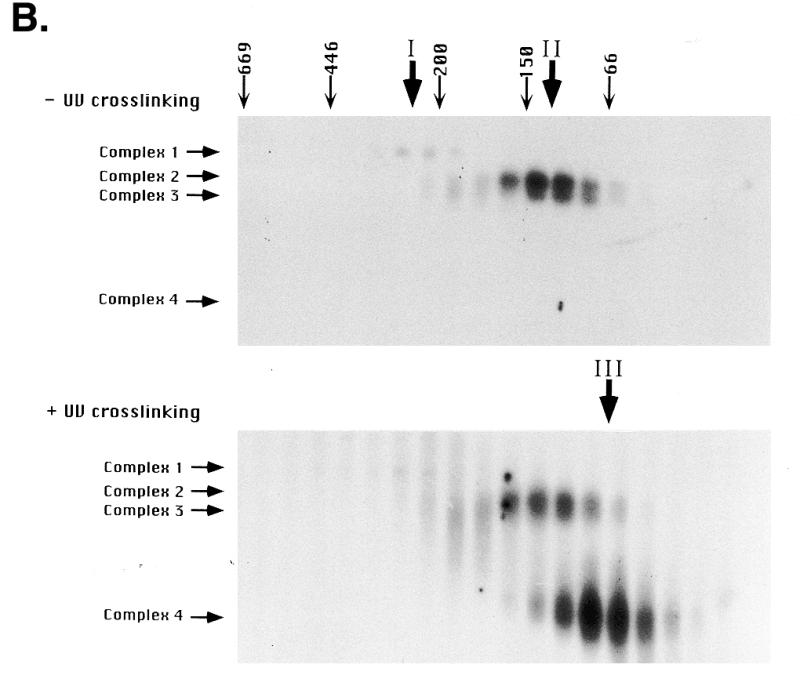
Type I element binding activities in Tetrahymena cell extracts. (A) Electrophoretic mobility shift assay of type I elements with vegetative S100 (HMG buffer) cell extracts. An aliquot of 4 µg protein was incubated with 0.1 pmol radiolabeled type IB element oligonucleotide (Fig. 1, wild-type C3 rDNA allele) and DNA–protein complexes (complexes 1–4) were resolved on 5% Tris–glycine/EDTA gels. The first two lanes (–/+UV crosslinking) are binding reactions performed in the absence of competitor DNAs. The remaining lanes (+UV crosslinking) are binding reactions performed in the presence of a molar excess of cold specific (type IB A37 and type IB A33; Fig. 1) or non-specific competitor DNAs (coding region, AACAGTACTAGCGTGTTGCG; 3′-NTS, CCAAAAGAATTCAAGATTTGATTTAAAA; data not shown). Unbound oligonucleotides were run off the gel in order that different DNA–protein complexes were resolved. In some instances complex 4 separated into two discrete species. (B) Gel shift analysis (+/–UV crosslinking) of S100 extract proteins following fractionation on a Superose 6 gel filtration column (Amersham/Pharmacia). The migrations and masses of protein molecular weight markers (kDa) are indicated. Peak binding fractions for gel shift complexes 1 (peak I), 2 and 3 (peak II) and 4 (peak III) are indicated.
Competition assays with unlabeled substrates indicated that all four complexes result from sequence-specific DNA–protein interactions. Whereas the unlabeled type IB 37mer competed efficiently for DNA binding, non-specific oligonucleotides from the coding region (Fig. 2A, non-specific) and 3′-NTS (data not shown) did not. The binding specificity for the UV-dependent species was slightly lower than the other species, in that a 100-fold excess of non-specific oligonucleotide diminished binding by ~30%. Furthermore, a 33 nt competitor which spans the entire type IB element, but lacks downstream sequences, competed well for binding to the UV-dependent species, but had little effect on the other binding activities. This difference suggests that the protein(s) responsible for complex 4 formation contacts the DNA differently than the other type I element binding activities. Previous experiments with purified ssA-TIBF demonstrated a 100-fold preference for binding to the 37mer type IB element oligonucleotide, which contains additional 3′-flanking sequences and lacks the poly(T) stretch at the 5′-end of the type I element, over the 33 nt type IB element oligonucleotide (28).
Gel filtration was performed as a first step to characterize the proteins that generate the four gel shift complexes. Three peaks of DNA binding activity were observed when the S100 extract was fractionated on an analytical Superose 6 FPLC column (Amersham/Pharmacia) (Fig. 2B, activity peaks I–III). The first peak had an apparent mass of ~250 kDa and was responsible for the slowest migrating gel shift complex (Fig. 2B, complex 1). The second activity peak (native mol. wt ~100 kDa) generated the two intermediate migrating gel shift complexes (complexes 2 and 3), whereas the final activity peak (native mol. wt ~65 kDa) generated the fast migrating, UV-dependent species (complex 4). Glutaraldehyde and UV crosslinking experiments with the purified 24 kDa protein, ssA-TIBF, revealed that it is responsible for gel shift complexes 2 and 3 and binds DNA as a homotetramer (4 × 24 kDa) (29).
To identify the DNA binding component in gel shift complexes 1 and 4, protein–DNA crosslinking was performed. Crude S100 extracts were incubated with the radiolabeled type I element substrate, ssA37, crosslinked with UV light, and covalent protein–DNA complexes resolved by 2D gel electrophoresis. Non-denaturing gel electrophoresis was performed in the first dimension to separate the four gel shift complexes (Fig. 3, upper). SDS–PAGE was then used to disrupt protein–protein interactions. Three unique radiolabeled proteins were observed (Fig. 3, lower). Taking into account the size of the bound oligonucleotide (12 kDa), the labeled protein in gel shift complex 4 has a predicted molecular weight of ~35 kDa. The mass of this protein is distinct from that observed in gel shift complexes 2 and 3 (mol. wt ~25 kDa; ssA-TIBF mol. wt 24 kDa), suggesting that it contains a DNA binding protein that is distinct from ssA-TIBF. Higher molecular weight species were also detected in crosslinked complexes 2 and 3, possibly due to the binding of additional proteins. The signals for these species were detected in crosslinking studies with ssA-TIBF fractions purified to apparent homogeneity, suggesting that more than one ssA-TIBF subunit may bind to DNA (data not shown; 29). Finally, crosslinking of neither ssA-TIBF (24 kDa) nor the ~35 kDa species was detected in the slowest migrating gel shift complex (complex 1). Instead, crosslinking of an ~110 kDa protein and no additional polypeptides was observed, suggesting that a third polypeptide was responsible for DNA binding in this complex.
Figure 3.
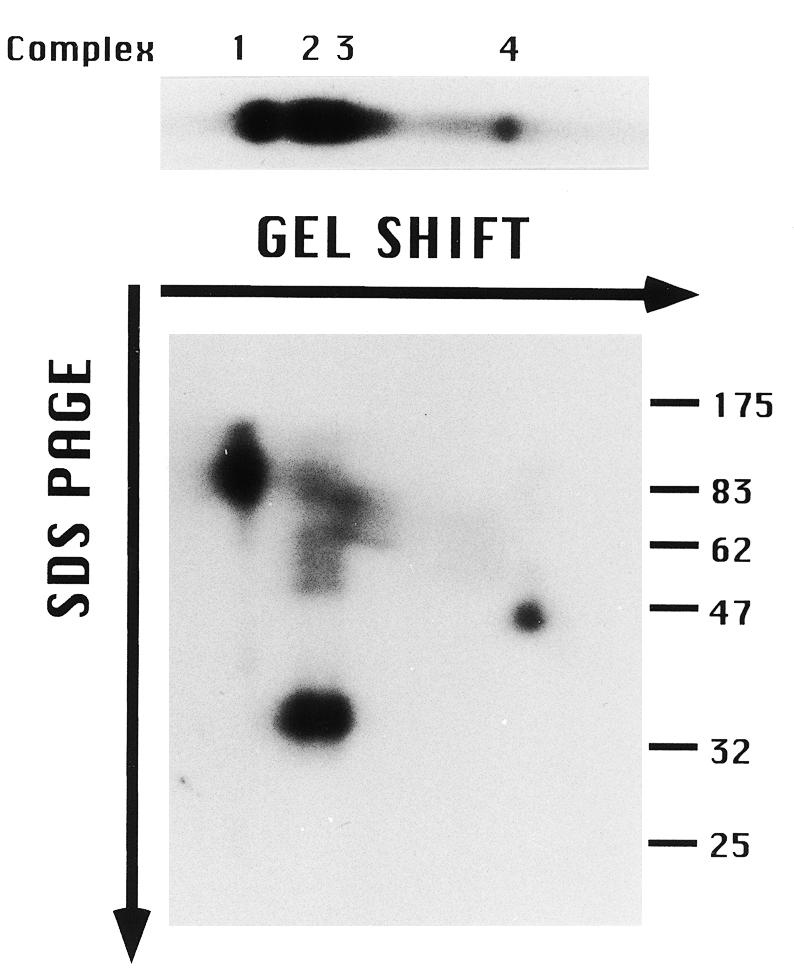
Two-dimensional gel analysis of type I element DNA–protein complexes. An aliquot of 0.1 pmol of 5′-end-labeled oligonucleotide (C3 type IB 37mer) was incubated with 4 µg S100 extract (in HMG buffer), crosslinked with UV light and proteins that were covalently bound to substrate DNA were resolved sequentially under native (TGE gel shift) and denaturing (SDS–PAGE) conditions. (Upper) First dimension (gel shift) only; (lower) 2D gel analysis. The migration positions of protein molecular weight markers (kDa) are shown on the right.
In that the two new activities that were fractionated by gel filtration overlapped with the ssA-TIBF activity peak (Fig. 2), it was possible that ssA-TIBF might play some role in regulating their ability to bind to DNA. To address this issue, we attempted to fractionate these activities by conventional and affinity chromatography. Whereas the 250 kDa binding activity was lost during chromatography on double-stranded DNA cellulose (data not shown), ssA-TIBF and a 32 kDa protein were each purified to apparent homogeneity. Following oligonucleotide affinity chromatography, these two proteins were eluted as distinct, but overlapping, peaks (Fig. 4A). To determine whether ssA-TIBF and the 32 kDa protein bound DNA independently, gel shift analyses were performed on fractions containing ssA-TIBF or 32 kDa protein alone, or both. As expected, the purified 24 kDa fraction generated gel shift complexes 2 and 3 (Fig. 4B). Similar to the 65 kDa binding activity detected by gel filtration, the fraction containing just the 32 kDa protein produced a UV-dependent complex corresponding to gel shift complex 4. We conclude that the 32 kDa protein can bind DNA independently of ssA-TIBF. When protein–DNA complexes formed with the 32 kDa protein were treated sequentially with UV and glutaraldehyde, all of the radiolabeled protein–DNA complexes migrated as a diffuse band at ~100 kDa (Fig. 4C). Taking into account the predicted mass of the native complex 4 binding activity (60–70 kDa), we suggest that all of the active 32 kDa protein exists in a higher molecular weight complex, as a homodimer that binds one or possibly more DNA substrate molecules.
Figure 4.
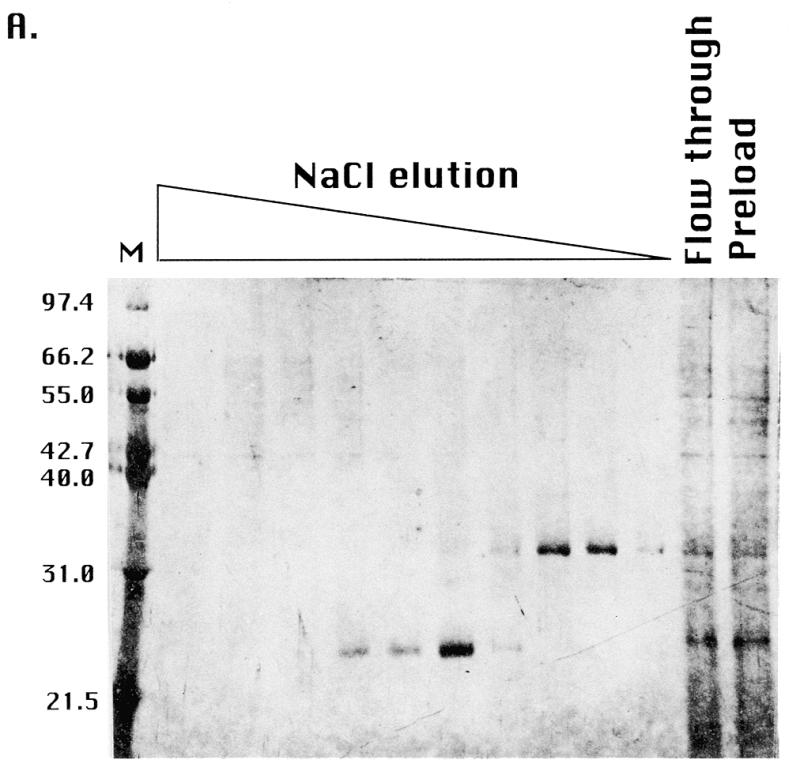
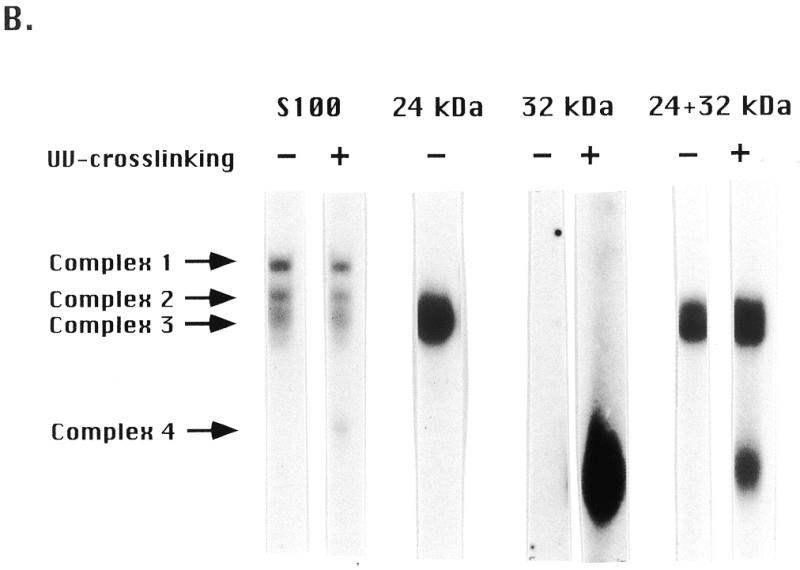
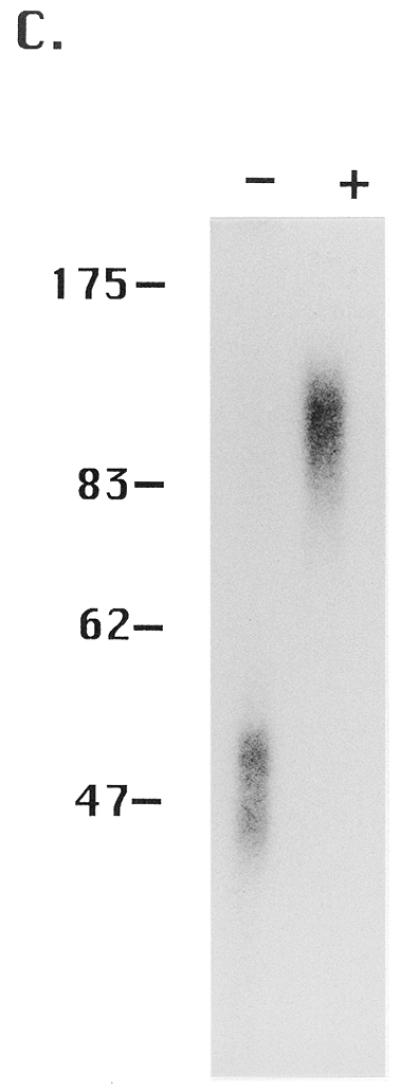
Purification of 24 (ssA-TIBF) and 32 kDa type I element binding proteins. (A) SDS–PAGE analysis of purified type I element binding proteins. Silver staining of preload, flow-through and fractions eluted from a type I element oligonucleotide affinity column (see Materials and Methods). The migration positions of protein molecular weight markers (kDa) are shown on the left. (B) Gel shift analysis of crude S100 and oligonucleotide affinity chromatography column fractions containing ssA-TIBF (24 kDa), 32 kDa polypeptide, or both (+/–UV crosslinking). Binding substrate, C3 type IB element (37mer; Fig. 1). (C) Gel shift analysis of UV crosslinked 32 kDa protein fractions with or without glutaraldehyde crosslinking.
Recognition of type I elements by different type I element binding proteins
Genetic and molecular studies strongly suggest that all four type I elements (types IA–ID) are involved in rDNA replication control (5–8; G.M.Kapler, unpublished results). However, only the promoter-proximal type I elements appear to be required for rRNA transcription (11,12). ssA-TIBF shows a preference for C3 rDNA over B rDNA, suggesting a possible role for this protein in DNA replication control. Footprinting studies indicate that purified ssA-TIBF contacts the wild-type C3 rDNA type IB element differently than the B rDNA type IB allele (42 bp deletion immediately 3′ to the type IB element). The sequences 3′ to the type I element contribute preferentially to C3 rDNA binding (29); however, no significant difference in binding affinity was observed. To address whether the 250 and 65 kDa activities bind more tightly to specific type I elements, gel shift studies were performed in the presence of increasing concentrations of unlabeled competitors. The competitors included 37mer oligonucleotides corresponding to the wild-type C3 type IA, IB and ID elements and mutant alleles that affect replication [B rDNA type IB element (5) and C3-rmm8 type ID element (7)] and transcription [C3 rDNA type ID(+G) element (12)] (Fig. 1). Since the type IB and IC elements and their 3′-flanking sequences are identical, binding to the type IC element was not assessed.
Competitive binding studies were performed under conditions in which the oligonucleotide substrate was present in excess relative to the three DNA binding activities. ssA-TIBF elicited no significant preference for the C3 rDNA type IB element, relative to the B rDNA type IB allele [Fig. 5, type IB (C3 versus B), complexes 2 and 3; graph, (96 kDa) average of three binding experiments for complexes 2+3; 29]. A similar result was obtained for the 250 kDa activity [Fig. 5, type IB (C3 versus B), complex 1; graph, (250 kDa) average of three binding experiments for complex 1]. As previously noted, these results differ from a previous study on ssA-TIBF, in which a 25-fold preference for the C3 type IB allele was reported (28). Gel shift analysis of the UV-dependent DNA–protein complex 4 revealed that the 32 kDa protein showed a slightly more pronounced preference for the C3 type IB allele [Fig. 5, type IB, (C3 versusB), complex 4 (+UV)]. Similar to the previous report for ssA-TIBF, all three DNA binding activities displayed a higher (3- to 6-fold) relative affinity for the promoter-proximal type ID element compared to the origin-proximal ones [type IA (data not shown) and type IB]. Furthermore, all three binding activities bound more tightly to the wild-type type ID element relative to alleles that affect DNA replication (C3-rmm8; 7) and transcription (type ID +G; 12) (Fig. 5). However, binding to both replication- and transcription-defective alleles was similar for the respective activities. We conclude that the three DNA binding activities recognize all four type I elements with similar affinity and do not strongly discriminate at the level of binding affinity between wild-type and mutant alleles that are compromised for rDNA replication or rRNA transcription.
Figure 5.

Competitive binding studies with type I element oligonucleotides. An aliquot of 4 µg of S100 extract (HMG buffer) was incubated with 0.1 pmol radiolabeled type IB element (wild-type C3 allele) in the absence (0) or presence (0.1- to 100-fold molar excess) of cold type I element oligonucleotide competitors and subjected to gel shift analysis. Cold competitors included the wild-type alleles of the type IA, IB and ID elements, and mutant alleles that affect replication (type IB B rDNA allele, type ID C3-rmm8 allele) and transcription (type ID +G allele) (see Fig. 1 for oligonucleotide sequences). Competition profiles without (complexes 1–3) and with (complex 4) UV crosslinking are shown. Graphs correspond to the averages from data obtained from three separate competition experiments with the C3 and B rDNA alleles of the type IB element. The 250 kDa binding activity is responsible for gel shift complex 1. ssA-TIBF is responsible for gel shift complexes 2 and 3. Relative binding is defined as the binding signal in the presence of a defined amount of cold competitor divided by the binding signal in the absence of competitor DNA.
Regulation of type I element binding activities
The experiments described above identified three distinct type I element binding activities in log phase vegetative cell extracts. To assess whether these proteins might compete with one another in vivo, the regulation of these activities was examined in extracts prepared from cells synchronized either in S phase or at the G1/S boundary. In that the synchronous release of starved (G1-arrested) cells into vegetative S phase is rather poor, S phase extracts were prepared instead from cells undergoing rDNA gene amplification. Nuclear and cytoplasmic fractions were examined to study subcellular localization (33; see Materials and Methods). Whole cell HMG lysates contained appreciable, but highly variable, amounts of the different type I element binding proteins, suggesting that the recovery of nuclear proteins was not quantitative. Extracts prepared in this way have high levels of the enzyme telomerase, which clearly resides in the nuclear compartment (35). Therefore, nuclear and cytoplasmic fractions were prepared instead, using an alternative lysis and extraction protocol (see Materials and Methods).
Equal amounts of nuclear and cytoplasmic proteins were incubated with radiolabeled DNA substrate under conditions of substrate excess (Fig. 6A). In all examined extracts, the 250 kDa activity (complex 1) and ssA-TIBF (complexes 2 and 3) were detected primarily in the cytoplasmic fraction. Since the nuclear extracts contained 2- to 3-fold less total protein on a per cell basis, we conclude that most of the ssA-TIBF and 250 kDa activities partitioned to the cytoplasmic fraction during isolation. In contrast, the 65 kDa activity (complex 4) partitioned predominantly into the nuclear fraction, indicating that it was more tightly associated with DNA or other nuclear components.
Figure 6.
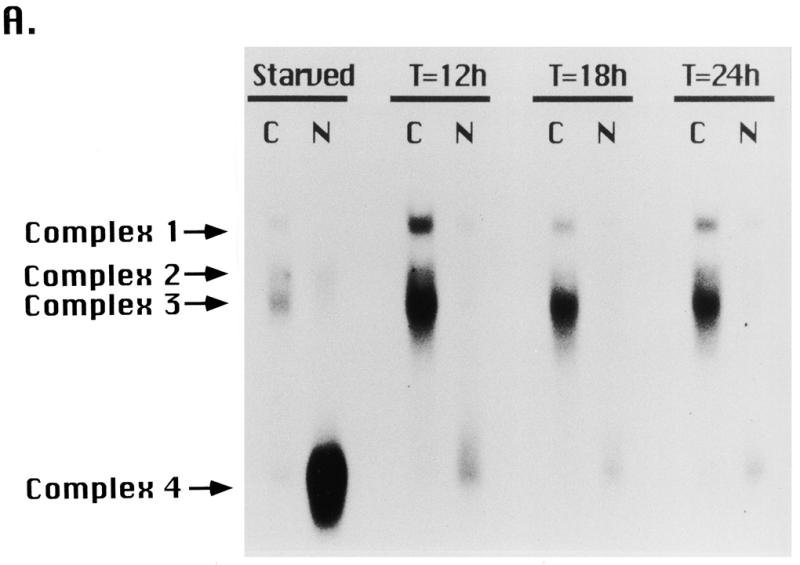
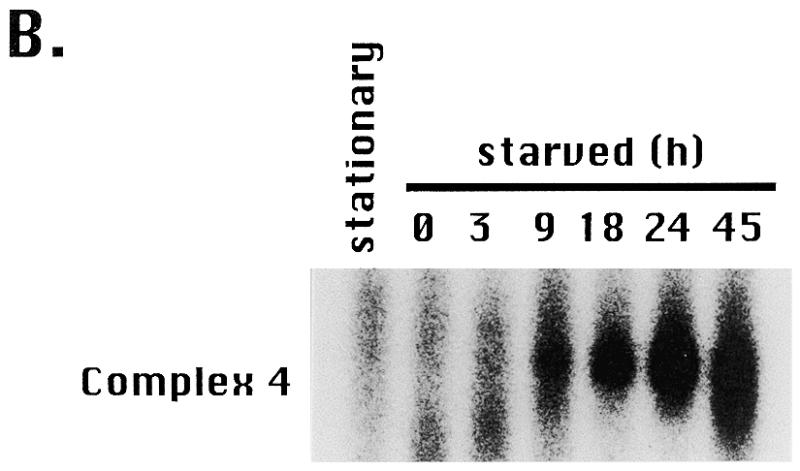
Fractionation of type I element binding activities. (A) Nuclear (N) and cytoplasmic (C) extracts were prepared from starved cultures or mated cells at various time points during new macronuclear development (T = 12, 18 or 24 h after initiating pair formation). Mated cells were starved for 16 h prior to pair formation and starved cells were harvested 40 h after initiating starvation (16 + 24 h). The rDNA copy number increases from ~50 at T = 12 h to ~10 000 at T = 24 h of macronuclear development. Protein concentrations were quantitated by the Bradford method. Aliquots of 2 µg of cytoplasmic or nuclear extracts were incubated with radiolabeled C3 type IB element and subjected to UV crosslinking prior to native gel electrophoresis. DNA–protein complexes were resolved on a TGE gel and visualized by autoradiography. (B) Profile of gel shift complex 4 (65 kDa binding activity) as a function of starvation time (h), where T = 0 designates the beginning of the starvation period.
The gel shift profiles of starved and mating cells indicated that the three type I element binding activities are differentially regulated. Complex 4 increased greatly upon starvation (Fig. 6B) as cells prepared for conjugation. The abundance of this activity declined rapidly following refeeding (data not shown) and in starved, mated cells undergoing rDNA gene amplification (Fig. 6A, T = 12, 18 and 24 h after pairing). The signal for complex 4 was elevated ~12-fold in starved cells, under conditions in which rDNA replication and transcription are repressed (4/4 extracts examined). The modest activity in mating cultures probably corresponds to proteins derived from the subpopulation of non-mating, starved cells. In comparison, the abundance of the ssA-TIBF and 250 kDa-dependent complexes (complexes 1–3) increased slightly (~3 times) in cells undergoing macronuclear development (T = 12, 18 and 24 h), when rDNA amplification and new rRNA transcription occur (250 kDa, 4/4 extracts examined; 100 kDa, 3/4 extracts examined) (36,37).
DISCUSSION
The Tetrahymena rDNA minichromosome has been a useful paradigm for studying the regulation of eukaryotic DNA replication. Molecular and genetic approaches have identified cis-acting determinants for amplification in the developing macronucleus and propagation of the rDNA in vegetatively growing cells (8,22; reviewed in 23). These studies have implicated the type I element as a critical determinant for replication initiation (5,8,24), replication fork arrest (9) and transcription of the rRNA genes (11,12; R.E.Pearlman, personal communication).
In this report we used gel mobility shift assays and protein purification to identify three distinct DNA binding activities that interact with the A-rich strand of type I elements. One of these activities corresponds to the previously characterized type I element binding factor, ssA-TIBF (27). The remaining activities contain new type I element binding proteins. As these three activities recognize the same genetic determinant in vitro, it is plausible that they may compete for DNA binding in vivo. A previously uncharacterized 32 kDa protein mediates DNA binding in the fastest migrating gel shift complex (Fig. 2A, gel shift complex 4). We purified this protein to homogeneity by conventional and affinity chromatography and showed that it binds DNA as a dimer. In that binding to type I elements is sequence-specific, the 32 kDa protein is not the functional equivalent of human RPA (replication protein A; human single-strand binding protein, HSSB) (38). In contrast to the other type I element binding activities, the 32 kDa protein partitioned primarily to the nucleus during fractionation. Whether this reflects additional interactions with components of the nuclear architecture (i.e. scaffold proteins) or DNA is not known.
The DNA–protein complexes formed between the 32 kDa protein and the 37 nt type IB element substrate are less stable than those formed with the other type I element binding activities and could only be detected by UV crosslinking. This observation suggests that the affinity of the 32 kDa protein for type I elements may be lower. However, several lines of evidence suggest that the 32 kDa protein may be physiologically important. First, this protein appears to be quite abundant, raising the possibility that it could compete in vivo for DNA binding. Roughly equivalent amounts of purified ssA-TIBF and 32 kDa protein were obtained from log phase extracts. Since the pooled column fractions from each purification step were only assayed for ssA-TIBF activity, the 32 kDa protein is likely to be more abundant. Second, steady-state levels of the 32 kDa binding protein (complex 4) increase in starved cells while the abundances of the other activities decline under these conditions. This results in an ~30-fold increase in the relative concentration of the 32 kDa protein. Since this increase occurs under conditions where rDNA replication and transcription are repressed, it is possible that the 32 kDa protein negatively regulates one or both of these processes.
The slowest migrating gel shift complex (Fig. 2A, gel shift complex 1, native mol. wt ~250 kDa) corresponds to binding of another novel type I element binding protein. Unlike ssA-TIBF and the 32 kDa protein, this binding activity was lost during purification on double-stranded DNA cellulose. Binding was retained, however, following gel filtration of crude S100 extracts. The 250 kDa activity contains a novel DNA binding protein with an estimated mass of 110 kDa. We propose that this activity corresponds to a multiprotein complex in which one component has now been identified. Other than its protein subunit composition, the 250 kDa activity was similar biochemically to ssA-TIBF. Both activities partitioned primarily into the cytoplasmic fraction and had comparable relative affinities for type I elements. They showed a modest preference for the C3 type IB element compared to the B rDNA IB allele. Furthermore, they were not induced by starvation, instead increasing slightly (3–5 times) in developing macronuclei. Whether this increase reflects true developmental regulation or cell cycle regulation (developing macronuclei are all in S phase) remains to be determined. Regardless of the mechanism, there is a positive correlation between these two type I element binding activities and periods of active DNA replication.
The remaining gel shift complexes that we detected in S100 extracts (Fig. 2A, complexes 2 and 3) were mediated by ssA-TIBF. The crude S100 activity and affinity-purified ssA-TIBF each chromatograph as a single ~100 kDa peak during gel filtration and generated both gel shift complexes. Two-dimensional gel studies and glutaraldehyde crosslinking confirmed that DNA binding is mediated by a homotetramer of ssA-TIBF in each gel shift complex (data not shown; 29). In that UV-crosslinked polypeptides in both complexes are of identical molecular weight, the faster migrating gel shift species (complex 3) is probably not due to partial proteolysis of ssA-TIBF. In addition, no evidence for differential protein phosphorylation (28) or heterogeneity in molecular weight was observed in purified protein preparations (SDS–PAGE, Fig. 3A; mass spectroscopy, L.Dangott, S.Saha and G.M.Kapler, unpublished results).
We examined the ability of type I element binding proteins to interact with wild-type C3 rDNA and mutant alleles that manifest defects in rDNA replication [type IB (B) and type ID (rmm8)] and rRNA transcription (type ID +G mutation). The B rDNA mutation produces an rDNA ‘maintenance’ phenotype, whereby B rDNA minichromosomes are lost from the macronucleus of heterozygous B/C3 cells during prolonged vegetative growth (5). This was proposed to result from a decreased ability of B rDNA to compete in vivo for trans-acting replication factors. Similar to ssA-TIBF, neither the 65 nor 250 kDa activities displayed a strong preference for the C3 type IB allele. All three binding activities showed no strong (>5-fold) preference for wild-type type I elements relative to point mutant alleles [type IB-rmm1 (28); type ID-rmm8 (this study)] that replicate comparably or worse than B rDNA (reviewed in 23).
Recent footprinting experiments with purified ssA-TIBF revealed that its modes of binding to C3 and B rDNA are dissimilar (29). This subtle form of allele specificity is not surprising, as genetic studies indicate that very small differences in replication competence can account for the observed C3 > B replication advantage (5). Modeling studies suggest that as little as a 10% decrease in replication efficiency can generate the maintenance phenotype observed in heterozygous B/C3 cells. In contrast, transcription from rDNA plasmids bearing the type ID +G mutation is undetectable and the in vivo footprint over the rRNA promoter is ablated, indicating a gross defect in rRNA transcription (12). Our in vitro studies detected only a slight decrease in the affinity of all three DNA binding activities for the type ID +G element, suggesting that they probably do not play a primary role in regulating gene expression.
As type I elements are required for rDNA replication, ssA-TIBF or other type I element binding activities are candidate replication initiator proteins. Type I elements are dispersed throughout the 5′-NTS. Two copies are proximal to the sites for replication initiation (Domains 1 and 2, respectively) (26), whereas the remaining copies map to the rRNA promoter. Mutations in promoter-proximal type I elements affect rDNA replication (7; D.L.Dobbs and E.H.Blackburn, personal communication), suggesting that initiation is controlled in part by long range DNA–protein/protein–protein interactions. Consequently, we would predict that a protein that regulates the initiation of replication would bind well to all four type I elements. The three proteins described here display this characteristic. Future studies will decipher whether these proteins cooperate through protein–protein interactions or act antagonistically towards one another.
The initiation of DNA replication involves unwinding of the DNA duplex, recruitment of replication enzymes and polymerization of nascent DNA strands. Prior to polymerization, the origin is in a single-stranded state. The best characterized eukaryotic initiator protein, SV40 large T antigen, binds to the double-stranded form of its target recognition sequence, melts the DNA duplex and remains bound to the single-strand DNA form (reviewed in 1). T antigen, then functioning as a helicase, continually unwinds duplex DNA at the elongating replication fork. In contrast to the T antigen binding site, Tetrahymena type I elements are very A+T-rich (85% A+T versus 20% A+T). Consequently, type I elements might naturally alternate between double-stranded and partially single-stranded forms. Although none of the proteins that we have identified display significant double-strand binding activity (S.Saha, M.Mohammad and G.M.Kapler, unpublished results), they may still gain access to the origin either by binding to partially unwound (melted) DNA or through recruitment by other proteins.
Binding to single-strand type I elements could also play a role in regulating replication fork movement. Pausing of replication forks occurs 50–100 bp upstream of type I elements. Genetic studies indicate that type I elements are cis-acting determinants responsible for arresting the movement of replication forks (9). During DNA replication, the DNA duplex is unwound by a replicative DNA helicase. This process generates single-stranded DNA segments, transiently creating binding sites for the in vitro binding activities described here. Whether DNA binding by one or more of these proteins is facilitated in vivo by interactions with the replication machinery awaits future studies.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Drena Dobbs for communicating unpublished results, including the use of hydroxyapatite and double-stranded DNA cellulose chromatography for the purification of ssA-TIBF. We thank Dorothy Shippen and Gary Kunkel for advice and use of equipment. We also thank Dorothy Shippen for critical reading of this manuscript and comments. This work was support by NIH grant GM56572.
REFERENCES
- 1.Fanning E. and Knippers,R. (1992) Annu. Rev. Biochem., 61, 55–85. [DOI] [PubMed] [Google Scholar]
- 2.Marahrens Y. and Stillman,B. (1992) Science, 255, 817–823. [DOI] [PubMed] [Google Scholar]
- 3.Shore D. and Nasmyth,K. (1987) Cell, 51, 721–732. [DOI] [PubMed] [Google Scholar]
- 4.Johnson A.D., Poteete,A.R., Lauer,G., Sauer,R.T., Ackers,G.K. and Ptashne,M. (1981) Nature, 294, 217–223. [DOI] [PubMed] [Google Scholar]
- 5.Larson D.D., Blackburn,E.H., Yaeger,P.C. and Orias,E. (1986) Cell, 47, 229–240. [DOI] [PubMed] [Google Scholar]
- 6.Yaeger P.C., Orias,E., Shaiu,W.-L., Larson,D.D. and Blackburn,E.H. (1989) Mol. Cell. Biol., 9, 452–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gallagher R.C. and Blackburn,E.H. (1998) Mol. Cell. Biol., 18, 3021–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reischmann K.P., Zhang,Z. and Kapler,G.M. (1999) Nucleic Acids Res., 27, 3079–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacAlpine D.M., Zhang,Z. and Kapler,G.M. (1997) Mol. Cell. Biol., 17, 4517–4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yue M., Reischmann,K.P. and Kapler,G.M. (1998) Nucleic Acids Res., 26, 4635–4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyahara K., Hashimoto,T., Higashinakagawa,T. and Pearlman,R.E. (1993) Gene, 127, 209–213. [DOI] [PubMed] [Google Scholar]
- 12.Pan W.-J., Gallagher,R.C. and Blackburn,E.H. (1995) Mol. Cell. Biol., 15, 3372–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bielinsky A.-K. and Gerbi,S. (1998) Science, 279, 95–98. [DOI] [PubMed] [Google Scholar]
- 14.Newlon C.S. (1996) In DePamphilis,M.L. (ed.), DNA Replication in Eukaryotic Cells. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 873–913.
- 15.Bell S.P. and Stillman,B. (1992) Nature, 357, 128–134. [DOI] [PubMed] [Google Scholar]
- 16.Delidakis C. and Kafatos,F.C. (1989) EMBO J., 8, 891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heck M.M.S. and Spradling,A.C. (1990) J. Cell Biol., 110, 903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitsberg D., Selig,S. and Cedar,H. (1993) Nature, 366, 588–590. [DOI] [PubMed] [Google Scholar]
- 19.Caddle M.S. and Calos,M.P. (1994) Mol. Cell. Biol., 14, 1796–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aladjem M.I., Groudine,M., Brody,L.L., Dieken,E.S., Fournier,R.E., Wahl,G.M. and Epner,E.M. (1995) Science, 270, 815–819. [DOI] [PubMed] [Google Scholar]
- 21.Austin R.J., Orr-Weaver,T.L. and Bell,S.P. (1999) Genes Dev., 13, 2639–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blomberg P., Randolph,C., Yao,C.-H. and Yao,M.-C. (1997) Mol. Cell. Biol., 17, 7237–7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kapler G.M., Dobbs,D.L. and Blackburn,E.H. (1996) In DePamphilis,M.L. (ed.), DNA Replication in Eukaryotic Cells. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 915–932.
- 24.Orias E. and Bradshaw,A.D. (1992) Dev. Genet., 13, 87–93. [DOI] [PubMed] [Google Scholar]
- 25.Palen T.E. and Cech,T.R. (1984) Cell, 36, 933–942. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Z., MacAlpine,D.M. and Kapler,G.M. (1997) Mol. Cell. Biol., 17, 6147–6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Umthun A.R., Hou,Z., Sibenaller,Z.A., Shiau,W.-L. and Dobbs,D.L. (1994) Nucleic Acids Res., 22, 4432–4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou Z., Umthum,A.R. and Dobbs,D.L. (1995) Biochemistry, 34, 4583–4592. [DOI] [PubMed] [Google Scholar]
- 29.Saha S. and Kapler,G.M. (1999) J. Mol. Biol., in press. [Google Scholar]
- 30.Orias E. and Hamilton,E.P. (1979) Genetics, 91, 657–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaertig J., Gu,L., Hai,B. and Gorovsky,M.A. (1994) Nucleic Acids Res., 3, 5391–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kapler G.M. and Blackburn,E.H. (1994) Genes Dev., 8, 84–95. [DOI] [PubMed] [Google Scholar]
- 33.Abmayr S.M. and Workman,J.L. (1987) In Ausubel,F.M. et al. (ed.), Current Protocols in Molecular Biology. John Wiley & Son, New York, NY, p. 12.1.1–12.1.8.
- 34.Bradford M.M. (1976) Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 35.Greider C.W. and Blackburn,E.H. (1985) Cell, 43, 405–413. [DOI] [PubMed] [Google Scholar]
- 36.Allis C.D., Colavito-Shepanski,M. and Gorovsky,M.A. (1987) Dev. Biol., 124, 469–480. [DOI] [PubMed] [Google Scholar]
- 37.Weiske-Benner A. and Eckert,W.A. (1985) Differentiation, 28, 225–236. [Google Scholar]
- 38.Wobbe C.R., Weissbach,J.A., Boroviec,J.A., Dean,F.B., Murkami,Y., Bullock,P. and Hurwitz,J. (1987) Proc. Natl Acad. Sci. USA, 84, 1834–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Challoner P.B., Amin,A.A., Pearlman,R.E. and Blackburn,E.H. (1985) Nucleic Acids Res., 13, 2661–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]