


Abstract
Mos1 and other mariner/Tc1 transposons move horizontally during evolution, and when transplanted into heterologous species can transpose in organisms ranging from prokaryotes to protozoans and vertebrates. To further develop the Drosophila Mos1 mariner system as a genetic tool and to probe mechanisms affecting the regulation of transposition activity, we developed an in vitro system for Mos1 transposition using purified transposase and selectable Mos1 derivatives. Transposition frequencies of nearly 10–3/target DNA molecule were obtained, and insertions occurred at TA dinucleotides with little other sequence specificity. Mos1 elements containing only the 28 bp terminal inverted repeats were inactive in vitro, while elements containing a few additional internal bases were fully active, establishing the minimal cis-acting requirements for transposition. With increasing transposase the transposition frequency increased to a plateau value, in contrast to the predictions of the protein overexpression inhibition model and to that found recently with a reconstructed Himar1 transposase. This difference between the ‘natural’ Mos1 and ‘reconstructed’ Himar1 transposases suggests an evolutionary path for down-regulation of mariner transposition following its introduction into a naïve population. The establishment of the cis and trans requirements for optimal mariner transposition in vitro provides key data for the creation of vectors for in vitro mutagenesis, and will facilitate the development of in vivo systems for mariner transposition.
INTRODUCTION
The Drosophila mauritiana transposable element mariner (Mos1 or Dmmar1) is one of the defining members of the mariner/Tc1 family of transposons (1,2). These transposons in turn belong to the larger D,D(35)E transposon superfamily, which occur sporadically in all kingdoms of living organisms (3,4). Mariner-like elements (MLEs) typically contain short terminal inverted repeats (28 bp for Mos1) flanking a single gene encoding the ~40 kDa transposase polypeptide (4–6). Transposition occurs by a cut and paste mechanism, mediated by recognition of the terminal inverted repeats by transposase (1).
Phylogenetic studies of MLE families frequently show patterns typical of horizontal gene transfer, suggesting that these elements function autonomously with minimal requirements for host cell functions (2,4,6,7). This has now been proven in vitro and in vivo. First, the purified Tc1 and Himar1 transposases can mediate transposition in vitro (8,9). Second, Drosophila Mos1 has been shown to transpose when introduced into other insects and vertebrates, and even in protozoans and bacteria, when appropriate steps to ensure transposase expression were taken (10–14; R.Groger, K.Fan, E.Brown, S.Goyard, L.R.O.Tosi and S.M.Beverley, unpublished data). Similar results have been reported with the related horn fly mariner element Himar1, the nematode Tc1 and Tc3 elements and the reconstructed salmonid element Sleeping Beauty (15–20). In several of these studies insertional inactivation mutants were obtained, and/or active gene fusions using modified elements were generated. These data thus establish the potential of this class of genetic elements as a tool for genetic manipulation.
Given its broad host range in vivo, the mariner Mos1 element is especially well suited for use as a tool for genomic research. Further development of this system would benefit greatly from the availability of an in vitro system for the study of Mos1 transposition. Here we describe the purification of Mos1 transposase, establish its ability to mediate transposition efficiently in vitro and characterize cis- and trans-acting requirements for mariner transposition in vitro. These data have also provided key parameters for the design of useful and efficient modified mariner elements for application in the emerging field of functional genomics. Additionally, comparisons of the properties of the ‘natural’ Mos1 transposase with those of a ‘reconstructed’ Himar1 transposase provide a new perspective on pathways leading to MLE inactivation during evolution.
MATERIALS AND METHODS
Plasmid constructs
The Mos1 transposase was obtained by a PCR protocol with Stratagene Pfu DNA polymerase (10 cycles of 30 s at 94° C, 1 min at 60° C and 2 min at 72° C), using the primers SMB620 (5′-gcgcccgggatccatATGTCGAGTTTCGTGCCGAATAAAGAGC) and SMB621 (5′-cgcggatccTTATTCAAAGTATTTGCCGTCGCTAGC; lower case letters represent bases not present in Mos1) and 100 ng of linearized denatured pBluescribe M13+/Mos1 (21; Beverley laboratory strain B3077). The 1 kb product was digested with NdeI + BamHI and inserted into the Escherichia coli expression vector pET3a (22) cut with the same enzymes, yielding pET3a-TPase (strain B3297), which was transformed into E.coli BL21(DE3)/pLysS (yielding strain B3315).
To make the transposon donor plasmid pMD13-mosK (strain B3351), the 1.3 kb BsaAI–BsaBI fragment of pBluescribe M13+/Mos1 (strain B3077; 21) was ligated to the 3.7 kb SacI–Tth111I fragment (after end repair with T4 DNA polymerase) from the plasmid pMD13 (strain B2058), yielding pMD13-Mos1 (strain B3350). pMD13 is a modification of pJM703.1 (23) bearing a tetracycline resistance marker and R6K origin of replication. The Tn903 kanamycin resistance (Kanr) gene was obtained from pEV2 (strain B1932; 24) by digestion with DraIII and NlaIV, and the 1.1 kb fragment was ligated into the SacI site of pMD13-Mos1 (after end repair) to yield pMD13-mosK. The deletions pMD13-mosKΔCD (strain B3571), pMD13-mosKΔCS (strain B3572) and pMD13-mosKΔDS (strain B3573) contained the Kanr cassette ligated into the Mos1-internal ClaI/DraI, ClaI/SacI or DraI/SacI sites of pMD13-Mos1, respectively. A deletion containing only the terminal TA dinucleotides and 28 bp inverted repeats was derived from plasmid pLew100hyg1 (S. Leal, Rockefeller University). In this plasmid, the inverted repeats were separated by a 6 bp PvuII site, into which the Kanr gene was inserted (strain B3613). An SpeI–BglII fragment containing the transposon segment was ligated to the 3.7 kb SacI–Tth111I fragment described above, yielding pMD13-mmosK (strain B3614).
Expression and purification of Mos1 transposase from E.coli
Escherichia coli strain B3315 was grown at 37°C to an OD600 of 0.6, and Mos1 transposase expression was induced by addition of isopropyl β-d-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM. After 1 h, cells were harvested and resuspended in 1/100 of the original volume of 20 mM Tris–HCl pH 7.6, 2 mM MgCl2, 25% sucrose, 0.6 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM benzamidine (BZA) and 1 mM dithiothreitol (DTT), and then 50 µl aliquots were quick frozen in liquid nitrogen. Frozen cells were thawed at room temperature and then incubated for 5 min with 0.25 mg of lysozyme, then for 15 min after the addition of 1 ml of lysis buffer [20 mM Tris–HCl pH 7.6, 4 mM EDTA, 200 mM NaCl, 1% deoxycholate, 1% nonylphenoxy polyethoxy ethanol (NP-40), 0.6 mM PMSF, 1 mM BZA, 1 mM DTT], and then for 20 min after the addition of 60 µg DNase I and MgCl2 to 10 mM. All subsequent steps were carried out at 4°C. Inclusion bodies were pelleted at 14 000 g in a microcentrifuge and washed three times with 1 ml of 100 mM Tris–HCl pH 7.6 containing 4 M deionized urea. They were resuspended in 0.5 ml of column buffer (20 mM Tris–HCl pH 7.6, 4 M guanidine–HCl 50 mM NaCl, 1 mM PMSF, 1 mM BZA, 5 mM DTT) and applied to a 10 ml DEAE–Sephadex column equilibrated with column buffer. Fractions of 0.5 ml were eluted with column buffer and checked by SDS–PAGE for the Mos1 transposase. Four fractions containing transposase were pooled, diluted to 12 ml with column buffer and dialyzed against 10% glycerol, 25 mM Tris–HCl pH 7.6, 50 mM NaCl, 5 mM MgCl2 and 2 mM DTT for 6 h. The buffer was replaced with buffer containing 0.5 mM DTT for 15 h. After dialysis, the sample was spun at 10 000 g to remove precipitated material and 100 µl aliquots were stored at –80°C. Protein concentration was determined using a micro-BCA method (Pierce) or by UV absorbance (280 nm) using a calculated extinction coefficient of 76 989 M–1 cm–1. The UV-determined protein concentration was nearly three times higher than that obtained with the BCA method.
Transposition assay
Transposition reactions were carried out for 1 h at 25°C, in a 20 µl reaction containing 10% glycerol, 25 mM HEPES pH 7.9, 250 µg acetylated BSA, 2 mM DTT and 5 mM MgCl2. Standard reactions contained ~10 fmol target DNA, 32 fmol transposon donor DNA (150 ng for pMD13-mosK) and 100 ng of purified Mos1 transposase (2.5 pmol). Reactions were stopped by incubation with 80 µl of 50 mM Tris–HCl pH 7.6, 0.5 mg/ml proteinase K, 10 mM EDTA and 250 mg/ml yeast tRNA for 30 min at 30°C. Transposition products were purified by phenol extraction and ethanol precipitation and resuspended in 10 µl of 10 mM Tris–HCl pH 7.4, 1 mM EDTA. Aliquots of 2 µl were transformed into electrocompetent E.coli DH10B, and aliquots plated on medium containing antibiotics appropriate for the target marker (ampicillin or hygromycin) to determine transformation efficiency, or the same drug plus kanamycin for identification of transpositions.
DNA preparation and sequencing
DNA sequences were obtained with an ABI PRISM Model 2.1.1 automated sequencer, using primers SMB686 (5′-GGTTGACACTTCACAAGGTC) on the left side of the transposon (as defined by the transposase ORF within Mos1) and SMB687 (5′-CCGAGAGATGGGAAAAATG) on the right side of the element. Sequencing templates were prepared using a QIAprep spin miniprep kit (Qiagen Inc.).
RESULTS
Purification and assay of Mos1 transposase
We expressed the Mos1 transposase under the control of an inducible T7 promoter in E.coli. Like other mariner/Tc1 transposases, the overexpressed protein formed inclusion bodies (8,9). These were recovered, washed in 4 M urea, solubilized in 4 M guanidine–HCl and subjected to batch ion exchange chromatography on DEAE–Sephadex. The denatured transposase was refolded by dialysis, and SDS–PAGE gel electrophoresis of these preparations revealed substantial purification of the 40 kDa transposase polypeptide, with minor protein contaminants (Fig. 1). Transposase activity was stable when the enzyme was stored at –80°C in the presence of 10% glycerol. Preliminary experiments employing gel filtration chromatography or membrane filtration suggested that the native transposase has a molecular weight in excess of 100 kDa (data not shown).
Figure 1.
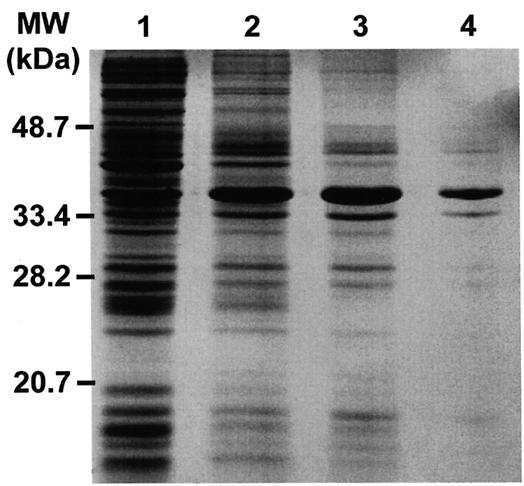
Purification of the Mos1 transposase from E.coli. A Coomassie blue stained 12% SDS–PAGE gel containing samples from the various steps involved in Mos1 transposase purification is shown. Lane 1, bacterial lysate before IPTG induction; lane 2, bacterial lysate after induction; lane 3, washed inclusion bodies; lane 4, purified Mos1 transposase after chromatography and refolding steps.
To assay transposase activity, we used a modified mariner element (mosK) containing a Tn903 Kanr marker inserted in the central Mos1 SacI site (Fig. 2A), carried on a ‘suicide’ donor plasmid bearing an R6K replication origin, which requires the pir gene product (25). In a typical in vitro reaction, donor pMD13-mosK was incubated for 1 h in the presence of transposase and a target plasmid bearing a drug resistance marker and a ColE1 replication origin. The products were transformed into a pir– E.coli, allowing selection against the transposon donor and detection of transpositions by the occurrence of doubly resistant colonies (Kanr + Ampr or Kanr + Hygr; Fig. 2B). Restriction analysis of recovered plasmids confirmed the presence of transpositions in all cases. The optimum amount of pMD13-mosK donor plasmid in a standard transposition reaction was 150 ng (1.6 nM; Fig. 3B). Analysis of a wide variety of targets revealed a transformation efficiency of from 10–4 to 10–3 (transposition insertions/target DNA molecule; Table 1).
Figure 2.

In vitro mariner transposition assay. (A) Organization of the modified Mos1 element, mosK. The Tn903 kanamycin resistance marker (Kanr) was inserted into the unique SacI (S) site. The gray arrowheads and the black arrow represent the inverted terminal repeats and the transposase ORF, respectively; ClaI (C) and DraI (D) sites are indicated. (B) The donor plasmid, pMD13-mosK, contains a tetracycline resistance marker (Tcr), an R6K replication origin (oriR6K) and mosK. In this example, the target DNA is carried in the vector pEL-HYG (24), which contains a hygromycin resistance marker (Hygr) and a minimal colE1 replication origin (oriColE1).
Figure 3.

Effect of different conditions on Mos1 transposase activity. In vitro transposition was performed as described in Materials and Methods, using the donor pMD13-mosK and the target pELHYG-H2. (A) Effect of enzyme concentration and (B) amount of donor plasmid on transposition efficiency. Transposition efficiency values were calculated from the ratio of Kanr + Hygr to Hygr colonies. In (A) the efficiency for the Himar1 transposase is shown as dotted lines; the peak height has been normalized to the maximal peak height seen for Mos1 and corresponds to an efficiency of 6.3 × 10–3 (26). Mos1 transposase protein concentration was determined by the BCA method; if the UV absorbance method was used, the X-axis for Mos1 should be multiplied by a factor of 3 (Materials and Methods).
Table 1. In vitro transposition using mosK.
| Target DNAa | Size (kb) | Transposition efficiencyb (×10–4) |
|---|---|---|
| pELHYG-H2c | 14 | 7.2 |
| pELHYG-H2 (Mn2+) | 14 | 0.5 |
| pUC-H3d | 7 | 3.2 |
| pUC-H4be | 7 | 5.7 |
| pUC-H4cf | 7 | 4.1 |
| pSNAR-c76g | 17 | 7.0 |
| cLHYG-BT1h | 22 | 4.0 |
| cLHYG-AX21i | 22 | 2.5 |
| cLHYG-B1j | 50 | 4.5 |
aAll targets were Leishmania spp. genomic DNA inserted in the vector indicated.
bEfficiency is given by the ratio of KanR to total transformants.
cA 12 kb EcoRI fragment of the H region inserted into the BglII site of pELHYG.
dA 4.8 kb HindIII fragment of the H region inserted into the HindIII site of pUCπ.
eA 5.2 kb SalI fragment of the H region inserted into the SalI site of pUCπ.
fA 4.8 kb SalI fragment of the H region inserted into the SalI site of pUCπ.
gA 11 kb EcoRI fragment inserted into EcoRI site of pSNAR.
hA 12 kb DNA fragment cloned into cLHYG.
iA 12 kb DNA fragment cloned into cLHYG.
jA 40 kb genomic DNA fragment cloned into cLHYG.
During refolding of the enzyme, ~50% of the protein remained soluble, yielding a preparation with a concentration of 50 µg/ml and a yield of 6 mg/l induced culture. We tested other methods for refolding the enzyme, including rapid dilution of the protein solubilized at low pH or dialysis of protein solubilized by detergent treatment. None yielded active enzyme. We tested the effects of removing the batch ion exchange step, as this seemingly yielded no purification (Fig. 1, lanes 3 and 4). These preparations were 80-fold less active, suggesting that this step removed some inhibitory factor. Lastly, omission of the dilution step prior to dialysis greatly diminished the yield, with only 1% of the protein being soluble.
Effect of transposase concentration
Transposition efficiency increased linearly with increasing quantities of Mos1 transposase, plateauing around 100 nM enzyme, and remaining elevated at concentrations up to 600 nM (Fig. 3A). In this respect Mos1 transposase differs greatly from the reconstructed Himar1 transposase, which shows a peak of activity at 10 nM and then declines dramatically at higher enzyme concentrations (26). Since both transposases were obtained following refolding of denatured E.coli-expressed protein, it was possible that renaturation-associated differences conferred artificially divergent properties. To test this, we examined all of the active preparations described above, arising from different purification and/or renaturation protocols. Without exception, all yielded a profile similar to that shown in Figure 3A, with peak activity occurring around 100 nM and remaining high thereafter (data not shown). This suggested that the quantitative difference between Mos1 and Himar1 transposases is intrinsic to the enzymes.
Randomness of Mos1 insertion in vitro
Restriction mapping and DNA sequencing of 132 insertion sites was used to characterize the target site specificity of mosK transposition (Fig. 4 and data not shown). Restriction mapping revealed little regional specificity for insertion (an example for 22 insertions in a 14 kb target is shown in Fig. 4A; P > 0.05, χ2 test). Transposition was reduced ~15-fold when Mn2+ was substituted for Mg2+ (Table 1). Transposition in the presence of Mn2+ yielded a different spectrum than observed with Mg2+, with a somewhat more pronounced regional specificity (an example of 21 insertions in a 14 kb target is shown in Fig. 4B; P < 0.05, χ2 test). Similar results were obtained with a number of other targets (data not shown).
Figure 4.

Randomness of mosK insertion and target specificity in the presence of Mg2+ or Mn2+. (A and B) Insertion of mosK into the 12 kb insert of pELHYG-H2 in reactions containing Mg2+ or Mn2+, respectively. The vertical arrows represent individual insertions; those above or below the map represent insertions with the Kanr marker in the forward or reverse orientation, respectively. (C and D) Target site analysis of 111 and 21 mosK insertions, obtained in the presence of Mg2+ and Mn2+, respectively. The consensus was determined and displayed using the Sequence Logo algorithm (49). The height of each base corresponds to its prevalence at that position.
The target sites of 111 insertions obtained in the presence of Mg2+ showed the expected requirement for insertion into a TA dinucleotide (Fig. 4C). Consensus analysis of the flanking sequences showed only minor deviations from random in the 20 bp surrounding the TA insertion site, with a weak preference for purines at position –8 (Fig. 4C). Sequence analysis of 21 insertions obtained in the presence of Mn2+ showed that 58% did not insert into a TA dinucleotide (Fig. 4D), as seen with the Himar1 transposase (8). Only minor sequence preferences for the target site were observed, and these differed from those found in the presence of Mg2+ (Fig. 4C and D).
Transposition into different targets
Many target DNAs contained genomic DNA from Leishmania, which typically has a GC content of ~62% (27). Tests of eight plasmid targets ranging in size from 7 to 50 kb showed transposition efficiencies with the pMD13-mosK donor ranging from 10–4 to 10–3; these values were not strongly dependent upon target size (Table 1). Tests with plasmid pELHYG-H2 showed that relaxation of supercoiled plasmid with DNA topoisomerase I reduced the transposition frequency by 16-fold (data not shown).
We compared the transposition efficiency with GC-rich and AT-rich targets. pUC-LPG1 contained a 4.5 kb fragment of Leishmania major DNA that is 63% GC, inserted into the cloning vector pUC19, while pBS-P62 contained a 3.5 kb Plasmodium falciparum PP2C DNA fragment that is 22.7% GC, inserted into the pBS cloning vector (28). The transposition efficiency with the pMD13-mosK donor into pBS-P62 was 1.4 × 10–3, nearly 3-fold higher than that seen with pUC-LPG1, 4.4 × 10–4. Thus, the effect of target base composition was relatively modest.
cis requirements for transposition
We examined several deletions of the mosK element to evaluate the cis-acting sequence requirements for transposition (Fig. 5). These lacked most of the left (725 bp, mosKΔCS), right (462 bp, mosKΔDS) or both halves (mosKΔCD) of the Mos1 element. This latter ‘symmetrical’ deletion retained 38 and 5 bp internal to the left and right inverted repeats, respectively. In these experiments, the transposition efficiency of mosK was 7.2 × 10–4, and a slightly higher efficiency was obtained with the symmetrical deletion mosKΔCD (8 × 10–4; Fig. 5). Surprisingly, the asymmetrical deletions showed less efficient transposition, with the left and right deletions showing activity of only 7 and 40% of that of mosK (Fig. 5). Similar results were obtained in multiple experiments, and reversing the orientation of the Kanr marker had no effect (data not shown). The decreased efficiency of the asymmetrical deletions cannot be explained by the size of the element, as mosK is 2.3 kb, versus 1.2 kb for the symmetrical and 1.7–1.9 kb for the asymmetrical deletions (Fig. 5).
Figure 5.

cis requirements for transposition. Deleted versions of pMD13-mosK were used as transposon donors in a standard in vitro transposition reaction, using the target plasmid pELHYG-H2. The terminal inverted repeats are indicated as arrows; restriction sites are: C, ClaI; S, SacI; D, DraI. The transposon sizes are: mosK, 2.4 kb; mosKΔCD, 1.2 kb; mosKΔCS, 1.7 kb; mosKΔDS, 1.9 kb; mmosK, 1.1 kb.
The mmosK deletion contained the flanking TA dinucleotides and 28 bp inverted repeats, but no other internal Mos1 sequences. This element was completely inactive in our assay (transposition efficiency <10–7). This argues that the few bases remaining internal to the inverted repeats in the mosKΔCD transposon are essential for activity.
DISCUSSION
We have purified the transposase encoded by the autonomous Mos1 mariner element, and shown its ability to carry out transposition in a defined in vitro system. These studies provide information relevant to the design and application of new transposable elements for use in functional genetic analysis. Additionally, our studies provide some information relevant to the study of the evolution of this large and widespread family of transposable elements.
Factors affecting mariner transposition
The presence of the terminal 28 bp inverted repeats alone was insufficient for transposition, while a construct retaining 38 and five internal base pairs, respectively, was fully active (Fig. 5). In contrast, only 26 bp of the outer portion of the Tc1 inverted repeats is sufficient for transposition (9). The results with Mos1 are in good agreement with those obtained with the related Himar1 transposase, as DNA footprinting studies showed that Himar1 transposase additionally protects several base pairs internal to the inverted repeats (8). Interestingly, a class of MLEs containing the inverted repeats plus a small number (<20 bp) of internal nucleotides is prevalent in the human genome (29,30). The resemblance of these elements to the active deletion mosKΔCD (Fig. 5) suggests that these deleted MLEs are competent for trans-mobilization by active transposase.
Remarkably, removal of either the ‘left’ or ‘right’ halves of the Mos1 element led to reductions in transposition efficiency (7 and 40% for mosKΔCS and mosKΔDS, respectively), but when combined, these deletions yielded a slightly more active transposon (110%, mosKΔCD; Fig. 5). Since the two asymmetrical deletions are similar in size, this implies that there are interactions involving internal sequences that can affect the rate of transposition. These interactions could involve specific interactions between the left and right internal portions of Mos1, or of these regions with the left and/or right inverted repeats. This behavior may have implications for the spread of defective transposons during evolution, and the design of modified transposons.
Transposition in vitro increased with increasing Mos1 transposase, plateauing at concentrations >100 nM (Fig. 3A). In this regard, Mos1 differs from the Himar1 transposase, which shows peak activity at 10 nM in vitro and then declines severely at higher concentrations (26). This phenomenon, termed ‘overproduction inhibition’, may be mediated by transposase subunit interactions occurring at high concentrations (31). Our preliminary data suggest that the Mos1 transposase occurs as a multimeric complex, and other MLE transposases exist as multimers which would be conducive to subunit interactions and/or regulation (32,33).
Why do Mos1 and Himar1 transposases differ? This could reflect intrinsic properties of the transposase or arise from differences in transposase preparation. Both transposases were obtained by renaturation of insoluble proteins expressed in E.coli (8,26), which potentially could yield a misfolded or improperly modified (albeit active) transposase. We tested several methods for renaturing and/or purifying the Mos1 transposase and, although the yields and/or specific activities differed, all preparations showed a profile similar to that shown in Figure 3A. Another possibility is that Himar1 preparations contain an inhibitor, possibly an inhibitory truncated form of the transposase, as seen in other transposition systems (34,35). At present we favor the view that the differences observed arise from the transposases themselves, but additional studies will be required to confirm this.
Implications for evolutionary spread and loss of mariner elements
While Mos1 transposase was taken from an element active in Drosophila (21), Himar1 transposase was ‘resurrected’ from defective elements in the horn fly genome by an insightful assembly of a consensus open reading frame (8). Potentially, Himar1 and other reconstructed transposases may represent artificial proteins whose properties do not mirror those of their evolutionary ancestors. Alternatively, the reconstructed Himar1 may be a faithful picture of the ancestral element, taken at a particularly interesting time in the evolutionary history of this transposon family.
Assuming that the differences between Mos1 and Himar1 transposases reflect intrinsic properties, these two proteins may represent different stages of evolutionary processes affecting the spread and loss of MLEs. It is generally accepted that following introduction into a naïve lineage, an active autonomous MLE can undergo a period of relatively unrestrained spread through transposition and sexual exchange, until regulatory and/or mutational inactivation results in dampening of transposition activity and its associated deleterious effects (31,36). Overproduction inhibition is one example of a regulatory mechanism postulated for the down-modulation of MLE transposition (2,31). Curiously, the autonomous Mos1 transposase element shows little evidence of overproduction inhibition, while the reconstructed Himar1 transposase exhibits this behavior strongly (Fig. 3A; 26). Although the data are limited, it is tempting to speculate that the Mos1 element is an example of an evolutionary ‘starting’ point for a freely transposing autonomous MLE, while the Himar1-type transposase may represent a ‘down-regulated’ intermediate in the evolutionary path leading to decreased transposition. Through protein–protein interactions, expression of a Himar1-type transposase mutant could serve to dampen the activity of dispersed Mos1-type transposases. An analogous ‘dominant negative’ interaction has been described previously in studies of defective Mos1 transposases interacting with the wild-type enzyme (37,38).
Since Himar1 and Mos1 arise from distantly related MLE subfamilies (4), it is not presently possible to test this hypothesis by direct tests of these two transposases. Future studies of the interactions between other natural and reconstructed transposases, including ones from the same subfamily, will be necessary in order to evaluate the merits of this proposal.
Application of the mariner system to genetic analysis in vivo and in vitro
Due to their ability to function in foreign milieus, mariner/Tc1 transposons show great potential for use in functional genomic studies in vivo. Indeed, several modified MLEs have been adapted for use as insertional mutagens and for the recovery of gene fusions (10,17–19). In vivo, mariner has a particularly broad host range, ranging from prokaryotes to protozoans to vertebrates, making it a particularly attractive element for such studies (10,11,14,16,17; R.Groger, K.Fan, E.Brown, S.Goyard, L.R.O.Tosi and S.M.Beverley, unpublished data). Thus, studies of mariner transposition in vitro have great potential to provide information essential for the optimization of transposition recoveries in vivo. High levels of Mos1 transposase yielded increased transposition (Fig. 3A), suggesting that the optimal strategy for efficient transposition of this element in vivo is to maximize transposase expression. Elements lacking internal sequences were inactive, but retention of 43 bp of internal mariner sequence yielded a fully active transposon (mosKΔCD; Fig. 5). Notably, the sequences of these flanking sequences contain several open reading frames and thus are compatible with the design of gene fusion vectors. With this information, we are now developing transposons suitable for the generation of transcriptional and translational gene fusions in vivo and in vitro (S.Goyard, L.R.O.Tosi and S.M.Beverley, unpublished data). Importantly, the in vitro system allows one to test the activity of new transposon constructs rapidly, rather than through more laborious tests available in the heterologous eukaryotic systems.
The in vitro mariner system also has utility as a tool for genetic analysis in its own right. The insertion site specificity for the Mos1 transposase appears to be effectively random and lacking in significant ‘hot spots’, both in vivo (10) and in vitro (Fig. 4), even with GC-rich templates. In contrast, the Tc1 transposase shows significant site preferences in vivo and in vitro (9,39,40). The randomness of mariner insertion in the presence of Mg2+ has enabled us to perform rapid primer-island sequencing of several DNA templates (data not shown). This was facilitated by the use of vectors such as pEL-HYG, a small 2.1 kb plasmid which preferentially yields insertions into the target DNA (24). In a shuttle mutagenesis approach, we have mapped genes implicated in Leishmania virulence borne on a plasmid shown to affect the biosynthesis of the major cell surface glycoconjugate, lipophosphoglycan (D.Dobson, K.Valdez, A.Hubel, B.Mengeling, S.J.Turco and S.M.Beverley, in preparation). Undoubtedly, many more applications will be forthcoming, including the generation and characterization of gene fusions (41–44) via in vitro transposition-based shuttle mutagenesis.
Efficient in vitro transposition systems have been developed for other transposons including Ty1, Tn7 and Tn5, which lack the TA target site preference of mariner and other MLEs (45–47). However, the ability of mariner to function broadly in vivo provides the opportunity for complementary and/or combined applications with the elements of the in vitro system described here. For example, we have recently found that co-injection of the Mos1 mariner transposase with appropriately constructed mariner elements into embryos of the mosquito Aedes aegypti can increase the recovery of transformed progeny nearly 10-fold (14,48).
The major task for genome scientists in the future will be to understand genes and their function, using the array of information generated by genome mapping and sequencing projects. Transposable elements offer a powerful tool for attacking these questions, and the tractable mariner system in vivo and in vitro promises to become a useful part of our molecular tool kit for functional genomics.
Acknowledgments
ACKNOWLEDGEMENTS
We thank M. Cunningham for help in estimating the size of the Mos1 transposase, S. Leal and G. Cross for providing pLew100hyg1, C.B. Mamoun for pBS-P62, G. Späth for pUC-LPG1, E. Brown, D. Dobson, K. Fan, S. Goyard, R. Groger, A. Hubel and K. Valdez for permission to mention unpublished results, S. Goyard, F. Gueiros-Filho, D. Lampe and H. Robertson for discussions, and M. Cunningham, D. Dobson, R. Groger and S. Goyard for comments on this manuscript. This work was supported by NIH grant AI29646 (S.M.B.) and a Pew Foundation Award (L.R.O.T.).
REFERENCES
- 1.Plasterk R.H., Izsvak,Z. and Ivics,Z. (1999) Trends Genet., 15, 326–332. [DOI] [PubMed] [Google Scholar]
- 2.Hartl D.L., Lohe,A.R. and Lozovskaya,E.R. (1997) Annu. Rev. Genet., 31, 337–358. [DOI] [PubMed] [Google Scholar]
- 3.Doak T.G., Doerder,F.P., Jahn,C.L. and Herrick,G. (1994) Proc. Natl Acad. Sci. USA, 91, 942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robertson H.M. (1995) J. Insect Physiol., 41, 99–105. [Google Scholar]
- 5.Jacobson J.W., Medhora,M.M. and Hartl,D.L. (1986) Proc. Natl Acad. Sci. USA, 83, 8684–8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plasterk R.H. (1996) Curr. Topics Microbiol. Immunol., 204, 125–143. [DOI] [PubMed] [Google Scholar]
- 7.Capy P., Langin,T., Bigot,Y., Brunet,F., Daboussi,M.J., Periquet,G., David,J.R. and Hartl,D.L. (1994) Genetica, 93, 161–170. [DOI] [PubMed] [Google Scholar]
- 8.Lampe D.J., Churchill,M.E. and Robertson,H.M. (1996) EMBO J., 15, 5470–5479. [PMC free article] [PubMed] [Google Scholar]
- 9.Vos J.C., De Baere,I. and Plasterk,R.H. (1996) Genes Dev., 10, 755–761. [DOI] [PubMed] [Google Scholar]
- 10.Gueiros-Filho F.J. and Beverley,S.M. (1997) Science, 276, 1716–1719. [DOI] [PubMed] [Google Scholar]
- 11.Fadool J.M., Hartl,D.L. and Dowling,J.E. (1998) Proc. Natl Acad. Sci. USA, 95, 5182–5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garza D., Medhora,M., Koga,A. and Hartl,D.L. (1991) Genetics, 128, 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lohe A.R. and Hartl,D.L. (1996) Genetics, 143, 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coates C.J., Jasinskiene,N., Miyashiro,L. and James,A.A. (1998) Proc. Natl Acad. Sci. USA, 95, 3748–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L., Sankar,U., Lampe,D.J., Robertson,H.M. and Graham,F.L. (1998) Nucleic Acids Res., 26, 3687–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akerley B.J., Rubin,E.J., Camilli,A., Lampe,D.J., Robertson,H.M. and Mekalanos,J.J. (1998) Proc. Natl Acad. Sci. USA, 95, 8927–8932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rubin E.J., Akerley,B.J., Novik,V.N., Lampe,D.J., Husson,R.N. and Mekalanos,J.J. (1999) Proc. Natl Acad. Sci. USA, 96, 1645–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schouten G.J., van Luenen,H., Verra,N.C.V., Valerio,D. and Plasterk,R.H.A. (1998) Nucleic Acids Res., 26, 3013–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raz E., van Luenen,H.G., Schaerringer,B., Plasterk,R.H.A. and Driever,W. (1998) Curr. Biol., 8, 82–88. [DOI] [PubMed] [Google Scholar]
- 20.Ivics Z., Hackett,P.B., Plasterk,R.H. and Izsvak,Z. (1997) Cell, 91, 501–510. [DOI] [PubMed] [Google Scholar]
- 21.Medhora M., Maruyama,K. and Hartl,D.L. (1991) Genetics, 128, 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Studier F.W., Rosenberg,A.H., Dunn,J.J. and Dubendorff,J.W. (1990) Methods Enzymol., 185, 60–89. [DOI] [PubMed] [Google Scholar]
- 23.Miller V.L. and Mekalanos,J.J. (1988) J. Bacteriol., 170, 2575–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garraway L.A., Tosi,L.R., Wang,Y., Moore,J.B., Dobson,D.E. and Beverley,S.M. (1997) Gene, 198, 27–35. [DOI] [PubMed] [Google Scholar]
- 25.Kolter R., Inuzuka,M. and Helinski,D.R. (1978) Cell, 15, 1199–1208. [DOI] [PubMed] [Google Scholar]
- 26.Lampe D.J., Grant,T.E. and Robertson,H.M. (1998) Genetics, 149, 179–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chance M.L. (1972) Trans. R. Soc. Trop. Med. Hyg., 66, 352. [DOI] [PubMed] [Google Scholar]
- 28.Mamoun C.B., Sullivan,D.J.,Jr, Banerjee,R. and Goldberg,D.E. (1998) J. Biol. Chem., 273, 11241–11247. [DOI] [PubMed] [Google Scholar]
- 29.Robertson H.M. and Zumpano,K.L. (1997) Gene, 205, 203–217. [DOI] [PubMed] [Google Scholar]
- 30.Morgan G.T. (1995) J. Mol. Biol., 254, 1–5. [DOI] [PubMed] [Google Scholar]
- 31.Hartl D.L., Lozovskaya,E.R., Nurminsky,D.I. and Lohe,A.R. (1997) Trends Genet., 13, 197–201. [DOI] [PubMed] [Google Scholar]
- 32.Lohe A.R., Sullivan,D.T. and Hartl,D.L. (1996) Genetics, 144, 1087–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Pouderoyen G., Ketting,R.F., Perrakis,A., Plasterk,R.H. and Sixma,T.K. (1997) EMBO J., 16, 6044–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rio D.C. (1991) Trends Genet., 7, 282–287. [DOI] [PubMed] [Google Scholar]
- 35.de la Cruz N.B., Weinreich,M.D., Wiegand,T.W., Krebs,M.P. and Reznikoff,W.S. (1993) J. Bacteriol., 175, 6932–6938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hartl D.L., Lohe,A.R. and Lozovskaya,E.R. (1997) Genetica, 100, 177–184. [PubMed] [Google Scholar]
- 37.Lohe A.R. and Hartl,D.L. (1996) Mol. Biol. Evol., 13, 549–555. [DOI] [PubMed] [Google Scholar]
- 38.Lohe A.R., De Aguiar,D. and Hartl,D.L. (1997) Proc. Natl Acad. Sci. USA, 94, 1293–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ketting R.F., Fischer,S.E.J. and Plasterk,R.H. (1997) Nucleic Acids Res., 25, 4041–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Luenen H.G. and Plasterk,R.H. (1994) Nucleic Acids Res., 22, 262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seifert H.S., Chen,E.Y., So,M. and Heffron,F. (1986) Proc. Natl Acad. Sci. USA, 83, 735–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morgan B.A., Conlon,F.L., Manzanares,M., Millar,J.B., Kanuga,N., Sharpe,J., Krumlauf,R., Smith,J.C. and Sedgwick,S.G. (1996) Proc. Natl Acad. Sci. USA, 93, 2801–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burns N., Grimwade,B., Ross-Macdonald,P.B., Choi,E.Y., Finberg,K., Roeder,G.S. and Snyder,M. (1994) Genes Dev., 8, 1087–1105. [DOI] [PubMed] [Google Scholar]
- 44.Chun K.T. and Goebl,M.G. (1996) Genetics, 142, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gwinn M.L., Stellwagen,A.E., Craig,N.L., Tomb,J.F. and Smith,H.O. (1997) J. Bacteriol., 179, 7315–7320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goryshin I.Y. and Reznikoff,W.S. (1998) J. Biol. Chem., 273, 7367–7374. [DOI] [PubMed] [Google Scholar]
- 47.Devine S.E. and Boeke,J.D. (1994) Nucleic Acids Res., 22, 3765–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coates C.J., Jasinskiene,N., Morgan,D., Tosi,L.R.O., Beverley,S.M. and James,A.A. (2000) Insect Mol. Biol. Biochem., submitted for publication. [DOI] [PubMed] [Google Scholar]
- 49.Schneider T.D. and Stephens,R.M. (1990) Nucleic Acids Res., 18, 6097–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]