

Abstract
Neuropilin-1 (NRP1) which is a main transmembrane cell surface receptor acts as a host cell mediator resulting in increasing the SARS-Cov-2 infectivity and also plays a role in neuronal development, angiogenesis and axonal outgrowth. The goal of this study is to estimate the impact of single nucleotide polymorphisms (SNPs) in the NRP1 gene on the function, structure and stabilization of protein as well as on the miRNA-mRNA binding regions using bioinformatical tools. It is also aimed to investigate the changes caused by SNPs in NRP1 on interactions with drug molecule and spike protein. The missense type of SNPs was analyzed using SIFT, PolyPhen-2, SNAP2, PROVEAN, Mutation Assessor, SNPs&GO, PhD-SNP, I-Mutant 3.0, MUpro, STRING, Project HOPE, ConSurf, and PolymiRTS. Docking analyses were conducted by AutoDock Vina program. As a result, a total of 733 missense SNPs were determined within the NRP1 gene and nine SNPs were specified as damaging to the protein. The modelling results showed that wild and mutant type amino acids had some different properties such as size, charge, and hydrophobicity. Additionally, their three-dimensional structures of protein were utilized for confirmation of these differences. After evaluating the results, nine polymorphisms rs141633354, rs142121081, rs145954532, rs200028992, rs200660300, rs369312020, rs370117610, rs370551432, rs370641686 were determined to be damaging on the structure and function of NRP1 protein and located in conserved regions. The results of molecular docking showed that the binding affinity values are nearly the same for wild-type and mutant structures support that the mutations carried out are not in the focus of the binding site, therefore the ligand does not affect the binding energy. It is expected that the results will be useful for future studies.
Keywords: Neuropilin-1 (NRP1), SARS-CoV-2, In silico analysis, Single nucleotide polymorphism (SNP)
Introduction
The spike protein of SARS-CoV-2 is main protein responsible for binding to ACE2 (angiotensin converting enzyme 2) receptor on the host cell. The spike protein is required to be activated and degraded by transmembrane protease, serine 2 (TMPRSS2) and FURIN which are host cell proteases (Kermani et al. 2021). In addition to the role of ACE2, neuropillin 1 (NRP1) which is an essential transmembrane cell surface receptor acts as a host cell mediator result in increasing of the SARS-Cov-2 infectivity (Kyrou et al. 2021; Davies et al. 2020).
Neuropilin-1, one of the signalling and catalytic proteins, has two isoforms as a secreted form and a transmembrane form that interacts with SARS-CoV-2. NRP1 may act as an entry factor, accelerating the transmission of SARS-CoV-2. In particular, recent studies have reported that the protein of the virus binds to the NRP1 receptor in addition to ACE2. It was reported that NRP1 expression was suppressed in cells responsible for ACE2 expression, and SARS-CoV-2 infection was significantly reduced. The use of NRPs as entry factors may be due to their high expression in the surrounding epithelium and their ability to induce cell, vascular and tissue penetration (Cantuti-Castelvetri et al. 2020; Mayi et al. 2021; Klaewkla et al. 2021; Raaben et al. 2017).
Neuropilin-1 is encoded by NRP1 gene which is located in 10p11.22 (https://www.genecards.org/cgi-bin/carddisp.pl?gene=NRP1). Previous studies investigated the association of variants in NRP1 gene with some diseases such as colorectal, breast, gastric, and pancreatic cancer, hepatocellular carcinoma, and migraine (Seo et al. 2020; Lin et al. 2018; Napolitano and Tamagnone 2019; Morin et al. 2018; Staton et al. 2013; Ansari et al. 2020; Pollock et al. 2018; Seifi-Alan et al. 2018). In addition, due to the role of this gene in the development of COVID-19 disease, it is important to determine the possible effects of variants in NRP1 gene.
SNP is an alteration in the human genome which founds commonly. In some cases, the SNPs may have the ability to increase genetic susceptibility to disorders. The identification of SNPs that are associated with the diseases is achieved by genotyping of SNPs in patients and controls and determaning the frequency differences between them (Harley and Narod 2009), (Özkan et al. 2015). In order to identify disease-related SNPs, one of the preferred approaches is to determine the possible harmful effects of SNPs by using in silico tools before planning genotyping studies, recently (Özkan Oktay et al. 2019). In addition, miRNAs have important roles in various biological functions such as development, cell differentiation, viral pathogenesis, proliferation, and progression of human diseases (Sun et al. 2009). For this reason, the aim of this study is to investigate the effect of SNPs on the stability, structure and function of neuropilin-1, to understand the effetcts of the variants on the ligand–protein interactions via molecular docking, to estimate the impacts of SNPs on miRNA binding sites, and to investigate the protein–protein interactions via different bioinformatics tools.
Methods
Training Data
The accession number of the human NRP1 gene (NCBI Gene ID:8829), missense SNPs, amino acid alterations were provided using the NCBI dbSNP (https://www.ncbi.nlm.nih.gov/snp/) database in October, 2021. The FASTA format sequence of the protein, UniProt entry name (NRP1_HUMAN) UniProtKB number (O14786) of neuropilin-1 was provided from the UniProt (https://www.uniprot.org/) database.
Freely available online software tools were used to investigate whether an amino acid alteration affects the targeted protein as well as to determine deleterious/damaging SNPs and three-dimensional models of the mutant protein (Fig. 1) (Kaman et al. 2019; Mustafa et al. 2020; Murthy et al. 2021; Özkan Oktay et al. 2019). Seven software tools were used for functional analysis of missense SNPs. The SIFT (https://sift.bii.a-star.edu.sg/www/SIFT_dbSNP.html) estimates the amino acid effects on the protein function utilizing some features of amino acids and homology (Ng and Henikoff 2001; Vaser et al. 2016). PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) characterizes amino acid substitutions in the sequence and gives ideas on their phylogenetic and structural information (Adzhubei et al. 2010). PROVEAN (http://provean.jcvi.org/index.php) gives information about the possible effect of an amino acid alteration on the protein function based on sequence homology (Choi et al. 2012). SNPs&GO (https://snps-and-go.biocomp.unibo.it/snps-and-go/) estimates if a variation may be identified as associated with disease or neutral (Calabrese et al. 2009). SNAP2 (https://rostlab.org/services/snap2web/) server predicts the functional effects of mutations based on a “neural network” which is a machine learning device. Then, it classifies SNPs into two categories (effect or neutral) (Hecht et al. 2015). PhD-SNP (https://snps.biofold.org/phd-snp/phd-snp.html) categorizes SNPs as disease-associated or as neutral based on SVM (support vector machine) method (Capriotti et al. 2006). Mutation Assessor (http://mutationassessor.org/r3/) is a server based on the evolutionary conservation of amino acids in protein homologs and estimates the functional impact of amino acid alterations (Reva et al. 2007).
Fig. 1.

Workflow diagram shows the prediction of high-risk SNPs, SNPs in miRNA target sites and protein–protein interactions
Prediction of Protein Stabilization Alteration
SVM based predictors, I-Mutant 3.0 (http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi) and MUpro (http://mupro.proteomics.ics.uci.edu/) were employed for prediction of protein stability changes (Capriotti et al. 2005), (Cheng et al. 2006).
Prediction of Amino Acid Properties and Modelling of Protein
Project HOPE (https://www3.cmbi.umcn.nl/hope/method/) was used to form 3D models of both wild and mutant type proteins and their amino acid sequences to compare the differences such as size, charge, hydrophobicity, etc. between these proteins (Venselaar et al. 2010).
Determination of Protein–Protein Associations
The STRING database (https://string-db.org) were used to predict the protein–protein association network. The obtained network contains both physical and functional interactions because all publicly available data of protein–protein interaction information is collected, scored and integrated by STRING (Szklarczyk et al. 2019). The prediction was limited to the top ten most interactive proteins.
Prediction of Conservation Profiles
The evolutionary conservation of residues in neurpillin-1 protein was estimated via the ConSurf server (https://consurf.tau.ac.il/). The conservation scores are divided on a nine-colour grade scale. Most conserved positions are located in grade 9 whereas most variable positions are located in grade 1. In addition, exposed or buried and functional or structural residues are predicted by ConSurf server, too (Ashkenazy et al. 2016, Ashkenazy et al. 2010; Celniker et al. 2013; Berezin et al. 2004).
Prediction of SNPs and miRNA Associations
PolymiRTS (https://compbio.uthsc.edu/miRSNP/) database were used to predict 3’ UTR SNPs in miRNA target sites. It calculates whether two alleles of SNPs give rise to other miRNA target sites or not. The results are presented by assigning the SNPs in one of the four classes (D, N, C, O). “D” and “N” classes represent the disruption of conserved and non-conserved miRNA sites, respectively. The creation of a new miRNA site is abbreviated as the “C” class. Finally, the “O” class is used for other cases when the ancestral allele cannot be determined definitely. Among them, the “C” and “D” classes are probably to have functional impacts (Bhattacharya et al. 2014).
Molecular Docking
Hesperidine molecules (Fig. 2) was selected for ligand–protein docking studies. The most stable conformer and the optimized structures were obtained from selected ligand for chelation studies in Spartan’16 program (Kong et al. 2000) by semi-experimental PM6 method (Stewart 2008) (Stewart 2009). Pdb id:2qq1 coded structure with 1.90 Å resolution was selected from the Protein Data Bank (https://www.rcsb.org/) database as the crystal structure of NRP1 in the docking processes performed for this study. Before starting the docking process, the protein structure was kept tight, while the number of rotatable bonds in the ligand molecule is released. The H2O molecules in the crystalline structure were deleted, H atoms were added to the structure, and the Kollman charge was calculated. The Thr316, Asp320, Ser346, Thr349, and Tyr353 aminoacids were selected as active side for the docking study between NRP1 and hesperidin molecule (Vique-S´anchez 2021). A grid box with dimensions of 40 Å × 40 Å × 40 Å was selected and the grid spacing of 0.375 Å was determined, and molecular docking was performed with the Lamarckian Genetic algorithm in 100 working steps. Docking studies were performed with the AutoDock Vina program (Trott and Olson 2009).
Fig. 2.
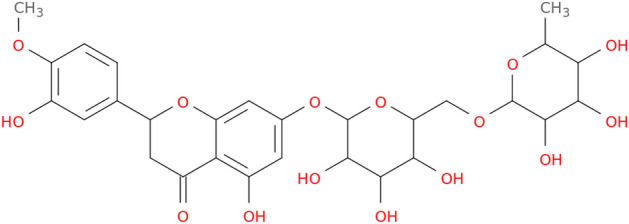
Hesperidine 2D structure
On the other hand, the changes in the interactions of the G101E, G366R, L464R, S416F, S432F and T337R mutations in NRP1 with the SARS-CoV-2 spike protein (protein–protein interaction: ppi) were investigated. First of all, mutant structures were obtained with BIOVIA Discovery Studio Visualizer (Dassault Syst`emes BIOVIA, Discovery Studio Modeling Environment, Release 2017, San Diego: Dassault Syst`emes, 2016), pdb id:6xra coded crystal structure was selected from pdb data bank as SARS-CoV-2 spike protein, and while the docking studies between NRP1 and hesperidine was conducted with Auto Dock Vina, docking analyses between NRP1 and spike protein was performed with ClusPro2.0 web server (Desta et al. 2020). Following Eq. 1 (by calculated ClusPro2.0) was used to perform cluster scores as well as to estimate the lowest binding energy.
| 1 |
The repulsive (rep), attractive (att), electrostatic (elec) energies and interactions taken from the decoys as the natural state (DARS), are calculated using molecular docking study. All imaging operations for mutant ppi were computed with the PYMOL program (The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC).
Results
Results of Deleterious/Damaging SNPs and Protein Stabilization Analysis
A total of 57,562 variants including 733 missense SNPs were retrieved from the dbSNP database in the NRP1 gene.
Prediction of deleterious or disease-related SNPs was carried out by using SIFT, PolyPhen-2, Mutation Assessor, PhD-SNP, SNAP2, PROVEAN, and SNPs&GO software tools. SNPs that were predicted to be deleterious or disease-related in all bioinformatics tools were determined as high-risk SNPs (rs141633354, rs142121081, rs145954532, rs200028992, rs200660300, rs369312020, rs370117610, rs370551432, rs370641686) and selected for further analysis. Detailed information on the results of the functional analysis is given in Tables 1 and 2. Protein stabilization predictions for deleterious SNPs from I-Mutant 3.0 and MUpro software were given in Table 3.
Table 1.
Functional analysis results of the NRP1 gene
| SNP ID | Amino acid alteration | SIFT | Score | PolyPhen-2 HumDiv | Score | PolyPhen-2 HumVar | Score | PROVEAN | Score | SNAP2 | Score | Expected Accuracy |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs141633354 | G28R |
D (Warning Low Confidence) |
0.043 | PD | 1.000 | PD | 1.000 | D | − 3.905 | E | 83 | 91% |
| rs142121081 | T337R | D | 0.009 | PD | 0.973 | PoD | 0.863 | D | − 3.640 | E | 56 | 75% |
| rs145954532 | S432F | D | 0 | PD | 1.000 | PD | 0.990 | D | − 5.411 | E | 14 | 59% |
| rs200028992 | G101E | D | 0 | PD | 1.000 | PD | 1.000 | D | − 4.043 | E | 69 | 80% |
| rs200660300 | L464R | D | 0 | PD | 1.000 | PD | 1.000 | D | − 4.525 | E | 77 | 85% |
| rs369312020 | S416F | D | 0.002 | PD | 0.985 | PD | 0.926 | D | − 3.103 | E | 69 | 80% |
| rs370117610 | G791D | D | 0.001 | PD | 0.998 | PD | 0.982 | D | − 3.213 | E | 78 | 85% |
| rs370551432 | G366R | D | 0.014 | PD | 0.999 | PoD | 0.969 | D | − 6.614 | E | 72 | 85% |
| rs370641686 | G760D | D | 0.015 | PD | 0.981 | PD | 0.948 | D | − 2.854 | E | 57 | 75% |
D Deleterious, PD Probably damaging, PoD Possibly damaging, E Effect
Table 2.
Results of disease relationship and pathological effects of the NRP1 gene
| SNP ID | Amino Acid Alteration | SNPs&GO | RI | PhD-SNP | RI | Mutation Assessor | FI score |
|---|---|---|---|---|---|---|---|
| rs141633354 | G28R | Disease | 9 | Disease | 7 | High | 3.71 |
| rs142121081 | T337R | Disease | 9 | Disease | 6 | Medium | 2.445 |
| rs145954532 | S432F | Disease | 9 | Disease | 2 | Medium | 2.635 |
| rs200028992 | G101E | Disease | 9 | Disease | 7 | Medium | 2.705 |
| rs200660300 | L464R | Disease | 10 | Disease | 9 | High | 4.205 |
| rs369312020 | S416F | Disease | 9 | Disease | 4 | Medium | 3.135 |
| rs370117610 | G791D | Disease | 9 | Disease | 6 | Medium | 3 |
| rs370551432 | G366R | Disease | 9 | Disease | 6 | Medium | 2.88 |
| rs370641686 | G760D | Disease | 9 | Disease | 9 | Medium | 2.765 |
FI Functional impact
Table 3.
Results of protein stabilization analysis of NRP1
| SNP ID | Amino Acid Alteration | I-Mutant 3.0 | MUpro | ||
|---|---|---|---|---|---|
| Result | RI | Result | MUpro DDG |
||
| rs141633354 | G28R | Decrease | 7 | Decrease | − 0.929 |
| rs142121081 | T337R | Decrease | 5 | Decrease | − 0.661 |
| rs145954532 | S432F | Decrease | 0 | Decrease | − 0.196 |
| rs200028992 | G101E | Decrease | 5 | Decrease | − 0.774 |
| rs200660300 | L464R | Decrease | 8 | Decrease | − 2.341 |
| rs369312020 | S416F | Increase | 5 | Decrease | − 0.203 |
| rs370117610 | G791D | Decrease | 6 | Decrease | − 0.887 |
| rs370551432 | G366R | Decrease | 7 | Decrease | − 0.995 |
| rs370641686 | G760D | Decrease | 6 | Decrease | − 0.5380 |
DDG Delta Delta G, RI Reliability Index
Results of Amino Acid Properties and Models
Results of the Project HOPE give schematic structures of the mutant protein showing the amino acid substitutions as well as their specific sizes, charges, hydrophobicity values, and location of each focussed variant. Three-dimensional modelling of protein for variants was structured and shown in Table 4, except three of them (G101E, G791D and G760D) due to the lack of structural information.
Table 4.
Project HOPE results of the models of the NRP1 protein (Venselaar et al. 2010) (Color figure online)
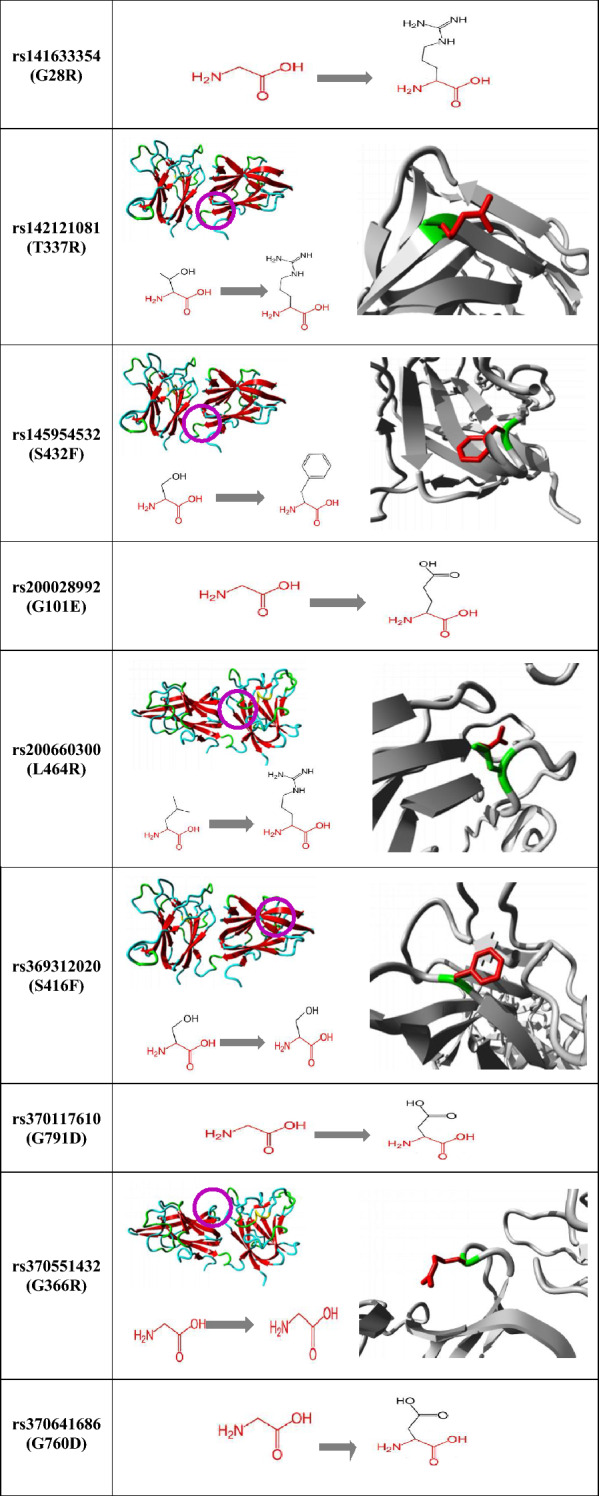
Polymorphism site, wild and mutant type residues represented by pink, green and red colours, respectively
Project HOPE results showed amino acid features such as charge, size, hydrophobicity and domains. The size of mutant type residues of G28R, T337R, S432F, G101E, L464R, S416F, G791D, G366R and G760D are larger than wild-type residues. Mutant type residues of G28R, T337R, L464R, and G366R have a positive charge and G101E, G791D, and G760D have a negative charge while wild-type residues of them were neutral. Wild-type residues at positions G28R, T337R, G101E, L464R, G791D, G366R, and G760D are more hydrophobic than mutant residues, while mutant residues at positions S432F and S416F are more hydrophobic than wild-type residues. In addition, HOPE results showed that the G28R and G101E polymorphisms are found in the CUB 1 domain, G760D and G791D polymorphisms are located in the MAM domain, T337R, S416F, G366R, L464R, and S432F polymorphisms are located in the FV/VIII domain. Those polymorphisms present amino acids with different properties that can disrupt these domains and damage their function (Venselaar et al. 2010).
Results of Conservation Analysis
The ConSurf server was used to estimate the conserved regions of neuropilin-1 as well as to predict exposed/buried and functional/structural residues. The ConSurf results showed that 189 residues predicted to be functional and 110 residues to be structural residue in the neuropilin-1. According to the ConSurf results of SNPs predicted to be high risk via in silico tools; 6 SNPs (G28R, G101E, G366R, S432F, L464R, G791D) are located in highly conserved regions, 2 SNPs (T337R and S416F) are located in relatively conserved regions and 1 SNP (G760D) is located in intermediately conserved regions in neuropillin-1. Furthermore, G101E, G366R, S432F and G791D are estimated to have functional impact whereas G28R and L464R are estimated to have structural roles. Figure 3 reveals detailed results of conservation analysis.
Fig. 3.

ConSurf result of conservation analysis. Note: In the first row, there are the residues of the query sequence (numbered 1-923). The second row shows the predicted burial state of the residues (‘b’: buried: ‘e’: exposed). The bottom row indicates the structural or functional importance of the residues (‘s’: structurally important. ‘f’: functionally important). Regions of 9 SNPs are boxed in red.
Results of SNPs and miRNA Associations
PolymiRTS results are presented in Table 5 which shows SNPs in miRNA target sites (dbSNP ID), the ancestral allele, two alleles of the SNP in the mRNA transcript, miR ID, miRSite (sequence context of the miRNA site), function class, context + scores.
Table 5.
PolymiRTS results of SNPs of NRP1 (Color figure online)
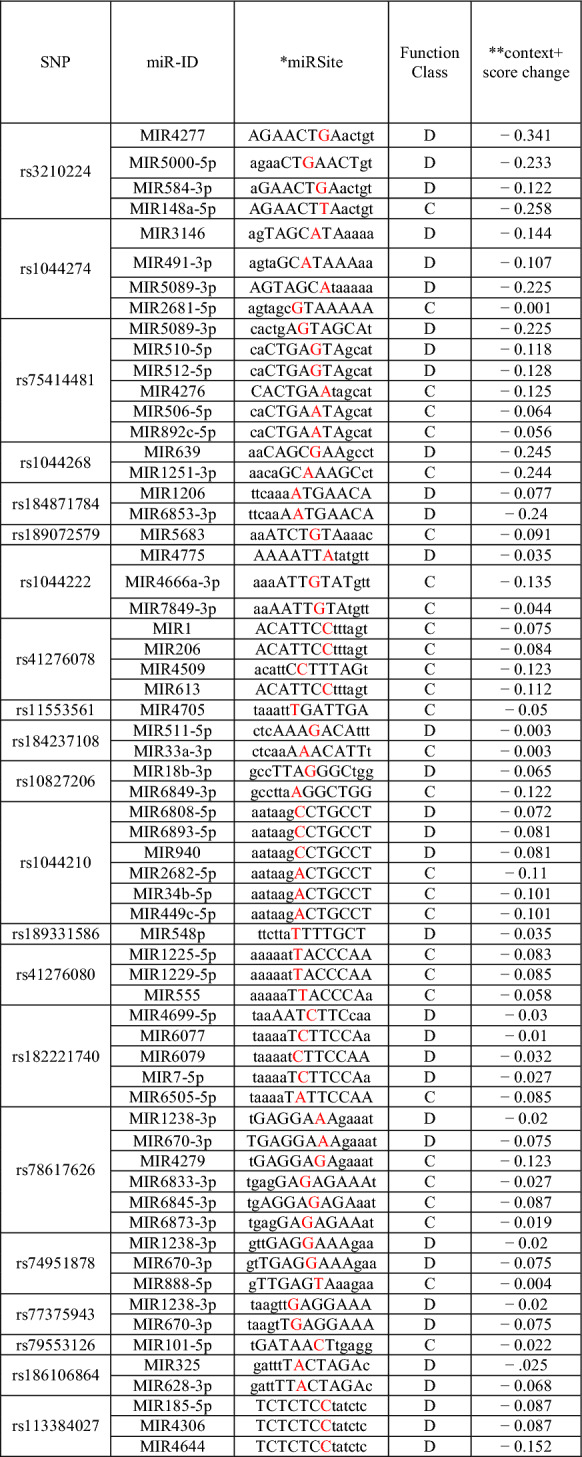
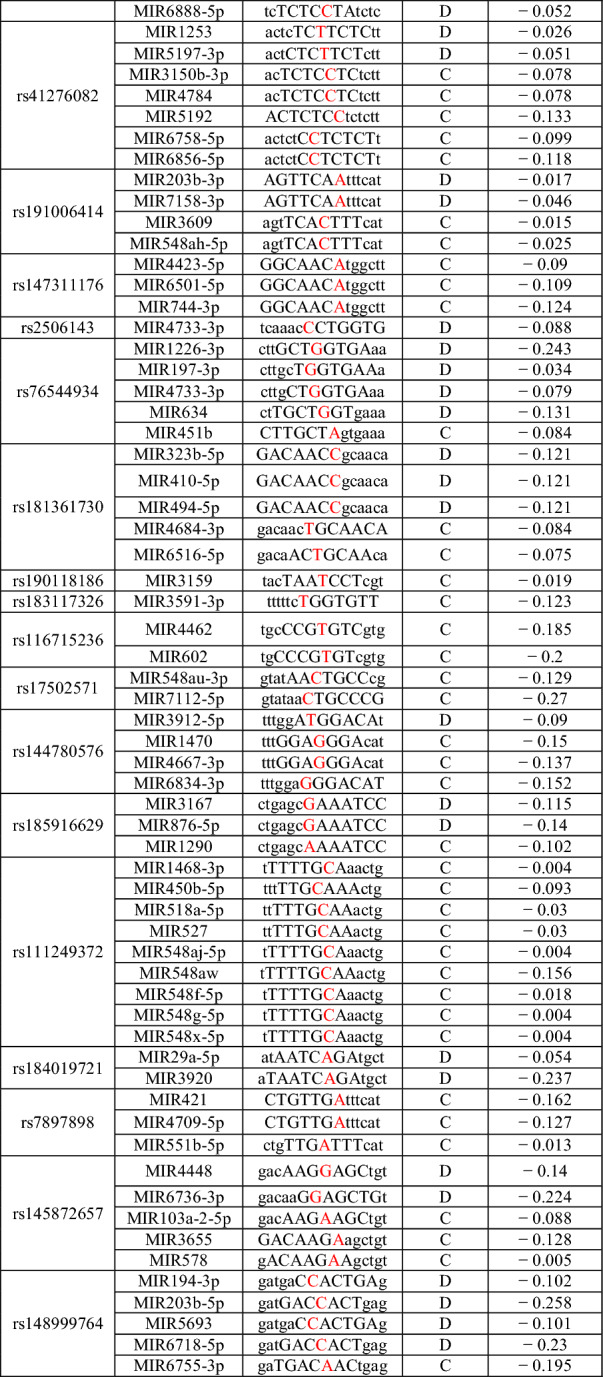
*Capital letters represent bases complementary to the seed region and SNPs are shown in red
**A more negative value of the context + score difference shows increased possibility of disruption or newly creation of miRNA targeting by the mutation
Determination of Protein–Protein Interactions
The protein–protein interaction results show that neuropillin-1 interacts with ten proteins including vascular endothelial growth factor receptor 2 (KDR), semaphorin-3A (SEMA3A), vascular endothelial growth factor receptor 1 (FLT1), Plexin-A1 (PLXNA1), vascular endothelial growth factor A (VEGFA), semaphorin-3F (SEMA3F), semaphorin-3C (SEMA3C), Plexin-D1 (PLXND1), Plexin-A2 (PLXNA2), Semaphorin-3B (SEMA3B). The details are presented in Fig. 4.
Fig. 4.
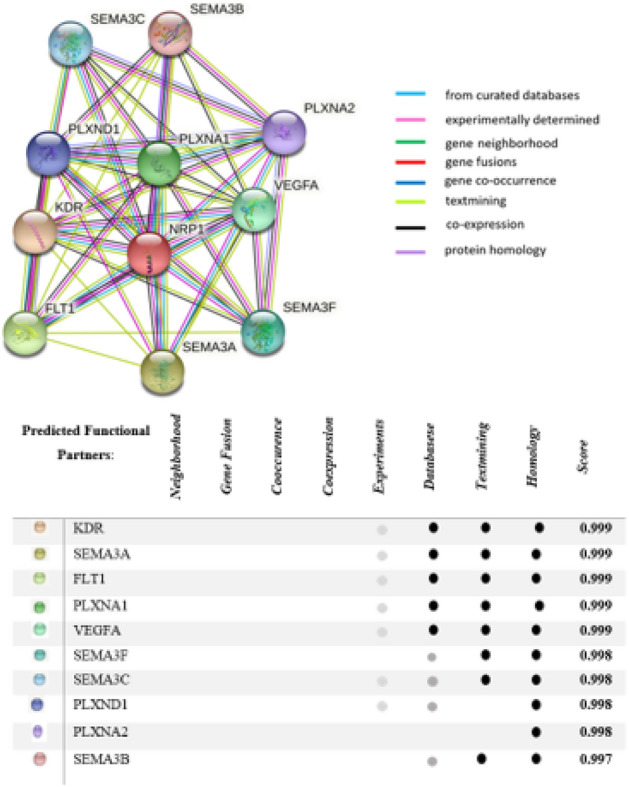
Protein–protein association network of NRP1 obtained from STRING database
Results of Molecular Docking
In a part of this study, the changes caused by possible mutations in NRP1 on interactions with drug molecule and spike protein were investigated. When the literature is examined, the hesperidin molecule was chosen as the drug molecule in the coupling studies of NRP1, therefore hesperidin was chosen as the drug reference in this study, and for the Wild Type (WT) and generated G101E, G366R, L464R, S416F, S432F and T337R mutations in Fig. 5 below. 2D interaction maps are given. When the 2D maps were examined, the binding affinity for WT-hesperidin chelating was − 9.7 kcal mole−1, and the interacted amino acids were consistent with the literature (Seadawy et al. 2020). On the other hand, when the docking interaction maps of the mutant structures were examined, it was calculated as − 9.9 kcal mole−1, − 9.7 kcal mole−1, 9.8 kcal mole−1, − 9.7 kcal mole−1, − 9.7 kcal mole−1 and − 9.7 kcal.mole−1 for the G101E, G366R, L464R, S416F, S432F and T337R mutations, respectively.
Fig. 5.

Representation of the 2D interactions map of the best docked pose of hesperidine molecule with the amino acids of NRP1 binding site and binding affinities
The list of amino acids with which WT and mutant protein structures interact in the active site as a result of docking with the hesperidin molecule is given in Table 6 below. Another important data that can be deduced from the table below is that there is no interaction with the mutant structures created.
Table 6.
List of amino acids with which WT and mutant protein constructs interact
| WT | G101E | G366R | L464R | S416F | S432F | T337R | |
|---|---|---|---|---|---|---|---|
| ILE13 | * | * | * | * | * | ||
| SER14 | * | * | * | * | * | * | * |
| ARG16 | * | * | * | * | * | ||
| ALA17 | * | * | |||||
| ASP24 | * | * | * | ||||
| ILE25 | * | * | |||||
| SER26 | * | * | * | * | * | * | * |
| GLY77 | * | ||||||
| THR78 | * | * | * | * | * | * | |
| ASP84 | * | * | * | ||||
| SER109 | * | * | * | ||||
| GLN112 | * | * | * | * | * | * | |
| VAL113 | * | * | |||||
| THR115 | * | * | |||||
| ALA116 | * | ||||||
| GLN121 | * | * | * | ||||
| GLY136 | * | * | * | ||||
| PRO135 | * | * | * | * | |||
| LYS137 | * | * | |||||
| SER140 | * |
The obtained interaction map and binding energies (kcal.mole-1) as a result of protein–protein docking between the Human NRP-1 receptor and the created mutant structures with the SARS CoV-2 spike protein fragment are given in Fig. 6 below. As can be seen from Fig. 6, no significant difference was detected between the protein–protein binding energies obtained. This result is also consistent with the ligand–protein docking results.
Fig. 6.

The 2D ppi maps between SARS CoV-2 spike protein fragment-NRP1 and SARS CoV-2 spike protein fragment-NRP1 mutant structures and binding affinities values
Discussion
It is necessary to investigate the possible effects of SNPs causing amino acid alterations on NRP1 due to the important roles of NRP1. Here, we attempted bioinformatical analysis to predict damaging SNPs on the structure, stabilization and function of NRP1. As a result, among 733 missense SNPs within NRP1 gene, nine SNPs rs141633354 (G28R), rs142121081 (T337R), rs145954532 (S432F), rs200028992 (G101E), rs200660300 (L464R), rs369312020 (S416F), rs370117610 (G791D), rs370551432 (G366R), rs370641686 (G760D) were identified as high-risk SNPs by using bioinformatical analysis tools as shown in workflow in Fig. 1 in this study (Tables 1 and 2). The changes in the interactions of the G101E, G366R, L464R, S416F, S432F and T337R mutations in NRP1 with the SARS-CoV-2 spike protein (protein–protein interaction: ppi) were investigated via AutoDock Vina. The fact that the binding affinity values are approximately the same for wt and mutant structures supports that the mutations carried out are not in the focus of the binding site, therefore the ligand does not affect the binding energy. Amino acid substitutions caused by missense SNPs were also investigated in terms of charge, hydrophobicity, and size differences by the Project HOPE server and those 9 SNPs were estimated how they affect the structure and/or function of the protein.
The protein stabilization results of the amino acid substitutions due to SNPs showed that eight amino acid substitution would have a decreasing effect on protein stabilization by both I-Mutant 3.0 and MUpro servers. S416F variant (rs369312020) is predicted to increase protein stability by the I-Mutant 3.0 server while it is predicted to decrease protein stability by the MUpro server (Table 3). Single amino acid substitutions caused by nsSNPs generally affect protein function by altering the structure and/or stability of the protein (Bromberg and Rost 2009). Protein stability alteration is a known mechanism by which amino acid substitutions result in human disease (Teng et al. 2010). Wang and Moult (2001) have reported that majority of disease-causing missense mutations (83%) are found to affect protein stability (Wang and Moult 2001). Moreover, Teng et al (2009) suggested that disease-causing mutations are inclined to destabilize protein–protein interactions (Teng et al. 2009). Therefore, STRING server was used to predict functional interactions pattern of NRP1 with other proteins.
Evolutionary conservation of a residue in the sequence of protein is very important to find if a mutation has any adverse effects on the host (Hossain et al. 2020). The ConSurf tool was used to obtain evolutionary conservation analysis for these nine amino acid substitutions in NRP1. As a result, all of those SNPs are predicted to be situated in conserved regions in varying proportions (highly/relatively/intermediately conserved).
SNPs in miRNA genes or their target cites have been reported to be associated with human diseases due to their key regulatory roles in gene expression (Gong et al. 2012). Therefore, in this study, we focused on the possible effects of SNPs on NRP1 using the PolymiRTS software tool. As a result, PolymiRTS predicted that 41 SNPs affects 164 target sites of the miRNA of NRP1 (Table 5).
Conclusion
In this study, the possible effects of SNPs in the NRP1 gene on the protein and miRNA target sites were investigated using various bioinformatics tools. In silico studies may provide an opportunity to identify the possible effects of functional SNPs in genes associated with various diseases and to understand the potential effects of SNPs. Further wet laboratory studies are recommended to confirm the results.
Author Contributions
EÖO, TK and ÖFK designed the study, collected the data, performed in silico SNP analysis. VEA performed molecular docking analysis. All authors wrote the draft of the manuscript and proof-reading of the manuscript.
Funding
None.
Declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari T, Lagman-Bartolome AM, Monsour D, Lay C. Management of menstrual migraine. Curr Neurol Neurosci Rep. 2020 doi: 10.1007/s11910-020-01067-x. [DOI] [PubMed] [Google Scholar]
- Ashkenazy Haim, Erez Elana, Martz Eric, Pupko Tal, Ben-Tal Nir. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38(suppl_2):W529–33. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, Ben-Tal N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44(W1):W344–W350. doi: 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezin C, Glaser F, Rosenberg J, Paz I, Pupko T, Fariselli P, Casadio R, Ben-Tal N. ConSeq: The identification of functionally and structurally important residues in protein sequences. Bioinformatics. 2004;20(8):1322–1324. doi: 10.1093/bioinformatics/bth070. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, Ziebarth JD, Cui Y (2014) PolymiRTS Database 3.0: linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic acids research, 42(D1):D86–D91. [DOI] [PMC free article] [PubMed]
- Bromberg Y, Rost B. Correlating protein function and stability through the analysis of single amino acid substitutions. BMC Bioinform. 2009;10(SUPPL. 8):1–9. doi: 10.1186/1471-2105-10-S8-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum Mutat. 2009;30(8):1237–1244. doi: 10.1002/humu.21047. [DOI] [PubMed] [Google Scholar]
- Cantuti-Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S, van der Meer F, et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020 doi: 10.1126/science.abd2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capriotti E, Fariselli P, Calabrese R, Casadio R. Predicting protein stability changes from sequences using support vector machines. Bioinformatics. 2005;21(SUPPL. 2):54–58. doi: 10.1093/bioinformatics/bti1109. [DOI] [PubMed] [Google Scholar]
- Capriotti E, Calabrese R, Casadio R. Predicting the Insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics. 2006;22(22):2729–2734. doi: 10.1093/bioinformatics/btl423. [DOI] [PubMed] [Google Scholar]
- Celniker G, Nimrod G, Ashkenazy H, Glaser F, Martz E, Mayrose I, Pupko T, Ben-Tal N. ConSurf: using evolutionary data to raise testable hypotheses about protein function. Isr J Chem. 2013;53(3–4):199–206. doi: 10.1002/ijch.201200096. [DOI] [Google Scholar]
- Cheng Jianlin, Randall Arlo, Baldi Pierre. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins. 2006;62(4):1125–32. doi: 10.1002/prot.20810. [DOI] [PubMed] [Google Scholar]
- Choi Yongwook, Sims Gregory E, Murphy Sean, Miller Jason R, Chan Agnes P. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012 doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J, Randeva HS, Chatha K, Hall M, Spandidos DA, Karteris E, Kyrou I. Neuropilin-1 as a new potential SARS-CoV-2 infection mediator implicated in the neurologic features and central nervous system involvement of COVID-19. Mol Med Rep. 2020 doi: 10.3892/mmr.2020.11510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desta IT, Porter KA, Xia B, Kozakov D, Vajda S. Performance and Its limits in rigid body protein-protein docking. Structure. 2020;28(9):1071–1081.e3. doi: 10.1016/j.str.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J, Tong Y, Zhang HM, Wang K, Tao Hu, Shan Ge, Sun J, Guo AY. Genome-wide identification of SNPs in MicroRNA genes and the SNP effects on MicroRNA target binding and biogenesis. Hum Mutat. 2012 doi: 10.1002/humu.21641. [DOI] [PubMed] [Google Scholar]
- Harley IJG, Narod SA. Single nucleotide polymorphisms - variation on a theme. BJOG. 2009 doi: 10.1111/j.1471-0528.2009.02352.x. [DOI] [PubMed] [Google Scholar]
- Hecht M, Bromberg Y, Rost B. Better prediction of functional effects for sequence variants. BMC Genomics. 2015;16(8):S1. doi: 10.1186/1471-2164-16-S8-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaman T, Karasakal ÖF, Özkan Oktay E, Ulucan K, Konuk M. In Silico approach to the analysis of snps in the human APAF1 gene. Turk J Biol. 2019 doi: 10.3906/biy-1905-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kermani NZ, Song WJ, Badi Y, Versi A, Guo Y, Sun K, Bhavsar P, et al. Sputum ACE2, TMPRSS2 and FURIN gene expression in severe neutrophilic asthma. Respir Res. 2021;22(1):1–11. doi: 10.1186/s12931-020-01605-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaewkla M, Charoenwongpaiboon T, Mahalapbutr P. molecular basis of the new COVID-19 target neuropilin-1 in complex with SARS-CoV-2 S1 C-End rule peptide and small-molecule antagonists. J Mol Liq. 2021 doi: 10.1016/j.molliq.2021.116537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong J, White Christopher A, Krylov Anna I, Sherrill D, Adamson Ross D, Furlani Thomas R, Lee Michael S, Lee Aaron M et al (2000) Q-Chem 2.0: a high-performance ab initio electronic structure program package. J Comput Chem 21(16):1532–48
- Kyrou I, Randeva HS, Spandidos DA, Karteris E. Not Only ACE2—the quest for additional host cell mediators of SARS-CoV-2 infection: neuropilin-1 (NRP1) as a Novel SARS-CoV-2 host cell entry mediator implicated in COVID-19. Signal Transduct Target Ther. 2021 doi: 10.1038/s41392-020-00460-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Zhang Y, Jiemin Wu, Li Li, Chen N, Ni P, Song L, Liu X. Neuropilin 1 (NRP1) Is a novel tumor marker in hepatocellular carcinoma. Clin Chim Acta. 2018 doi: 10.1016/j.cca.2018.06.046. [DOI] [PubMed] [Google Scholar]
- Mayi BS, Leibowitz JA, Woods AT, Ammon KA, Liu AE, Raja A. The role of neuropilin-1 in COVID-19. PLoS Pathog. 2021 doi: 10.1371/journal.ppat.1009153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin E, Sjöberg E, Tjomsland V, Testini C, Lindskog C, Franklin O, Sund M, et al. VEGF Receptor-2/Neuropilin 1 trans-complex formation between endothelial and tumor cells is an independent predictor of pancreatic cancer survival. J. Pathol. 2018 doi: 10.1002/path.5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy ASN, Suresh RV, Nallur BR (2021) Comprehensive in silico mutational-sensitivity analysis of PTEN establishes signature regions implicated in pathogenesis of autism spectrum disorders. Genomics. 10.1016/j.ygeno.2020.10.035 [DOI] [PubMed]
- Mustafa MI, Murshed NS, Abdelmoneim AH, Makhawi AM. In Silico analysis of the functional and structural consequences of SNPs in human ARX gene associated with EIEE1. Inform Med Unlocked. 2020 doi: 10.1016/j.imu.2020.100447. [DOI] [Google Scholar]
- Napolitano V, Tamagnone L. Neuropilins controlling cancer therapy responsiveness. Int J Mol Sci. 2019;20(8):1–14. doi: 10.3390/ijms20082049.o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11(5):863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özkan Oktay E, Kaman T, Karasakal ÖF, Ulucan K, Konuk M, Tarhan N (2019) “Alzheimer Hastalığı Ile İlişkilendirilen APH1A Genindeki Zararlı SNP’lerin In Silico Yöntemler Ile Belirlenmesi. Süleyman Demirel Üniversitesi Fen Bilimleri Enstitüsü Dergisi. 10.19113/sdufenbed.522738
- Özkan E, Erdemir A,Törer BD, Taşçi Aİ, Baskin Y, ElliDokuz H, and Turgut Balik D (2015). Genotyping and Analysis of rs7501939 Polymorphism For Prostate Cancer. Sigma J Eng Nat Sci 2015; 6: 101-107
- Pollock CE, Sutherland HG, Maher BH, Lea RA, Haupt LM, Alison Frith E, MacGregor A, Griffiths LR. The NRP1 migraine risk variant shows evidence of association with menstrual migraine. J Headache Pain. 2018 doi: 10.1186/s10194-018-0857-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaben M, Jae LT, Herbert AS, Kuehne AI, Stubbs SH, Chou YY, Blomen VA, et al. NRP2 and CD63 are host factors for lujo virus cell entry. Cell Host Microbe. 2017 doi: 10.1016/j.chom.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva Boris, Antipin Yevgeniy, Sander Chris. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007 doi: 10.1186/gb-2007-8-11-r232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seadawy M, Shamel M, Ahmed A, Zekri ARN (2020) In Silico Docking for Inhibition Neuropilin-1 (SARS-CoV-2 Receptor) by Some Natural and Approved Drugs. 10.2139/ssrn.3735823
- Seifi-Alan M, Shams R, Bandehpour M, Mirfakhraie R, Ghafouri-Fard S. Neuropilin-I expression is associated with lymph node metastasis in breast cancer tissues. Cancer Manag Res. 2018 doi: 10.2147/CMAR.S169533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo HS, Hyeon J, Song IH, Lee HH. Relationship between neuropilin-1 expression and prognosis, according to gastric cancer histology. J Mol Histol. 2020 doi: 10.1007/s10735-020-09870-z. [DOI] [PubMed] [Google Scholar]
- Hossain MS, Roy AS, Islam MS. In Silico analysis predicting effects of deleterious SNPs of human RASSF5 gene on its structure and functions. Sci Rep. 2020;10(1):1–14. doi: 10.1038/s41598-020-71457-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staton CA, Koay I, Wu JM, Hoh L, Reed MWR, Brown NJ. Neuropilin-1 and neuropilin-2 expression in the adenoma-carcinoma sequence of colorectal cancer. Histopathology. 2013 doi: 10.1111/his.12098. [DOI] [PubMed] [Google Scholar]
- Stewart JJP. Application of the PM6 method to modeling the solid state. J Mol Model. 2008;14(6):499–535. doi: 10.1007/s00894-008-0299-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart JJP. Application of the PM6 method to modeling proteins. J Mol Model. 2009;15(7):765–805. doi: 10.1007/s00894-008-0420-y. [DOI] [PubMed] [Google Scholar]
- Sun G, Yan J, Noltner K, Feng J, Li H, Sarkis DA, Sommer SS, Rossi JJ. SNPs in human MiRNA genes affect biogenesis and function. RNA. 2009 doi: 10.1261/rna.1560209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, et al. STRING V11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng S, Madej T, Panchenko A, Alexov E. Modeling effects of human single nucleotide polymorphisms on protein-protein interactions. Biophys J. 2009;96(6):2178–2188. doi: 10.1016/j.bpj.2008.12.3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng S, Srivastava AK, Schwartz CE, Alexov E, Wang L. Structural assessment of the effects of amino acid substitutions on protein stability and protein-protein interaction. Int J Comput Biol Drug Des. 2010 doi: 10.1504/IJCBDD.2010.038396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott Oleg, Olson Arthur J. AutoDock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2009 doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaser R, Adusumalli S, Leng SN, Sikic M, Pauline CN. SIFT missense predictions for genomes. Nat Protoc. 2016;11(1):1–9. doi: 10.1038/nprot.2015.123. [DOI] [PubMed] [Google Scholar]
- Venselaar Hanka, te Beek Tim A.H., Kuipers Remko K.P., Hekkelman Maarten L, Vriend Gert. “Protein structure analysis of mutations causing inheritable diseases an e-science approach with life scientist friendly interfaces. BMC Bioinform. 2010 doi: 10.1186/1471-2105-11-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vique-Sánchez JL. Potential inhibitors interacting in neuropilin-1 to develop an adjuvant drug against COVID-19, by molecular docking. Bioorg Med Chem. 2021;33:1106040. doi: 10.1016/j.bmc.2021.116040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Moult J. SNPs, protein structure, and disease. Hum Mutat. 2001;17(4):263–270. doi: 10.1002/humu.22. [DOI] [PubMed] [Google Scholar]