


Abstract
We have screened a human cDNA expression library with a digoxygenin-labelled protein phosphatase 1 (PP1) probe to identify novel PP1 interacting proteins. Eleven cDNA clones were isolated, which included genes encoding two previously characterised and six novel PP1 binding proteins. Three of the cDNAs encoded a protein called host cell factor (HCF), which is an essential component of the cellular complex required for the transcription of the herpes simplex virus (HSV) immediate-early (IE) genes. We demonstrate that HCF and PP1 exist as a complex in nuclear extracts and that this complex is distinct from the form of HCF that associates with HSV VP16. The data suggest novel roles for HCF and PP1, which may be relevant to their functions in transcription and cell cycle progression.
INTRODUCTION
The transcription of the immediate-early (IE) genes of herpes simplex virus (HSV) involves the formation of a multi-protein complex containing a structural component of HSV (VP16), and the cellular proteins oct1 and host cell factor (HCF) (1,2). HCF is a family of related polypeptides ranging in size from 110 to 300 kDa. These polypeptides are all encoded by a single gene and are generated by processing of a 300 kDa-precursor protein by site-specific cleavages located within a repetitive peptide sequence of about 20 amino acids (3,4). HCF is highly conserved (5–9) but its cellular function is not yet clear. The protein’s conservation and its interaction with cellular and viral transcription factors indicates a role in the regulation of expression of cellular genes (10–12). Furthermore, HCF has also been implicated in the regulation of the G0/G1 phase of the cell cycle (13).
Cellular transcription factors are frequently modified by phosphorylation, resulting in a diverse range of effects including alteration in subcellular localisation and the modulation of both the DNA binding and transcriptional regulatory domains. For example, VP16, the viral protein that interacts with oct1 and HCF to form the TRF-C complex (3,14–19) is phosphorylated in virally infected cells (20,21) and this phosphorylation may be directed by cellular kinases (22–25). Mutation of a phosphorylation site (serine 375) of VP16 to alanine abolishes complex assembly with Oct1 and HCF (25).
The reversible phosphorylation of proteins is an important mechanism for the regulation of various cellular metabolic pathways. The phosphorylation status of each protein substrate is maintained by complex regulatory interactions involving specific kinases and phosphatases. In some instances these interactions may involve the targeting of kinases and phosphatases to specific subcellular locations in the cell (26,27). Protein phosphatase 1 (PP1) is an important member of the family of serine/threonine phosphatases and had been shown to be involved in the regulation of several cellular processes and pathways in mammalian cells such as glycogen metabolism, muscle contraction, protein synthesis, cell division and calcium transport (28,29). PP1 is also thought to regulate nuclear processes such as pre-mRNA splicing (30) and the progression and exit from mitosis in mammalian cells (31).
Members of the serine/threonine protein phosphatase 1 family have a highly conserved catalytic subunit that shows a broad substrate specificity in vitro (28). Several cytosolic (26,32,33) and nuclear (34–39) proteins have been identified that may play a role in the subcellular location and the determination of substrate specificity of this enzyme. These interacting proteins that regulate PP1 activity have been named targeting subunits (26).
In order to identify new PP1 targeting subunits, we have screened a HeLa cDNA expression library using the digoxygenin (DIG)-labelled catalytic subunit of PP1. In addition to previously characterised PP1 regulatory proteins, several novel PP1 binding proteins were detected, including HCF. We show that HCF exhibits characteristics of a PP1 regulatory or targeting protein.
MATERIALS AND METHODS
Labelling of protein phosphatase 1 with digoxigenin
The human PP1 catalytic subunit (gamma isoform) (40), was labelled with digoxigenin-3-O-methylcarbonyl-aminocaproic-acid-N-hydroxy-succinamide ester (Boehringer-Mannheim, Germany) (41). Approximately 30 µg of PP1 were incubated with 5.5 µl of digoxigenin ester (1 mg/ml) in a total volume of 200 µl of phosphate buffered saline (PBS), pH 8.5, with constant shaking for 1 h. The mixture was dialysed against a buffer containing 60% glycerol, 20 mM Tris–Cl pH 7.6 and 1 mM DTT.
Screening of a HeLa cDNA expression library with digoxigenin-labelled PP1
A HeLa cell cDNA expression library (prepared by G. Banting and K. Stanley) in the Escherichia coli expression vector pUEX1 (42) was plated on eight 24 cm × 24 cm plates or 10 cm Petri dishes (Life Technologies) so that each large plate contained about 5 × 104 colonies. The plates were then incubated at 30°C for ~16–20 h until the colonies were ~1–2 mm in diameter. The colonies were transferred to Hybond-C extra nitrocellulose filters (Amersham) and incubated at 42°C for 2 h to induce expression of the fusion proteins. The colonies were then lysed on the filters by placing on 3MM Whatman paper impregnated with 5% SDS in an oven at 80°C for 25 min. The filters were washed in PBS containing 10% fat-free dry milk and 1 mM benzamidine (Sigma) for 5 min. A second wash was performed in the same buffer containing 10 µg/ml DNAse I (Sigma) for 5 min. The filters were washed once more with the same buffer but without DNase. The filters were incubated in PBS buffer containing 10% fat-free milk for 1 h to block any remaining protein adsorption sites. The wash buffer was discarded and the filters incubated in fresh binding buffer containing 250 mM NaCl, 20 mM Tris–Cl pH 7.6 and 1 mg/ml BSA. Digoxigenin-labelled PP1-γ was added to the binding buffer to give a final concentration of 1 µg of enzyme per millilitre. The binding reaction was performed at room temperature for 2–17 h. After the binding incubation, the filters were washed six times for 25 min with shaking at room temperature in binding buffer without BSA. The filters were then incubated in PBS containing 2% fat-free milk and mouse anti-digoxigenin antibody (Roche) for 1 h at room temperature according to the manufacturer’s instructions. The filters were washed four times for 5 min each in PBS and then a further incubation in 2% milk in PBS containing horseradish peroxidase conjugated anti-mouse IgG was performed for 1 h at room temperature. The nitrocellulose membranes were finally washed four times for 5 min each in PBS. The filters were developed by enhanced chemiluminescence (ECL) (Amersham) and exposed to X-ray film (Kodak) according to the manufacturers’ instructions. Spots on the films corresponding to positive colonies were marked and the appropriate colonies were picked from the plates and subjected to several rounds of plating and reprobing until distinct and well separated positive colonies could be obtained.
Characterisation of PP1-binding colonies
Colonies were grown in LB medium containing 50 µg/ml of ampicillin and plasmid DNA prepared from the bacteria using standard methods. The DNA obtained was sequenced using the T7 DNA polymerase sequencing kit (Amersham-Pharmacia) or on the Applied Biosystems 377 automated DNA sequencer using the Taq dye terminator cycle sequencing according to the manufacturer’s instructions. Sequence data obtained were used for homology database searches against either the NRdb, a comprehensive non-redundant database or dbEST, an EST database using the BLAST algorithm.
Expression of the HCF PP1-binding region of clone 17 in E.coli
The HCF PP1 binding fragment in clone 17 (Table 1) was subcloned by PCR into the BamHI/EcoRI sites of the E.coli expression vector pGEX-4T1 (Pharmacia-LKB) to generate the plasmid pGEX-HCF17. pGEX-HCF17 was transformed into E.coli BL21(DE3). The expression and purification of the recombinant protein was carried out as described previously (43).
Table 1. Protein phosphatase 1 binding proteins identified from a HeLa cDNA expression library screen.

The clone number column designates the 11 positive clones identified in the screen. This number represents the reference to the positive colony after the primary screen. EST accession numbers have only been shown for genes whose full-length cDNAs are not yet available in the databases.
NA, information is not yet available.
*Putative PP1 binding domain motifs were identified by comparative sequence analyses.
In vitro protein binding assay of GST-HCF17 and PP1γ
The in vitro binding assays were carried out in 50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1 mM EGTA, 5% glycerol, 0.02% Brij-35, 0.1% 2-mercaptoethanol and 0.1 mg/ml bovine serum albumin (buffer C). Approximately 3 nmol of each protein were incubated in 100 µl of the above buffer for 1 h at 4°C. Glutathione–Sepharose beads (20 µl) were added and incubation at 4°C was continued with shaking for 30 min. After centrifugation at ~1500 g for 1 min the supernatant was removed and the pellet was washed three times with 1 ml of Buffer C (lacking BSA), then denatured by heating in SDS–PAGE loading buffer at 95°C for 5 min and subjected to SDS–PAGE. Proteins in the gel were transferred to nitrocellulose membranes and probed with 0.1 µg/ml anti-PP1 or anti-GST (Amersham-Pharmacia) according to the manufacturer’s instructions. Secondary probing was performed using anti-sheep IgG antibodies conjugated to horseradish peroxidase (Roche). The nitrocellulose membranes were developed by enhanced chemiluminescence (Amersham) according to the manufacturer’s instructions.
Production and affinity purification of peptide antisera to HCF
Two peptides were designed essentially as described in (4) and synthesised by Immune Systems. The peptides were conjugated to BSA by glutaraldehyde using standard methods (44). A mixture of the conjugated peptides was used to immunise sheep (SAPU) for the production of polyclonal antisera. The antiserum was affinity purified against the peptides coupled by EDC [1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide] to diaminodipropylamine immobilised on the azlactone-activated support, 3 M Emphaze Biosupport Medium AB1 (Pierce) according to the manufacturer’s recommendations. Affinity purified antibody (named HC2) was dialysed against PBS containing 0.02% sodium azide and stored at –80°C.
Immunoprecipitation and western blotting
HeLa nuclear extracts were obtained commercially from Computer Cell Culture Centre (Mons, Belgium). Immunoprecipitations of the HCF complex from nuclear extract were performed using either anti-rHCF antibody (7) or HC2. 100 µl of nuclear extract (4–5 mg/ml) was precleared for 1 h at 4°C on 25 µl of settled protein G Sepharose (Amersham-Pharmacia) or protein G agarose beads (Roche) that had been preincubated with ~10 µg of sheep pre-immune IgG. The precleared nuclear extract was diluted 10 times with PBS buffer containing 0.5% Triton X-100 before adding protein-G Sepharose or agarose beads that had been preincubated with ~5 µg of antibody HC2 for 1 h at 4°C. Immunoprecipitations were carried out at 4°C for 2–16 h. The immunoprecipitates were washed with 1 ml PBS containing 0.5% Triton X-100 three times at 4°C. The protein G beads carrying the immune complexes were collected after each wash by centrifugation at 1500 g for 1 min. For immunoblotting, the washed immunoprecipitates were incubated with 50 µl of 2× SDS–PAGE loading buffer at 95°C for 5 min. The supernatant (15 µl) was used for SDS–PAGE and the separated proteins transferred to nitrocellulose membranes by electroblotting. The membranes carrying the transferred proteins were probed with antibodies to PP1 (33) and HCF or digoxigenin-labelled PP1 (41). The anti-HCF antibody (HC2) and the anti-PP1 antibody were each used at a concentration of 0.1 µg/ml. Protein bands were detected by probing the nitrocellulose blots with the appropriate secondary antibodies conjugated to horseradish peroxidase (Pierce, Roche) according to the manufacturer’s recommendations and developing by ECL (Amersham).
Assay of protein phosphatase 1 activity in HCF immunoprecipitates
To 20 µl of the immune pellet, 10 µl of a buffer containing 50 mM Tris–HCl pH 7.5, 0.1 mM EGTA, 0.03% Brij-35 and 0.1% 2-mercaptothanol was added. The immune precipitates were then digested with trypsin to release any bound phosphatase activity. The digestion reaction was started by the addition of 1 µl of 0.1 mg/ml trypsin (11 000 U/mg, Sigma) and after incubation at 30°C for 5 min, the reaction was stopped by the addition of 1 µl (1 mg/ml) soybean trypsin inhibitor (Sigma). This resulted in the release of free active, C-terminally nicked PP1 catalytic subunit. The released phosphatase activity was assayed by using 32P-labelled rabbit skeletal muscle phosphorylase (1.0 mol phosphate/mol subunit) as substrate as described in (45).
For inhibition studies, the immunoprecipitates were preincubated with the inhibitor (okadaic acid or inhibitor-2) for 15 min at 30°C prior to the assays being carried out as above. Okadaic acid was a kind gift from Dr Y. Tsukitani, Fujisawa Pharmaceutical Company, Tokyo, Japan.
Peptide release of PP1 activity from the HCF immune pellet complex
The HCF immune pellets obtained as described above were incubated with a 32 residue peptide (10 µM final concentration) (38) from the PP1-binding protein p53BP2 (33) containing a ‘canonical’ PP1-binding motif. The control samples contained a 16 residue peptide (10 µM final concentration) designed for a region located outside the PP1 binding domain motif (38). Both control and test samples were incubated at 30°C for 15 min. After the incubation, the pellets were collected by centrifugation and the supernatants assayed for phosphatase activity.
Gel retardation assay
The electrophoretic gel retardation assays were performed essentially as described in (25). The reactions contained ~10 µg of HeLa nuclear extract and were carried out in a buffer containing 25 mM HEPES, pH 7.9, 50 mM KCl, 5 mM DTT, 1 mM EDTA, 0.05% NP-40 and 10% glycerol. GST-VP16 was added as described in the figure legend. End-labelled oligonucleotide probe, TAAT24, containing the octamer–GARAT motif from HSV IE 110Kda promoter was added after pre-incubation of extracts and non-specific competitor DNA (10 µg of poly dIdC) for 5 min at 30°C. The reaction samples were further incubated at 30°C for 30 min after addition of TAAT24. The samples were then resolved by electrophoresis on a 4% non-denaturing polyacrylamide gel and detected by autoradiography.
RESULTS
Identification of novel protein phosphatase 1 binding proteins
A human cDNA/β-GAL-fusion plasmid expression library was screened using digoxigenin (DIG)-labelled PP1 as a probe (Materials and Methods). Eleven cDNAs were identified that express polypeptides that bind DIG-PP1. Each clone was purified through at least three rounds of expression screening until all colonies were positive for binding DIG–PP1. The cDNA inserts from each positive clone were then sequenced. The validity of this screening method was confirmed when two of the 11 positive clones were found to encode previously characterised PP1 binding proteins, i.e. NIPP1 and FB-19/P99/PNUTS (35,38,39). The other clones encoded six separate genes (Table 1). One of these is a novel gene for which we have retrieved EST (expressed sequence tag) sequences in the database of expressed sequence tags (dBEST) corresponding to this cDNA. For the remaining five clones, we were able to retrieve full-length cDNA sequences that code for each of these proteins in the non-redundant (NR) database of Genebank (Table 1). None of these proteins have previously been shown to interact with PP1. Interestingly, three of the 11 positive clones expressed fragments of the nuclear protein HCF (clones 1, 15 and 17; Fig. 1). Furthermore, all three clones encoded overlapping C-terminal fragments of HCF indicating that the PP1 binding site is likely present in the region of overlap. HCF is known to interact with cellular and viral transcription factors and is proposed to have possible roles in gene expression and cell cycle progression. It was not known to bind or regulate PP1. We decided therefore to characterise the interaction of PP1 and HCF because it may shed light on the normal cellular role of this factor.
Figure 1.

Linear representation of HCF showing the three overlapping clones that interact with PP1-γ. The diagram is based on the HCF described in (7) with some modifications. The numbers above the rectangles representing clones 1, 15 and 17 show the start and end positions of the amino acid sequence of the HCF fragments expressed by the cDNAs obtained from the library screen.
Analysis of the interaction between HCF and PP1
Clone 17 contains the smallest cDNA fragment of HCF of the three cDNAs obtained in this screen. To confirm the interaction of this HCF fragment with DIG–PP1, protein lysates from bacteria either expressing the fusion protein, a non-expressing control or a positive control corresponding to cells expressing a β-GAL–NIPP1 fusion, were separated by SDS–PAGE and transferred to a nitrocellulose membrane. The membrane was incubated with the DIG–PP1 probe, which confirmed that PP1 interacts directly with the C-terminus of HCF (residues 1604–1956) (Fig. 2A).
Figure 2.
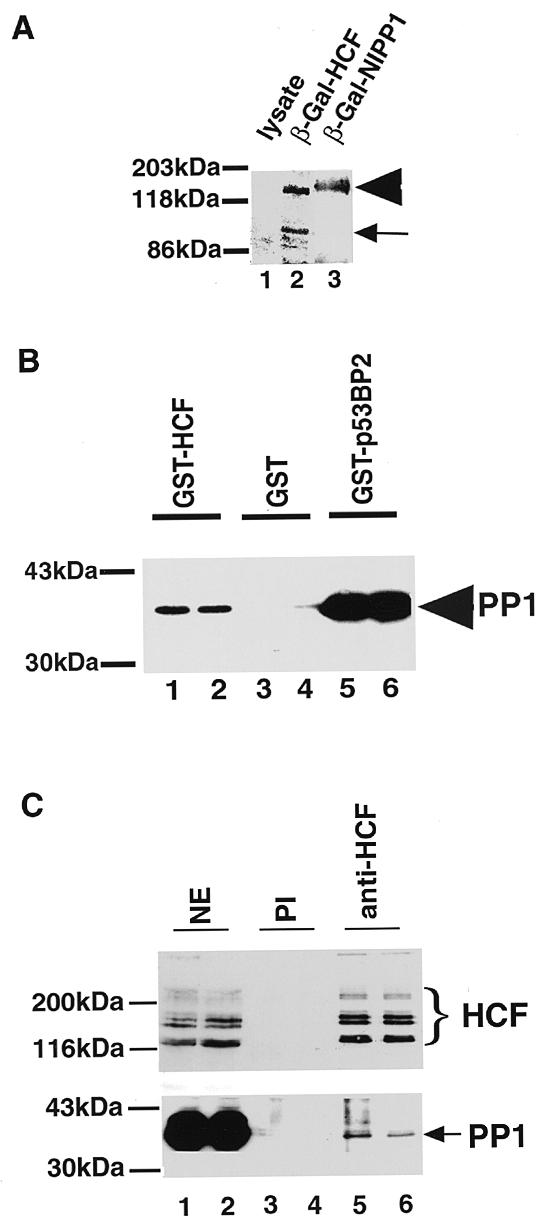
Recombinant HCF binds PP1. The HCF cDNA fragment in clone 17 was expressed as a β-galactosidase (β-Gal) and GST fusions. (A) Western blot of an SDS–PAGE gel probed with digoxigenin-labelled PP1-γ. The arrowhead indicates recombinant fusion protein. The small arrow indicates a proteolytic product of β-Gal–HCF. Escherichia coli cells were grown to an OD600 of 0.9–1. Each lane contained 10 µl of a 25 times concentrated E.coli lysate. Lane 1 contains bacterial lysate of cells expressing β-Gal alone. Lanes 2 and 3 contain bacterial lysates of cells expressing HCF (clone 17) and NIPP1 fusions respectively. (B) The GST–HCF fusion protein was incubated with purified PP1 (~3 nmol of each protein) and glutathione–Sepharose beads. Each GST-fusion pull-down was done in duplicate. After several washes the samples were resolved by SDS–PAGE and immunoblotted with anti-PP1. Lanes 1 and 2 contained the binding assay between the GST–HCF fragment in clone 17 and PP1-γ. Lanes 3 and 4 contained GST and PP1 and lanes 5 and 6 contained a positive control: GST-p53BP2 (p53BP2 is known to bind PP1 avidly) (33). The arrow indicates bands corresponding to the PP1 pulled down. (C) HCF was immunoprecipitated from 0.4–0.5 mg of HeLa nuclear extract using ~5 µg of anti-HCF antibody and the beads resuspended in ~50 µl of SDS–PAGE loading buffer. Fifteen microlitres of the immunoprecipitates of HCF from HeLa nuclear extract were electroblotted as above and probed with anti-HCF antibody (upper panel) and anti-PP1 antibody (lower panel). Each immunoprecipitation experiment contained duplicate samples. Lanes 1 and 2 contained nuclear extract. Lanes 3 and 4 had immunoprecipitates using pre-immune IgG (PI) and lanes 5 and 6 represent immunoprecipitates using anti-HCF antibody. The brace indicates the major HCF polypeptides immunoprecipitated by anti-HCF antibody. The arrow indicates the PP1 co-immunoprecipitated with the HCF polypeptides.
The library screening and far-western analyses were performed under denaturing conditions. We therefore determined if PP1 and HCF would interact in solution using a GST pull-down assay. The HCF fragment of clone 17 (Fig. 1) was cloned in frame with GST (Materials and Methods) and the resulting GST–HCF fusion protein purified and immobilized on glutathione–Sepharose beads. Bacterially expressed PP1 was then incubated with either the GST–HCF beads or control GST alone beads. As a positive control, PP1 binding to GST–p53BP2 beads (33) was also tested. After extensive washing, the bound proteins were released with 1% SDS then resolved by electrophoresis, transferred to a membrane and PP1 detected by immunoblotting with an anti-PP1 antibody (Fig. 2B). This shows that PP1, when bacterially expressed, binds directly to GST–HCF and to GST–p53BP2, but not to GST alone (lanes 1 and 2). Taken together with the library screening data, we conclude that PP1 interacts in vitro with HCF and this interaction depends upon a sequence contained within HCF between amino acids 1604 and 1956.
We next examined whether endogenous HCF and PP1 exist in a stable complex in HeLa nuclear extract (Fig. 2C). HCF was immunoprecipitated from a HeLa nuclear extract and the immune complexes resolved by SDS–PAGE. After transfer to a membrane, HCF and PP1 were each detected by immunoblotting with the respective antibodies. This shows that PP1 is specifically immunoprecipitated along with HCF, indicating that a common complex containing both HCF and PP1 is present in a crude nuclear extract (Fig. 2C). However, we note that only a small amount of the total PP1 in nuclear extract is associated with HCF. This is not surprising because major forms of nuclear PP1 are known which have the catalytic PP1 subunit bound to either of the nuclear regulators NIPP1 or FB19/p99/pNUTS (38 and references therein).
Native HCF complex contains a protein phosphatase activity
Since HCF and PP1 can co-exist as a complex in nuclear extract, we next tested whether the anti-HCF antibody can co-immunoprecipitate protein phosphatase activity. Initial protein phosphatase assays, using 32P-labelled rabbit skeletal muscle phosphorylase a as substrate, showed only background levels of phosphatase activity in anti-HCF immunoprecipitates (data not shown and Fig. 3A, lane 3). One possible reason for this is that the catalytic activity of the PP1 in this complex is inhibited as seen for forms of PP1 bound to other regulatory subunits. Tryptic cleavage can be used to remove inhibitory proteins from a complex with PP1 because the catalytic activity of PP1 is not affected by cleavage of the protein with trypsin. Treatment of the anti-HCF immunoprecipitates with trypsin resulted in a >20-fold increase in protein phosphatase activity (Fig. 3A, compare lanes 3 and 4). These results indicate that the PP1 activity in the complex with HCF is inhibited, at least towards standard assay substrates such as phosphorylase a. In this regard HCF behaves similarly to the other known nuclear PP1 regulators, NIPP1 and p99 (34,38,39). We observe that increasing the amount of anti-HCF antibody used for immunoprecipitation results in a parallel increase in both the protein phosphatase activity (Fig. 3B) and the amount of HCF polypeptides recovered in the immune complex (data not shown). This provides further evidence that a form of nuclear PP1 that contains HCF is present in HeLa nuclei.
Figure 3.

The HCF complex in nuclear extract contains a PP1 activity. Immunoprecipitates of HCF were assayed as described in the methods section for protein phosphatase activity using 32P-labelled skeletal muscle phosphorylase a as substrate. The results of the assay are represented as shaded bars in the figure. (A) Columns 1 and 2 represent pre-immune IgG (40 µg) immunoprecipitates assayed for protein phosphatase activity with (+) or without (–) trypsinization. Columns 3 and 4 indicate anti-HCF (40 µg) immunoprecipitates with column 4 assayed after trypsinization. Column 5 shows a control containing the enzyme trypsin, soybean trypsin inhibitor and substrate. (B) Protein phosphatase activity was assayed in anti-HCF immunoprecipitates after trypsin treatment using increasing amounts of anti-HCF antibody or pre-immune IgG. The graph with the squares represent protein phosphatase activity in immunoprecipitates using anti-HCF antibody while the graph with the diamond shapes shows the enzyme’s activity in immunoprecipitates using pre-immune IgG. (C) Inhibition of protein phosphatases in trypsin treated anti-HCF immunoprecipitates indicates the presence of a PP1 activity in the HCF complex. Column 1 shows phosphatase activity in pre-immune IgG immunoprecipitate. Column 2 represents the protein phosphatase activity obtained from assaying the anti-HCF immunoprecipitate while column 3 shows phosphatase activity after treatment of the anti-HCF immunoprecipitate with nanomolar amounts okadaic acid. Column 4 indicates the protein phosphatase activity after treatment with micromolar amounts of okadaic acid and column 5 represents the phosphatase activity of the anti-HCF immunoprecipitate after incubation in 0.5 µM inhibitor 2, a specific inhibitor of PP1.
HeLa nuclear extracts have been shown to contain both endogenous PP1 and PP2A activities (46). To confirm that the protein phosphatase activity released by trypsinization of the HCF immunoprecipitate is PP1 and not PP2A, the samples were treated with specific protein phosphatase inhibitors. PP2A is inhibited by nanomolar concentrations of okadaic acid, whereas PP1 is less sensitive to this inhibitor, requiring micromolar amounts of okadaic acid for complete inhibition. Conversely, inhibitor 2 is a potent inhibitor of PP1 but has no effect on PP2A activity (28). Figure 3C shows that 5 µM but not 2 nM okadaic acid was able to inhibit the phosphatase activity released by trypsin treatment of an anti-HCF immunoprecipitate. In addition, 0.5 µM inhibitor 2 completely abolished the phosphatase activity associated with HCF. Taken together, these data suggest that most or all of the phosphatase activity associated with HCF in these assays is a form of PP1.
Effect of a peptide containing a consensus PP1-binding motif on the HCF–PP1 complex in nuclear extract
The PP1 binding region of HCF (clone 17) shares no apparent sequence similarity with any of the previously characterised PP1 binding proteins (47). To investigate whether there was any higher order structural conservation in the PP1 binding domain between HCF and the other PP1 binding proteins, we performed a competition experiment. A synthetic peptide containing the putative PP1 binding domain of p53BP2 has previously been used to characterise the PP1 binding domain of the nuclear PP1 regulator p99 (33,38). By competition, this peptide is able to disrupt the p99–PP1 complex, thus releasing free PP1 activity. Other studies have also shown that peptides containing this motif can bind to PP1 and are capable of disrupting or attenuating the effects of PP1-binding proteins (48–51). Thus, endogenous HCF was immunoprecipitated from HeLa nuclear extract, challenged with p53BP2 peptide and then assayed for PP1 activity (Fig. 4A). This showed that the p53BP2 peptide caused release of PP1 activity. However, HCF immune complexes treated with either buffer or control peptide (38) did not release any phosphatase activity. To determine the extent of competitive PP1 release by the peptide, after the treatment with the p53BP2 or control peptides, the immune complexes were subsequently trypsinised. After challenge with either buffer or control peptide, trypsin treatment released comparable levels of PP1 activity (Fig. 4B, lanes 4 and 5). However, the anti-HCF immunoprecipitate challenged with the p53BP2 peptide showed as expected less increase in PP1 activity upon trypsin treatment (lane 6) because much of the PP1 had already been removed by treatment with the p53BP2 peptide. We estimate that ~65% of the PP1 activity in the HCF complex was released by the p53BP2 peptide, with the remaining 35% released by trypsinising the HCF–PP1 complex. Despite the absence of an obvious PP1 binding motif in HCF, the peptide competition data suggest that HCF interacts with PP1 through either the same or else a closely overlapping binding site to that recognised by p53BP2, p99 and other PP1 regulators. In summary, we conclude that a peptide containing a consensus PP1-binding motif can disrupt the HCF–PP1 interaction.
Figure 4.

Release of PP1 activity from the HCF complex by a peptide containing the consensus PP1-binding motif. HCF immunoprecipitates were incubated with 10 µM of the peptide containing the putative PP1 binding domain of p53BP2 with sequence: GKRTNLRKTGSERIAMGMRVKFNPLALLLDSC or with a control peptide lacking this domain (38). (A) Columns 1, 2 and 3 represent pre-immune IgG immunoprecipitates incubated with buffer (O), control peptide (C) and p53BP2 peptide (P) respectively. Columns 4, 5 and 6 show the PP1 activities obtained when the anti-HCF immune precipitates were incubated with buffer (O), control peptide (C) and p53BP2 peptide (P) respectively. (B) The immunoprecipitates described in (A) that had been incubated with or without PP1-binding peptides were further treated with trypsin to reveal remaining PP1 activity after incubation with the peptides. Columns marked 1, 2 and 3 were the same samples as in (A) except that the immunoprecipitates were treated with trypsin and then assayed for PP1 activity. The columns labelled 4, 5 and 6 represent the same samples as corresponding columns in (A) but with the anti-HCF immunoprecipitates trypsinised and then assayed for PP1 activity not released by peptides.
The HCF–PP1 complex and the HCF–VP16 induced complex
HCF was originally identified as a key cellular component involved in the recruitment of HSV VP16 to the TAATGARAT sequences located upstream of the IE genes of HSV. Indeed, HCF can be partially purified due to its ability to bind directly to VP16. We next investigated if there was any relationship between the PP1-binding activity of HCF and the role of HCF in VP16-mediated transcription complex formation. HCF was isolated by either GST-VP16 affinity chromatography or immunoprecipitation with anti-HCF antibodies (Fig. 5A). The isolated proteins were separated by SDS–PAGE, transferred to a membrane and analysed by immunoblotting with either anti-HCF antibody (upper panel) or anti-PP1 antibody (lower panel). Surprisingly, PP1 was only associated with the HCF isolated by immunoprecipitation, and not in the HCF fraction that was isolated through its binding to VP16. To characterise further the VP16–HCF and PP1–HCF interactions, we performed electrophoretic mobility shift assays to analyse the VP16-induced complex at a TAATGARAT DNA element (Fig. 5B, lanes 2–4). Incubation of HeLa cell nuclear extract with labelled probe produced a complex corresponding to the previously described interaction of Oct 1 with this sequence. Addition of GST-VP16 caused formation of a complex of lower mobility. This complex required HCF because it is disrupted by the addition of anti-HCF antibody (data not shown). In contrast, neither anti-PP1 nor anti-PP2A antibodies had any effect on the VP16-induced complex (data not shown). Addition of excess active bacterially expressed PP1 had no effect on the VP16-induced complex (Fig. 5B, lanes 6–8). Also we observed that the p53BP2 peptide had no effect on the ability of GST-VP16 to form a complex with HCF in nuclear extract (data not shown). This suggests that PP1 and VP16 are not competing for the same binding site in HCF and is consistent with the absence of PP1 in the HCF complexed with VP16; i.e. PP1 does not form part of the HCF-containing VP16-induced transcription complex.
Figure 5.
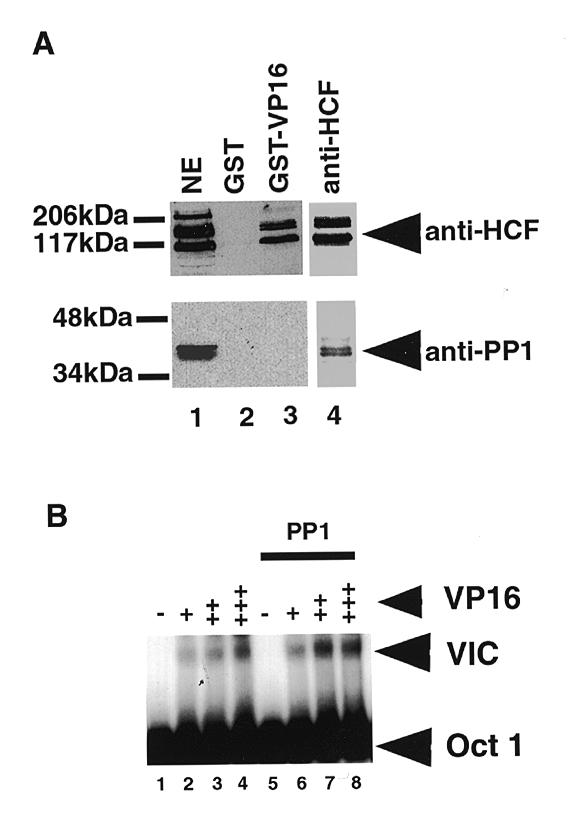
The PP1–HCF and VP16–HCF complexes in HeLa nuclear extracts. (A) About 10 µg GST-VP16 was used to pull down HCF from 0.4 to 0.5 mg of HeLa nuclear extract. Bound HCF on the beads was resuspended in 50 µl of SDS–PAGE buffer and 15 µl of the HCF pull-down probed with anti-PP1 antibody (bottom panel). The panel at the top shows a western blot of the pull-down probed with anti-HCF antibody. Lane 1 contained nuclear extract. Lanes 2 and 3 had GST and GST-VP16 pull-downs respectively. Lane 4 contained an immunoprecipitation from HeLa nuclear extract with anti-HCF antibody. The arrowheads indicate the major HCF polypeptides and PP1 respectively. Because of the high levels of PP1 in nuclear extract (Fig. 2C), the blot in the lower panel probed with anti-PP1 antibody contained ~15% of the amount of nuclear extract used in the corresponding lane in the top panel western blot probed with anti-HCF antibody. (B) The effect of adding active bacterially expressed PP1 on the VP16 induced complex. No PP1 was added to assays in lanes 1–4. The samples in lanes 5–8 contained ~250 ng of PP1. Varying the quantity of PP1 in the assays to levels below or above 250 ng did not have any significant effect on the formation of the VP16-induced complex. The + signs represent increasing amounts of VP16 while the – signs indicate the absence of VP16. +, 2 ng VP16; ++, 10 ng of VP16; and +++, 50 ng of the protein in the assay. The Oct-1-TAATGARAT complex (Oct 1) and VP16-induced complex (VIC) are indicated.
DISCUSSION
In this study we report the identification of eight human genes encoding PP1 binding proteins by screening a HeLa cDNA expression library in E.coli using digoxigenin-labelled PP1 as a probe. Two of the genes isolated encoded the previously described PP1 regulatory proteins NIPP1 and FB19/p99. One of the genes has not been previously reported and only its EST sequences are available in the sequence databases. The other five have full-length cDNA sequences in the databases but have never been shown to interact with or regulate the activity of any protein phosphatases. One of these, for which we isolated three independent cDNA clones encoded the protein HCF, a cellular factor implicated in transcription and cell cycle progression mechanisms. We characterised further the interaction between HCF and PP1 because this may offer new clues about the cellular role of HCF.
We detected an endogenous complex in HeLa nuclear extract that contains both HCF and PP1. However, in this complex PP1 activity towards phosphorylase a is inhibited and had to be released by treatment of the complex with trypsin. This indicates that the complex may contain a regulated form of nuclear PP1 activity. Because we also show that a fragment of HCF including amino acids 1604–1956 binds directly to PP1 in vitro, it is likely that HCF may act as a regulator of PP1 activity. These data suggest a new cellular function for HCF. It remains to be determined if HCF inhibits the activity of PP1 towards all substrates or if it, instead, restricts its substrate specificity and/or directs PP1 activity towards a specific substrate(s). Indeed, this is a feature of many PP1 regulatory proteins. Given the role of both HCF and PP1 in the cell cycle, one possibility is that their association might be involved in regulating cell cycle progression. HCF cDNA codes for a high molecular mass protein (~300 kDa) and attempts to express the full-length protein in E.coli have been unsuccessful. We observed that the recombinant C-terminal fragments of HCF that bind PP1 were not sufficient to inhibit the enzyme’s activity towards phosphorylase a. It is possible that the recombinant fragment is inactive in regulating the activity of PP1 because it lacks some post-translational modification or the regulatory domain is located at a different region of the HCF molecule. Alternatively, the HCF–PP1 complex may contain a third component that suppresses PP1 activity.
Previous studies of HCF have concentrated on its interaction with the viral protein VP16 and its involvement in the expression of the IE genes of HSV (5,9,18). Interestingly, we did not detect PP1 within the HCF complex that associates with VP16 even though HCF purified by VP16-affinity chromatography was capable of binding PP1 (data not shown). Furthermore, PP1 was not present in the HCF-containing complex that forms at the TAATGARAT DNA element. One possible explanation for these data is that distinct forms of the HCF complex exist and that the form that associates with VP16 is devoid of PP1 or the binding of VP16 to HCF may inhibit the protein’s binding to PP1. The latter possibility could not be investigated further due to problems in expressing full-length HCF in bacteria. Also, neither DIG–PP1 binding to membrane bound HCF nor the HCF–PP1 complex in immunoprecipitates were affected by addition of VP16 (data not shown). This means that if VP16 displaced PP1 from HCF in vivo, this may involve interactions with other proteins. We found that PP1 associates with the C-terminus of HCF, whereas the N-terminus of HCF has been shown to be sufficient for VP16-induced complex (VIC) formation and transcriptional activation of HSV IE genes in vitro (52). We also found that the p53BP2 peptide was unable to dissociate the VP16–HCF complex in a GST-VP16 pull-down assay (data not shown) although it released PP1 from the HCF–PP1 complex in HeLa nuclear extract. Taken together, these data would likely rule out simple competition by VP16 and PP1 for binding to the same site in HCF. It has been reported that treatment of purified HCF with potato acid phosphatase disrupts the interaction with VP16 (19). It is possible that PP1-bound HCF is dephosphorylated (possibly even by PP1 itself) and is therefore not a substrate for the VP16-induced complex. Another possibility is that VP16 displaces the PP1 from HCF by causing a conformational change that would disrupt interaction with other regions of HCF. This latter possibility is intriguing because, although the N-terminus of HCF is sufficient to interact with VP16, a role for the C-terminus of HCF in modulating the interaction has been reported (9). Thus, it is possible that VP16 can disrupt the interaction between HCF and PP1 in vivo, thereby deregulating PP1 activity.
Interestingly, herpes simplex virus 1 (HSV1) expresses a protein called γ134.5 that binds avidly to PP1 and it has been suggested that this interaction redirects PP1 to dephosphorylate elF-2α to enable continued protein synthesis in infected cells (53). It is possible that an HSV1 PP1-binding protein, such as γ134.5, may recruit PP1 from other binding proteins in virally infected cells, thus making more phosphorylated HCF available to interact with VP16. Increased levels of phosphorylated HCF will favour the activation of transcription of the viral IE genes. It would be of interest to further investigate if there is any difference in the phosphorylation status of HCF in the HCF–PP1 and the HCF–VP16 complexes. Alternatively, HCF–PP1 may regulate the dephosphorylation of VP16 thus rendering the protein inactive and unable to interact with HCF in the formation of the VP16-induced complex. Indeed, it has been shown that a single serine residue at position 375 of VP16 is a target for casein kinase II phosphorylation and that phosphorylation at this site is critical for the assembly of VP16 in a complex with Oct-1 and HCF. A mutation of serine 375 to alanine abolishes the activity of VP16 in vitro and in vivo (25). This may account for the absence of PP1 in the VP16–HCF complex in HeLa nuclear extract. Given the role of PP1 in cell cycle progression, such a model would be consistent with the changes in cell cycle observed upon HSV infection. Perhaps, in addition to exploiting HCF to target VP16 to the HSV IE promoter, VP16 deregulates normal HCF cellular functions in a manner akin to other viral transcription factors such as adenovirus E1a. For example E1a targets the CBP/p300 transcription factors to disrupt their normal cellular activities (54). Further experiments will be required to test these possibilities.
The sequence of the HCF binding region contained in our smallest HCF clone does not contain the consensus PP1 binding domain motif that has been observed in most of the previously identified PP1 regulatory subunits (47). Of the five other novel PP1-binding proteins identified in the cDNA expression library screen, four either have the R/K-V/I-x-F/W motif or amino acid sequences that are closely related to the PP1-binding consensus domain motif (55). Our attempts to identify a simple linear sequence motif that may be responsible for the binding of PP1 by HCF have so far not been successful although the peptide competition data indicate that HCF may bind to either the same or else a closely overlapping site on PP1 as that bound by other regulatory proteins. However, the previously identified PP1 binding protein inhibitor-2 does not contain the consensus PP1 binding motif as described above (56). It is possible that there may be some structural similarity between the PP1-binding domains of these proteins that is not apparent at the level of primary amino acid sequence. Further structural studies of PP1-interacting proteins should shed light on the nature of the PP1-binding domain.
In summary, we have identified HCF as a likely nuclear PP1 regulatory protein. This suggests several novel possibilities concerning the role of HCF in transcription and cell cycle control. These findings prompt new lines of investigation for future studies into the normal cellular function of HCF and its role upon HSV infection.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Martin Lowe for providing us with a HeLa cDNA expression library. We thank Winship Herr for his gift of antibodies to HCF, and VP16ΔC and HCF plasmids. We also thank Chris Preston and Ben Luisi for providing us with their VP16 and Oct 1 plasmids and Laura Trinkle-Mulcahy for a gift of GST-NIPP1. We are grateful to Philip Cohen for helpful suggestions and critical comments on the manuscript. The Wellcome Trust and the Medical Research Council funded this work. G.J.B. was supported by a postgraduate Studentship from the Royal Society (London). N.A.H. has a Research Studentship from the Biotechnology and Biological Sciences Research Council (BBSRC). S.G.E.R. is a Career Development Fellow of the Wellcome Trust. A.I.L. is a Wellcome Trust Principal Research Fellow.
REFERENCES
- 1.Thompson C.C. and McKnight,S.L. (1992) Trends Genet., 8, 232–236. [Google Scholar]
- 2.O’Hare P. (1993) In Davison,A.J. (ed.), The Alpha-Herpes Viruses. Saunders Scientific Publications, Philadelphia, PA, pp. 145–155.
- 3.Wilson A.C., Cleary,M.A., Lai,J.S., LaMarco,K., Peterson,M.G. and Herr,W. (1993) Cold Spring Harbor Symp. Quant. Biol., 58, 167–178. [DOI] [PubMed] [Google Scholar]
- 4.Kristie T.M., Pomeranz,J.L., Twomey,T.C., Parent,S.A. and Sharp,P.A. (1995) J. Biol. Chem., 270, 4387–4394. [DOI] [PubMed] [Google Scholar]
- 5.Kristie T.M., LeBowitz,J.H. and Sharp,P.A. (1989) EMBO J., 8, 4229–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kristie T.M. and Sharp,P.A. (1990) Genes Dev., 4, 2383–2396. [DOI] [PubMed] [Google Scholar]
- 7.Wilson A.C., LaMarco,K., Peterson,M.G. and Herr,W. (1993) Cell, 74, 115–125. [DOI] [PubMed] [Google Scholar]
- 8.Kristie T.M. (1997) J. Biol. Chem., 272, 26749–26755. [DOI] [PubMed] [Google Scholar]
- 9.LaBoissiere S., Walker,S. and O’Hare,P. (1997) Mol. Cell. Biol., 17, 7108–7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freiman R. and Herr,W. (1997) Genes Dev., 11, 3122–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu R., Yang,P., O’Hare,P. and Misra,V. (1997) Mol. Cell. Biol., 17, 5117–5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu R., Yang,P., Padmakumar,S. and Misra V. (1998) J. Virol., 72, 6291–6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goto H., Motomura,S., Wilson,A.C., Freiman,R.N., Nakabeppu,Y., Fukushima,K., Fujishima,M., Herr,W. and Nishimoto,T. (1997) Genes Dev., 11, 726–737. [DOI] [PubMed] [Google Scholar]
- 14.Kristie T.M. and Roizman,B. (1987) Proc. Natl Acad. Sci. USA, 84, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerster T. and Roeder,R.G. (1988) Proc. Natl Acad. Sci. USA, 85, 6347–6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Hare P. and Goding,C.R. (1988) Cell, 52, 435–445. [DOI] [PubMed] [Google Scholar]
- 17.Stern S., Tanaka,M. and Herr,W. (1989) Nature, 341, 624–630. [DOI] [PubMed] [Google Scholar]
- 18.Katan M., Haigh,A., Verrijer,C.P., van der Vliet,P.C. and O’Hare,P. (1990) Nucleic Acids Res., 18, 6871–6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kristie T. and Sharp,P. (1993) J. Biol. Chem., 268, 6525–6534. [PubMed] [Google Scholar]
- 20.Wilcox K.W., Kohn,A., Sklyanskaya,E. and Roizman,B. (1980) J. Virol., 33, 167–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moss H. (1989) J. Gen. Virol., 70, 1579–1585. [DOI] [PubMed] [Google Scholar]
- 22.Purves F.C., Spector,D. and Roizman,B. (1991) J. Virol., 65, 5757–5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Purves F.C. and Roizman,B. (1992) Proc. Natl Acad. Sci. USA, 89, 7310–7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coulter L.J., Moss,H.W.M., Lang,J. and McGeoch,D.J. (1993) J. Gen. Virol., 74, 387–395. [DOI] [PubMed] [Google Scholar]
- 25.O’Reilly D., Hanscombe,O. and O’Hare,P. (1997) EMBO J., 16, 2420–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hubbard M.J. and Cohen,P. (1993) Trends Biochem. Sci., 18, 172–177. [DOI] [PubMed] [Google Scholar]
- 27.Faux M.C. and Scott,J.D. (1996) Trends Biochem. Sci., 21, 312–315. [PubMed] [Google Scholar]
- 28.Cohen P. (1989) Annu. Rev. Biochem., 58, 453–508. [DOI] [PubMed] [Google Scholar]
- 29.Bollen M. and Stalmans,W. (1992) Crit. Rev. Biochem. Mol. Biol., 27, 227–281. [DOI] [PubMed] [Google Scholar]
- 30.Mistelli T. and Spector,D.L. (1997) Trends Cell Biol., 7, 135–138. [DOI] [PubMed] [Google Scholar]
- 31.Fernandez A., Brautigan,D.L. and Lamb,N.J.C. (1992) J. Cell Biol., 116, 1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Y.H., Chen,M.X., Alessi,D.R., Campbell,D.G., Shanahan,C., Cohen,P. and Cohen,P.T.W. (1994) FEBS Lett., 356, 51–55. [DOI] [PubMed] [Google Scholar]
- 33.Helps N.R., Barker,H.M., Elledge,S.J. and Cohen,P.T.W. (1995) FEBS Lett., 377, 295–300. [DOI] [PubMed] [Google Scholar]
- 34.Beullens M., Eynde,A.V., Stalmans,W. and Bollen,M. (1992) J. Biol. Chem., 267, 16538–16544. [PubMed] [Google Scholar]
- 35.Jagiello I., Beullens,M., Stalmans,W. and Bollen,M. (1995) J. Biol. Chem., 270, 17257–17263. [DOI] [PubMed] [Google Scholar]
- 36.Eynde A.V., Wera,S., Beullens,M., Torrekens,S., Leuven,F.V., Stalmans,W. and Bollen,M. (1995) J. Biol. Chem., 270, 28068–28074. [DOI] [PubMed] [Google Scholar]
- 37.Kawabe T., Muslin,A.J. and Korsmeyer,S.J. (1997) Nature, 385, 454–458. [DOI] [PubMed] [Google Scholar]
- 38.Kreivi J.-P., Trinkle-Mulcahy,L., Lyon,C.E., Morrice,N.A., Cohen,P. and Lamond,A.I. (1997) FEBS Lett., 420, 57–62. [DOI] [PubMed] [Google Scholar]
- 39.Allen P.B., Kwon,Y.-G., Nairn,A.C. and Greengard,P. (1998) J. Biol. Chem., 273, 4089–4095. [DOI] [PubMed] [Google Scholar]
- 40.Barker H.M., Craig S.P., Spurr N.K. and Cohen P.T. (1993) Biochim. Biophys. Acta, 1178, 228–233. [DOI] [PubMed] [Google Scholar]
- 41.Alessi D., MacDougall,L.K., Sola,M.M., Ikebe,M. and Cohen,P. (1992) Eur. J. Biochem., 210, 1023–1035. [DOI] [PubMed] [Google Scholar]
- 42.Bressan G.M. and Stanley,K.K. (1987) Nucleic Acids Res., 15, 10056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin Y.-S. and Green,M.R. (1991) Cell, 64, 971–981. [DOI] [PubMed] [Google Scholar]
- 44.Harlow E. and Lane,D. (1988) Antibodies, A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 45.Cohen P., Alemany,S., Hemmings,B.A., Resink,T.J., Stralfors,P. and Tung,H.Y.L. (1988) Methods Enzymol., 159, 390–408. [DOI] [PubMed] [Google Scholar]
- 46.Mermoud J.E., Cohen P. and Lamond,A.I. (1992) Nucleic Acids Res., 20, 5263–5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He B., Gross,M. and Roizman,B. (1998) J. Biol. Chem., 273, 20737–20743. [DOI] [PubMed] [Google Scholar]
- 48.Johnson D.F., Moorhead,G., Caudwell,F.B., Cohen,P., Chen,Y.H., Chen,M.X. and Cohen,P.T.W. (1996) Eur. J. Biochem., 239, 317–325. [DOI] [PubMed] [Google Scholar]
- 49.Zhao S. and Lee,E.Y.C. (1997) J. Biol. Chem., 272, 28368–28372. [DOI] [PubMed] [Google Scholar]
- 50.Kwon Y.G., Huang,H.B., Desdouits,F., Girault,J.A., Greengard,P. and Nairn,A.C. (1997) Proc. Natl Acad. Sci. USA, 94, 3536–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Egloff M.P., Johnson,D.F., Moorhead,G., Cohen,P.T., Cohen,P. and Barford,D. (1997) EMBO J., 16, 1876–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilson A.C., Freiman,R.N., Goto,H., Nishimoto,T. and Herr,W. (1997) Mol. Cell. Biol., 17, 6139–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.He B., Gross,M. and Roizman,B. (1997) Proc. Natl Acad. Sci. USA, 94, 843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shikama N., Lyon,J. and LaThangue,N.B. (1997) Trends Cell Biol., 7, 230–236. [DOI] [PubMed] [Google Scholar]
- 55.Zhoa S., Xia,W. and Lee,E.Y.C. (1996) Arch. Biochem. Biophys., 325, 82–90. [DOI] [PubMed] [Google Scholar]
- 56.Helps N.R., Street,A., Elledge,S. and Cohen,P.T.W. (1994) FEBS Lett., 340, 93–98. [DOI] [PubMed] [Google Scholar]