

Abstract
The 16p11.2 BP4 and BP5 region, is a recurrent ~600 kb copy number variant (CNV), and deletions are one of the most frequent etiologies of neurodevelopmental disorders and autism spectrum disorder with an incidence of approximately 1/2000. Deletion carriers have delays in early neurodevelopment that most specifically impair speech, phonology and language in 70%. Intelligence quotient is shifted 1.8 standard deviations lower than family controls without the deletion. Other common neurobehavioral conditions include motor coordination difficulties (60%) and autism (20–25%). Unprovoked seizures are common (24%) and readily treated and resolve with age in many. Obesity evolves throughout childhood and by adulthood 75% are obese. Congenital anomalies are more common than the general population. The deletion is associated with an increase in brain volumes across all areas of the brain, changes in the white matter microstructural properties, and early electrophysiological cortical responses from auditory cortex. Studies of genetically defined conditions, particularly CNVs that are not associated with profound disabilities, provide homogeneity to study genetic impact on brain development, structure, and function to better understand complex neurobehavioral phenotypes such as autism.
Introduction
The recurrent ~600 kb copy number variant (CNV) at 16p11.2 BP4 and BP5 deletion is flanked by highly homologous blocks of low-copy repeats (Figure 1) and is one of the most frequent etiologies of neurodevelopmental disorders. Reciprocal deletions and duplications are observed with a prevalence of approximately 1/2000 and 1/1100 in the general population, respectively [1]. Most of this review will focus on 16p11.2 deletion carriers although we will briefly compare and contrast characteristics of duplication carriers. The deletion is most often de novo but is inherited in approximately 7% of probands. It is one of the most common known genetic etiologies of autism spectrum disorders (ASD) (~0.5%) [2–6]. To better understand the 16p11.2 deletion on the brain structure, cognition, and behavior, we created the Simons Variation in Individuals Project VIP consortia to recruit and study a large number of individuals with this deletion [7] and have combined our data with a similar European consortia [7–10].
Figure 1.
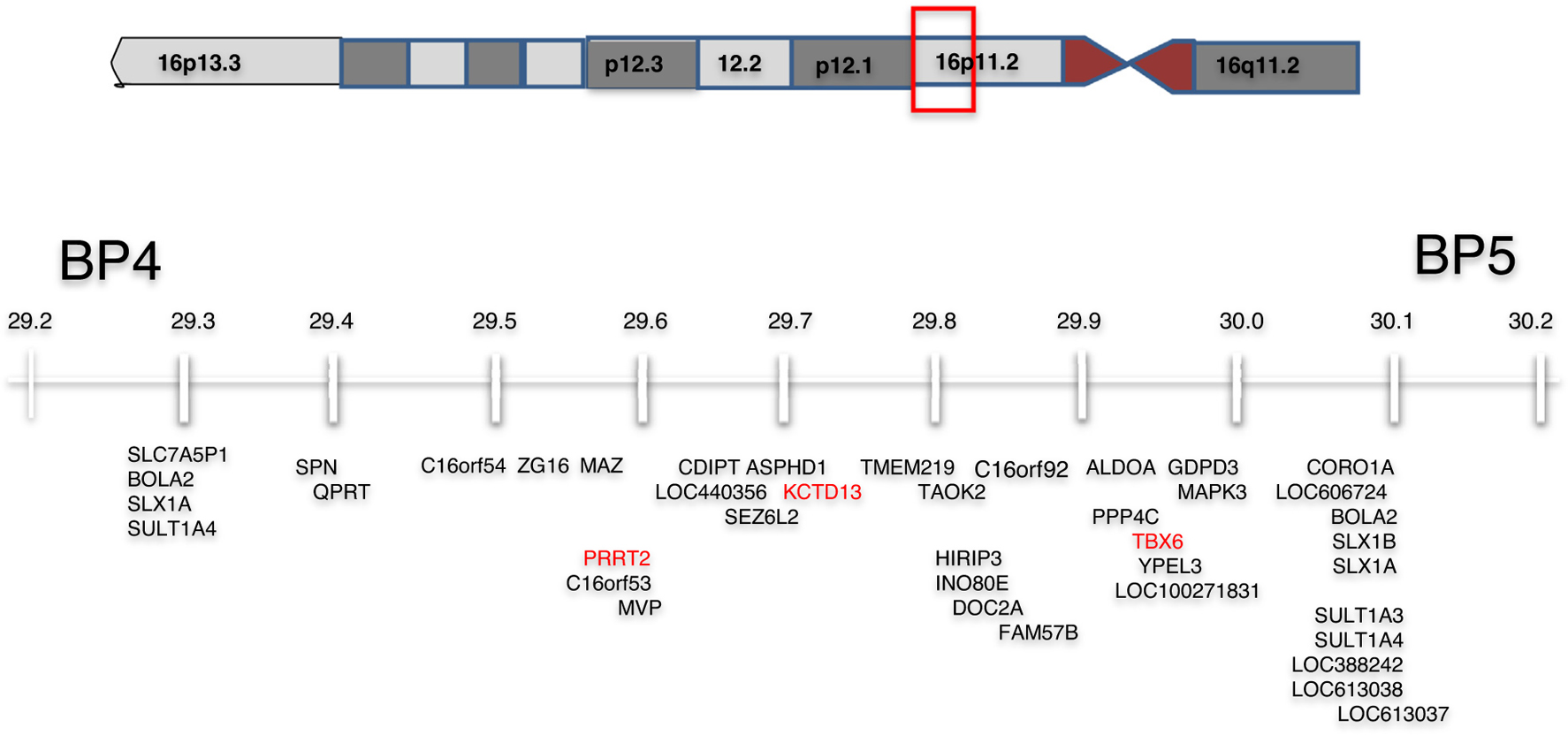
The gene content of the BP4-BP5 region of 16p11.2 with genes of interest potentially responsible for portions of the phenotype highlighted in red.
The Simons VIP project, launched in 2010 and funded by the Simons Foundation Autism Research Initiative (SFARI), is based upon a ‘genetics first’ approach to increase the homogeneity of genetic subtypes of ASD and to study a large number of individuals who share highly penetrant CNVs and monogenic conditions, recruited without regard to clinical diagnosis. A key aim is to study clinical phenotypes longitudinally and to assess similarities and differences between genetic etiologies.
Phase 1 of the project focused on direct, detailed phenotyping of individuals with deletions and duplications of 16p11.2. Individuals diagnosed clinically were recruited through internet searches, clinical testing laboratories, and physicians. Cascade testing in families was also performed to identify additional carriers to address the ascertainment bias of those seeking a clinical diagnosis. Evaluations involved neurologic evaluation, psychometric testing, and structural brain imaging for those who could cooperate without sedation. A subset of the carriers returned for detailed functional imaging (fMRI) and MEG. Data were also analyzed in combination with a European 16p11.2 cohort, recruited by the referring physician [9].
Phase 2 of the project, which has been renamed ‘Simons Searchlight’ (https://www.sfari.org/resource/simons-searchlight/) was designed to greatly expand the cohort size and number of conditions studied by converting the study to remote online and telephonic caregiver report and medical records review. Blood from a subset of individuals is stored at a central repository, and the project is starting to distribute iPS cells made from these samples to the research community. Importantly, the cohort is designed to be recontactable, so investigators can request access to 179 individuals for additional research questions.
Medical features
Anthropometrics are affected by the 16p11.2 deletion. Although head circumference is smaller at birth, there is an overall increase in head circumference by 2 years of age and macrocephaly (Z score ≥2) is present in 17% of carriers (Figure 2a) [10]. Obesity and increased body mass index (BMI) emerge, usually during childhood. Birth weight is below average (Z score = −0.61), but Z scores for BMI are significantly higher by the age of 10 years (Z score = 1.0). Longitudinal data show that this increase in BMI can be sudden and dramatic. Among adults, ~75% are obese and among all adult obese patients 45% are morbidly obese, associated with hyperphagia [11]. Malformations or major medical problems are present in just over half of deletion carriers and most have only a single medical problem although most individuals studied are children or young adults. No specific recurrent malformation sequence or multisystemic involvement is observed, and the 16p11.2 deletion should not be regarded primarily as a malformation syndrome. Infrequent malformations include cerebro-cortical malformations, craniosynostosis, coloboma, microphthalmia, hypospadias, cryptorchidism, vesico-ureteral reflux, pyloric stenosis, congenital heart disease, and cleft palate/velopharyngeal insufficiency. The most common anomaly is vertebral abnormalities (hemivertebrae or kyphoscoliosis affect ~20% of carriers) [10]. Facial dysmorphism are present in half of carriers, but the features are not striking or easily recognized.
Figure 2.
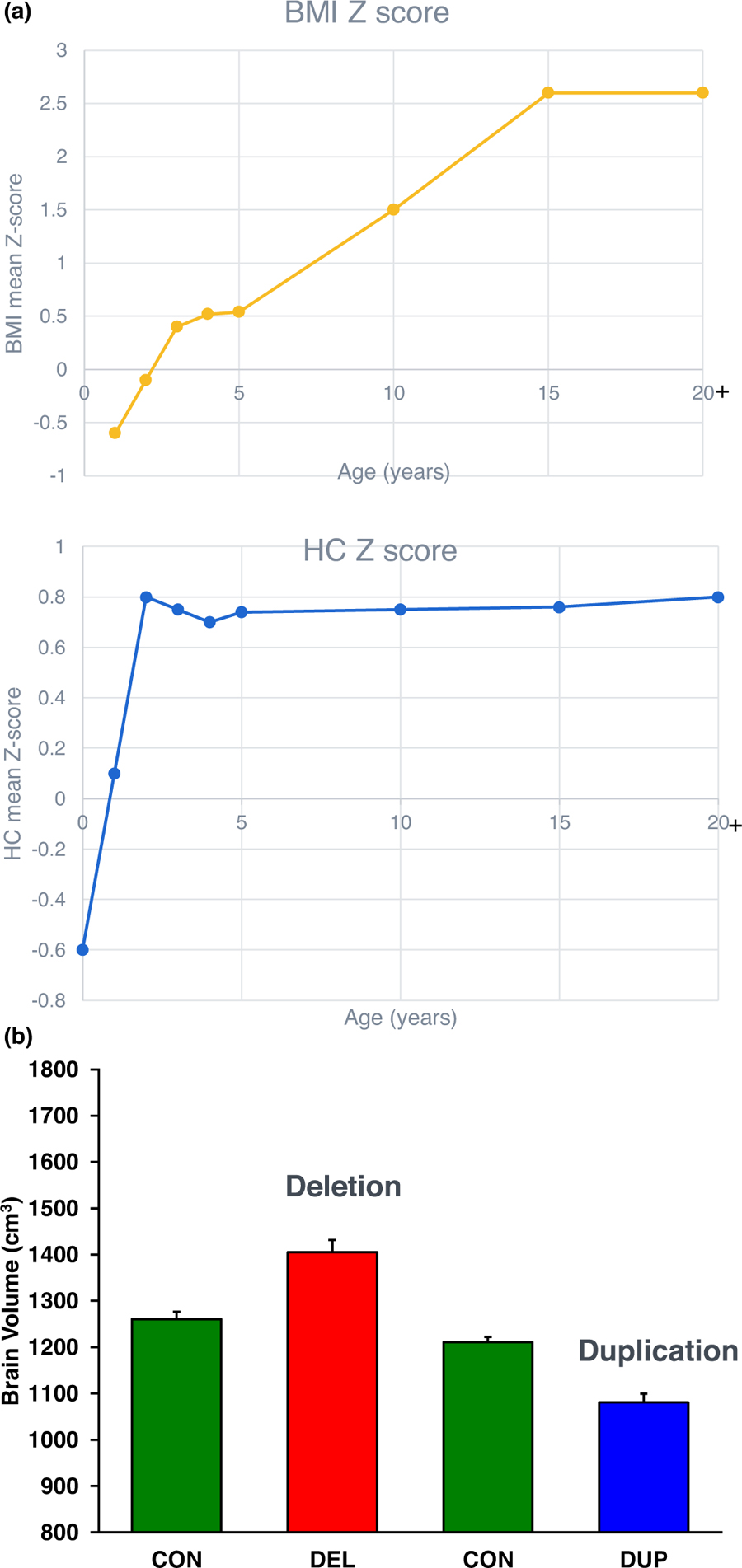
(a) Anthropometric measures of 16p11.2 carriers over time. Body mass index (BMI) increases over childhood, and head circumference increases over the first two years of age. (b) Brain volume as determined by brain MRI of 16p11.2 deletion carriers (DEL) relative to controls (CON) and duplication carriers (DUP) compared to controls (CON) shows relatively larger brain volumes in deletion carriers and smaller brain volumes in duplication carriers [29].
Behavioral features
Children with 16p11.2 deletion have a neurobehavioral profile dominated by pervasive speech and language impairment (>70%) and motor coordination difficulties (~60%) in the majority of individuals, with learning and intellectual disabilities (20%), autism (20–25%), and other behavioral/psychiatric issues (Figure 3) observed in a minority of individuals [6,10,12–17]. Intellectual ability shows a predictable decrement relative to family members (Figure 4a) [18]. The effect of the 16p11.2 deletion is characterized by specific disruption of speech, phonology and language, and less overall intellectual impairment [19–22]. As they age, young children with the deletion improve in verbal abilities, and have persistent delays and deficits in motor coordination and social skills, which require ongoing support [23].
Figure 3.
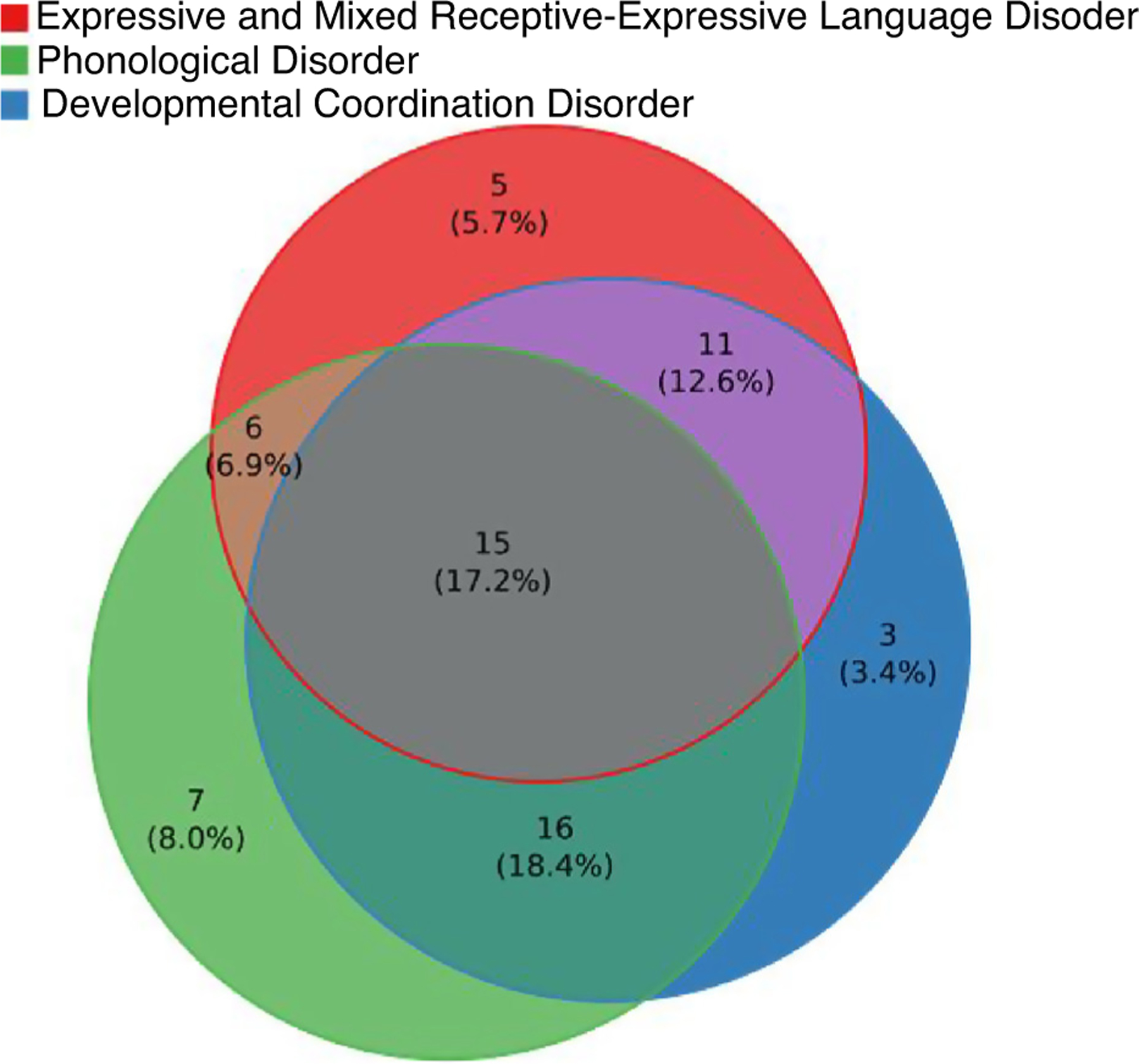
Diagnostic behavioral profile of 16p11.2 deletion carriers. Most individuals have more than one diagnosis.
Figure 4.
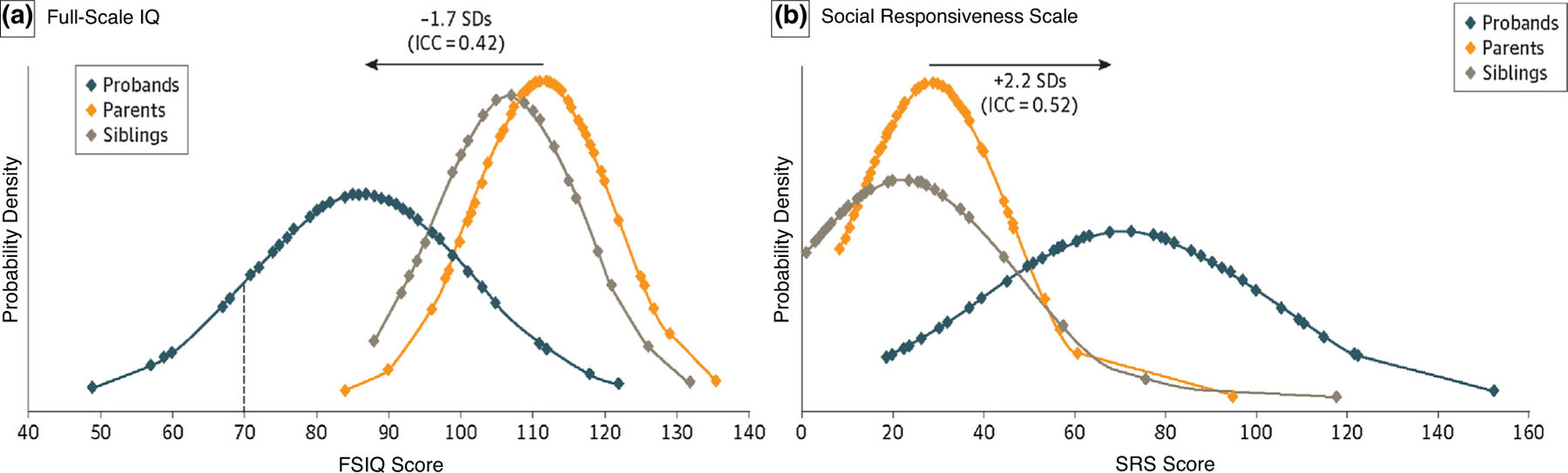
(a) 16p11.2 deletion carriers have full scale intelligence quotient (FSIQ) of 1.8 standard deviations lower than their non-carrier siblings and parents [18]. (b) 16p11.2 deletion carriers have social responsiveness scale score of 1.7 standard deviations higher (worse) than their non-carrier siblings [18].
Detailed examination of language strongly associates the 16p11.2 deletion with childhood apraxia of speech, which is seen in a majority (77%) of children and half of adults [24,25], as well as articulation deficits and clinical dysarthria. Furthermore, impairments to language production, including syntax and higher level pragmatics, persist even after controlling for ASD diagnosis and cognitive impairment [26].
In terms of behavioral and psychiatric issues, a recent report from the U.K. IMAGINE-ID study confirmed a broad range of neurobehavioral features in 16p11.2 deletion, with the most frequent traits relating to hyperactivity and ASD [27]. Taken with the ECHO and European 16p11.2 Consortium cohorts, disinhibited behavior, attention deficit hyperactivity disorder (ADHD) and ASD diagnoses were again the most common (20–25%) [8]. Even in children without a diagnosis of autism, traits such as repetitive behaviors and poor social skills are common, and result in a significant shift in Social Responsiveness Scale scores from familial controls (Figure 4b) [18]. However, early concerns regarding risk of schizophrenia have not been borne out in more recent studies.
Neurological features
We conducted a comprehensive chart review and in person examination by a child neurologist of 136 deletion carriers and identified features including symmetric hypotonia (50%), abnormal agility or clumsiness (47%), tremor (equivalent to essential tremor-25%) and increased reflexes/clonus (13%) in deletion carriers [28]. Sacral dimples were noted in 34%, but without an associated tethered cord when assess by imaging. Unprovoked seizures were initially reported in 22% of deletion carriers and a more comprehensive study of epilepsy found that among 129 individuals with 16p11.2 deletions, 24% had at least one seizure and 21 of them (16%) went on to have recurrent unprovoked seizures. Nearly half of the individuals with epilepsy could be classified as having benign infantile epilepsy (likely due to deletion of PRRT2). Most individuals with epilepsy were well controlled or outgrew their seizures and only a minority had intractable epilepsy (Christelle Moufawad El Achkar, personal communication).
Brain imaging and comparisons between deletion and duplication carriers
A comprehensive battery of neuroimaging techniques (magnetic resonance imaging (MRI) and magnetoen-cephalography (MEG)) was used to assess structural and functional differences in the brain. The 16p11.2 deletion was associated with increased volumetric measures of brain structures (as well as total brain size and intracranial volume, ICV) (Figure 2b) [29]
In addition to the research imaging studies of higher functioning 16p11.2 carriers, we also centrally assessed clinical brain MRIs with the hypothesis that the greater range of clinical impairment would enable us to detect a greater range of structure/function relationships [30]. Indeed, we found that deletion carriers had a thicker corpus callosum compared to controls and that 30% of the deletion carriers had cerebellar tonsillar ectopia and 9% had radiologically defined Chiari I malformations [31]. A thicker corpus callosum, as assessed by neuroradiologists blinded to genetic status was found to correlate with scores on adaptive behavioral assessments for these deletion carriers (p < 0.003).
When considering brain white matter microstructural properties derived from diffusion tensor imaging (DTI), 16p11.2 deletion was associated with widespread increases in diffusion ‘fractional anisotropy’ [32], suggesting atypical white matter maturation, with implications for brain connectivity and large-scale network function. In MEG studies, assessing neural function on a millisecond scale, early cortical responses from auditory cortex (specifically, the ~100 ms ‘M100’ response) showed response latency delays of ~20 ms in 16p11.2 deletion carriers compared to age-matched controls [33]. Whether this delay arises from impaired propagation along the auditory pathway (from ear to brainstem to cortex) or whether it relates to cortico-cortical delay remains unknown. Similarly, whether the delays can be attributed to white matter conduction versus synaptic transmission remains unresolved. Finally, magnetic mismatch field (MMF), a signature of the detection of a ‘change’ in the auditory stream, showed delays in 16p11.2 deletion carriers [34]. Interestingly, these delays were associated with both general cognitive (nonverbal IQ) and language-domain specific (CELF-5) clinical assessments. As such, while the structural and microstructural imaging, and early electrophysiological responses show sensitivity to gene dosage, the MMF delays demonstrate correlation with clinical presentation and behavioral phenotype.
Comparisons of 16p11.2 deletion and duplication carriers
Carriers of the 16p11.2 duplication are relatively common and inherited in 76% of probands and provide an excellent opportunity to study genes dosage effects in comparison with deletion carriers. Duplication carriers are clinically less severely impacted than deletion carriers, and many duplications carriers identified from population-based screening or cascade testing in clinically ascertained families are neurotypical using standardized measures of cognition and behavior and through review of medical records [1,28,35]. Reciprocal to the deletion carriers with larger head circumference, duplication carriers have relatively smaller head circumference. On brain MRI, there are ‘mirrored’ association of generalized increased brain volume in deletion carriers and reduced brain volume in duplication carriers. The DTI findings in deletion carriers directly oppose observations in 16p11.2 duplication carriers, echoing the brain structural findings and again implicating a gene-dosage influence on microstructural properties of brain white matter, and ultimately, brain structural connectivity. In assessing early cortical responses, delayed M100 responses observed in deletion carriers were not seen in 16p11.2 duplication carriers. This may not be surprising given the evidence linking such auditory cortex evoked response latencies to the microstructural properties of auditory pathway white matter [36]. Finally, and in contradistinction to the above opposing effects observed in structural and microstructural imaging as well as early electrophysiological responses, magnetic mismatch field (MMF), a signature of the detection of a ‘change’ in the auditory stream, showed delays in both 16p11.2 deletion and duplication carriers [34]. Resolution of the primary and secondary effects of gene dosage on brain structure and function is only possible with the comprehensive and detailed paradigmatic approaches applied in parallel to comparable groups of deletion and duplication carriers.
What accounts for variability among deletion carriers?
There is significant phenotypic variability among carriers of 16p11.2 deletions, and the etiology is still unclear; but some preliminary analyses suggest that perinatal complications and additional CNVs may be factors in predicting greater severity in features [37,38]. The 16p11.2 deletion is contained within the context of the rest of the genome, with other rare and common genetic variants contributing to these medical and neurobehavioral phenotypes. More complete genomic characterization and understanding of the genome may allow us to identify some of these additional contributing factors over time.
The 16p11.2 deletion is associated with somewhat non-specific phenotypes, and the question of penetrance is frequently raised. Penetrance is age-dependent and can be considered for each of the individual associated features (head size, brain volume, ASD, intellectual disability, language and coordination disorders, seizures, obesity, or congenital anomalies). However, it is important to also consider how the deletion shifts the distribution of quantitative traits: on average as a group, IQ tends to be shifted downward from parental IQ by 1.7 standard deviations and head circumference is shifted upward by 0.6 standard deviations, although there are individual exceptions.
In terms of discrete phenotypes such as medically recognizable congenital anomalies, the majority of these abnormalities are infrequent, suggesting either fortuitous associations or low penetrance. We have evaluated the original Simons VIP cohorts for possible unmasking of recessive mutations in the deletion interval, but we have found no support for this mechanism. Of the 29 genes within the interval, three (MAZ, TAOK2, CORO1A) are dosage sensitive to loss of function with a pLI >0.9 and could be major contributors to the phenotype. TBX6, which maps within the interval, is a candidate gene for vertebral malformations since it is associated with auto-somal dominant and recessive spondylocostal dysostosis 5 (OMIM #122600), and mice homozygous for a Tbx6 mutation showed rib and vertebral body anomalies [39,40]. Mutations in PRRT2 have been identified in patients diagnosed with epilepsy and paroxysmal dyskinesia (OMIM # 128200, 602066, 605751) [41]. A 118 kb deletion that encompasses MVP, CDIPT, SEZ6L2, ASPHD1, and KCTD13 segregated with ASD and other neurodevelopmental abnormalities but not epilepsy in a single three generation family [42]. Experimentally reducing expression of the KCTD13 ortholog in zebrafish produced macrocephaly and knock out in mice resulted in an increase of proliferating cells [43].
Future directions
Diagnoses of all CNVs, including 16p11.2 deletion are increasing as access to clinical chromosome microarrays is increasing in the prenatal and pediatric setting for indications including congenital anomalies, developmental delay/intellectual disability, ASD, and epilepsy. Individuals are being diagnosed earlier, even prenatally, offering the opportunity for early intervention for neurobehavioral conditions including ASD, early recognition and treatment of seizures, and even control of body weight. Clinical Research Associates will soon be launching a clinical trial for the use of arbaclofen for the 16p11.2 deletion based partly upon data from three different mouse models of 16p11.2 (syntenic 7qF3 region; 3 different background strains) demonstrating that in many cases arbaclofen had a normalizing effect on spontaneous open-field activity as well as contextual fear memory deficits [44]. With this and other trials, we will test the ability to develop treatments for ASD and related neurodevelopmental disorders based upon a genetics first strategy.
We are continuing to expand the cohort of 16p11.2 deletion and duplication carriers and follow them longitudinally over time. To expand access to study participation, data are collected remotely online and by telephone interviews. We are exploring additional methods to collect online phenotyping data and use of wearables and sensors in a home environment to collect more naturalistic quantitative data about behaviors including sleep that may provide insights into the brain and behavior. Studies of genetically more homogeneous groups of individuals with reciprocal deletions and duplications who are able to cooperate with detailed neuropsychometric and neuroimaging protocols longitudinally over time provide an invaluable opportunity to understand primary and secondary effects of genes on brain structure and function and complex human behavior. Large numbers of individuals are necessary to robustly interpret results and support the strategy of studying relatively common genetic variants. If sufficient numbers of individuals with the same CNV can be assessed, it may also be possible to study additional genetic, familial, environmental, and/or developmental factors that contribute to the complexity and variability of the phenotype among some individuals carrying the same CNV.
Acknowledgements
This work was supported by the Simons Foundation Autism Research Initiative. We are grateful to all of the families at the participating Simons Searchlight sites, as well as the Simons VIP (Simons Searchlight) Consortium. We appreciate obtaining access to the phenotypic, genetic and imaging data on SFARI Base. Approved researchers can obtain the Simons Searchlight population dataset described in this study by applying at https://base.sfari.org.
Footnotes
Conflict of interest statement
TR declares equity positions in Prism Clinical Imaging and Proteus Neurodynamics. He also declares consulting/advisory board relationships with CTF, Ricoh, Spago Nanomedicine, Avexis and Acadia Pharmaceuticals.
LGS and JES are employees of the Simons Foundation.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
- 1.Mannik K, Magi R, Mace A, Cole B, Guyatt AL, Shihab HA, Maillard AM, Alavere H, Kolk A, Reigo A et al. : Copy number variations and cognitive phenotypes in unselected populations. JAMA 2015, 313:2044–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh KM, Bracken MB: Copy number variation in the dosage-sensitive 16p11.2 interval accounts for only a small proportion of autism incidence: a systematic review and meta-analysis. Genet Med 2011, 13:377–384. [DOI] [PubMed] [Google Scholar]
- 3.Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, Gilliam TC, Nowak NJ, Cook EH Jr, Dobyns WB, Christian SL: Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet 2008, 17:628–638. [DOI] [PubMed] [Google Scholar]
- 4.Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y et al. : Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet 2008, 82:477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J et al. : Strong association of de novo copy number mutations with autism. Science 2007, 316:445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T et al. : Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med 2008, 358:667–675. [DOI] [PubMed] [Google Scholar]
- 7.Simons VIP Consortium: Simons Variation in Individuals Project (Simons VIP): a genetics-first approach to studying autism spectrum and related neurodevelopmental disorders. Neuron 2012, 73:1063–1067. [DOI] [PubMed] [Google Scholar]
- 8.Niarchou M, Chawner S, Doherty JL, Maillard AM, Jacquemont S, Chung WK, Green-Snyder L, Bernier RA, Goin-Kochel RP, Hanson E et al. : Correction: psychiatric disorders in children with 16p11.2 deletion and duplication. Transl Psychiatry 2019, 9:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin-Brevet S, Rodriguez-Herreros B, Nielsen JA, Moreau C, Modenato C, Maillard AM, Pain A, Richetin S, Jonch AE, Qureshi AY et al. : Quantifying the effects of 16p11.2 copy number variants on brain structure: a multisite genetic-first study. Biol Psychiatry 2018, 84:253–264. [DOI] [PubMed] [Google Scholar]
- 10.Zufferey F, Sherr EH, Beckmann ND, Hanson E, Maillard AM, Hippolyte L, Mace A, Ferrari C, Kutalik Z, Andrieux J et al. : A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. J Med Genet 2012, 49:660–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gill R, Chen Q, D’Angelo D, Chung WK: Eating in the absence of hunger but not loss of control behaviors are associated with 16p11.2 deletions. Obesity (Silver Spring) 2014, 22:2625–2631. [DOI] [PubMed] [Google Scholar]
- 12.Jacquemont S, Reymond A, Zufferey F, Harewood L, Walters RG, Kutalik Z, Martinet D, Shen Y, Valsesia A, Beckmann ND et al. : Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature 2011, 478:97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bijlsma EK, Gijsbers AC, Schuurs-Hoeijmakers JH, van Haeringen A, Fransen van de Putte DE, Anderlid BM, Lundin J, Lapunzina P, Perez Jurado LA, Delle Chiaie B et al. : Extending the phenotype of recurrent rearrangements of 16p.2: deletions in mentally retarded patients without autism and in normal individuals. Eur J Med Genet 2009, 52:77–87. [DOI] [PubMed] [Google Scholar]
- 14.Rosenfeld JA, Coppinger J, Bejjani BA, Girirajan S, Eichler EE, Shaffer LG, Ballif BC: Speech delays and behavioral problems are the predominant features in individuals with developmental delays and 16p11.2 microdeletions and microduplications. J Neurodev Disord 2010, 2:26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA, Probst FJ, Craigen WJ, Graham BH, Pursley A et al. : Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet 2010, 47:332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K, Arnarsdottir S, Bjornsdottir G, Walters GB, Jonsdottir GA, Doyle OM et al. : CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 2014, 505:361–366. [DOI] [PubMed] [Google Scholar]
- 17.Hanson E, Nasir RH, Fong A, Lian A, Hundley R, Shen Y, Wu BL, Holm IA, Miller DT: p11.2 study group C: cognitive and behavioral characterization of 16p11.2 deletion syndrome. J Dev Behav Pediatr 2010, 31:649–657. [DOI] [PubMed] [Google Scholar]
- 18.Moreno-De-Luca A, Evans DW, Boomer KB, Hanson E, Bernier R, Goin-Kochel RP, Myers SM, Challman TD, Moreno-De-Luca D, Slane MM et al. : The role of parental cognitive, behavioral, and motor profiles in clinical variability in individuals with chromosome 16p11.2 deletions. JAMA Psychiatry 2015, 72:119–126. [DOI] [PubMed] [Google Scholar]
- 19.Hanson E, Bernier R, Porche K, Jackson FI, Goin-Kochel RP, Snyder LG, Snow AV, Wallace AS, Campe KL, Zhang Y et al. : The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biol Psychiatry 2015, 77:785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Green Snyder L, D’Angelo D, Chen Q, Bernier R, Goin-Kochel RP, Wallace AS, Gerdts J, Kanne S, Berry L, Blaskey L et al. : Autism spectrum disorder, developmental and psychiatric features in 16p.2 duplication. J Autism Dev Disord 2016, 46:2734–2748. [DOI] [PubMed] [Google Scholar]
- 21.D’Angelo D, Lebon S, Chen Q, Martin-Brevet S, Snyder LG, Hippolyte L, Hanson E, Maillard AM, Faucett WA, Mace A et al. : Defining the effect of the 16p.2 duplication on cognition, behavior, and medical comorbidities. JAMA Psychiatry 2016, 73:20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hippolyte L, Maillard AM, Rodriguez-Herreros B, Pain A, Martin-Brevet S, Ferrari C, Conus P, Mace A, Hadjikhani N, Metspalu A et al. : The number of genomic copies at the 16p11.2 locus modulates language, verbal memory, and inhibition. Biol Psychiatry 2016, 80:129–139. [DOI] [PubMed] [Google Scholar]
- 23.Bernier R, Hudac CM, Chen Q, Zeng C, Wallace AS, Gerdts J, Earl R, Peterson J, Wolken A, Peters A et al. : Developmental trajectories for young children with 16p11.2 copy number variation. Am J Med Genet B Neuropsychiatr Genet 2017, 174:367–380. [DOI] [PubMed] [Google Scholar]
- 24.Fedorenko E, Morgan A, Murray E, Cardinaux A, Mei C, Tager-Flusberg H, Fisher SE, Kanwisher N: A highly penetrant form of childhood apraxia of speech due to deletion of 16p11.2. Eur J Hum Genet 2016, 24:302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mei C, Fedorenko E, Amor DJ, Boys A, Hoeflin C, Carew P, Burgess T, Fisher SE, Morgan AT: Deep phenotyping of speech and language skills in individuals with 16p11.2 deletion. Eur J Hum Genet 2018, 26:676–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim SH, Green-Snyder L, Lord C, Bishop S, Steinman KJ, Bernier R, Hanson E, Goin-Kochel RP, Chung WK: Language characterization in 16p11.2 deletion and duplication syndromes. Am J Med Genet B Neuropsychiatr Genet 2020, 183:380–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chawner S, Owen MJ, Holmans P, Raymond FL, Skuse D, Hall J, van den Bree MBM: Genotype-phenotype associations in children with copy number variants associated with high neuropsychiatric risk in the UK (IMAGINE-ID): a case-control cohort study. Lancet Psychiatry 2019, 6:493–505. [DOI] [PubMed] [Google Scholar]
- 28.Steinman KJ, Spence SJ, Ramocki MB, Proud MB, Kessler SK, Marco EJ, Green Snyder L, D’Angelo D, Chen Q, Chung WK et al. : 16p11.2 deletion and duplication: characterizing neurologic phenotypes in a large clinically ascertained cohort. Am J Med Genet A 2016, 170:2943–2955. [DOI] [PubMed] [Google Scholar]
- 29.Qureshi AY, Mueller S, Snyder AZ, Mukherjee P, Berman JI, Roberts TP, Nagarajan SS, Spiro JE, Chung WK, Sherr EH et al. : Opposing brain differences in 16p11.2 deletion and duplication carriers. J Neurosci 2014, 34:11199–11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Owen JP, Bukshpun P, Pojman N, Thieu T, Chen Q, Lee J, D’Angelo D, Glenn OA, Hunter JV, Berman JI et al. : Brain MR imaging findings and associated outcomes in carriers of the reciprocal copy number variation at 16p.2. Radiology 2018, 286:217–226. [DOI] [PubMed] [Google Scholar]
- 31.Ejarque I, Millan-Salvador JM, Oltra S, Pesudo-Martinez JV, Beneyto M, Perez-Aytes A: Arnold-Chiari malformation in Noonan syndrome and other syndromes of the RAS/MAPK pathway. Rev Neurol 2015, 60:408–412. [PubMed] [Google Scholar]
- 32.Chang YS, Owen JP, Pojman NJ, Thieu T, Bukshpun P, Wakahiro ML, Marco EJ, Berman JI, Spiro JE, Chung WK et al. : Reciprocal white matter alterations due to 16p11.2 chromosomal deletions versus duplications. Hum Brain Mapp 2016, 37:2833–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jenkins J 3rd, Chow V, Blaskey L, Kuschner E, Qasmieh S, Gaetz L, Edgar JC, Mukherjee P, Buckner R, Nagarajan SS et al. : Auditory evoked M100 response latency is delayed in children with 16p.2 deletion but not 16p11.2 duplication. Cereb Cortex 2016, 26:1957–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuzaki J, Berman JI, Blaskey L, Kuschner ES, Gaetz L, Mukherjee P, Buckner RL, Nagarajan SS, Chung WK, Sherr EH et al. : Abnormal auditory mismatch fields in children and adolescents with 16p.2 deletion and 16p.2 duplication. Biol Psychiatry Cogn Neurosci Neuroimaging 2020, 5:942–950. [DOI] [PubMed] [Google Scholar]
- 35.Kendall KM, Rees E, Escott-Price V, Einon M, Thomas R, Hewitt J, O’Donovan MC, Owen MJ, Walters JTR, Kirov G: Cognitive performance among carriers of pathogenic copy number variants: analysis of 152,000 UK biobank subjects. Biol Psychiatry 2017, 82:103–110. [DOI] [PubMed] [Google Scholar]
- 36.Roberts TP, Khan SY, Blaskey L, Dell J, Levy SE, Zarnow DM, Edgar JC: Developmental correlation of diffusion anisotropy with auditory-evoked response. Neuroreport 2009, 20:1586–1591. [DOI] [PubMed] [Google Scholar]
- 37.Hudac CM, Bove J, Barber S, Duyzend M, Wallace A, Martin CL, Ledbetter DH, Hanson E, Goin-Kochel RP, Green-Snyder L et al. : Evaluating heterogeneity in ASD symptomatology, cognitive ability, and adaptive functioning among 16p.2 CNV carriers. Autism Res 2020, 13:1300–1310. [DOI] [PubMed] [Google Scholar]
- 38.Pizzo L, Jensen M, Polyak A, Rosenfeld JA, Mannik K, Krishnan A, McCready E, Pichon O, Le Caignec C, Van Dijck A et al. : Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease-associated variants. Genet Med 2019, 21:816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bruno MK, Hallett M, Gwinn-Hardy K, Sorensen B, Considine E, Tucker S, Lynch DR, Mathews KD, Swoboda KJ, Harris J et al. :Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia: new diagnostic criteria. Neurology 2004, 63:2280–2287. [DOI] [PubMed] [Google Scholar]
- 40.Chen WJ, Lin Y, Xiong ZQ, Wei W, Ni W, Tan GH, Guo SL, He J, Chen YF, Zhang QJ et al. : Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet 2011, 43:1252–1255. [DOI] [PubMed] [Google Scholar]
- 41.Crepel A, Steyaert J, De la Marche W, De Wolf V, Fryns JP, Noens I, Devriendt K, Peeters H: Narrowing the critical deletion region for autism spectrum disorders on 16p11.2. Am J Med Genet B Neuropsychiatr Genet 2011, 156:243–245. [DOI] [PubMed] [Google Scholar]
- 42.Crepel A: Structurele chromosoomherschikkingen in autisme spectrum stoornissen. Structural Chromosome Rearrangements in Autism Spectrum Disorders 2010.
- 43.Golzio C, Willer J, Talkowski ME, Oh EC, Taniguchi Y, Jacquemont S, Reymond A, Sun M, Sawa A, Gusella JF et al. : KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p.2 copy number variant. Nature 2012, 485:363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luo A et al. : abstract. Gordon Research Conference: Fragile X and autism-related disorders 2018. [Google Scholar]