

Abstract
Background
O’Donnell-Luria-Rodan syndrome (ODLURO) is an autosomal-dominant neurodevelopmental disorder caused by pathogenic, mostly truncating variants in KMT2E. It was first described by O’Donnell-Luria et al in 2019 in a cohort of 38 patients. Clinical features encompass macrocephaly, mild intellectual disability (ID), autism spectrum disorder (ASD) susceptibility and seizure susceptibility.
Methods
Affected individuals were ascertained at paediatric and genetic centres in various countries by diagnostic chromosome microarray or exome/genome sequencing. Patients were collected into a case cohort and were systematically phenotyped where possible.
Results
We report 18 additional patients from 17 families with genetically confirmed ODLURO. We identified 15 different heterozygous likely pathogenic or pathogenic sequence variants (14 novel) and two partial microdeletions of KMT2E. We confirm and refine the phenotypic spectrum of the KMT2E-related neurodevelopmental disorder, especially concerning cognitive development, with rather mild ID and macrocephaly with subtle facial features in most patients. We observe a high prevalence of ASD in our cohort (41%), while seizures are present in only two patients. We extend the phenotypic spectrum by sleep disturbances.
Conclusion
Our study, bringing the total of known patients with ODLURO to more than 60 within 2 years of the first publication, suggests an unexpectedly high relative frequency of this syndrome worldwide. It seems likely that ODLURO, although just recently described, is among the more common single-gene aetiologies of neurodevelopmental delay and ASD. We present the second systematic case series of patients with ODLURO, further refining the mutational and phenotypic spectrum of this not-so-rare syndrome.
INTRODUCTION
Large cohort studies of individuals with intellectual disability (ID) or autism spectrum disorder (ASD) first reported isolated individuals with de novo variants in the KMT2E gene starting in 2012.1–5 Due to the nature of these studies, association of KMT2E variants with human neurodevelopmental disorders (NDDs) remained unclear until additional individuals could be identified and described in detail. In 2019, 38 patients (including previously described individuals) with a syndromic neurodevelopmental phenotype and heterozygous variants disrupting KMT2E were jointly reported in the seminal paper by O’Donnell-Luria and colleagues.6 In this large cohort of patients, a recurring phenotype of generally mild ID (ranging from low-normal intelligence to moderate ID), macrocephaly (55%), ASD (26%) and epilepsy (15%) was observed. Further common features were a subtle facial gestalt and gastrointestinal symptoms, including vomiting or reduced bowel motility.
The phenotype was generally more severe in patients with missense changes compared with patients with null variants (including those predicted to escape nonsense-mediated decay (NMD)) and whole gene deletions. Rather than macrocephaly, two out of three patients carrying missense variants with a known head circumference showed microcephaly, the third had a low-normal head circumference. The authors hypothesised that the detected missense variants in KMT2E may cause this discordant phenotype due to a gain-of-function effect, rather than the assumed loss-of-function effect of the other described variants, but noted more data were needed.6 Interestingly, there was a statistically significant sex bias in the subgroup of patients carrying putative loss-of-function variants, with 24 out of 34 (71%) being male. Subsequently to this first systematic case series, another single family with three affected individuals carrying a frameshift variant has been reported in the literature with a detailed description of the patients’ brain MRI findings. All three patients displayed a phenotype within the described clinical spectrum, but falling on the lower end of cognitive function with moderate and borderline severe ID.7 Two patients with de novo missense variants in KMT2E were reported in a separate work. Both had microcephaly and moderate to severe ID, one had seizures.8 Recently, two more patients were described, one carrying a de novo missense variant, the other carrying a synonymous variant predicted to impair normal splicing. The patient carrying the missense variant had microcephaly, profound ID and seizures. The patient carrying the predicted splice variant had mild ID, ASD and also suffered from seizures.9
The KMT2E-associated neurodevelopmental disorder has since been named O’Donnell-Luria-Rodan syndrome (ODLURO, OMIM #618512).
The KMT2E gene (lysine methyltransferase 2E) on chromosome 7q22.3 encodes an enzyme of the mixed lineage leukaemia/lysine N-methyltransferase 2 (KMT2) family. This gene family is involved in the transcriptional regulation of various genes through H3K4 methylation and chromatin remodelling, but KMT2E differs structurally from the other members of this family and seems to lack any methyltransferase activity.10 Functional evidence suggests that the KMT2E protein is able to recognise and selectively bind to H3K4me3 and H3K4me2 methylated histones,11 where it may regulate transcription factor binding or otherwise regulate transcription. Although it has been experimentally linked to the regulation of cell cycle progression and genomic stability,10 and has been suggested to interact with NCOR2 and TBL1X in a repressor complex regulating cytokinesis-associated genes,12 the precise function of KMT2E remains unclear to date. Many interactors of the NCOR1/2 nuclear corepressor complex, such as MECP2 (associated with Rett syndrome; OMIM #312750) or TBL1X/TBL1XR1 (associated with Pierpont syndrome and autosomal-dominant intellectual disability type 41; OMIM #602342 and #616944), among others, are known, when mutated, to be causative of ID and ASD.13–18
We report the second systematic case series of patients with ODLURO. With this study, we add an additional 18 multinational patients to the literature and increase the total number of known individuals with ODLURO to more than 60 patients worldwide. We confirm and further refine the phenotypic and mutational spectrum of ODLURO.
MATERIALS AND METHODS
Patient recruitment
The research in this study conforms to the Helsinki Declaration of ethical principles for medical research involving human subjects. Written informed consent for study participation and publication was obtained by the attending geneticist or the referring physician from the patients’ legal guardian(s).
The cases included in this study were ascertained through GeneMatcher19 and through personal correspondence between May 2018 and September 2020. All patients with the molecular genetic diagnosis of a KMT2E-associated neurodevelopmental disorder were included in this analysis, regardless of variant type.
Genetic testing
In all patients, variants and deletions disrupting KMT2E were detected either by chromosome microarray (CMA) or next-generation sequencing (NGS). For a description of CMA and NGS methodology, see the online supplemental methods.
Phenotypic features comparison
Phenotypic features were compared between this study’s cohort and the cohort described by O’Donnell-Luria and colleagues6 as reported in either that publication’s main text or online supplemental table 2. Patients for whom specific feature information was missing (eg, no MRI was done) were not included in the respective totals, hence the group sizes differ between features.
Statistical analyses
Statistical tests were performed using Microsoft Excel. A p value of ≤0.05 was considered statistically significant.
Prediction algorithms
Splice predictions were calculated using the MaxEntScan algorithm.20
Variant reporting
All variants reported in this study refer to the KMT2E transcript NM_182931.2. Genomic coordinates refer to the hg19 human reference genome.
RESULTS
Molecular findings
In the 18 reported patients with heterozygous deleterious variants affecting KMT2E, four nonsense variants, seven frameshift variants, two single nucleotide substitutions affecting the canonical splice site, one multi-nucleotide deletion spanning the canonical splice site and one apparent synonymous variant that is predicted to generate a novel splice donor site were detected (see figure 1A). Two siblings carried the identical canonical splice site variant. Additionally, two microdeletions (one with a size of 711 kb encompassing ~98% of KMT2E’s coding sequence and the other one with a size of 61 kb encompassing ~75% of KMT2E’s coding sequence; figure 1B) were found. The larger heterozygous deletion contained two other known disease-associated genes, PUS7 (OMIM*616261) and RINT1 (OMIM*610089), both of which are currently associated exclusively with disorders that are inherited in an autosomal-recessive manner. Of all 17 variants, 13 were confirmed de novo, 2 were proven to be not inherited maternally and paternal samples were unavailable, one variant in two brothers and another variant in an unrelated patient were inherited paternally. The fathers did not participate in clinical phenotyping and were not included in this study. But the father of patients 14 and 15 stated that he has difficulties in social interaction and the father of patient 6 attended a school for disabled children. Both fathers were not formally assessed for neurodevelopmental features. Only one of the detected variants had been previously described (c.4829dupT, reported in previous work6), all others were novel and all 17 were classified as likely pathogenic or pathogenic according to the revised ACMG (American College of Medical Genetics and Genomics) recommendations for the interpretation and reporting of sequence variations21 or the ACMG/ClinGen technical standards for the interpretation of constitutional CNVs.22
Figure 1.
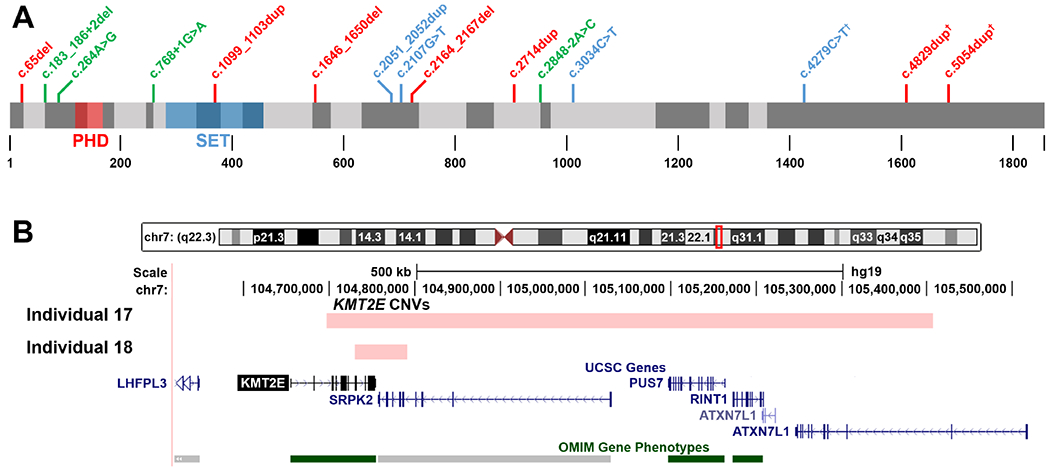
(A) Localisation of KMT2E variants reported in this study at the protein level. Exon regions are depicted with alternating grey shading, the first coding exon in NM_182931.2 is exon 3. Frameshifting variants are written in red, nonsense variants in blue and (putative) splice variants in green. The c.65del (p.(Gly22Valfs*7)) variant represents the earliest truncating variant in KMT2E published to date. PHD, PHD zinc finger domain; SET, SET domain; †, variants predicted to escape nonsense-mediated decay. (B) Genomic locations of the CNVs reported in individuals 17 and 18.
Individual 13 carried a seemingly synonymous heterozygous variant c.264A>G (the translation impact is predicted as p.(Glu88=) but will subsequently be given as p.?) that is predicted to activate a cryptic donor splice site. The donor site MaxEntScore at this position was calculated to increase from 3.93 to 8.99, signifying a very strong splice site prediction. The SpliceAI23 prediction algorithm similarly predicted a donor site gain with a score of 0.74. The variant was confirmed de novo in the patient.
For a more detailed description of the detected pathogenic variants, see table 1.
Table 1.
KMT2E mutational findings in the study cohort
| Individual | Variant | Translation impact | Variant type | Inheritance | ACMG |
|---|---|---|---|---|---|
| 1 | c.2051_2052dup | p.(Glu685*) | Nonsense | De novo | P |
| 2 | c.2107G>T | p.(Glu703*) | Nonsense | De novo | P |
| 3 | c.3034C>T | p.(Gln1012*) | Nonsense | De novo | P |
| 4 | c.4279C>T | p.(Gln1427*) | Nonsense† | De novo | LP |
| 5 | c.65del | p.(Gly22Valfs*7) | Frameshift | n/a* | LP |
| 6 | c.1099_1103dup | p.(Glu369Serfs*25) | Frameshift | Paternal | LP |
| 7 | c.1646_1650del | p.(Ile549Argfs*6) | Frameshift | De novo | P |
| 8 | c.2164_2167del | p.(Lsy722Valfs*17) | Frameshift | De novo | P |
| 9 | c.2714dup | p.(Met906Tyrfs*15) | Frameshift | De novo | P |
| 10 | c.4829dup | p.(Leu1610Phefs*259) | Frameshift† | De novo | LP |
| 11 | c.5054dup | p.(Pro1686Serfs*183) | Frameshift† | De novo | LP |
| 12 | c.183_186+2del | p.? | Splice | De novo | LP |
| 13 | c.264A>G | p.? | Splice | De novo | LP |
| 14 | c.768+1G>A | p.? | Splice | Paternal | LP |
| 15 | c.768+1G>A | p.? | Splice | Paternal | LP |
| 16 | c.2848-2A>C | p.? | Splice | n/a* | LP |
| 17 | arr(hg19)7q22.3 (104,696,686–105,407,628) x1 | p.? | Gross deletion | De novo | P |
| 18 | arr(hg19)7q22.3 (104,730,300–104,791,108) x1 | p.? | Gross deletion | De novo | P |
The mother was tested negative, the father not available for testing.
Variant predicted to escape NMD.
ACMG, American College of Medical Genetics and Genomics classification; LP, likely pathogenic; NMD, nonsense-mediated decay; P, pathogenic.
Phenotypic findings
From the 18 patients in our cohort carrying putative loss-of-function variants in KMT2E, 14 are boys and 4 are girls. The age at the most recent clinical assessment ranged from 1 to 16 years. Pregnancy and birth history were generally uncomplicated for these patients, except for the youngest patient of our cohort (patient 2). The boy was born prematurely at 26 weeks of gestation. He had a large ventricular septal defect which was closed operatively, developed a retinopathy of prematurity and shows recurrent apparent life-threatening events and brief resolved unexplained events.
All but one patient (94%) showed speech developmental delay and 13 (72%) presented with global developmental delay. In one boy and one girl, developmental regression was observed regarding speech development with a loss of previously learnt vocabulary starting at 14 months in the girl, and at 18 months in the boy. These two patients were also diagnosed with ASD. It is unclear if the observed language regression occurred in the context of their ASD.
Intellectual function was assessed in 12 patients, with 6 having intellectual disability. In two patients, the degree of ID could be specified; their respective IQ measurements lay in the upper range of mild ID to low normal intelligence at 65/71 (performance and verbal IQ; patient 7) and 63/71 (performance and verbal IQ; patient 17). One boy (patient 10) had a below average IQ of 83, while both parents were tested to have above average IQs of 125 and 134. A speech disorder with dysphasia, but without intellectual disability was present in another boy (patient 3).
Of the tested patients, 41% (7/17) met the criteria of ASD based on the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5) criteria and assessment by an expert clinician, with 6 (86%) of these individuals being male. Two of the patients (patient 14 and 15) with high-functioning ASD are brothers and inherited the familial KMT2E canonical splice site variant from their father. In more than half of our cohort, other behavioural problems were reported, especially sleep disturbances (47%) and less frequently repetitive (33%) and self-injurious (11%) behaviour.
The reported sleep disturbances in our cohort consisted of frequent awakening and difficulty falling asleep with prolonged sleep latency. Sleep disorders are commonly seen in patients with ASD,24 although in our cohort, sleep disorders were also reported in 3 out of 10 patients (30%) without an ASD diagnosis, the oldest of whom being 9 years and 4 months when last examined.
One patient was diagnosed with attention deficit hyperactivity disorder (ADHD) and another one with sensory integration disorder.
Clinically recurrent dysmorphic features were macrocephaly (10 patients, up to +3.5 SD) and subtle facial features, especially a large forehead, deep-set eyes and full cheeks (see figure 2). In our cohort, the more common presentation of macrocephaly was that of a high-normal occipitofrontal circumference (OFC) at birth, crossing the 97th percentile within the first year of life, although this was not universal and we also observed macrocephaly in infancy normalising to a high-normal OFC by age six in one patient (patient 17).
Figure 2.
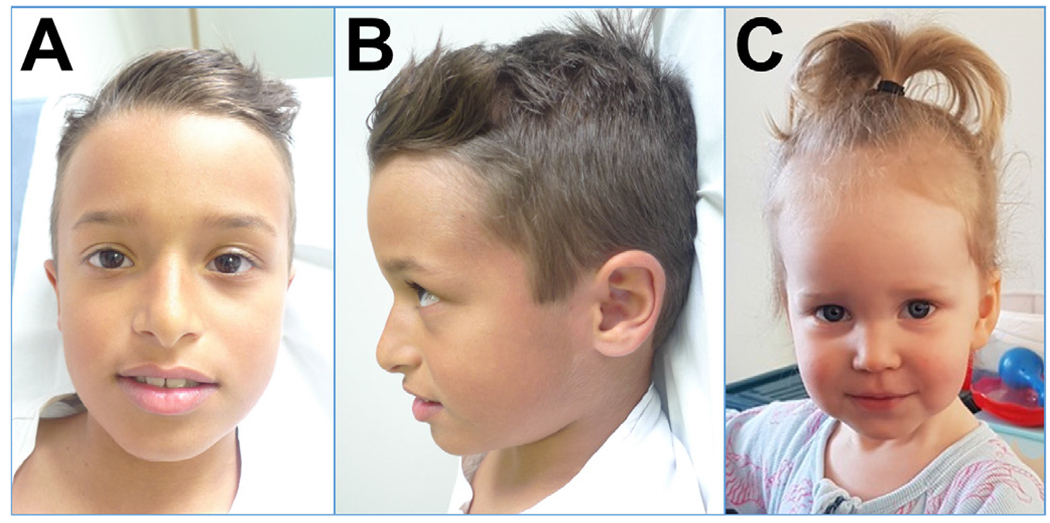
Facial photographs of two patients reported in this study: (A) patient 17 frontal; (B) patient 17 profile; (C) patient 8 frontal.
Muscular hypotonia (eight patients; 44%) and non-specific radiographic signs on brain MRI were also seen frequently. Fourteen patients had a brain MRI, seven (50%) with non-specific findings. The described recurrent central nervous system features were corpus callosum hypoplasia (patients 2, 4, 9), ventriculomegaly (patients 1, 9), cerebral cysts (patients 2, 3, 17) and delayed myelination (patients 1, 2, 4, 6). The following changes on brain MRI were described for only single patients, respectively: mega cisterna magna, reduced brain volume, pachygyria, and multiple ependymal nodules and small pituitary (patient 2, born prematurely).
Half of the patients reported gastrointestinal symptoms, most frequently constipation (eight individuals). Interestingly, no patient in our cohort was diagnosed with epilepsy so far. Only two unrelated patients had recurrent febrile seizures without complications.
For a summary of the phenotypical findings and a comparison with the original ODLURO publication, see table 2. A detailed phenotypical description for each patient listing additional non-recurrent findings is available in online supplemental table S1.
Table 2.
Cohort overview and summary of phenotypic findings
| This study | O’Donnell-Luria et al,6 excl. missense variants | Combined | |
|---|---|---|---|
| Sex | |||
| Male | 14/18 (78%) | 24/34 (71%) | 38/52 (73%) |
| Female | 4/18 (22%) | 10/34 (29%) | 14/52 (27%) |
| Age at last examination | 7.2±4.5 years | 8.9±5.7 years | – |
| (Mean±SD (range)) | (1–16 years) | (1.5–24 years) | |
| Dysmorphic features | |||
| Macrocephaly | 10/18 (56%) | 18/33 (55%) | 28/51 (55%) |
| Dolichocephaly | 2/17 (12%) | n/q | – |
| Large forehead | 10/17 (59%) | n/q | – |
| Epicanthal folds | 7/17 (41%) | n/q | – |
| Deep-set eyes | 7/17 (41%) | n/q | – |
| Prominent nasolabial folds | 3/17 (18%) | n/q | – |
| Full cheeks | 9/17 (53%) | n/q | – |
| Small ears | 4/17 (24%) | n/q | – |
| Clinodactyly | 4/17 (24%) | n/q | – |
| Neurodevelopmental features | |||
| Motor delay | 13/18 (72%) | 18/27 (67%) | 31/45 (69%) |
| Speech delay | 17/18 (94%) | 21/28 (75%) | 38/46 (83%) |
| Regression | 2/18 (11%) | 4/33 (12%) | 6/51 (12%) |
| Autism spectrum disorder | 7/17 (41%) | 8/31 (26%) | 15/48 (31%) |
| Intellectual disability | 6/12 (50%) | 17/20 (85%) | 23/32 (72%) |
| Mild ID | 2/6 (33%) | 6/17 (35%) | 8/23 (35%) |
| Moderate ID | 0/6 (0%) | 6/17 (35%) | 6/23 (26%) |
| Not specified | 4/6 (67%) | 5/17 (29%) | 9/23 (39%) |
| Epilepsy | 0/18 (0%) | 7/30 (23%) | 7/48 (15%) |
| Febrile seizures | 2/18 (11%) | n/s | – |
| Muscular hypotonia | 8/18 (44%) | 15/27 (56%) | 23/45 (51%) |
| Behavioural features | |||
| ADHD | 1/18 (6%) | 2/34 (6%) | 3/52 (6%) |
| Stereotypical behaviour | 6/18 (33%) | 3/34 (9%) | 9/52 (17%) |
| Self-injurious behaviour | 2/18 (11%) | 1/34 (3%) | 3/52 (6%) |
| Aggressive behaviour | 1/18 (6%) | 2/34 (6%) | 3/52 (6%) |
| Sleep disturbances | 8/17 (47%) | n/s | – |
| CNS features | |||
| Corpus callosum hypoplasia | 3/14 (21%) | 4/22 (18%) | 7/36 (19%) |
| Ventriculomegaly | 2/14 (14%) | 3/22 (14%) | 5/36 (14%) |
| Cerebral cysts | 3/14 (21%) | 4/22 (18%) | 7/36 (19%) |
| Delayed myelination | 4/14 (29%) | 1/22 (5%) | 5/36 (14%) |
| GI features | |||
| Constipation | 8/18 (44%) | 5/27 (19%) | 13/45 (29%) |
| Gastro-oesophageal reflux | 1/18 (6%) | 2/27 (7%) | 3/45 (7%) |
| Vomiting | 3/18 (18%) | 1/27 (4%) | 4/45 (9%) |
Features that were present in more than one individual or were previously reported to be associated with ODLURO are listed. From the original publication, only patients with putative loss-of-function variants (including those predicted to escape NMD) and microdeletions are compared, as the patients carrying missense variants displayed a notably different phenotype, suggesting a different mutational pathomechanism.
ADHD, attention deficit/hyperactivity disorder; CNS, central nervous system; GI, gastrointestinal; ID, intellectual disability; NMD, nonsense-mediated decay; n/q, feature noted but not quantified; n/s, feature not specified.
Together with the original cohort,6 eight patients have been described carrying nonsense and frameshifting variants that are predicted to escape NMD. Regarding quantitative traits like OFC and IQ, and the prevalence of recurring KMT2E-associated features, we detected no significant phenotypic differences between these patients and patients carrying variants that are predicted to lead to NMD.
Additional molecular findings
In one patient (patient 3), exome sequencing additionally identified the maternally inherited heterozygous variant c.2021G>A; p.(Arg674Gln) in MYH8 (NM_002472.2), causative of a known familial trismus-pseudocamptodactyly syndrome (distal arthrogryposis type 7, DA7; OMIM #158300) as an independent genetic condition. Patients with DA7 may also present with macrocephaly and difficulties with speech articulation; however, developmental delay and cerebral cysts, as observed in this patient, are not typically associated with this condition. Whether the patient’s dysphasia is caused by ODLURO or DA7 remains unclear, we assume that both pathologies cause a hybrid phenotype in this case.
In another patient (patient 4), the heterozygous nonsense variant c.3340G>T; p.(Glu1114*) in the CHD8 gene (NM_001170629.1) in addition to a heterozygous nonsense variant in KMT2E was detected. Both variants were confirmed de novo (maternity and paternity confirmed). Nonsense variants in CHD8 have been robustly associated with mild to moderate ID and ASD susceptibility.25 26 Additionally, a large proportion of patients display overgrowth with increased height and/or macrocephaly.27 Again, corpus callosum hypoplasia, delayed myelination and developmental regression—as present in patient 4—are not typical features of the CHD8-associated neurodevelopmental phenotype and likely represent an added phenotypical component caused by disruption of KMT2E.
In patient 14, the homozygous well-known pathogenic28 variant c.109G>A; p.(Val37Ile) in GJB2 (NM_004004.5) was detected, causative of the patient’s mild to moderate sensorineural hearing impairment. This variant is exceedingly common in Southeast Asia (carrier frequency of 8.4%) and the most common cause of monogenic mild hearing impairment in this population. It is not associated with a neurodevelopmental disorder.
DISCUSSION
In this case series, we present 18 individuals with ODLURO, refining the phenotypic spectrum of this recently described6 7 9 syndrome.
As in the initial cohort, we also see a male bias in our cohort (male/female 14:4). Combined with the other patients published so far,6 7 9 this sex distribution of 41 males to 15 females with putative loss-of-function variants (excluding 7 patients carrying missense variants with a suspected gain-of-function6 8 9) is significantly biased towards affected males (binomial test: p<0.001 vs ~1:1 sex distribution in the general population). A male preponderance has been described in various NDDs and especially in ASD.29 Even in monogenic NDD caused by pathogenic variants in autosomal genes, a sex-dependent penetrance has been observed,30 31 being for some genes lower in males, and lower in females for others. Intriguingly in the context of KMT2E, post-translational histone modification profiles in the brain show sex differences in mice32 and humans,33 and may influence differences in brain development and behaviour, although we reiterate that the precise nature of the interaction between histones and KMT2E is still uncertain. Another gene in the KMT2 family, KMT2A (coding for a functional histone lysine methyltransferase) shows a higher expression in the female prefrontal adult human brain.34 In summary, many studies on the topic of sexual dimorphism in neurodevelopmental disorders exist and suggest complex (epigenetic) developmental interactions as one important factor; however, the precise mechanisms for the sex-specific variable penetrance in some disorders remain unclear at present.
Our study confirms the originally described ODLURO phenotype with mostly speech developmental delay and variable, but generally mild, ID and/or ASD. In the cohort of O’Donnell-Luria et al,6 8 out of 31 patients (26%) carrying loss-of-function variants were diagnosed with ASD, while in our cohort, we observed a somewhat higher rate at 7 out of 17 (41%). This difference is not significant (Fisher’s exact test: p=0.34). In both studies, one additional patient, respectively, showed characteristics of a sensory integration disorder. The additionally reported behavioural concerns were similar in both case series. However, we report a high prevalence (47%) of sleep disturbances among affected individuals. Sleep disturbances have so far not been explicitly reported to be associated with ODLURO, although one patient carrying a missense KMT2E variant in the original publication was described as lacking normal sleep (patient 34 from previous work6). We propose that sleep disturbances, including frequent awakening and difficulties falling asleep, may be a recurring feature in ODLURO.
We observed previously described features like macrocephaly, muscular hypotonia and gastrointestinal symptoms like constipation at comparable frequencies to the original ODLURO publication.6
Patients presented with a subtle facial gestalt including a prominent forehead, epicanthal folds, full cheeks and deep-set eyes. It remains to be seen if, combined with other phenotypic features, ODLURO can be specifically recognised in the clinical setting.
In both ours and the original case series, various non-specific, but partly recurring abnormalities were detected in more than half of the conducted brain MRIs. About 28% of studied patients had corpus callosum hypoplasia, cerebral cysts or both. These findings substantiate the important role of KMT2E in structural brain development. Interestingly, it has been shown that KMT2E is highly expressed in the brain during fetal development35 and remains highly expressed even in the adult brain (Human Protein Atlas36; https://www.proteinatlas.org—accessed 28 July 2020).
In the subgroup of patients carrying intragenic truncating variants in KMT2E described in the original ODLURO publication,6 only 4 out of 26 (15%) had epilepsy, but 3 out of 4 (75%) of the patients with microdeletions and 4 out of 4 (100%) of the patients carrying missense variants did. Three out of the four microdeletion carriers from the original cohort had large deletions affecting multiple genes, between 1.9 and 2.9 megabases in size. No patients in our cohort— including two patients with microdeletions at 5 and 9 years old—have been diagnosed with epilepsy so far. Larger cohorts will be needed to elucidate the suggested, but presently unclear genotype–phenotype correlation regarding intragenic KMT2E truncating variants and microdeletions. As only two patients of our cohort and four patients of the originally described cohort carry microdeletions containing the KMT2E gene either partially or fully, the information regarding this subgroup of patients is still very limited. It is worth noting that those two patients in our cohort with microdeletions show a generally similar phenotype to those with intragenic loss-of-function variants, consistent with haploinsufficiency as a common disease mechanism.
When comparing the symptoms of our patients carrying null variants in KMT2E with the previously reported patients carrying missense variants,6 8 9 we again see a notably different and generally milder phenotype in our patients with null variants.
We did not detect a significant phenotypic difference between patients carrying variants predicted to lead to NMD, and patients carrying nonsense and frameshift variants in the last exon of KMT2E predicted to escape NMD.
The additional phenotypic features that are described only in one or two ODLURO patients to date will need to be studied in future larger cohorts in order to correctly recognise their association, or lack thereof, with ODLURO. Identifying additional clinical hints could be helpful in the diagnosis of patients and eventually in optimising the clinical care of affected patients. Since there is so far only a smaller number of reports on teenagers and adults (10 patients in total, including the father/daughter/son trio in previous work7), conclusions about patients’ long-term development and possible late-onset health problems in adulthood remain speculative at best.
Three patients in our cohort carried pathogenic variants in one other gene, respectively, associated with a monogenic disorder. Two of these conditions (distal arthrogryposis type 7 and the CHD8-associated neurodevelopmental disorder) had some clinical overlap with ODLURO. As such, the respective contribution of these variants to the phenotypes in these two patients remains unclear. But it seems likely that the patients have two genetic conditions causing blended, partly overlapping phenotypes.
With the description of ODLURO, all members of the KMT2 gene family have now been associated with autosomal-dominant NDDs (see table 3). Common features of these disorders appear to be variable ID, seizures, ASD and gastrointestinal symptoms, but these show incomplete penetrance to varying degrees. The ODLURO phenotypic spectrum has a considerable overlap with these disorders, which may suggest an underlying pathomechanistic similarity.
Table 3.
Disorders associated with genes in the KMT2 gene family and common recurring phenotypic features
| Gene | OMIM phenotype | DD | ID | ASD | Sleep | Sz | GI | SS | Scol | OFC |
|---|---|---|---|---|---|---|---|---|---|---|
| KMT2A | Wiedemann-Steiner syndrome | + | + | + | + | + | + | ↓ | ||
| KMT2B | Dystonia 28, childhood-onset | + | + | + | + | + | ↓ | |||
| KMT2C | Kleefstra syndrome 2 | + | + | + | + | + | + | + | + | ↓ |
| KMT2D | Kabuki syndrome 1 | + | + | + | + | + | + | ↓ | ||
| KMT2E | O’Donnell-Luria-Rodan syndrome | + | + | + | + | + | + | ↑ | ||
| SETD1A | NDD with speech impairment and dysmorphic facies | + | + | + | + | + | + | – | ||
| SETD1B | ID with seizures and language delay | + | + | + | + | – | ||||
| ASH1L | Autosomal dominant ID 52 | + | + | + | + | + | + | ↑ |
ASD, autism spectrum disorder; DD, developmental delay; GI, gastrointestinal symptoms (eg, constipation, reflux, gastrointestinal malformation); ID, intellectual disability; NDD, neurodevelopmental disorder; OFC, occipitofrontal circumference; Scol, scoliosis; Sleep, sleep disturbances; SS, short stature; Sz, seizures.
In the STRING database of protein–protein association networks,37 among KMT2E’s top 20 highest-confidence interactors are KMT2A, KMT2B, KMT2C, KMT2D, SETD1A and SETD1B, but also the histone acetyltransferase EP300, and SIN3A, which is part of a histone deacetylase complex. These proteins are associated with the autosomal-dominant phenotypes of Rubinstein-Taybi syndrome type 2 (OMIM #613684) and Witteveen-Kolk syndrome (OMIM #613406), respectively. Both phenotypes are not dissimilar to the KMT2-associated phenotypes summarised above (table 3) with variable ID, ASD, seizures, short stature and constipation. Future research will need to examine whether these phenotypic parallels have a biological basis, representing a ‘histone modification disease network’, or are coincidental.
A weakness of our study is the missing formal intelligence tests in most patients, partly due to the young age of many patients. A midterm or long-term follow-up with formal development testing in the future could further substantiate our observations in this cohort so far.
Shortly after the systematic description of ODLURO in 2019, our report now brings the number of known patients to 63.6–9 This suggests that among the more than 2500 currently known or suspected ID and ASD-associated genes,38–41 deleterious variants in KMT2E are a more common aetiology for developmental delay, ID and ASD than presently thought. We recommend that KMT2E be included in the routine NGS testing in patients with global developmental delay, ID or ASD, especially in the presence of macrocephaly.
Supplementary Material
Acknowledgements
We thank the patients and families for their willingness to contribute to this project. BC, MCYC and JL-FF acknowledge the Autism Sequencing Consortium for sequencing and sharing of raw exome data.
Funding
AHO-L, EME and ES: Sequencing and analysis for some of the patients were provided by the Broad Institute of MIT and Harvard Centre for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1 HG008900 and in part by the National Human Genome Research Institute grant R01 HG009141. BHYC, MCYC, and JL-FF: Funding and support of the Society for the Relief of Disabled Children contributed to this project.
Footnotes
Excerpts from this study have been previously presented at the European Society of Human Genetics conference 2020.
Additional supplemental material is published online only. To view, please visit the journal online (http://dx.doi.org/10.1136/jmedgenet-2020-107470).
Competing interests None declared.
Patient consent for publication Written informed consent for study participation and publication was obtained by the attending geneticist or the referring physician from the patients’ legal guardian(s).
Ethics approval The study was approved by the Institutional Review Board of the Baylor College of Medicine (H-34578).
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). it has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
REFERENCES
- 1.Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee Y-H, Narzisi G, Leotta A, Kendall J, Grabowska E, Ma B, Marks S, Rodgers L, Stepansky A, Troge J, Andrews P, Bekritsky M, Pradhan K, Ghiban E, Kramer M, Parla J, Demeter R, Fulton LL, Fulton RS, Magrini VJ, Ye K, Darnell JC, Darnell RB, Mardis ER, Wilson RK, Schatz MC, McCombie WR, Wigler M. De novo gene disruptions in children on the autistic spectrum. Neuron 2012;74:285–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Girirajan S, Dennis MY, Baker C, Malig M, Coe BP, Campbell CD, Mark K, Vu TH, Alkan C, Cheng Z, Biesecker LG, Bernier R, Eichler EE. Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am J Hum Genet 2013;92:221–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang T, Guo H, Xiong B, Stessman HAF, Wu H, Coe BP, Turner TN, Liu Y, Zhao W, Hoekzema K, Vives L, Xia L, Tang M, Ou J, Chen B, Shen Y, Xun G, Long M, Lin J, Kronenberg ZN, Peng Y, Bai T, Li H, Ke X, Hu Z, Zhao J, Zou X, Xia K, Eichler EE. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun 2016;7:13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo H, Wang T, Wu H, Long M, Coe BP, Li H, Xun G, Ou J, Chen B, Duan G, Bai T, Zhao N, Shen Y, Li Y, Wang Y, Zhang Y, Baker C, Liu Y, Pang N, Huang L, Han L, Jia X, Liu C, Ni H, Yang X, Xia L, Chen J, Shen L, Li Y, Zhao R, Zhao W, Peng J, Pan Q, Long Z, Su W, Tan J, Du X, Ke X, Yao M, Hu Z, Zou X, Zhao J, Bernier RA, Eichler EE, Xia K. Inherited and multiple de novo mutations in autism/developmental delay risk genes suggest a multifactorial model. Mol Autism 2018;9:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong S, Walker MF, Carriero NJ, DiCola M, Willsey AJ, Ye AY, Waqar Z, Gonzalez LE, Overton JD, Frahm S, Keaney JF, Teran NA, Dea J, Mandell JD, Hus Bal V, Sullivan CA, DiLullo NM, Khalil RO, Gockley J, Yuksel Z, Sertel SM, Ercan-Sencicek AG, Gupta AR, Mane SM, Sheldon M, Brooks AI, Roeder K, Devlin B, State MW, Wei L, Sanders SJ. De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell Rep 2014;9:16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Donnell-Luria AH, Pais LS, Faundes V, Wood JC, Sveden A, Luria V, Abou Jamra R, Accogli A, Amburgey K, Anderlid BM, Azzarello-Burri S, Basinger AA, Bianchini C, Bird LM, Buchert R, Carre W, Ceulemans S, Charles P, Cox H, Culliton L, Currò A, Demurger F, Dowling JJ, Duban-Bedu B, Dubourg C, Eiset SE, Escobar LF, Ferrarini A, Haack TB, Hashim M, Heide S, Helbig KL, Helbig I, Heredia R, Héron D, Isidor B, Jonasson AR, Joset P, Keren B, Kok F, Kroes HY, Lavillaureix A, Lu X, Maas SM, Maegawa GHB, Marcelis CLM, Mark PR, Masruha MR, McLaughlin HM, McWalter K, Melchinger EU, Mercimek-Andrews S, Nava C, Pendziwiat M, Person R, Ramelli GP, Ramos LLP, Rauch A, Reavey C, Renieri A, Rieß A, Sanchez-Valle A, Sattar S, Saunders C, Schwarz N, Smol T, Srour M, Steindl K, Syrbe S, Taylor JC, Telegrafi A, Thiffault I, Trauner DA, van der Linden H, van Koningsbruggen S, Villard L, Vogel I, Vogt J, Weber YG, Wentzensen IM, Widjaja E, Zak J, Baxter S, Banka S, Rodan LH, McRae JF, Clayton S, Fitzgerald TW, Kaplanis J, Prigmore E, Rajan D, Sifrim A, Aitken S, Akawi N, Alvi M, Ambridge K, Barrett DM, Bayzetinova T, Jones P, Jones WD, King D, Krishnappa N, Mason LE, Singh T, Tivey AR, Ahmed M, Anjum U, Archer H, Armstrong R, Awada J, Balasubramanian M, Banka S, Baralle D, Barnicoat A, Batstone P, Baty D, Bennett C, Berg J, Bernhard B, Bevan AP, Bitner-Glindzicz M, Blair E, Blyth M, Bohanna D, Bourdon L, Bourn D, Bradley L, Brady A, Brent S, Brewer C, Brunstrom K, Bunyan DJ, Burn J, Canham N, Castle B, Chandler K, Chatzimichali E, Cilliers D, Clarke A, Clasper S, Clayton-Smith J, Clowes V, Coates A, Cole T, Colgiu I, Collins A, Collinson MN, Connell F, Cooper N, Cox H, Cresswell L, Cross G, Crow Y, D’Alessandro M, Dabir T, Davidson R, Davies S, de Vries D, Dean J, Deshpande C, Devlin G, Dixit A, Dobbie A, Donaldson A, Donnai D, Donnelly D, Donnelly C, Douglas A, Douzgou S, Duncan A, Eason J, Ellard S, Ellis I, Elmslie F, Evans K, Everest S, Fendick T, Fisher R, Flinter F, Foulds N, Fry A, Fryer A, Gardiner C, Gaunt L, Ghali N, Gibbons R, Gill H, Goodship J, Goudie D, Gray E, Green A, Greene P, Greenhalgh L, Gribble S, Harrison R, Harrison L, Harrison V, Hawkins R, He L, Hellens S, Henderson A, Hewitt S, Hildyard L, Hobson E, Holden S, Holder M, Holder S, Hollingsworth G, Homfray T, Humphreys M, Hurst J, Hutton B, Ingram S, Irving M, Islam L, Jackson A, Jarvis J, Jenkins L, Johnson D, Jones E, Josifova D, Joss S, Kaemba B, Kazembe S, Kelsell R, Kerr B, Kingston H, Kini U, Kinning E, Kirby G, Kirk C, Kivuva E, Kraus A, Kumar D, Kumar VKA, Lachlan K, Lam W, Lampe A, Langman C, Lees M, Lim D, Longman C, Lowther G, Lynch SA, Magee A, Maher E, Male A, Mansour S, Marks K, Martin K, Maye U, McCann E, McConnell V, McEntagart M, McGowan R, McKay K, McKee S, McMullan DJ, McNerlan S, McWilliam C, Mehta S, Metcalfe K, Middleton A, Miedzybrodzka Z, Miles E, Mohammed S, Montgomery T, Moore D, Morgan S, Morton J, Mugalaasi H, Murday V, Murphy H, Naik S, Nemeth A, Nevitt L, Newbury-Ecob R, Norman A, O’Shea R, Ogilvie C, Ong K-R, Park S-M, Parker MJ, Patel C, Paterson J, Payne S, Perrett D, Phipps J, Pilz DT, Pollard M, Pottinger C, Poulton J, Pratt N, Prescott K, Price S, Pridham A, Procter A, Purnell H, Quarrell O, Ragge N, Rahbari R, Randall J, Rankin J, Raymond L, Rice D, Robert L, Roberts E, Roberts J, Roberts P, Roberts G, Ross A, Rosser E, Saggar A, Samant S, Sampson J, Sandford R, Sarkar A, Schweiger S, Scott R, Scurr I, Selby A, Seller A, Sequeira C, Shannon N, Sharif S, Shaw-Smith C, Shearing E, Shears D, Sheridan E, Simonic I, Singzon R, Skitt Z, Smith A, Smith K, Smithson S, Sneddon L, Splitt M, Squires M, Stewart F, Stewart H, Straub V, Suri M, Sutton V, Swaminathan GJ, Sweeney E, Tatton-Brown K, Taylor C, Taylor R, Tein M, Temple IK, Thomson J, Tischkowitz M, Tomkins S, Torokwa A, Treacy B, Turner C, Turnpenny P, Tysoe C, Vandersteen A, Varghese V, Vasudevan P, Vijayarangakannan P, Vogt J, Wakeling E, Wallwark S, Waters J, Weber A, Wellesley D, Whiteford M, Widaa S, Wilcox S, Wilkinson E, Williams D, Williams N, Wilson L, Woods G, Wragg C, Wright M, Yates L, Yau M, Nellåker C, Parker M, Firth HV, Wright CF, FitzPatrick DR, Barrett JC, Hurles ME. Heterozygous variants in KMT2E cause a spectrum of neurodevelopmental disorders and epilepsy. Am J Hum Genet 2019;104:1210–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conforti R, Iovine S, Santangelo G, Capasso R, Cirillo M, Fratta M, Caranci F. ODLURO syndrome: personal experience and review of the literature. Radiol Med 2021;126:316–22. [DOI] [PubMed] [Google Scholar]
- 8.Sharawat IK, Panda PK, Dawman L. Clinical characteristics and genotype-phenotype correlation in children with KMT2E gene-related neurodevelopmental disorders: report of two new cases and review of published literature. Neuropediatrics 2021;52:98–104. [DOI] [PubMed] [Google Scholar]
- 9.Li Y, Fan L, Luo R, Yang Z, Yuan M, Zhang J, Gan J. Case report: de variants of KMT2E cause O’Donnell-Luria-Rodan syndrome: additional cases and literature review. Front Pediatr 2021;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, Novera W, Zhang Y, Deng L-W. Mll5 (KMT2E): structure, function, and clinical relevance. Cell Mol Life Sci 2017;74:2333–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lemak A, Yee A, Wu H, Yap D, Zeng H, Dombrovski L, Houliston S, Aparicio S, Arrowsmith CH. Solution NMR structure and histone binding of the PHD domain of human MLL5. PLoS One 2013;8:e77020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kittler R, Pelletier L, Heninger A-K, Slabicki M, Theis M, Miroslaw L, Poser I, Lawo S, Grabner H, Kozak K, Wagner J, Surendranath V, Richter C, Bowen W, Jackson AL, Habermann B, Hyman AA, Buchholz F. Genome-Scale RNAi profiling of cell division in human tissue culture cells. Nat Cell Biol 2007;9:1401–12. [DOI] [PubMed] [Google Scholar]
- 13.Kong Y, Zhou W, Sun Z. Nuclear receptor corepressors in intellectual disability and autism. Mol Psychiatry 2020;25:2220–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MeCP2, encoding methyl-CpG-binding protein 2. Nat Genet 1999;23:185–8. [DOI] [PubMed] [Google Scholar]
- 15.Heinen CA, Jongejan A, Watson PJ, Redeker B, Boelen A, Boudzovitch-Surovtseva O, Forzano F, Hordijk R, Kelley R, Olney AH, Pierpont ME, Schaefer GB, Stewart F, van Trotsenburg ASP, Fliers E, Schwabe JWR, Hennekam RC. A specific mutation in TBL1XR1 causes Pierpont syndrome. J Med Genet 2016;53:330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laskowski RA, Tyagi N, Johnson D, Joss S, Kinning E, McWilliam C, Splitt M, Thornton JM, Firth HV, Wright CF, DDD Study. Integrating population variation and protein structural analysis to improve clinical interpretation of missense variation: application to the WD40 domain. Hum Mol Genet 2016;25:927–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riehmer V, Erger F, Herkenrath P, Seland S, Jackels M, Wiater A, Heller R, Beck BB, Netzer C. A heritable microduplication encompassing TBL1XR1 causes a genomic sister-disorder for the 3q26.32 microdeletion syndrome. Am J Med Genet A 2017;173:2132–8. [DOI] [PubMed] [Google Scholar]
- 18.Saitsu H, Tohyama J, Walsh T, Kato M, Kobayashi Y, Lee M, Tsurusaki Y, Miyake N, Goto Y-I, Nishino I, Ohtake A, King M-C, Matsumoto N. A girl with West syndrome and autistic features harboring a de novo TBL1XR1 mutation. J Hum Genet 2014;59:581–3. [DOI] [PubMed] [Google Scholar]
- 19.Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting Investigators with an interest in the same gene. Hum Mutat 2015;36:928–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 2004;11:377–94. [DOI] [PubMed] [Google Scholar]
- 21.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee A, ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet Med 2015;17:405–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, Raca G, Ritter DI, South ST, Thorland EC, Pineda-Alvarez D, Aradhya S, Martin CL. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of medical genetics and genomics (ACMG) and the clinical genome resource (ClinGen). Genet Med 2020;22:245–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, Kosmicki JA, Arbelaez J, Cui W, Schwartz GB, Chow ED, Kanterakis E, Gao H, Kia A, Batzoglou S, Sanders SJ, Farh KK-H. Predicting splicing from primary sequence with deep learning. Cell 2019;176:535–48. [DOI] [PubMed] [Google Scholar]
- 24.Devnani PA, Hegde AU. Autism and sleep disorders. J Pediatr Neurosci 2015;10:304–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O, Witherspoon K, Gerdts J, Baker C, Vulto-van Silfhout AT, Schuurs-Hoeijmakers JH, Fichera M, Bosco P, Buono S, Alberti A, Failla P, Peeters H, Steyaert J, Vissers LELM, Francescatto L, Mefford HC, Rosenfeld JA, Bakken T, O’Roak BJ, Pawlus M, Moon R, Shendure J, Amaral DG, Lein E, Rankin J, Romano C, de Vries BBA, Katsanis N, Eichler EE. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 2014;158:263–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.An Y, Zhang L, Liu W, Jiang Y, Chen X, Lan X, Li G, Hang Q, Wang J, Gusella JF, Du Y, Shen Y. De novo variants in the Helicase-C domain of CHD8 are associated with severe phenotypes including autism, language disability and overgrowth. Hum Genet 2020;139:499–512. [DOI] [PubMed] [Google Scholar]
- 27.Ostrowski PJ, Zachariou A, Loveday C, Beleza-Meireles A, Bertoli M, Dean J, Douglas AGL, Ellis I, Foster A, Graham JM, Hague J, Hilhorst-Hofstee Y, Hoffer M, Johnson A, Josifova D, Kant SG, Kini U, Lachlan K, Lam W, Lees M, Lynch S, Maitz S, McKee S, Metcalfe K, Nathanson K, Ockeloen CW, Parker MJ, Pierson TM, Rahikkala E, Sanchez-Lara PA, Spano A, Van Maldergem L, Cole T, Douzgou S, Tatton-Brown K. The CHD8 overgrowth syndrome: a detailed evaluation of an emerging overgrowth phenotype in 27 patients. Am J Med Genet C Semin Med Genet 2019;181:557–64. [DOI] [PubMed] [Google Scholar]
- 28.Oza AM, DiStefano MT, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, Shen J, Chapin A, Boczek NJ, Schimmenti LA, Murry JB, Hasadsri L, Nara K, Kenna M, Booth KT, Azaiez H, Griffith A, Avraham KB, Kremer H, Rehm HL, Amr SS, Abou Tayoun AN, ClinGen Hearing Loss Clinical Domain Working Group. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum Mutat 2018;39:1593–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferri SL, Abel T, Brodkin ES. Sex differences in autism spectrum disorder: a review. Curr Psychiatry Rep 2018;20:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Douzgou S, Liang HW, Metcalfe K, Somarathi S, Tischkowitz M, Mohamed W, Kini U, McKee S, Yates L, Bertoli M, Lynch SA, Holder S, Banka S, Deciphering Developmental Disorders Study. The clinical presentation caused by truncating CHD8 variants. Clin Genet 2019;96:72–84. [DOI] [PubMed] [Google Scholar]
- 31.Turner TN, Wilfert AB, Bakken TE, Bernier RA, Pepper MR, Zhang Z, Torene RI, Retterer K, Eichler EE. Sex-Based analysis of de novo variants in neurodevelopmental disorders. Am J Hum Genet 2019;105:1274–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsai H-W, Grant PA, Rissman EF. Sex differences in histone modifications in the neonatal mouse brain. Epigenetics 2009;4:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qureshi IA, Mehler MF. Genetic and epigenetic underpinnings of sex differences in the brain and in neurological and psychiatric disease susceptibility. Prog Brain Res 2010;186:77–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang H-S, Matevossian A, Whittle C, Kim SY, Schumacher A, Baker SP, Akbarian S. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J Neurosci 2007;27:11254–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, Sousa AMM, Pletikos M, Meyer KA, Sedmak G, Guennel T, Shin Y, Johnson MB, Krsnik Z, Mayer S, Fertuzinhos S, Umlauf S, Lisgo SN, Vortmeyer A, Weinberger DR, Mane S, Hyde TM, Huttner A, Reimers M, Kleinman JE, Sestan N. Spatio-Temporal transcriptome of the human brain. Nature 2011;478:483–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sjöstedt E, Zhong W, Fagerberg L, Karlsson M, Mitsios N, Adori C, Oksvold P, Edfors F, Limiszewska A, Hikmet F, Huang J, Du Y, Lin L, Dong Z, Yang L, Liu X, Jiang H, Xu X, Wang J, Yang H, Bolund L, Mardinoglu A, Zhang C, von Feilitzen K, Lindskog C, Ponteén F, Luo Y, Hökfelt T, Uhlén M, Mulder J. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 2020;367. doi: 10.1126/science.aay5947. [Epub ahead of print: 06 03 2020]. [DOI] [PubMed] [Google Scholar]
- 37.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Mering Cvon. String v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47:D607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Erger F, Schaaf CP, Netzer C. Which genes to assess in the NGS diagnostics of intellectual disability? the case for a consensus database-driven and expert-curated approach. Mol Cell Probes 2019;45:84–8. [DOI] [PubMed] [Google Scholar]
- 39.Jamra R. Genetics of autosomal recessive intellectual disability. Med Genet 2018;30:323–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wieczorek D. Autosomal dominant intellectual disability. Med Genet 2018;30:318–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An J-Y, Peng M, Collins R, Grove J, Klei L, Stevens C, Reichert J, Mulhern MS, Artomov M, Gerges S, Sheppard B, Xu X, Bhaduri A, Norman U, Brand H, Schwartz G, Nguyen R, Guerrero EE, Dias C, Cook EH, Gallagher L, Gill M, Sutcliffe JS, Thurm A, Zwick ME, Børglum AD, State MW, Cicek AE, Talkowski ME, Cutler DJ, Devlin B, Sanders SJ, Roeder K, Daly MJ, Buxbaum JD, Autism Sequencing Consortium, iPSYCH-Broad Consortium. Large-Scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 2020;180:568–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data relevant to the study are included in the article or uploaded as supplementary information.