


Abstract
Nucleic acid triple helices have provoked interest since their discovery more than 40 years ago, but it remains unknown whether such structures occur naturally in cells. To pursue this question, it is important to determine the stabilities of representative triple helices at physiological temperature and pH. Previous investigations have concluded that while both DNA and RNA can participate in the pyrimidine triplex motif under mildly acidic conditions, these structures are often relatively unstable at neutral pH. We are now explorin g the stability of intrastrand DNA and RNA pyrimidine motif triplexes at physiological temperature and pH. Duplex and triplex formation were monitored by thermal denaturation analysis, circular dichroism spectroscopy and gel shift experiments. Short intrastrand triplexes were observed to form in the pyrimidine motif in both DNA and RNA. In the presence of physiological concentrations of Mg2+ and at physiological pH, all detected triplexes were sufficiently stable to persist at physiological temperature. If sequences specifying such intrastrand triplexes are encoded in genomes, the potential exists for the formation of stable structures in RNA or DNA in vivo.
INTRODUCTION
DNA and RNA are both known to form double helices. In addition to these well-documented forms, nucleic acids have also been found to form three- and four-stranded complexes under some circumstances (1–5). Formation of triple helices requires the third strand to hydrogen bond to consecutive purine bases present in the underlying Watson–Crick duplex. Depending on the sequence composition and relative orientation of this third strand, triplex structures can be divided into two categories or motifs (4). In the pyrimidine (Y) motif, the third strand is composed of pyrimidine bases bound parallel to the purine strand of the Watson–Crick duplex by Hoogsteen hydrogen bonds and is stabilized under slightly acidic conditions that favor cytosine protonation (6). Isomorphous T•A•T and C+•G•C base triplets predominate. In the purine (R) motif, the third strand is homopurine (typically G-rich), binds antiparallel to the purine strand of the duplex via reverse-Hoogsteen hydrogen bonds and its binding is relatively pH independent (7). Base triplets T• (or A•) A•T and G•G•C predominate.
Triplexes can also be subdivided into intramolecular versus intermolecular complexes. Intermolecular triplexes form when a third strand binds to a separate, pre-established double helix (1–5), or when a single strand is bound simultaneously by a Watson–Crick complement and third strand present on a separate molecule (8). Intramolecular triplexes form when the third domain is physically tethered to the underlying duplex (9,10). In principle, both intermolecular and intramolecular complexes can occur in both the purine and the pyrimidine motifs.
Intermolecular triple helices have been of great scientific interest due to their possible therapeutic uses (4,11). Traditional approaches to inhibiting the production of gene products include drugs or small molecules that can bind to a protein and either disrupt its function or signal its degradation. An alternative approach to altering gene expression might be the design of an oligonucleotide that can specifically bind to a particular sequence in the DNA double helix, forming an intermolecular triplex. Intermolecular triplexes have demonstrated the potential to modulate gene expression by altering DNA/protein interactions (12–14).
Several groups have studied intermolecular triplexes, with the majority of investigations focused on the ability of DNA to form these structures (reviewed in 4). Fewer studies have explored the abilities of RNA strands to participate in oligonucleotide-directed triple helix formation in the Y and R motifs (11,14–18). Based on this work, the emerging consensus is that both DNA and RNA can participate in the Y motif at low pH. However, only DNA can participate in the pH-independent R motif (11,17,18).
Although intramolecular triplexes lack obvious therapeutic applications, they might arise in natural systems. Intramolecular triplexes could occur through two processes. In one case, commonly known as H-DNA, local denaturation of homopurine–homopyrimidine mirror repeats in duplex DNA allows one DNA strand to fold back, forming base triplets with the adjacent native duplex DNA (9,10,19,20). Sequences thought to support such structures are over-represented in mammalian genomes (21,22). In H-DNA the triplex is effectively intramolecular although the participating domains are not covalently linked in sequence along one strand. We term this latter case, which is the subject of this paper, an intrastrand triplex to emphasize the fact that the three domains lie sequentially along one strand of DNA or RNA. The four possible classes of intrastrand triplexes (I–IV) are presented schematically in Figure 1A. Several studies have established that appropriate oligonucleotides can fold to form intramolecular intrastrand triplexes (23–32). However, these studies typically focused only on DNA. Within this limitation, it was ascertained that triplexes in the Y motif formed either at low pH in the absence of Mg2+ or at neutral pH in the presence of Mg2+ (23) whereas triplexes in the R motif were detected at neutral pH either with or without Mg2+ (26).
Figure 1.

Experimental design. (A) Strand orientations of the four possible classes of intrastrand triplexes. R, purine; Y, pyrimidine; H, Hoogsteen; RH, reverse Hoogsteen. Arrows indicate 5′ to 3′ direction. (B) Sequences of DNA oligonucleotides studied. (C) Sequences of RNA oligonucleotides studied (italics). Oligonucleotide domains were linked with hexaethylene glycol (–) spacers.
To extend these results, the present study systematically investigates intrastrand triplex formation in appropriate short DNA and RNA pyrimidine motif sequences at physiological temperature and pH. Because the third strand is held at high local concentration near the duplex in a single molecule, we considered it possible that such triplexes might form even when the corresponding intermolecular triplexes had not been detected. We reasoned that if any detected triplexes were sufficiently stable to persist at physiological temperatures and pH, the discovery of similar sequences in genomes could indicate potential structural and functional roles in vivo.
MATERIALS AND METHODS
Oligonucleotides
DNA oligonucleotides were synthesized by phosphoramidite methodology using an Applied Biosystems Model 380B DNA synthesizer. RNA oligonucleotides were synthesized using appropriately protected RNA phosphoramidites followed by the methylamine deprotection protocol suggested by the supplier (Glen Research, Sterling, VA). Hexaethylene glycol linker phosphoramidites were obtained from Glen Research and were introduced during synthesis as directed by the supplier. All oligonucleotides were purified by denaturing polyacrylamide gel electrophoresis, elution from gel slices and desalting by precipitation from ethanol or on C18 reverse phase cartridges (Waters). Oligonucleotides were characterized by electrospray ionization mass spectroscopy in the negative ion mode after on-line cation exchange to suppress metal adducts.
Thermal denaturation studies
Solutions of Y motif molecules for thermal denaturation studies contained 1 µM oligonucleotide in 50 mM MOPS, pH 7.1 in the absence or presence of the indicated concentrations of MgCl2 (similar results were obtained in cacodylate buffer).
Melting experiments were performed in teflon-stoppered, 1 cm path length quartz cells using a Varian Cary 300Bio UV-vis spectrophotometer equipped with a thermoprogrammer. Samples were heated to 85°C for 1 min and then cooled to 5°C over 30 min. UV absorbance (A) at 260 nm was monitored as the sample temperature was increased from 5 to 95°C at a rate of one half degree per min. Below 20°C, nitrogen was continuously flushed through the sample compartment to prevent moisture condensation on the cuvettes.
Melting temperatures (Tm) and free energy values (ΔG25) were derived by computer fitting theoretical models to the denaturation data using a previously described two-state approximation for melting (33). In cases where two transitions were observed for triplex denaturation, each transition was analyzed separately.
Circular dichroism spectroscopy
Circular dichroism (CD) studies were performed using a modified Jasco J720 spectrometer at ambient temperature. Selected DNA oligonucleotides (6 µM) were studied in 50 mM MOPS, pH 7.1 in the absence or presence of 5 mM MgCl2. Native polyacrylamide gel electrophoresis studies of the molecules at concentrations 40 nM–6 µM confirmed that the respective oligonucleotides did not aggregate under these conditions. Samples were heated to 90°C for 5 min and cooled to room temperature over 30 min before spectra were collected. Rectangular optical cells with path lengths of 0.1 cm were used for DNA samples.
Gel shift assay
Oligonucleotides were labeled by treatment with T4 polynucleotide kinase in the presence of [γ-32P]ATP. Labeled oligonucleotides were subsequently purified by gel filtration chromatography. For the gel shift assay, ~0.4 pmol labeled oligonucleotide, 1 µM unlabeled oligonucleotide and 1 µg yeast tRNA were incubated in 50 mM MOPS buffer, pH 7.1 either in the absence or presence of 5 mM MgCl2. Samples were heated to 90°C for 5 min and then cooled to room temperature over 30 min. Samples were then analyzed on 20% native polyacrylamide gels (19:1 acrylamide/ bisacrylamide) prepared in 50 mM MOPS buffer, pH 7.1. Electrophoresis was performed in this buffer at room temperature overnight. (Samples containing MgCl2 were also analyzed on gels that contained Mg2+, and similar results were obtained.) The resulting gel was imaged and analyzed by storage phosphor technology using a Molecular Dynamics Storm 840 phosphorimager.
RESULTS AND DISCUSSION
Experimental design
To systematically investigate the stabilities of representative short pyrimidine (Y) motif intrastrand triple helices at physiological temperature and pH, we created a set of DNA (1–6) and RNA (7–10) molecules (Fig. 1B and C). In this design, all three strands (Watson, Crick and Hoogsteen) are present in a single molecule that can fold back on itself to form an intramolecular triplex. Flexible tethers, composed of hexaethylene glycol units, were used to link the oligonucleotide domains (Fig. 1). Previous studies have demonstrated that this class of hydrophilic linkers allows formation of duplexes and triplexes with conformations indistinguishable from non-linked molecules (28,34,35). Hexaethylene glycol linkers thus provided an economical means to create generic linkers. Thermal denaturation experiments comparing the stabilities of DNAs containing tetra-cytosine linkers in place of hexaethylene glycol established that the composition of the linkers did not significantly alter the melting temperature at physiological pH and MgCl2 concentrations (data not shown). The two possible folded isomers in the Y motif, differing in the 5′ to 3′ order of their domains were designed, thus creating representatives of triplex classes III and IV (Fig. 1A). The appropriate underlying duplexes were also synthesized as controls. A DNA molecule containing only the Hoogsteen domain terminating with hexaethylene glycol groups was synthesized for CD experiments (oligonucleotide 5; Fig. 1B). As a control for the gel shift experiments, a Y motif DNA molecule was also synthesized such that the Hoogsteen domain was constrained to lie anti-parallel to the purine strand, thereby precluding conventional Y motif triplex formation (oligonucleotide 6; Fig. 1B).
Y motif triplexes detected in both DNA and RNA at neutral pH
Using UV spectroscopy, triplex formation was assayed by monitoring thermal hyperchromicity at 260 nm for Y motif molecules in 50 mM MOPS buffer, pH 7.1 either in the absence or presence of 5 mM MgCl2.
Two melting transitions are typically expected if triplex formation occurs in molecules containing three domains. The first transition reflects dissociation of the Hoogsteen strand. The second transition reflects melting of the underlying duplex into a single strand. Because the two-domain control molecules represent only the underlying duplexes, they were expected to demonstrate single melting transitions comparable to the second transitions of the corresponding three-domain molecules.
Representative UV melting curves are shown in Figure 2A and B. In the Y motif in the absence of Mg2+, both DNA and RNA appear to form triplexes that dissociate in two transitions (Fig. 2A and B, open symbols). The melting temperatures and calculated free energies of the second transitions in these molecules are very similar to the melting transitions observed for their corresponding duplex derivatives, as expected (Fig. 2A and B, oligonucleotides 1–4, 7–10, Table 1). For both class III and class IV triplex–duplex transitions, DNA structures were found to be more stable than the corresponding RNA structures. For class III DNA 1, the first melting transition occurred at 30.1°C (ΔG25 = 0.4 kcal/mol) as compared to 15.6°C (ΔG25 = –1.5 kcal/mol) for class III RNA 7 (Table 1). Similar results were obtained for class IV isoforms in this motif (oligonucleotides 3–4, 8–9, Table 1). These stable triplexes are noteworthy because they occur at pH 7.1, well above the pKa for cytosine protonation commonly observed to stabilize the Y motif. The high stability of these triplexes at physiological pH is presumably attributable to their intrastrand character.
Figure 2.

Results of thermal denaturation and CD experiments for Y motif molecules. Intramolecular complexes were analyzed in solutions containing 50 mM MOPS, pH 7.1 in the absence (open symbols) or presence (filled symbols) of 5 mM Mg2+. (A) UV spectra of Y motif RNAs 8 (1 µM; circles) and 9 (1 µM, squares). (B) UV spectra of Y motif DNAs 1 (1 µM; circles) and 2 (1 µM, squares). (C) CD spectra of Y motif DNA 3 (6 µM; circles) versus the added spectra of 4 and 5 (each at 6 µM, squares) at 23°C. UV spectra recorded at 260 nm.
Table 1. Melting and thermodynamic parameters (± standard deviation) for DNA and RNA oligomers at pH 7.1 in the absence or presence of 5 mM Mg2+.

aDG25 refers to the unfolding process at 25°C.
bTm estimated as maximum of dA/dT plot.
cParameter could not be accurately estimated by fitting.
Y motif triplexes stabilized by Mg2+
The addition of Mg2+ stabilizes RNA triplex formation for both isoforms in the Y motif (filled symbols in Fig. 2A, oligonucleotides 7–10, Table 1). Addition of 5 mM Mg2+ effected an increase in the temperature of the first melting transition of ~23°C (oligonucleotides 7 and 9, Table 1). Thus, short intrastrand RNA triplexes of this kind could occur in vivo.
In contrast to RNA, addition of Mg2+ to both isoforms of DNA Y motif molecules altered their melting behavior such that the molecular structure no longer dissociated in two transitions upon heating (filled symbols in Fig. 2B). Moreover, the single melting transitions observed in these three-domain molecules occurred at temperatures similar to their corresponding duplexes (oligonucleotides 1–4, Table 1). This result for DNA is surprising, as Mg2+ is thought to stabilize Y motif triplexes. While unexpected, there is some precedent for this observation. A previous study also noted that the biphasic melting of Y motif triplexes disappeared as Mg2+ concentration was increased above 1 mM (28). Additional experiments employing lower concentrations of MgCl2 demonstrated the same melting behavior down to 1 mM (data not shown). In the presence of 0.1 mM MgCl2, the profile is slightly biphasic (data not shown). Possible explanations for the appearance of a single transition include: (i) Mg2+ ions somehow inhibit association of the Hoogsteen domain with the underlying duplex; (ii) the three-domain molecule forms a triplex that is at least as stable as the underlying duplex, such that the melting of the third strand is coincident with the duplex melting transition; (iii) the dissociation of the Hoogsteen domain does not produce a detectable change in the UV spectrum in the presence of Mg2+; (iv) the absorption properties of the triplex may be altered due to a change in extinction coefficient; or (v) the third strand dissociates with low cooperativity such that the melting transition is too broad to detect.
In order to clarify whether or not intrastrand triplexes were forming in DNA Y motif molecules in the presence of Mg2+, CD spectroscopy was employed. Spectra of the three-domain molecules, their corresponding duplexes and their isolated Hoogsteen (H) or reverse-Hoogsteen (RH) domains were collected separately. The spectra of the duplexes and the H or RH domains were then summed mathematically and compared to the spectra of the corresponding three-domain molecules. If triplexes occur, the interaction of the H (or RH) domain with the underlying duplex may cause the spectrum of the three-domain molecule to be different from the added spectra of the corresponding two-domain molecule and isolated third strand (36).
The spectrum of the three-domain DNA Y motif molecule 3 is characterized by a maximum at 281 nm and a minimum at 250 nm in the absence of Mg2+ (open circles in Fig. 2C). In contrast, the spectral sum of corresponding duplex 4 and third strand 5 is different, characterized by an increase in amplitude of the maximum at 281 nm, as well as a loss of a slight dip at ~268 nm (open squares in Fig. 2C). Durand and co-workers obtained similar results in their characterization of a Y motif triplex with T•A•T triads (28). Upon addition of 5 mM Mg2+, the shape of the spectra for both 3 and 4+5 did not change, although the amplitudes were slightly decreased (filled symbols in Fig. 2C). These results suggest that an alternative structure consistent with triplex formation occurs in DNA Y motif molecules both in the absence and presence of Mg2+, despite the fact that only a single melting transition is detected in the presence of Mg2+.
In order to further clarify these results, gel shift experiments were undertaken. The migration of DNA Y motif molecule 3 was compared to that of control DNA molecule 6 whose domains had been reordered such that a conventional Y motif triplex structure is impossible (Fig. 3A). Triplex formation is expected to generate compact structures that migrate more quickly through native polyacrylamide gels. In contrast, the unfolded molecule whose Hoogsteen domain cannot associate with the underlying duplex should exhibit retarded mobility. The electrophoretic mobilities of both molecules were examined either in the absence or presence of Mg2+.
Figure 3.
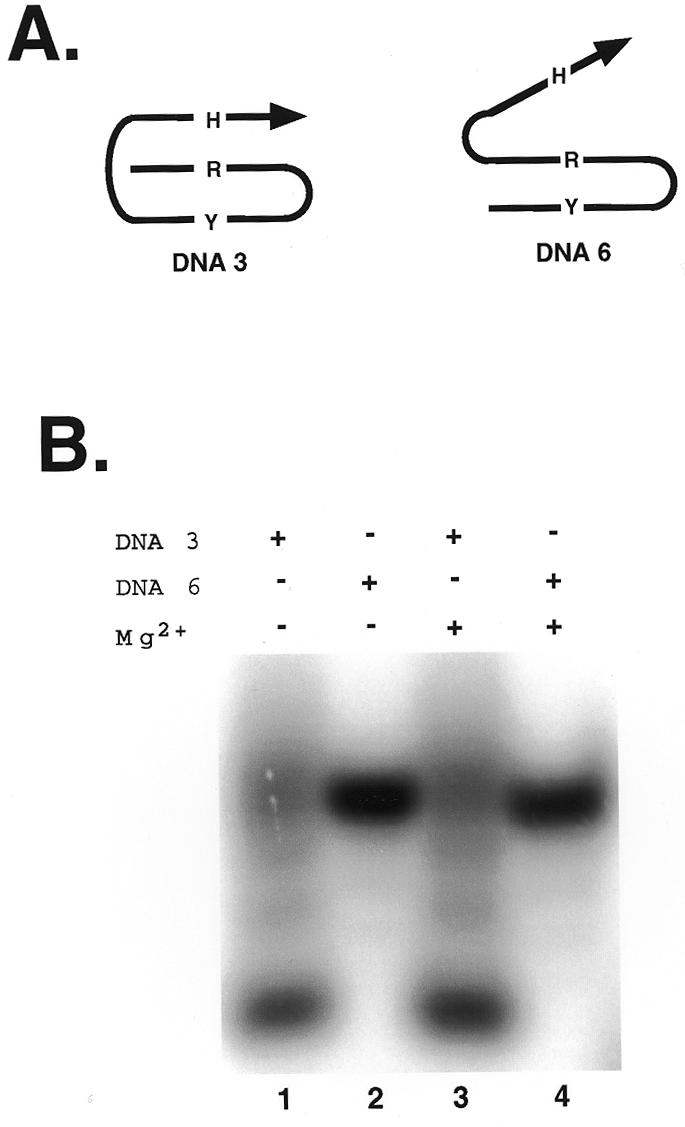
Gel shift experiments. Autoradiogram of 20% native polyacrylamide gel suggesting the formation of a compact intrastrand triplex by DNA 3 (lanes 1 and 3) but not by DNA 6 (lanes 2 and 4). Mg2+ (5 mM) was present during equilibrium of samples in lanes 3 and 4 prior to electrophoresis.
DNA molecule 3 migrated faster than control molecule 6, both in the absence and presence of Mg2+ in the loaded samples (Fig. 3B, lanes 2 and 4). Comparable results were obtained when the entire gel and electrophoresis buffer contained 5 mM Mg2+ (data not shown). This behavior indicates that stable Y motif triplex formation occurs for DNA 3 in the absence or presence of Mg2+, supporting the results obtained with CD spectroscopy.
Conclusions and implications
Our data for thermal denaturation, CD and gel shift analyses of intrastrand DNA and RNA triplexes are summarized in Table 2. In our UV studies we found that DNA Y motif triplexes melted in two transitions. In contrast to a previous report (23), we did not detect significant differences among the stabilities of the various triplex isoforms. For DNA in the Y motif, triplexes were observed in the absence of Mg2+. However, triplexes were not detected by UV melting analysis upon addition of 5 mM Mg2+. In order to determine whether this result was due to Mg2+-dependent triplex inhibition or masking of the transition by one of the mechanisms described above, CD spectroscopy was utilized. Employing this method, we detected evidence of triplex formation in DNA Y motif molecules in both the absence and presence of Mg2+ (Table 2, column 5). These CD results, together with the finding that the melting profile is still slightly biphasic in the presence of 0.1 mM Mg2+ (data not shown) support the notion that the presence of Mg2+ in concentrations above 1 mM masks the triplex transition in the melting profile. We suggest three possible mechanisms. First and most likely, Mg2+ may promote a triplex that is at least as stable as the underlying duplex so that the melting of the third strand becomes coincident with the duplex melting transition. We cannot rule out the possibility that Mg2+ eliminates a detectable change in the UV spectrum at 260 nm when the Hoogsteen domain associates with the duplex. It also remains possible that Mg2+ causes the DNA triplex to dissociate with low cooperativity, producing a broad and undetectable melting transition. The notion that the Y motif intrastrand DNA triplex is indeed stable in the presence of Mg2+ was further supported by our gel mobility assays (Fig. 3).
Table 2. Summary of results. Intrastrand triplex formation detected (+) or undetected (–) in DNA (D) and RNA (R) oligomers at pH 7.1 in the absence or presence of 5 mM Mg2+.
| Motif | Class | Backbone | Thermal denaturation(–Mg2+/+Mg2+) | CD Spectroscopy(–Mg2+/+Mg2+)a | Gel shift(–Mg2+/+Mg2+)a |
|---|---|---|---|---|---|
| III | D | +/– | ND | ND | |
| Pyrimidine | R | +/+ | ND | ND | |
| (Y) | IV |
D | +/– | +/+ | +/+ |
| R | +/+ | ND | ND |
aND, not determined.
For RNA, Y motif triplexes were detected in the absence of Mg2+ using UV spectroscopy. Addition of Mg2+ increased the stability of these triplexes. In both cases, RNA in Y motif complexes demonstrated two melting transitions.
In summary, all four possible DNA and RNA Y motif intrastrand triplexes were observed using thermal denaturation experiments, CD spectroscopy and gel shift experiments (Table 2). In the presence of physiological concentrations of Mg2+ and at physiological pH, all detected triplexes are sufficiently stable to persist at physiological temperatures. The data suggest that, if such sequences are encoded in genomes, the potential exists for the formation of stable intramolecular triple helix structures in single-stranded DNA and RNA in vivo.
It is interesting that while isolated base triplets occur in tRNA and ribozyme structures (37–39), no intrastrand triple helices with consecutive stacked triplets have yet been observed in the folded structures of natural RNAs. The present demonstration that short structures of this type readily occur in the Y motif (class III and IV triplexes) at physiological pH and temperature suggest that intrastrand Y motif triplex structures may eventually be discovered in natural RNAs. If detected, the possible functions of such structures would be intriguing.
An obvious limitation to the natural occurrence of intrastrand DNA triple helices is the requirement for all three domains to be available along a single DNA strand. Thus, natural DNA triplexes would be expected only at DNA sites where the complementary DNA strand had not yet been synthesized (40), where the complement was complexed with single-stranded DNA binding proteins, or where denaturation was favored by negative superhelical strain. Even if present only transiently, such triple-helical DNA structures might alter the fidelity or speed of replication or transcription, or serve as binding sites for specialized proteins. It will therefore be of interest to explore genomes for sequences with the potential to adopt or encode intrastrand triple helices.
Acknowledgments
ACKNOWLEDGEMENTS
We thank the past and present members of the Maher laboratory for helpful suggestions during the course of this research. We appreciate the excellent assistance of Maryjane Doerge (Mayo Foundation Molecular Biology Core Facility) and Linda Benson (Mayo Foundation Biomedical Mass Spectrometry Facility). We thank Tom Burghardt for providing the CD spectrophotometer and expert guidance. This work was supported by the Mayo Foundation and NIH grants GM47814 and GM54411 to L.J.M., and NRSA fellowship GM18926 to P.R.H.
REFERENCES
- 1.Felsenfeld G., Davies,D.R. and Rich,A. (1957) J. Am. Chem. Soc., 79, 2023–2024. [Google Scholar]
- 2.Chastain M. and Tinoco,I. (1992) Nucleic Acids Res., 20, 315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sen D. and Gilbert,W. (1990) Nature, 344, 410–414. [DOI] [PubMed] [Google Scholar]
- 4.Maher L.J. (1992) Bioessays, 14, 807–815. [DOI] [PubMed] [Google Scholar]
- 5.Guschlbauer W., Chantot,J.F. and Thiele,D. (1990) J. Biomol. Struct. Dyn., 8, 491–571. [DOI] [PubMed] [Google Scholar]
- 6.Moser H.E. and Dervan,P.B. (1987) Science, 238, 645–650. [DOI] [PubMed] [Google Scholar]
- 7.Beal P.A. and Dervan,P.B. (1991) Science, 251, 1360–1363. [DOI] [PubMed] [Google Scholar]
- 8.Kool E. (1996) Annu. Rev. Biophys. Biomol. Struct., 25, 1–28. [DOI] [PubMed] [Google Scholar]
- 9.Lyamichev V.I., Mirkin,S.M. and Frank-Kamenetski,M.D. (1986) J. Biomol. Struct. Dyn., 3, 667–669. [DOI] [PubMed] [Google Scholar]
- 10.Mirkin S.M., Lyamichev,V.I., Drushlyak,K.N., Dobrynin,V.N., Filippov,S.A. and Frank-Kamenetskii,M.D. (1987) Nature, 330, 495–487. [DOI] [PubMed] [Google Scholar]
- 11.Skoog J. and Maher,L.J. (1993) Nucleic Acids Res., 21, 2131–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maher L.J., Wold,B. and Dervan,P.B. (1989) Science, 245, 725–730. [DOI] [PubMed] [Google Scholar]
- 13.Young S.L., Krawczyk,S.H., Matteucci,M.D. and Toole,J.J. (1991) Proc. Natl Acad. Sci. USA, 88, 10023–10026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maher L.J., Dervan,P.B. and Wold,B. (1992) Biochemistry, 31, 70–81. [DOI] [PubMed] [Google Scholar]
- 15.Roberts R.W. and Crothers,D.M. (1992) Science, 258, 1463–1466. [DOI] [PubMed] [Google Scholar]
- 16.Han H. and Dervan,P. (1993) Proc. Natl Acad. Sci. USA, 90, 3806–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Escude C., Francois,J.C., Sun,J.S., Ott,G., Sprinzl,M., Garestier,T. and Helene,C. (1993) Nucleic Acids Res., 21, 5547–5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Semerad C.L. and Maher,L.J. (1994) Nucleic Acids Res., 22, 5321–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Htun H. and Dahlberg,J.E. (1988) Science, 241, 1791–1796. [DOI] [PubMed] [Google Scholar]
- 20.Htun H. and Dahlberg,J.E. (1989) Science, 243, 1571–1576. [DOI] [PubMed] [Google Scholar]
- 21.Schroth G.P. and Ho,P.S. (1995) Nucleic Acids Res., 23, 1977–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bucher P. and Yagil,G. (1991) DNA Seq., 1, 157–172. [DOI] [PubMed] [Google Scholar]
- 23.Haner R. and Dervan,P.B. (1990) Biochemistry, 29, 9761–9765. [DOI] [PubMed] [Google Scholar]
- 24.Sklenar V. and Feigon,J. (1990) Nature, 345, 836–838. [DOI] [PubMed] [Google Scholar]
- 25.Pilch D.S., Brousseau,R. and Shafer,R.H. (1990) Nucleic Acids Res., 18, 5743–5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen F.M. (1991) Biochemistry, 30, 4472–4479. [DOI] [PubMed] [Google Scholar]
- 27.Macaya R., Schultze,P. and Feigon,J. (1992) J. Am. Chem. Soc., 114, 781–783. [Google Scholar]
- 28.Durand M., Peloille,S., Thuong,N.T. and Maurizot,J.C. (1992) Biochemistry, 31, 9197–9204. [DOI] [PubMed] [Google Scholar]
- 29.Radhakrishnan I. and Patel,D.J. (1993) Structure, 1, 135–152. [DOI] [PubMed] [Google Scholar]
- 30.Volker J., Botes,D.P., Lindsey,G.G. and Klump,H.H. (1993) J. Mol. Biol., 230, 1278–1290. [DOI] [PubMed] [Google Scholar]
- 31.Gondeau C., Maurizot,J.C. and Durand,M. (1998) J. Biomol. Struct. Dyn., 15, 1133–1145. [DOI] [PubMed] [Google Scholar]
- 32.Gondeau C., Maurizot,J.C. and Durand,M. (1998) Nucleic Acids Res., 26, 4996–5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gacy A.M. and McMurray,C.T. (1998) Biochemistry, 37, 9426–9434. [DOI] [PubMed] [Google Scholar]
- 34.Durand M., Chevrie,K., Chassignol,M., Thuong,N.T. and Maurizot,J.C. (1990) Nucleic Acids Res., 18, 6353–6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maurizot J.C., Chevrie,K., Durand,M. and Thuong,N.T. (1991) FEBS Lett., 288, 101–104. [DOI] [PubMed] [Google Scholar]
- 36.Gray D.M., Hung,S. and Johnson,K.H. (1995) Methods Enzymol., 246, 19–34. [DOI] [PubMed] [Google Scholar]
- 37.Goddard J. (1977) Prog. Biophys. Mol. Biol., 32, 233–308. [PubMed] [Google Scholar]
- 38.Westhof E., Romby,P., Romaniuk,P.J., Ebel,J.P., Ehresmann,C. and Ehresmann,B. (1989) J. Mol. Biol., 207, 417–431. [DOI] [PubMed] [Google Scholar]
- 39.Michel F. and Westhof,E. (1990) J. Mol. Biol., 216, 585–610. [DOI] [PubMed] [Google Scholar]
- 40.Gacy A.M., Goellner,G.M., Spiro,C., Chen,X., Gupta,G., Bradbury,E.M., Dyer,R.B., Mikesell,M.J., Yao,J.Z., Johnson,A.J., Richter,A., Melancon,S.B. and McMurray,C.T. (1998) Mol. Cell, 1, 583–593. [DOI] [PubMed] [Google Scholar]