


Abstract
The sequence specificity of the ‘10–23’ RNA-cleaving DNA enzyme (deoxyribozyme) was utilised to discriminate between subtle differences in nucleic acid sequence in a relatively conserved segment of the L1 gene from a number of different human papilloma virus (HPV) genotypes. DNA enzymes specific for the different HPV types were found to cleave their respective target oligoribonucleotide substrates with high efficiency compared with their unmatched counterparts, which were usually not cleaved or cleaved with very low efficiency. This specificity was achieved despite the existence of only very small differences in the sequence of one binding arm. As an example of how this methodology may be applied to mutation analysis of tissue samples, type-specific deoxyribozyme cleavable substrates were generated by genomic PCR using a chimeric primer containing three bases of RNA. The RNA component enabled each amplicon to be cleavable in the presence of its matching deoxyribozyme. In this format, the specificity of deoxyribozyme cleavage is defined by Watson–Crick interactions between one substrate-binding domain (arm I) and the polymorphic sequence which is amplified during PCR. Deoxyribozyme-mediated cleavage of amplicons generated by this method was used to examine the HPV status of genomic DNA derived from Caski cells, which are known to be positive for HPV16. This method is applicable to many types of nucleic acid sequence variation, including single nucleotide polymorphisms.
INTRODUCTION
To effectively capitalise on the rapidly expanding nucleic acid sequence database, there is a need for convenient methods which can readily discriminate between closely related sequences. Conventional methods for analysing sequence variation such as restriction fragment length polymorphisms (RFLP) (1), single-strand conformation polymorphisms (SSCP) (2) and sequencing are either time consuming or limited by their dependence on recognition sequences for restriction endonucleases which are effected by the mutation. Gene-specific amplification techniques such as polymerase chain reaction (PCR) and ligase chain reaction (LCR) can detect very small amounts of nucleic acids but often lack the sensitivity to detect specific point mutations without ancillary technology such as SSCP or RFLP analysis (3). Allele-specific oligonucleotide hybridisation can also serve this purpose (4,5) with much greater flexibility, particularly in a microarray format which allows parallel verification or simultaneous screening of many targets in one test. Microarrays can effectively sequence small sections of polymorphic nucleic acids by providing a different probe for each alternative sequence (6,7). One of the challenges of this methodology is achieving the desired level of hybridisation specificity, particularly when discriminating between two sequences that differ by only a single base mutation. As these point mutations or single nucleotide polymorphisms (SNP) only generate small changes in the melting temperature of an oligonucleotide duplex, these systems require fine tuning in order to function effectively. For example, in a low stringency hybridisation it is very easy to record false positives, whereas if the stringency is set too high, a false negative may be indicated. Microarrays supporting thousands of different oligonucleotides can usually meet this challenge by brute force interrogation of a SNP at every possible sequence permutation and with the mutation aligned at all positions of the probe. In a further development of this strategy, the hybridisation stringency of an oligonucleotide array assembled on a semiconductor microchip is controlled electronically by altering the voltage at small electrodes embedded in the silicone wafer (8). Subtle differences in oligonucleotide melting temperature (Tm) can also be used to differentiate between SNPs by analysis of the melting curve of a probe during PCR which can be monitored in real time through fluorescence resonance energy transfer (9).
In this study we explore the potential of a catalytic DNA known as the ‘10–23’ RNA-cleaving DNA enzyme, as a means of discriminating between a range of closely related sequences. The 10–23 DNA enzyme or deoxyribozyme was derived by in vitro selection from a combinatorial library of oligonucleotides (10). It consists of a conserved catalytic domain flanked by two substrate binding domains, and has the potential to cleave RNA at any purine–pyrimidine junction (Fig. 1). The deoxyribozyme, as in the case of oligonucleotide hybridisation, achieves its target specificity by Watson–Crick interactions which occur via two substrate-binding domains formed by arms I and II. However, while oligonucleotide hybrids can tolerate a certain amount of base mismatch, efficient deoxyribozyme-mediated cleavage can usually only occur when the deoxyribozyme–substrate heteroduplex is perfectly matched (11,12). In addition to this level of specificity achieved through the substrate-binding domains, the 10–23 DNA enzyme can also discriminate by its requirement for an unpaired purine at the substrate cleavage site followed by a paired pyrimidine. This flexibility should enable deoxyribozyme-based sequence analysis to identify almost any polymorphism. To demonstrate the potential of this approach to sequence recognition we examined the selectivity of human papilloma virus (HPV) type-specific deoxyribozymes at a small polymorphic site within a relatively conserved region of the L1 gene. The specificity of each deoxyribozyme was determined by comparing the extent of cleavage on the matched substrate with cross-reactivity on mismatched substrates. As expected, only the matched deoxyribozymes were capable of generating substantial cleavage in the various substrates tested. In order to translate this RNA cleavage assay into a more accessible format, we designed a chimeric primer with a three base RNA sequence corresponding to the cleavage site core. When this generic primer was used to amplify the target DNA, it generated a specific cleavable site which enabled deoxyribozyme-mediated identification of the DNA sample.
Figure 1.

The 10–23 RNA-cleaving deoxyribozyme. This illustration shows the secondary structure of a generic enzyme–substrate complex formed by Watson–Crick interactions between the target RNA sequence and the deoxyribozyme binding domains (both represented by N). The conserved 10–23 catalytic motif is situated between the binding domains (arms I and II) and bridges the unpaired purine of the purine–pyrimidine cleavage site.
MATERIALS AND METHODS
Oligonucleotides
DNA/RNA chimeric oligonucleotides were synthesised and purified by Oligos etc. Other oligos were made by Oligos etc or Pacific Oligos. The name, sequence and origin of each oligonucleotide are given in Table 1.
Table 1. Substrate, deoxyribozymes and primer oligonucleotides.
| Name | Sequence | Target |
|---|---|---|
| Chimeric substratesa | ||
| DT148 | ACAGTAACAAAUAATTGATTA | HPV18 L1 |
| DT149 | ACAGTAACAAAUAGATGATTA | HPV11 L1 |
| DT150 | ACAGTAACAAAUAACTGATTG | HPV31 L1 |
| DT151 | ACAGTAACAAAUAGTTGATTA | HPV6 L1 |
| DT152 | ACAGTAACAAAUACCTGATTG | HPV33 L1 |
| DT153 | ACAGTAACAAAUAGTTGGTTA | HPV16 L1 |
| Deoxyribozymesb | ||
| DT160 | TAATCATCTAggctagctacaacgaTTGTTACTGT | HPV11 L1 |
| DT174 | TAATCAATTAggctagctacaacgaTTGTTACTGT | HPV18 L1 |
| DT175 | TAATCAACTAggctagctacaacgaTTGTTACTGT | HPV 6 L1 |
| DT176 | CAATCAGTTAggctagctacaacgaTTGTTACTGT | HPV31 L1 |
| DT177 | CAATCAGGTAggctagctacaacgaTTGTTACTGT | HPV33 L1 |
| DT178 | TAACCAACTAggctagctacaacgaTTGTTACTGT | HPV16 L1 |
| Primersc | ||
| DT184 | GTATCTACCACAGTAACAAAUA | HPV L1 |
| DT185 | AAYAATGGYATYTGYTGG | HPV L1 |
| DT280 | AATAATGGCATTTGTTGG | HPV16 L1 |
aThe RNA component of the chimeric substrates are indicated in bold.
bThe conserved 15 base motif of the 10–23 catalytic domain was denoted by lower case.
cPrimer DT185 contained degenerate pyrimidines at four positions such that Y = T+C. A non-degenerate version of this primer specific for HPV16 was also tested. All oligonucleotides were typed in the 5′→3′ direction.
Cleavage reactions
The specificity of deoxyribozyme-mediated cleavage was examined by comparing the extent of cleavage achieved (during a 1 h incubation) by the various matched and unmatched deoxyribozyme–substrate combinations. The reactions were performed under single turnover conditions with an 8-fold excess of deoxyribozyme. The chimeric substrate oligonucleotides (1 µM) were 5′-end-labelled prior to the cleavage reaction with 1 U of polynucleotide kinase (New England Biolabs) in 60 mM Tris–HCl (pH 7.5), 9 mM MgCl2, 10 mM dithiothreitol, and 10 µCi of [γ-32P]ATP (GeneWorks) at 37°C for 30 min and 75°C for 5 min. For each cleavage reaction the labelled substrate and deoxyribozyme were pre-equilibrated separately in the reaction buffer (50 mM Tris–HCl, pH 7.5, 10 mM MgCl2) at 37°C for 5 min before being combined to a final concentration of 50 and 500 nM, respectively. After 60 min the reaction was stopped by mixing samples with an equal volume of ice-cold buffer containing 90% formamide, 20 mM EDTA and loading dye. After reaction, the uncleaved substrate and products were resolved by electrophoresis on a 10% denaturing polyacrylamide gel and analysed using a phosphorimager (Molecular Dynamics).
Cell culture and DNA preparation
The HPV16-positive Caski cell line was cultured in DMEM supplemented with 10% fetal calf serum at 37°C. Genomic DNA was extracted from the cells using a DNA Extraction Kit (Stratagene) according to the manufacturer’s instructions.
Substrate amplification
A type-specific deoxyribozyme cleavable substrate was generated directly at high copy number by a generic HPV L1 PCR. The RNA component of the cleavage site was incorporated into the amplicon by a chimeric primer which contained three ribonucleotides (DT184). This was 5′-end-labelled prior to amplification with polynucleotide kinase (as described for the substrates above) and combined (2 pmol) with a degenerate L1 primer (DT185, 20 pmol) and 10 ng of Caski cell DNA in a mixture consisting of 50 mM KCl, 10 mM Tris–HCl (pH 8.3), 2.5 mM MgCl2, 1.5 U of AmpliTaq DNA polymerase (Perkin Elmer), 200 µM each dGTP, dATP, dTTP and dCTP. After 25 cycles at 95°C for 30 s, 50°C for 60 s and 72°C for 90 s, the PCR product was either purified by 6% native PAGE or used directly in cleavage reactions.
Cycle cleavage reaction
To maximise the cleavage extent in a double-stranded target we used multiple cycles of cleavage and denaturation. In this reaction purified or unpurified PCR product containing the cleavable substrate was split into six different tubes and supplemented with a type-specific deoxyribozyme (1 µM) and the MgCl2 concentration made up to 10 mM. The reaction was then carried out over 10 cycles of thermal denaturation at 80°C for 10 s followed by hybridisation and cleavage at 37°C for 5 min. As a control of this cycle cleavage reaction, a static incubation at 37°C was also carried out for the same duration (~80 min) after an initial pre-denaturation at 95°C for 2 min. After the cycle cleavage reaction each sample was mixed with an equal volume of formamide loading dye and electrophoresed on a 10% denaturing polyacrylamide gel. The respective amounts of cleaved and uncleaved substrate were revealed and analysed using a phosphorimager.
RESULTS AND DISCUSSION
Deoxyribozyme specificity
Six chimeric substrates with sequences derived from a relatively conserved region of the L1 gene in different HPV types were each challenged with a perfectly matched deoxyribozyme and five unmatched molecules designed to cleave the alternative substrates. The sequence of arm I for each type-specific deoxyribozyme was slightly different, so as to correspond to the polymorphism in each substrate (Fig. 2). The remaining binding domain (arm II) and catalytic component of each deoxyribozyme were identical between HPV types.
Figure 2.

HPV type-specific deoxyribozyme–substrate complexes. The secondary structure of six HPV type-specific deoxyribozymes with their corresponding substrate sequences. Each of the substrate sequences are derived from various HPV types (indicated above each complex) and differ from each other by polymorphisms contained within a small region corresponding to the 5′ binding domain of the deoxyribozyme.
In general agreement with earlier studies with other substrates (11) only the specifically matched deoxyribozyme was capable of achieving substantial cleavage (Fig. 3). The most intense reaction was observed in the HPV16 substrate (lane 42), with 24% cleavage achieved by the appropriately matched deoxyribozyme after 1 h. Some cross-reactivity with this substrate was observed with the deoxyribozyme specific for the HPV6 sequence, with 2% cleavage after 1 h. While this was not surprising considering that they only differ by a SNP, the difference in cleavage intensity was large enough for clear discrimination between the two reactions. Similarly, in addition to a strong reaction with its matched counterpart (20%), the HPV11 substrate also experienced a low level of cross-reaction (1%) with the HPV6-specific deoxyribozyme, which also differed by a SNP. Despite the similarity to both the HPV16 and HPV11 substrates, the HPV6 substrate, with 19% cleavage in 1 h by its specifically matched deoxyribozyme, was only cleaved to a very small extent by the unmatched HPV16-specific deoxyribozyme (1%). The other three substrates did not display any significant reaction with unmatched deoxyribozymes, including HPV31 and HPV 33, which only differ by a SNP. However, this may be due in part to the lower cleavage intensity observed with these reactions, with only 14 and 9% cleavage by the matched deoxyribozyme, respectively, after 1 h (Fig. 3, lanes 34 and 19).
Figure 3.

Deoxyribozyme cleavage-based sequence analysis. The image contains a 16% polyacrylamide sequencing gel used to resolve end-labelled cleavage product from the uncleaved substrate. Each synthetic substrate derived from one of six HPV types was incubated with its matching deoxyribozyme and the unmatched counterparts in the presence of magnesium. The substrate sequence origin for each divided reaction set is indicated at the top of the gel. The deoxyribozymes used in each set were numbered 1–7 and code for (1) no deoxyribozyme, (2) HPV11, (3) HPV18, (4) HPV6, (5) HPV31, (6) HPV33 and (7) HPV16.
The cleavage extents achieved by these HPV L1-specific deoxyribozymes, while being sufficient to produce an unambiguous signal, were in general lower than expected. This is perhaps partly due to the majority DNA–DNA homoduplex composition of the enzyme–substrate complex, which has already been shown to be the least stable hybrid and to have a lower cleavage efficiency compared with other duplex structures (13). Earlier investigations of the 10–23 deoxyribozyme have also shown that there is significant sequence-specific variation in cleavage efficiency. This variation could usually be reconciled in terms of the heteroduplex stability predicted by nearest neighbour analysis (10,11,14). In accordance with the general performance of the HPV L1 deoxyribozymes, the predicted duplex stability of the enzyme–substrate complexes was below average. However, the variation between different L1 substrates did not follow the predicted hybridisation stability pattern closely. These differences in activity must therefore be due to more subtle influences of the sequence polymorphism than gross helix stability.
Substrate sequence amplification
To effectively harness the capacity of the 10–23 deoxyribozymes to discriminate between chimeric substrates with similar sequences, we designed a chimeric primer with the aim of producing a deoxyribozyme-cleavable amplicon by a generic PCR. The primer introduces the fixed RNA component and extension produces the variable component as it traverses the template polymorphism (Fig. 4). We explored the potential of this system by generating a deoxyribozyme-cleavable amplicon from the L1 gene of HPV16-positive human cells, such that it contained the same chimeric HPV16 cleavage site examined earlier using oligonucleotides. This amplicon was challenged with the matching HPV16-specific deoxyribozyme and other unmatched analogues (as in the oligonucleotide cleavage experiment) to ensure that the activity and specificity of the reaction was maintained with the substrate in this format (Fig. 5). The results demonstrated this, with only the HPV16-specific deoxyribozyme showing cleavage activity. As the substrate was embedded in a double-stranded PCR product we found that cleavage extent could be maximised by cycling between 37 and 80°C. Under these reaction conditions, the thermal denaturation phases gave the deoxyribozyme multiple opportunities to compete with the template strand during hybridisation with the substrate-containing strand. This dynamic reaction scheme was found to produce more cleavage product than a static incubation at 37°C (Fig. 5).
Figure 4.

Substrate sequence amplification and cycle cleavage. Schematic representation of a method for generating a deoxyribozyme cleavable substrate from a small amount of genomic DNA using PCR and a cycle cleavage reaction. In the first three stages of this procedure, genomic DNA is thermally denatured and used as a template for amplification with generic HPV primers. These flank a polymorphic region which enables the production of type-specific amplicons. As the reverse primer contains a 3 bp stretch of RNA (upper case) these amplicons are cleavable in the presence of a deoxyribozyme with an arm sequence complementary to the polymorphic region. Polymorphic purine and pyrimidine bases are denoted by r and y, respectively. In the later stages of this scheme, the substrate cleavage efficiency in this double-stranded format is enhanced by thermal cycling.
Figure 5.
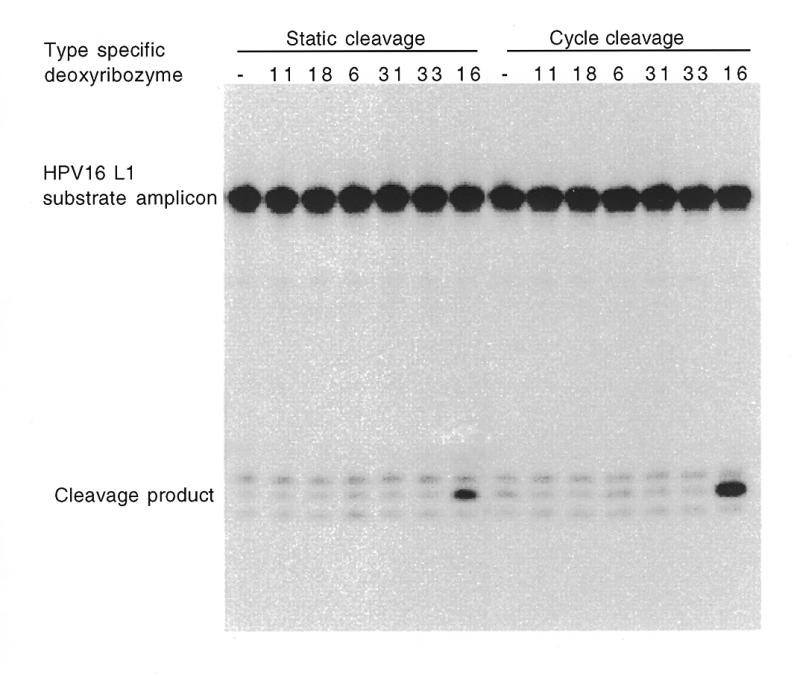
Substrate amplification and cleavage. This image was derived from a 10% sequencing gel containing purified PCR products generated in the presence of HPV16-positive Caski cell DNA followed by incubation with various HPV type-specific deoxyribozymes (indicated at the top of each lane). The relative positions of the cleaved and uncleaved substrate amplicons, which appear as intense low and high molecular weight bands, are indicated down the left side of the gel. Each cleavage reaction was carried out both at a constant 37°C (static) and by thermal cycling between 80 and 37°C (cycle cleavage) as indicated at the top of the gel.
The combination of amplification by PCR and deoxyribozyme cleavage analysis of polymorphisms provides a convenient means of discriminating between subtle differences in genomic DNA. As the 10–23 deoxyribozyme can cleave almost any purine–pyrimidine junction, this configuration will be able to accommodate most mutations, particularly SNPs, many of which would not have been amenable to conventional RFLP analysis due to the lack of appropriate restriction endonuclease recognition sequences.
In conclusion, the 10–23 deoxyribozyme was shown to be capable of discriminating between a series of closely related substrate sequences derived from the L1 gene of various HPV types. The utility of this deoxyribozyme, which has the potential for cleaving a broad spectrum of sequences with very high specificity under simulated physiological conditions, was then expanded by coupling it with PCR in a method described as substrate sequence amplification. This system should enable the deoxyribozyme cleavage approach to be accessible to almost any nucleic acid sequence, even those in low abundance, provided they are receptive to amplification by PCR.
REFERENCES
- 1.Jeffreys A.J. (1979) Cell, 18, 1–10. [DOI] [PubMed] [Google Scholar]
- 2.Orita M., Iwahana,H., Kanazawa,H., Hayashi,K. and Sekiya,T. (1989) Proc. Natl Acad. Sci. USA, 86, 2766–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mullis K.B. and Falloona,F.A. (1987) Methods Enzymol., 155, 335–350. [DOI] [PubMed] [Google Scholar]
- 4.Saiki R.K., Bugawan,T.L., Horn,G.T., Mullis,K.B. and Erlich,H.A. (1986) Nature, 324, 163–166. [DOI] [PubMed] [Google Scholar]
- 5.Malmgren H., Gustavsson,J., Turvemo,T. and Dahl,N. (1996) Clin. Genet., 50, 202–205. [DOI] [PubMed] [Google Scholar]
- 6.Hacia J.G., Brody,L.C., Chee,M.S., Fodor,S.P.A. and Collins,F.S. (1996) Nature Genet., 14, 441–447. [DOI] [PubMed] [Google Scholar]
- 7.Chee M., Yang,R., Hubbell,E., Berno,A., Huang,X.C., Stern,D., Winkler,J., Lockhart,D.J., Morris,M.S. and Fodor,S.P. (1996) Science, 274, 610–614. [DOI] [PubMed] [Google Scholar]
- 8.Gilles P.N., Wu,D.J., Foster,C.B., Dillon,P.J. and Chanock,S.J. (1999) Nature Biotechnol., 17, 365–370. [DOI] [PubMed] [Google Scholar]
- 9.Bernard P.S., Lay,M.J. and Wittwer,C.T. (1998) Anal. Biochem., 255, 101–107. [DOI] [PubMed] [Google Scholar]
- 10.Santoro S.W. and Joyce,G.F. (1997) Proc. Natl Acad. Sci. USA, 94, 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santoro S.W. and Joyce,G.F. (1998) Biochemistry, 37, 13330–13342. [DOI] [PubMed] [Google Scholar]
- 12.Kuwabara T., Warashina,M., Tanabe,T., Tani,K., Asano,S. and Taira,K. (1997) Nucleic Acids Res., 25, 3074–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ota N., Warashina,M., Hirano,K., Hatanaka,K. and Taira,K. (1998) Nucleic Acids Res., 26, 3385–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cairns M.J., Hopkins,T.M., Witherington,C., Wang,L. and Sun,L.Q. (1999) Nature Biotechnol., 17, 480–486. [DOI] [PubMed] [Google Scholar]