


Abstract
Expression of the BAP3 gene of Saccharomyces cerevisiae, encoding a branched chain amino acid permease, is induced in response to the availability of several naturally occurring amino acids in the medium. This induction is mediated via an upstream activating sequence (called UASaa) in the BAP3 promoter, and dependent on Stp1p, a nuclear protein with zinc finger domains, suggesting that Stp1p is a transcription factor involved in BAP3 expression. In this paper, we show that Stp2p, a protein with considerable similarity to Stp1p, is also involved in the induction of BAP3 expression. To gain more insight into the roles of STP1 and STP2, we have overexpressed both Stp1p and Stp2p in yeast cells. Gel shift assays with the UASaa as a probe show that the UASaa can form two major complexes. One complex is dependent on Stp2p overexpression and the other is formed independently of STP1 or STP2, suggesting that the UASaa is also bound by another factor. Here we show that the other factor is Abf1p, which binds specifically to the UASaa of BAP3.
INTRODUCTION
Saccharomyces cerevisiae can respond to the availability of several naturally occurring amino acids in its environment by inducing the expression of the corresponding amino acid uptake systems. It does so by sensing of the extracellular amino acids via a sensor, Ssy1p (1,2). The addition of amino acids results in a drastic induction of expression of several amino acid permease genes, e.g. the AGP1 gene, encoding a broad specificity amino acid permease (3), the TAT1 and TAT2 genes encoding the tyrosine and tryptophan permeases (4), the BAP2 gene encoding a branched chain amino acid permease (5) and the BAP3 (PAP1) gene, encoding a protein with high similarity to Bap2p (6). We have identified an element in the promoter of BAP3 that we called UASaa (amino acid-dependent upstream activator sequence), which is responsible for this induction (7). We have shown that this UASaa is both necessary and sufficient for induction of BAP3 expression by most l-amino acids found in proteins. Furthermore, we showed that Stp1p, a nuclear protein with zinc finger domains, plays an essential role in induction via the UASaa. Gel shift analysis showed that the UASaa can form a specific DNA–protein complex when incubated with total yeast extract. This complex is formed irrespective of whether the extract is isolated from cells grown in the presence or absence of amino acids in the medium, or from cells that contain or lack Stp1p. A limited mutational analysis of the UASaa showed that there is a strong correlation between the ability of the mutated UASaa to form a complex in vitro and its ability to function as an amino acid-dependent promoter element when fused to a CYC1–LacZ reporter. These observations led to the conclusion that the UASaa is constitutively bound by a factor different from Stp1p, and that this factor is also involved in the induction of transcription in response to amino acids in the medium.
In this paper we show that the factor that in vitro binds to the UASaa is Abf1p. This global transcription factor, encoded by ABF1 (also known as OBF, BAF1, TAF and GF1; 8), was originally identified as a protein binding to autonomously replicating sequences. Abf1p has been shown to take part also in the regulation of expression of a large number of genes involved in various cellular processes and with a variety of expression patterns. For example, Abf1p has been shown to be involved in the regulation of transcription of some of the ribosomal protein genes, as well as the COX6 gene (9), the ARO3 gene (10), the FOX3 gene (11), the CHA1 gene (12) and the HIS7 gene (13). We show that the UASaa harbours an Abf1p-binding site and that mutations within the UASaa that obliterate Abf1p binding also lead to a loss of the capacity of this DNA element to function in vivo as an amino acid-dependent UAS. However, we also show that the mere binding of Abf1p to a DNA element placed in front of a CYC1–LacZ reporter is not sufficient for induction of that reporter by amino acids.
Furthermore, we provide evidence that in addition to Stp1p, Stp2p, a protein with considerable similarity to Stp1p and having the same number and arrangement of zinc finger domains as Stp1p, is also involved in amino acid-induced transcription via the UASaa. Whereas amino acid-induced expression of BAP3 is severely compromised in Δstp1 as well as in Δstp2 mutant cells, induction is completely lost in a Δstp1/Δstp2 double mutant. To test whether Stp1p or Stp2p can bind to the UASaa, we have overexpressed both proteins in yeast cells. Using extracts from cells overexpressing Stp2p in band shift assays, we found formation of an Stp2p-dependent complex.
Taken together, we have shown that induction of BAP3 transcription in response to amino acids requires Stp1p and/or Stp2p, and that Abf1p is involved in the response.
MATERIALS AND METHODS
Strains, media and genetic methods
The S.cerevisiae strains used in this study are derived from M4054 (MATα, gap1-Δ101, ura3, gal2) as described by Grauslund et al. (5). For the extraction of protein extracts used in the band shift assays we used the protease minus strain BJ1991 (MATa, leu2, trp1, ura3-251, pbr1-1122, pep4-3, gal2; 14). Yeast cells were grown at 30°C, either in SD medium (2% glucose, 0.67% YNB with or without amino acids; Difco) or non-selectively in YPD medium (1% yeast extract, 2% bacto-peptone, 2% glucose). Yeast transformation was performed as described by Chen et al. (15). The Escherichia coli strain used in this study was SURE™ {el4-(mcrA), Δ(mcrCB-hsdSMR-mrr)171, sbcC, recB, recJ, umuC::Tn5 (kanr), uvrC, supE44, lac, gyrA96, relA1, thi-1, end A1 [F′ proAB, laclqZDM15, Tn10, (tetr)]}. Bacterial cells were grown in 2YT medium (1% yeast extract, 1.6% bacto-tryptone, 0.5% NaCl), to which ampicillin (100 µg/ml) was added if necessary. Bacterial transformation and plasmid DNA isolation were performed according to procedures described elsewhere (16,17).
STP1 and STP2 disruption
For the disruption of STP2 we used 5ΔXho3 (a plasmid encoding the STP2 gene, kindly provided by A. Hopper, Seattle, WA). First, the STP2 gene was subcloned into pUC19. Subsequently, a 626 bp HindII–SphI fragment encoding amino acids 13–225 of Stp2p was replaced by the KanMX4 gene (18), resulting in a STP2 disruption cassette. The disruption cassette was isolated and transformed to strain M4054. After 6 h of growth in liquid non-selective YPD medium, transformants were plated onto YPD containing G418 (200 mg/l; Calbiochem) in order to select for disruptants. To make an STP1 disruption mutant we made use of the URA3-based STP1 disruption cassette pBRstp1::URA3 as described by Wang and Hopper (19). Correct disruption of both STP1 and STP2 was confirmed by Southern blot analysis. The stp2Δ10 mutant M4270 and stp1Δ10/stp2Δ10 double mutant M4272 were constructed as follows. From plasmid Yep24 containing the STP2 ORF in a 2.8 kb SspI fragment 1150 bp were removed by digestion with BstXI followed by religation. The remaining ORF was inserted into plasmid pRS306. This construct was linearised with HindIII and introduced into strains M4054 and M4173. Subsequent selection on FOA plates yielded clones having lost STP2. Two clones, M4270 and M4272, were identified by PCR on chromosomal DNA to contain only stp2Δ10.
Overexpression of Stp1p and a HA epitope-tagged version of Stp2p
To overexpress Stp1p and Stp2p we placed the STP1 and STP2 genes under the control of the PGK1 promoter. The 850 bp upstream region of the PGK1 gene was isolated as a HindIII–EcoRI fragment from YEpR5 (a plasmid encoding the PGK1 gene and flanking regions; 20) and subcloned (EcoRI–HindIII) into pBluescript (pBSKS; Stratagene), resulting in pMB15. The STP1 gene was amplified from genomic DNA by PCR using the primers STP1-forward (5′-AAAGAATTCTGACATGAACTGTATCACTATTGACG-3′) and STP1-reverse (5′-TTTCTGCAGCTACGAATCGACTCTATGCGCTG-3′). The STP2 gene was amplified from pRS426 (a plasmid encoding the STP2 gene, generously supplied by A. Hopper, Seattle, WA) using the primers STP2-forward (5′-AA-AGAATTCATCATGGGCGGCCGCCCTATCTTATCACT-ATCTTCAACACGG-3′) and STP2-reverse (5′-TTTCTGC-AGGAGGAGAGGAGTTTTGGGG-3′). The forward primers introduced an EcoRI site (bold) just in front of the ATG start codon (underlined) and in the case of STP2 also a NotI site just after the ATG start codon (italic). The reverse primers introduced a PstI site at position +1987 relative to the ATG start codon for STP1 (255 bp after the coding region) and a PstI site at position +1877 relative to the ATG start codon for STP2 (250 bp after the open reading frame). The amplified genes were fused to the PGK1 promoter in pMB15 making use of the EcoRI site. The PGK1–STP1 fusion was cloned into the expression vectors YCplac33 (CEN, URA3) and YEplac195 (2µ, URA3) (21) resulting in the single and multiple copy vectors pMB70 (SCPGK1–STP1) and pMB71 (MCPGK1–STP1), respectively. The NotI site introduced into the STP2 gene was used to insert a DNA fragment encoding a triple HA epitope tag [(HA)3] (22). After this insertion, the PGK1–(HA)3–STP2 fusion was cloned into YCplac33 (CEN, URA3) and YEplac195 (2µ, URA3) resulting in the single and multiple copy vectors pMB73 [SCPGK1–(HA)3–STP2] and pMB74 [MCPGK1–(HA)3–STP2], respectively. To combine Stp1p and Stp2p overexpression, the PGK1–(HA)3–STP2 fusion was cloned into the expression vectors YCplac111 (CEN, LEU2) and YEplac181 (2µ, LEU2), resulting in the single and multiple copy vectors pMB79 [LEU2, SCPGK1–(HA)3–STP2] and pMB80 [LEU2, MCPGK1–(HA)3–STP2].
Expression of Abf1p in Escherichia coli
For the expression of Abf1p in E.coli we made use of the constructs described by Halfter et al. (23). These constructs encode the wild-type Abf1p and an Abf1p mutant lacking its N-terminal DNA-binding domain (Abf1pΔ33/240), both under the control of the isopropyl-β-d-1-thiogalactopyranoside (IPTG; Fluka)-inducible LacZ promoter. Both constructs were transformed into E.coli and pregrown overnight in 2YT with ampicillin. Subsequently, the cells were inoculated into fresh 2YT medium with ampicillin at an OD660 nm = 0.05 and grown to an OD660 nm = 0.4–0.5. Then IPTG was added to a final concentration of 1 mM and aliquots were taken at 0, 1, 2 and 3 h after IPTG addition. Protein extracts were made as described by Halfter et al. (23) and expression of the yeast proteins was monitored by subjecting the protein extracts to SDS–PAGE (10%); subsequently the separated proteins were electroblotted onto nitrocellulose membranes (as recommended by the manufacturer). The Abf1 proteins were visualised using a commercially available affinity-purified goat polyclonal primary antibody, yC-20 (diluted 1:500), raised against a peptide corresponding to an amino acid sequence mapping at the C-terminus of Abf1p, and with a horseradish peroxidase-conjugated secondary anti-goat antibody (diluted 1:5000). Both antibodies were purchased from Santa Cruz Biotechnology. Subsequently, proteins were visualised using the ECL detection kit from Amersham.
STP2 expression in E.coli
The STP2 gene was amplified from total S.cerevisiae DNA by PCR using primers 5′-CCCGCTAGCCCTATCTTATCACTATCTTCAACACG-3′ and 5′-CCCGAATTCTTAAAATTC TATCCCATAAGC-3′. The PCR product was digested with NheI and EcoRI and subcloned into the E.coli expression vector pET24a(+) (Novagen), which had been opened with NheI and EcoRI. The resulting plasmid was termed pVDH22. The cloned STP2 PCR product in pVDH22 was sequenced, and the sequence proved to be identical to the published STP2 sequence. For expression of the STP2 gene in E.coli, plasmids pVDH22 and pET24a(+) (as control) were transformed into strain BL21 (Novagen). Transformants were grown at 37°C in 100 ml LB cultures containing 50 µg/ml kanamycin to an OD600 nm of 0.5. IPTG was then added to a final concentration of 100 mM and the cultures were incubated overnight at 20°C. Subsequently, the cultures were centrifuged (15 min, 6000 r.p.m.) and the cells were resuspended in 4 ml binding buffer (20 mM Tris–HCl pH 8, 200 mM KCl, 5 mM MgCl2, 0.5 mM CaCl2, 0.1 mM EDTA, 0.5 mM DTT, 11% glycerol). PMSF was added to the suspensions to a concentration of 1 mM and, subsequently, the suspensions were sonicated five times for 30 s with cooling on ice. The resulting lysates were centrifuged (15 min, 15 000 g, 6°C) and the supernatants of this centrifugation were used for band shift assays.
Gel shift assays
Gel shift analyses using the double-stranded oligonucleotides described in Table 2 were carried out essentially as described earlier (7). As a control for Abf1p binding we used the S28A oligonucleotide. This oligonucleotide encodes part of the rpS28A promoter harbouring the Abf1p-binding consensus as described by Dorsman et al. (24). The putative Abf1p-binding site found in the UASaa of BAP3 is located in the reverse direction. Therefore, we also designed a double-stranded oligonucleotide harbouring the Abf1p-binding consensus in the reversed direction (S28Arev). As a negative control for Abf1p binding we used the S28An oligonucleotide, which contains a point mutation that is known to abolish Abf1p interaction (see Table 2, shown in bold; 24). Labelling, annealing and purification of oligonucleotides used for gel shift analyses were carried out as described earlier (7). Gel shift analyses using extracts from Abf1p-expressing E.coli mutants were performed as described by Halfter et al. (23). The Abf1p–DNA complexes were supershifted by adding the yC-20 anti-Abf1p antibody to the binding reaction after incubation of the binding reactions for 20 min on ice. The antibody-containing reactions were subsequently incubated for 10 min at room temperature.
Table 2. Oligonucleotides used in this studya.
| Name | Sequence | Abf1p | Stp2p-dependent | β-Galactosidase activityb (U/mg) |
|
|---|---|---|---|---|---|
| interaction | complex formation | –Leucine | +Leucine (2 mM) | ||
| oBAP3-WT | AGCTTAGCCGTGCATGCGGCTCCGCGAAAAGAGCTCTGCTATATTTG | + | + | 13 | 180 |
| ΔBAP3-A | AGCTTAGAAATGCATGCGGCTCCGCGAAAAGAGCTCTGCTATATTTG | – | + | 13 | 18 |
| ΔBAP3-B | AGCTTAGCCGTGCATGAAACTCCGCGAAAAGAGCTCTGCTATATTTG | – | – | 7 | 10 |
| ΔBAP3-C | AGCTTAGCCGTGCATGCGGCTCAAAGAAAAGAGCTCTGCTATATTTG | + | + | 9 | 166 |
| ΔBAP3-D | AGCTTAGCCGTGCATGCGGCTCCGCGAAAAG | + | + | 25 | 297 |
| ΔBAP3-E | AGCTTAGCCGTGCATGCGGCTG | + | – | 8 | 17 |
| ΔBAP3-F | AGCTTAGCCGTGCATGCGGCTCCGCG | + | + | 20 | 263 |
| ΔBAP3-G | AGCTTAGCCGTGCATGCGGCTACGCGAAAAG | + | – | 21 | 236 |
| ΔBAP3-H | AGCTTAGCCGTGCATGCGGCTCCGCAAAAAG | + | + | 17 | 256 |
| ΔBAP3-K | AGCTTAGCCATGCATGCGGCTCCGCGAAAAG | – | + | 17 | 67 |
| ΔBAP3-L | AGCTTAGCCGTGCATGCGACTCCGCGAAAAG | – | – | 10 | 11 |
| ΔBAP3-M | AGCTTAGACGTGCATGCGGCTCCGCGAAAAG | – | + | 19 | 25 |
| ΔBAP3-N | AGCTTAACCGTGCATGCGGCTCCGCGAAAAG | + | + | 30 | 83 |
| ΔBAP3-P | AGCTTAGCCGTGCATGCGGATCCGCGAAAAG | + | – | 12 | 26 |
| ΔBAP3-R | AGCTTAGCCGTGCATACGGCTCCGCGAAAAG | + | + | 18 | 266 |
| S28A | AGCTTTGCGTGGTCACTCTAGACGGCCGCG | ++ | – | 26 | 21 |
| S28An | AGCTTTGCGTGGTCACTCTAGAGGGCCGCG | – | – | 9 | 8 |
| S28Arev | AGCTTGCGGCCGTCTAGAGTGACCACGCG | ++ | – | 25 | 21 |
aOligonucleotide oBAP3-WT corresponds to the region between –418 and –376 of the BAP3 promoter. The Abf1p-binding site in the oligonucleotides (see also Table 1) is underlined. Oligonucleotides ΔBAP3-A, B and C correspond to the same fragment but contain a number of mutations, indicated in bold. Oligonucleotides D, E and F correspond to the wild-type BAP3 promoter sequence, but are shortened from the 3′-side in comparison to oBAP3-WT (7). Oligonucleotides G, H, K, L, M, N, P and R correspond to the ΔBAP3-D oligonucleotide but contain point mutations, indicated in bold. S28A and S28Arev contain sequences that comply with the Abf1p-binding consensus (see Materials and Methods) and S28An contains a point mutation in the Abf1p-binding consensus that abolishes Abf1p interaction (24). The ability to form in vitro Abf1p- or Stp2p-dependent complexes (as illustrated in Figs 1 and 4B) is indicated by: –, no; +, yes; ++, very strong. Only the coding strand of the fragments is shown and each fragment has at its 5′- and 3′-termini sequences which are complementary to the cohesive ends generated by the restriction enzymes HindIII and EcoRI, respectively (as described in ref. 7). This enabled easy insertion into the LacZ reporter construct.
bLacZ expression was measured in cells grown with or without 2 mM leucine (see Materials and Methods for further details). The specific β-galactosidase activity is given in U/mg protein extract (1 U = 1 nmol o-nitrophenol/min). All assays were carried out in triplicate using single copy integrants and individual measurements differed by <10%.
Miscellaneous techniques
Expression of LacZ fusions was measured by liquid β-galactosidase assay and BAP3 transcription was monitored by northern blot analysis as described earlier (7).
RESULTS
Abf1p binds in vitro to the UASaa
A thorough analysis of the previously identified UASaa (7) shows that it comprises a sequence having considerable similarity to the Abf1p-binding site present in, for example, the FOX3 promoter (11). This Abf1p-binding site does not completely conform to the consensus Abf1p-binding sequence as published by Dorsman et al. (24). Since then, however, several reports have been published in which it was shown that Abf1p can bind to alternative sequences found in the promoters of genes involved in distinct biochemical processes (see Table 1). Table 1 also illustrates that the putative Abf1p-binding site in the BAP3 promoter contains the fully conserved C nucleotide at position 3 and ACG nucleotides at positions 11–13. Based on the observation that the UASaa may comprise an Abf1p-binding site we set out to test whether indeed Abf1p is the protein that does bind to the UASaa in vitro in gel shift analyses. First, we carried out a gel shift experiment with a labelled double-stranded oligonucleotide containing the UASaa (ΔBAP3-D) and using excess of an unlabelled double-stranded oligonucleotide harbouring an Abf1p-binding site (i.e. the Abf1p-binding site as present in the promoter of the rpS28A gene) as competitor. We found that this latter oligonucleotide is an efficient competitor for protein binding to the UASaa, suggesting that both the UASaa and the Abf1p-binding site bind the same protein (data not shown). Next, we made mutations at two of the four fully conserved nucleotides present in the consensus Abf1p-binding sequence, i.e. at the conserved C at position 3 (ΔBAP3-L) or at the conserved C at position 12 (ΔBAP3-K; see also Table 2) and used these oligonucleotides as probes in a gel shift assay. From Figure 1 it is clear that the mutations in both ΔBAP3-L and K lead to a severe decrease in protein binding. We also found that a mutation at position 14 (ΔBAP3-M), which is less conserved, reduces protein binding. Point mutations as present in ΔBAP3-N, -P and -R which do not affect the Abf1p-binding consensus, have no effect on the formation of complex Y. Then we asked whether bacterially produced Abf1p is able to bind to the UASaa. Abf1p has been shown to contain two DNA-binding domains. The first is a zinc finger motif in the N-terminal region (amino acids 40–91) and the second a DNA-binding region located in the middle of the protein (25). Using two constructs described by Halfter and co-workers (23) we expressed both a full-length Abf1p and a shorter version from which the N-terminal zinc finger domain was deleted and which therefore could no longer bind to Abf1p-binding sites in E.coli. Production of bona fide Abf1p and Abf1pΔ33/240 was established by western blotting using an antiserum against Abf1p (results not shown). We then used extracts from E.coli producing these two proteins in gel shift analyses. Figure 2A illustrates that the complete recombinant Abf1p can form a complex with the UASaa with the same mobility as the complex it forms with the S28A probe (a double-stranded oligonucleotide harbouring an Abf1p-binding site as present in the promoter of the rpS28A gene). Furthermore, the complexes found with the recombinant Abf1p have similar mobility to the complexes observed using yeast extracts (Fig. 2A, compare lanes 2 and 4). When we use a protein extract from E.coli cells expressing Abf1pΔ33/240 no complex formation was observed (Fig. 2A, lanes 3 and 8). Finally, we studied the effect of antibodies directed against Abf1p in gel shift assays to reveal the nature of the protein(s) that binds to the UASaa. Using the double-stranded oligonucleotide oBAP3-WT, comprising the UASaa, and yeast extract in a gel shift assay we find two complexes, a weak complex X and a much stronger complex Y (Fig. 2B, lane 2). Addition of anti-Abf1p antibodies to such a gel shift experiment resulted in disappearance of the major complex, Y, and a concomitant increase in the intensity of complex X (Fig. 2B, lanes 3–6). This effect can either be explained by a supershift of the Y complex as a result of binding of the antibody to the complex or by the inability of antibody-bound Abf1p to bind to the Abf1p-binding site, which can then complex with another factor(s) to form complex X. To distinguish between these two possibilities we performed a similar series of gel shift assays using the S28A probe and yeast extract in the presence or absence of anti-Abf1p antibodies. Figure 2B (lanes 11–14) clearly shows that upon addition of anti-Abf1p antibodies the S28A–Abf1p complex is supershifted to a complex of similar mobility to that of complex X. Taken together, these experiments unambiguously demonstrate that complex Y is formed as a result of the interaction of oBAP3-WT with Abf1p, meaning that in vitro the UASaa can be bound by Abf1p. It must be noted, however, that this binding site is much weaker than the Abf1p-binding site in the S28A promoter (cf. Fig. 2A, lanes 4 and 9).
Table 1. Comparison of the putative Abf1p-binding sites found in the UASaa of BAP3 to known Abf1p-binding sites.
| Gene | Sequence | Reference |
|---|---|---|
| Abf1p-binding consensus | 5′-RTCRYYYNNNACG-3′ | 24 |
| RPS28A (–164/–152)a | 5′-GTCACTCTAGACG-3′ | 24 |
| BAP3 (–414/–402)a | 5′-AGCCGCATGCACG-3′ | This work |
| BAP2 (–416/–404) | 5′-GCCGCTTCTCACG-3′ | This work |
| FOX3 (–213/–201)a | 5′-AGCGCCTTACACG-3′ | 11 |
| HIS7 (–200/–188) | 5′-CTCTCTCTCCACG-3′ | 13 |
| HSP12 (–264/–252) | 5′-AGCACTCTAGACG-3′ | 29 |
aSequence as present on the non-coding strand.
Figure 1.
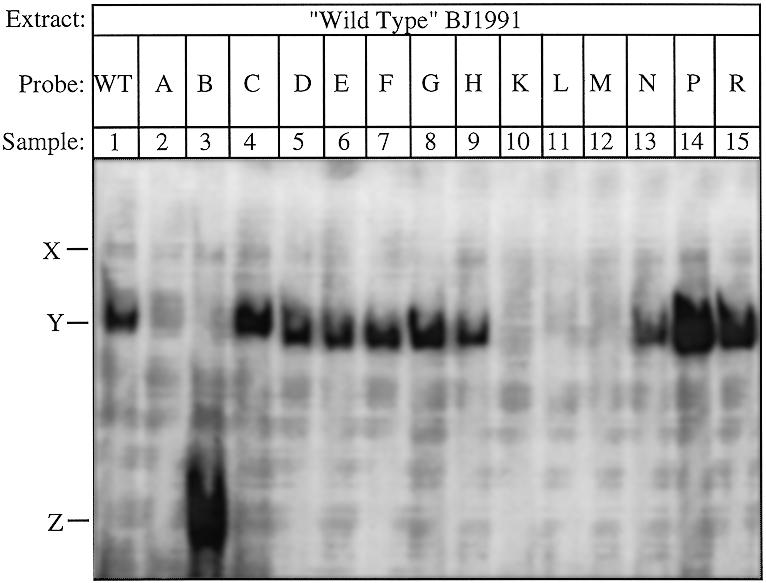
Gel shift analysis of the UASaa and mutated versions thereof. The oligonucleotides oBAP3-WT through ΔBAP3-R (see Table 2 for sequences) were radioactively labelled and used as probes in gel shift analysis. Each reaction contained 16 µg of yeast protein extract isolated from wild-type cells. The minor and major protein–DNA complexes are called X and Y, respectively. Z indicates the complex found using ΔBAP3-B as probe.
Figure 2.
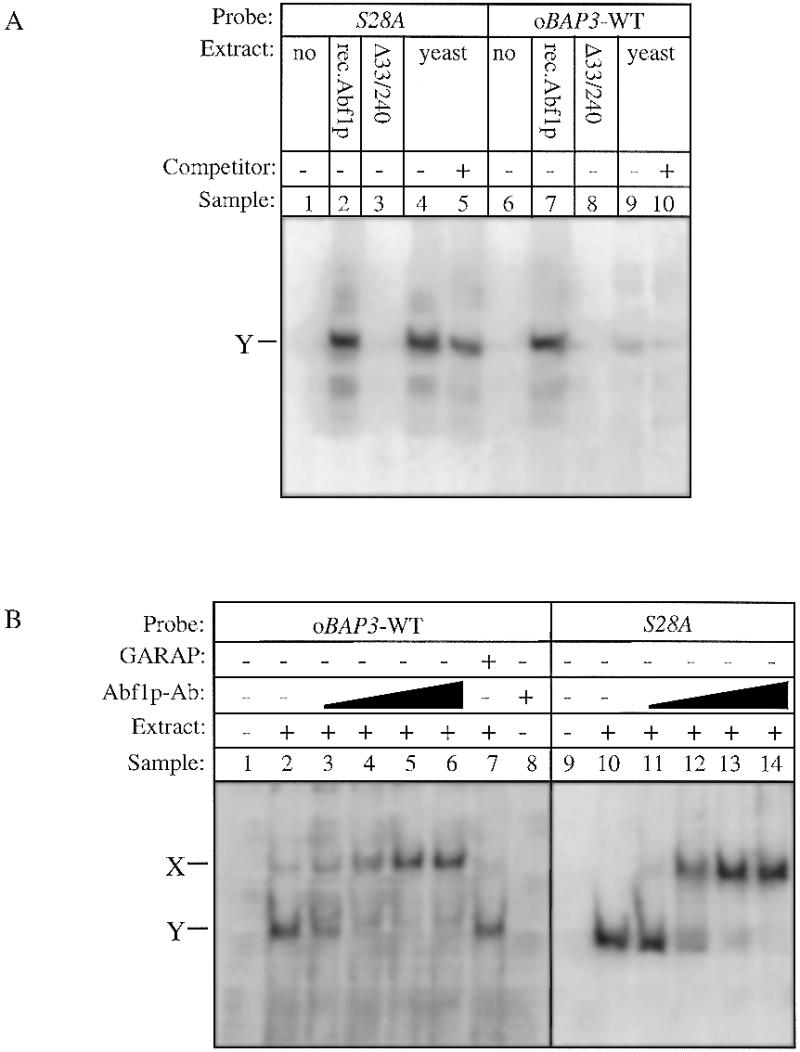
Identification of Abf1p complexes using recombinant Abf1p and Abf1p antibodies in gel shift assays. (A) The oBAP3-WT fragment and the rpS28A oligonucleotide were radioactively labelled and used as probes in gel shift assays. Whole cell protein extracts were prepared from wild-type yeast cells and from E.coli cells expressing the yeast Abf1 protein and a mutated Abf1 protein lacking the N-terminal DNA-binding domain (Abf1pΔ33/240) as described in Materials and Methods. Each reaction contained 16 µg of yeast protein extract (lanes 4, 5, 9 and 10) or 5 µg of total protein extract isolated from wild-type Abf1p-expressing E.coli cells (lanes 2 and 7) or from E.coli cells expressing the mutated Abf1p form (lanes 3 and 8). As negative controls the probes were incubated without protein extract (lanes 1 and 6). The position of the protein–DNA complex is marked Y. Complex formation was competed with 250-fold excess of unlabelled double-stranded oligonucleotide oBAP3-WT where indicated (lanes 5 and 10). (B) The oBAP3-WT and the S28A oligonucleotides were radioactively labelled and used as probes in gel shift assays. To show that Abf1p is involved in the formation of complex Y, we added increasing amounts of anti-Abf1p antibody to the binding reactions (in lanes 2–6 and 11–14, respectively). As negative control the probes were incubated without protein extract (lanes 1 and 9) or with only anti-Abf1p antibody in lane 8. As a negative control for the supershift we added a goat anti-rabbit antibody to the binding reaction (lane 7).
In addition to Stp1p, Stp2p is involved in the induction of BAP3
STP1 encodes a nuclear protein with zinc finger domains which is involved in pre-tRNA maturation (26) and which is also required for the induced expression of BAP2 (27). We previously reported that Stp1p is also involved in the induction of BAP3 transcription by leucine (7). Since the absence of Stp1p leads to a strongly reduced induction of BAP3 expression, rather than to a complete loss of induction, we looked for other proteins that might be involved in the induction process. Stp2p is such a protein. STP2 was found in a screen for genes that in higher copy number would confer growth on minimal proline medium containing the inhibitor of branched chain amino acid synthesis, methylsulfonyl urea (MM), plus isoleucine (0.2 mM) and valine (20 µM). On this medium cells grow only when the uptake of valine is increased. This may for instance be the result of an increased activity or an increased expression of one or more of the permeases Bap2p, Bap3p or Tat1p. To investigate the role of STP2 in BAP3 expression, we used northern blot analysis and monitored BAP3 expression in a Δstp2 as well as in a Δstp1/Δstp2 double disruption mutant in comparison to the BAP3 expression in isogenic wild-type cells after addition of leucine. Figure 3A shows that as in a Δstp1 mutant, BAP3 induced expression is severely diminished in a Δstp2 mutant. However, in either single disruption mutant we still find a low level of leucine-induced BAP3 expression. In a Δstp1/Δstp2 double mutant, however, leucine-induced BAP3 expression is completely lost. We have also monitored expression of BAP2 in a similar experiment and found that BAP2 expression follows a pattern similar to BAP3 (data not shown). These results indicate that both Stp1p and Stp2p are involved in the amino acid-induced expression of BAP2 and BAP3.
Figure 3.

The induction of BAP3 is dependent on both STP1 and STP2. (A) BAP3 expression was analysed by northern blot analysis in Δstp1, Δstp2 and Δstp1/Δstp2 mutants and in their isogenic wild-type (WT) after addition of leucine to a final concentration of 2 mM. Total RNA was isolated at 0, 5, 15, 30 and 60 min after leucine addition. Probes were used corresponding to parts of the genes encoding Bap3p and actin. The actin mRNA signal was used as a loading control. (B) Functionality of the STP1-overexpressing constructs was tested by monitoring BAP3 induction in wild-type and Δstp1 cells transformed with the corresponding STP1-overexpression constructs after addition of leucine to a final concentration of 2 mM. As a negative control the cells were transformed with the empty expression vector YCplac33. Total RNA was isolated at 0, 15 and 60 min after leucine addition. Probes were used corresponding to parts of the genes encoding Bap3p, Stp1p and actin. The actin mRNA signal was used as a loading control. (C) Functionality of the STP2-overexpressing constructs was monitored by northern blot analysis as described above using part of the gene encoding STP2 as probe.
Overexpression of Stp1p and Stp2p
To gain more insight into the functions of Stp1p and Stp2p, we sought to overexpress either or both these proteins by placing the corresponding genes under control of the PGK1 promoter and to express them from a multiple copy vector (see Materials and Methods). Stp1p was overexpressed as the wild-type protein, whereas the overexpressed Stp2p carried a triple HA epitope tag [(HA)3] at its N-terminus. Strong overexpression of either protein was clearly demonstrated in a western blot analysis using either an anti-Stp1p antibody (a kind gift of Dr A. Hopper, Seattle, WA) or the 12CA5 antibody directed against the HA epitope in Stp2p (results not shown). To find out whether the overproduced Stp1p or (HA)3–Stp2p is functional in leucine-induced BAP3 expression we tested the recovery of BAP3 induction in Δstp1 and Δstp1/Δstp2 disruption mutants with the respective overexpression constructs by northern blot analysis. Figure 3B shows that upon transformation of the Δstp1 mutant with either the single copy STP1 overexpression construct (SCPGK1–STP1) or the multiple copy STP1 overexpression vector (MCPGK1–STP1) leucine-induced BAP3 induction was restored. Figure 3C shows, likewise, that upon transformation of the Δstp1/Δstp2 mutant with either the single copy (HA)3–STP2 overexpression construct (SCPGK1–STP2) or the multiple copy (HA)3–STP2 overexpression vector (MCPGK1–STP2) leucine-induced BAP3 induction was restored. We find that overexpression of either STP1 or STP2 leads to an increase in BAP3 induction as compared to untransformed wild-type cells. Furthermore, strong overexpression of STP1 or STP2 leads to BAP3 expression without the addition of leucine (compare lanes 1 and 7 of Fig. 3B and lanes 1 and 16 of Fig. 3C). We conclude that we have successfully constructed vectors that enable us to overexpress functional Stp1p or Stp2p in yeast.
To see whether Stp1p or Stp2p directly interacts with the BAP3 promoter, we performed gel shift analysis with the –495 to –392 region of the BAP3 promoter involved in induction by amino acids (7). Figure 4A shows that the BAP3 promoter fragment forms several DNA–protein complexes when extracts from wild-type cells are used (lane 5). The weakest complexes with the slowest mobility (collectively called X) appear as one or two bands depending on the size of the UASaa fragment. Previously we have shown that these X complexes are formed by non-specific protein binding since their formation cannot be competed out by unlabelled fragments (7). The strongest complex, Y, is, as we have shown, formed by Abf1p. Finally, we found a very weak complex with a higher mobility than the Abf1p complex. Stp2p overexpression led to a significant increase in the formation of this weak DNA–protein complex, which we therefore called the Stp2p-dependent complex. The Stp2p-dependent complex is also formed using the ΔBAP3-D fragment as probe and using yeast extracts from cells overexpressing Stp2p (Fig. 4A, lane 8). Overexpression of Stp1p has no apparent effect on the gel shift pattern. We have monitored the sequence requirements for Stp2p-dependent complex formation by testing various mutated versions of the BAP3 UASaa in gel shift analyses (Fig. 4C; see Table 2 for the specific sequences). Stp2p-dependent complex formation is highly sequence specific; the complex is formed using the non-mutagenised UASaa version ΔBAP3-D (cf. Fig. 4A), but there is no complex formation with the mutagenised versions ΔBAP3-B, E, G, L and P. Loss of Stp2p-dependent complex formation with these latter oligonucleotides suggests that the sequence 5′-CGGCTC-3′ in the UASaa is important for protein binding.
Figure 4.

Gel shift analysis with extracts of cells overexpressing Stp1p and Stp2p. (A) The –495/–392 BAP3 promoter fragment and the double-stranded oligonucleotide ΔBAP3-D were radioactively labelled and used as a probe in gel shift assays. Each reaction contained 16 µg of yeast protein extract (except for lanes 1 and 6) isolated from either wild-type cells (lanes 5 and 7), cells overexpressing both Stp1p and Stp2p (lane 2), cells overexpressing Stp1p (lane 3) or from cells overexpressing Stp2p (lanes 4 and 8). (B) The radioactively labelled oBAP3-WT fragment was used as a probe in gel shift assays. Each reaction contains 5 µg of protein extract isolated from either wild-type E.coli cells (lane 2) or from E.coli cells expressing Stp2p (lane 3). As a negative control no protein extract was added in lane 1. (C) Gel shift analysis using the UASaa and mutated versions thereof (see Table 2) and protein extracts from cells overexpressing Stp2p (see also legend to Fig. 1).
To show that the Stp2p-dependent complex involves Stp2p, we have expressed Stp2p in E.coli and used such extracts in gel shift assays. Using these protein extracts in band shift analysis, an Stp2p-dependent complex is formed with the oBAP3-WT promoter fragment, which is absent when protein extracts from wild-type E.coli cells are used (Fig. 4B, compare lanes 2 and 3). We have also tested binding of the Stp2p protein, expressed in E.coli, to the mutated BAP3 fragments used in Figure 4C. We found a similar pattern of complex formation, indicating that in vitro produced Stp2p binds to the UASaa with the same sequence specificity as found for the tagged Stp2p version expressed in yeast (results not shown).
Expression of the LacZ fusions
To test whether the ability of a DNA fragment to bind Abf1p or/and to form the Stp2p-dependent complex in vitro correlates with its capacity to render a promoter inducible by amino acids we cloned the UASaa and several mutated versions thereof or the rpS28A elements (Table 2) in front of a LacZ reporter gene. The reporter constructs were integrated at the URA3 locus and single copy integrants were tested for induction of the fusion gene by measuring β-galactosidase activity after growing the mutants with or without 2 mM leucine in the medium.
Table 2 shows that mutations in the Abf1p-binding site of the UASaa result in a loss of induced LacZ expression in response to leucine (ΔBAP3-A, -B, -K, -L and -M). This loss of induction correlates well with the results obtained from the gel retardation assays. In other words, binding of Abf1p correlates with induction in response to leucine via the UASaa. However, when the Abf1p-binding site as found in the rpS28A promoter was fused to the LacZ reporter (S28A and S28Arev), no induction by the addition of leucine was observed. This means that Abf1p interaction is not enough for induction in response to leucine and that other elements and/or factors are required.
Since Stp2p overexpression results in the formation of an additional complex in the gel shift assay, it is possible that Stp2p plays a direct role in BAP3 transcription. For example, oligonucleotides E and P are still bound by Abf1p, but they do not form the Stp2p-dependent complex and have lost their capacity to induce the LacZ reporter in response to leucine. On the other hand, oligonucleotide ΔBAP3-G, which is still bound by Abf1p but does not form the Stp2p-dependent complex, is still able to induce the LacZ reporter in response to leucine. This would imply that, in addition to Abf1p and Stp2p, another protein(s) is also involved in the induction of BAP3 in response to amino acids (e.g. Stp1p).
DISCUSSION
We previously identified an upstream activating sequence in the BAP3 promoter that is responsible for the induced expression of BAP3 in response to the availability of amino acids in the medium (7). This paper describes the identification of trans-acting factors interacting with this cis-acting element.
We have found that the UASaa constitutes a weak binding site for the global transcription factor Abf1p. This Abf1p-binding site does not completely comply with the consensus sequence, but has considerable similarity to the Abf1p-binding site as found in the promoters of other yeast genes. We tested several mutated versions of the UASaa to establish the sequence specificity of the Abf1p interaction and found that point mutations in nucleotides known to be essential for Abf1p binding result in a severe decrease in complex formation. We previously showed that the UASaa is both necessary and sufficient for induction of a LacZ reporter in response to addition of amino acids. We also tested mutated UASaa versions that can no longer be bound by Abf1p and found that there is a correlation between Abf1p complex formation and ability to induce the LacZ reporter. In other words, binding of Abf1p to the UASaa may be involved in the induced expression in response to amino acids. To investigate whether Abf1p binding alone is sufficient to induce the LacZ reporter, we fused an Abf1p-binding site as present in the rpS28A promoter to the LacZ reporter. We found that this fusion does not induce induction of the LacZ reporter in response to amino acids. This implies that the binding of Abf1p to its cognate site by itself is not sufficient to support induction of the gene concerned in response to amino acids and that other elements and factors are involved in the specific induction of expression of UASaa-controlled genes.
One candidate involved in the transcriptional regulation of BAP3 might be the STP1 gene product (7). We found that deleting the STP1 gene leads to a severe decrease in BAP3 induction; however, there is still some remaining residual BAP3 induction. This implies that another factor(s) might play a role in leucine-induced BAP3 transcription. STP2 was found in a screen for genes that in higher copy number would confer increased activity of one or both of the permeases Bap2p and Bap3p. Comparison of STP1 and STP2 reveals that the STP2 gene encodes a protein with considerable similarity to the STP1 gene product. We constructed a Δstp2 mutant and found that this mutant, like Δstp1, also had a decreased level of leucine-induced BAP3 transcription. Not unexpectedly, upon deletion of both STP1 and STP2, expression of BAP3 was completely lost. Since the double deletion mutant had an aggravating effect in comparison to the single deletion mutants, we propose that the STP1 and STP2 gene products have similar functions in the regulation of BAP3 expression. To confirm this, we overexpressed Stp2p in a Δstp1/Δstp2 deletion mutant. We found that overexpression of Stp2p could restore BAP3 induction, even to higher levels than in wild-type cells. This means that indeed Stp1p and Stp2p have similar functions in BAP3 expression and can function independently of each other.
Stp1p and Stp2p are the most likely candidates for being the transcription factors that directly interact with the UASaa. Overexpression of Stp2p does result in formation of an additional UASaa–protein interaction in an in vitro gel shift experiment. We have also used protein extracts from yeast cells overexpressing Stp1p. However, we could not find any interaction of Stp1p with the BAP3 promoter. Furthermore, attempts to express Stp1p in E.coli have been unsuccessful. Both negative findings could result from differences in protein stability and sequence specificity between Stp1p and Stp2p. We found that formation of the Stp2p-dependent complex requires the sequence 5′-CGGCTC-3′ in the UASaa, which partially overlaps with the Abf1p-binding consensus. This suggests that Abf1p is either required to recruit other transcription factors, like Stp2p, for amino acid-induced transcription or that Abf1p competes with these factors for binding to the UASaa. Similar sequences appear to be present in the BAP2 and TAT1 promoters (see also ref. 7), which makes it interesting to note that we have found that the Δstp1 and Δstp2 mutations resulted in a similar expression pattern of BAP2 as reported here for BAP3 (data not shown).
Since overexpression of Stp2p does not lead to complexes that involve both Stp2p and Abf1p, it is not likely that there is concomitant binding of both Abf1p and Stp2p to the UASaa. For leucine-induced BAP3 expression we propose a model in which Abf1p acts as a general transcription factor interacting with the UASaa irrespective of whether leucine is present or not. Several reports have been published in which it has been shown that Abf1p is responsible for transcriptional regulation of genes involved in distinct processes. This functional diversity can only be explained if Abf1p plays a role in a general mechanism such as, for example, chromatin (re)modelling (28). Recent data from our laboratory show that Abf1p is indeed involved in bending of DNA and nucleosome positioning, thereby regulating expression of the rpS28 gene (Lascaris et al., submitted for publication). In the case of the BAP3 gene, Abf1p may also function by excluding nucleosomes or by bending the DNA, thereby rendering the UASaa accessible to the specific transcription factors Stp1p and Stp2p.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Sacha N. Coesel for skillful technical assistance and Anita K. Hopper (Seattle, WA) for providing us with the Stp1p antibodies and the constructs encoding STP1, the STP1 disruption cassette and STP2. We wish to thank Morten C. Kielland-Brandt for critical reading of the manuscript.
REFERENCES
- 1.Didion T., Regenberg,B., Jorgensen,M.U., Kielland-Brandt,M.C. and Andersen,H.A. (1998) Mol. Microbiol., 27, 643–650. [DOI] [PubMed] [Google Scholar]
- 2.Iraqui I., Vissers,S., Bernard,F., De Craene,J.-H., Boles,E., Urrestarazu,A. and André,B. (1999) Mol. Cell. Biol., 19, 989–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schreve J.L., Sin,J.K. and Garret,J.M. (1998) J. Bacteriol., 180, 2556–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmidt A., Hall,M.N. and Koller,A. (1994) Mol. Cell. Biol., 14, 6597–6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grauslund M., Didion,T., Kielland-Brandt,M.C. and Andersen,H.A. (1995) Biochim. Biophys. Acta, 1269, 275–280. [DOI] [PubMed] [Google Scholar]
- 6.Mai B. and Lipp,M. (1994) Gene, 143, 129–133. [DOI] [PubMed] [Google Scholar]
- 7.De Boer M., Bebelman,J.P., Gonçalves,P.M., Maat,J., van Heerikhuizen,H. and Planta,R.J. (1998) Mol. Microbiol., 30, 603–613. [DOI] [PubMed] [Google Scholar]
- 8.Rhode P.R., Elsasser,S. and Campbell,J.L. (1992) Mol. Cell. Biol., 12, 1064–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silve S., Rhode,P.R., Coll,B., Campbell,J. and Poyton,R.O. (1992) Mol. Cell. Biol., 12, 4197–4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Künzler M., Springer,C. and Braus,G.H. (1995) Mol. Microbiol., 15, 167–178. [DOI] [PubMed] [Google Scholar]
- 11.Einerhand A.W.C., Voorn-Brouwer,T.M., Erdmann,R., Kunau,W.-H. and Tabak,H.F. (1991) Eur. J. Biochem., 200, 113–120. [DOI] [PubMed] [Google Scholar]
- 12.Bornaes,C., Ignjatovic,M.W., Schjerling,P., Kielland-Brandt,M.C. and Holmberg,S. (1993) Mol. Cell. Biol., 13, 7604–7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Springer C., Krappmann,S., Künzler,M., Zmasek,C. and Braus,G.H. (1997) Mol. Gen. Genet., 256, 136–146. [DOI] [PubMed] [Google Scholar]
- 14.Jones E.W. (1977) Genetics, 85, 23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen D.C., Yang,B.C. and Kuo,T.T. (1992) Curr. Genet., 21, 83–84. [DOI] [PubMed] [Google Scholar]
- 16.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 17.Inoue H., Nojima,H. and Okayama,H. (1990) Gene, 96, 23–28. [DOI] [PubMed] [Google Scholar]
- 18.Wach A., Brachat,A., Pohlmann,R. and Philippsen,P. (1994) Yeast, 10, 1793–1808. [DOI] [PubMed] [Google Scholar]
- 19.Wang S.S. and Hopper,A.K. (1988) Mol. Cell. Biol., 8, 5140–5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoekema A., Kastelein,R.A., Vasser,M. and de Boer,H.A. (1987) Mol. Cell. Biol., 7, 2914–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gietz R.D. and Sugino,A. (1988) Gene, 74, 527–534. [DOI] [PubMed] [Google Scholar]
- 22.Tyers M., Tokiwa,G. and Futcher,B. (1993) EMBO J., 15, 1955–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Halfter H., Kavety,B., Vandekerckhove,J., Kiefer,F. and Gallwitz,D. (1989) EMBO J., 8, 4265–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dorsman J.C., van Heeswijk,W.C. and Grivell,L.A. (1990) Nucleic Acids Res., 18, 2769–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho G., Kim,J., Rho,H.M. and Jung,G. (1995) Nucleic Acids Res., 23, 2980–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang S.S., Stanford,D.R., Silverse,C.D. and Hopper,A.K. (1992) Mol. Cell. Biol., 12, 2633–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jörgensen M.U., Gjermansen,C., Andersen,H.A. and Kielland-Brandt,M.C. (1997) Curr. Genet., 31, 241–247. [DOI] [PubMed] [Google Scholar]
- 28.Wolffe A.P. (1994) Trends Biochem. Sci., 19, 240–244. [DOI] [PubMed] [Google Scholar]
- 29.Varela J.C.S., Praekelt,U.M., Meacock,P.A., Planta,R.J. and Mager,W.H. (1995) Mol. Cell. Biol., 15, 6232–6245. [DOI] [PMC free article] [PubMed] [Google Scholar]