

Abstract
Chimeric antigen receptor (CAR) T cells have not induced meaningful clinical responses in solid tumors. Loss of T cell stemness, poor expansion capacity, and exhaustion during prolonged tumor antigen exposure are major causes of CAR T cell therapeutic resistance. Single-cell RNA-sequencing analysis of CAR T cells from a first-in-human trial in metastatic prostate cancer identified two independently validated cell states associated with antitumor potency or lack of efficacy. Low expression of PRDM1, encoding the BLIMP1 transcription factor, defined highly potent TCF7 [encoding T cell factor 1 (TCF1)]–expressing CD8+ CAR T cells, whereas enrichment of HAVCR2 [encoding T cell immunoglobulin and mucin-domain containing-3 (TIM-3)]–expressing CD8+ T cells with elevated PRDM1 was associated with poor outcomes. PRDM1 knockout promoted TCF7-dependent CAR T cell stemness and proliferation, resulting in marginally enhanced leukemia control in mice. However, in the setting of PRDM1 deficiency, a negative epigenetic feedback program of nuclear factor of activated T cells (NFAT)–driven T cell dysfunction was identified. This program was characterized by compensatory up-regulation of NR4A3 and other genes encoding exhaustion-related transcription factors that hampered T cell effector function in solid tumors. Dual knockout of PRDM1 and NR4A3 skewed CAR T cell phenotypes away from TIM-3+CD8+ and toward TCF1+CD8+ to counter exhaustion of tumor-infiltrating CAR T cells and improve antitumor responses, effects that were not achieved with PRDM1 and NR4A3 single knockout alone. These data underscore dual targeting of PRDM1 and NR4A3 as a promising approach to advance adoptive cell immuno-oncotherapy.
One Sentence Summary:
Two transcription factors, BLIMP1 and NR4A3, reciprocally regulate T-cell stemness and exhaustion and can be exploited for CAR T-cell therapy.
Editor’s Summary:
Tuning Up CAR T-Cells. Despite their success at treating hematological malignancies, CAR T-cells have not been as effective in treating solid tumors. This is, in part, due to loss of stemness and development of an exhausted phenotype after infusion. Here, Jung et al. investigated a strategy to reverse this phenotype based on analysis of CAR T-cells employed in a clinical trial for metastatic prostate cancer. The authors identified two transcription factors, BLIMP1 and NR4A3, that regulated CAR T-cell dysfunction. Dual knockout of these two transcription factors reduced CAR T-cell exhaustion and enhanced their antitumor activity in multiple murine models, outperforming single knockouts. These data suggest that transcription factor modulation may be a key strategy to improve CAR T-cells for solid tumors.
Introduction
Chimeric antigen receptor (CAR) T-cells have induced unprecedented high rates of complete remission in relapsed and refractory B-cell malignancies. Despite these successes, a substantial portion of patients with B-cell leukemia, lymphoma, and myeloma fail to respond to CAR T-cell therapy and only small subsets of patients experience long-term durable responses (1–5). The efficacy of CAR T-cell therapy in solid tumor indications has been even more limited to date (6–8). Unlike the situation in hematologic malignancies, CAR T-cells must traffic to solid tumor sites and surmount stromal elements to infiltrate into the tumor bed and elicit antigen-directed cytotoxicity. Even if trafficking and infiltration are successful, CAR T-cells often become dysfunctional due to chronic antigen exposure and additional immunosuppressive mechanisms operative within the tumor microenvironment (TME).
CAR T-cells derived from naïve and early memory subsets have been shown to robustly expand in vivo and are long-lived with a self-renewal capacity (9–11). We and others have demonstrated that naïve or stem-like early memory T-cells genetically redirected with CARs have more durable engraftment and antitumor effector function compared to highly differentiated cells (12, 13). However, persistent tumor antigen stimulation in the setting of hematopoietic and non-hematopoietic cancers often leads to exhaustion. T-cell exhaustion is characterized by upregulation of multiple inhibitory receptors (10, 13–15), the inability to respond to homeostatic cytokines, loss of effector function (9, 16) and reduced survival (17). CAR T-cell exhaustion can also be facilitated by antigen-independent tonic signaling through the synthetic antigen receptor during ex vivo cell expansion or following infusion (18, 19).
We previously found that sustained remissions in CD19 CAR T-cell therapy of chronic lymphocytic leukemia (CLL) were associated with an increased peak expansion after infusion and relatively longer persistence. Cell products that were particularly effective showed greater proliferative capacity prior to and during treatment. Transcriptomic analysis suggested that remission correlated with early memory T-cell signatures as well as stemness (defined by the self-renewal capacity and the ability to differentiate into multiple downstream cell types), whereas gene expression profiles from non-responders were associated with terminal differentiation and exhaustion (20). Similarly, memory CD8+ T-cell transcriptional programs enriched in axicabtagene ciloleucel infusion products were strongly related to the long-term durability of response, whereas gene signatures of exhaustion correlated with early treatment failure in patients with large B-cell lymphomas (LBCL) (21). Importantly, it may be possible to overcome many of the above issues by using highly proliferative, “optimally programmed” CAR T-cells. However, the cellular and molecular basis for CAR T-cell-intrinsic resistance in failed therapy of solid tumors has not been elucidated. Thus, engineering approaches to generate CAR T-cells with optimal potency and other aforementioned desirable features informed by correlative analyses have been scant to date.
Here we analyzed dominant negative transforming growth factor β receptor II (TGF-βRII) ‘armored’ prostate-specific membrane antigen (PSMA) CAR T-cell infusion products from patients with metastatic castration-resistant prostate cancer (mCRPC) (clinicaltrials.gov NCT03089203) (22). Using single cell RNA-sequencing (scRNA-seq) analysis, we identified heterogeneity in the cellular and molecular features of CAR T-cells associated with variation in therapeutic product potency across multiple CAR T-cell trials. Based on these findings, we hypothesized that deletion of the PR/SET domain 1 (PRDM1) gene encoding the B-lymphocyte-induced maturation protein 1 (BLIMP1) transcription factor, which is upregulated in poorly functional Hepatitis A Virus Cellular Receptor 2 (HAVCR2; encoding T-cell immunoglobulin and mucin-domain containing-3, TIM-3)-expressing CD8+ T-cells and downregulated in highly potent Transcription Factor 7 (TCF7; encoding T cell factor 1, TCF1)-expressing CD8+ T-cells, may potentiate CAR T-cell stemness and antitumor function. This hypothesis is further supported by the results of a very recently published study showing that genetic knockout of PRDM1 in T-cells enhances persistence and therapeutic responses in cancer models of adoptive immunotherapy (23). Accordingly, PRDM1 single knockout (KO) CAR T-cells exhibited enrichment of the TCF7-expressing CD8+ phenotype in association with improved CAR T-cell persistence and expansion. However, genetic deletion of PRDM1 alone resulted in only marginal enhancement of CAR T-cell efficacy against B-cell leukemia and completely failed to rescue CAR+ tumor-infiltrating lymphocyte (TIL) function in animal models of high tumor burden. These results are consistent to some degree with previous reports of antigen-specific PRDM1 KO T-cells exhibiting comparable or even inferior control of viruses and cancer through mechanisms that remain largely elusive (24–27). We found that during prolonged tumor antigen exposure, several exhaustion-related transcription factors, including Nuclear Receptor Subfamily 4 Group A Member 3 (NR4A3), were reciprocally upregulated in PRDM1-deficient CAR T-cells and drove epigenetically programmed T-cell dysfunction in a Nuclear factor of activated T-cells (NFAT)-dependent manner. PRDM1 and NR4A3 dual-ablation counteracted the development of T-cell exhaustion, which was not observed with single KO of either gene. This strategy also led to a marked increase in the survival and expansion of TILs; increased the capacity of the CAR TILs to produce effector cytokines after stimulation; and reduced their expression of inhibitory factors. In immunodeficient mice bearing soft tissue or bone engrafted prostate cancer cells, pancreatic tumors or aggressive B-cell leukemia, a single intravenous infusion of PRDM1/NR4A3 dual KO CAR T-cells resulted in higher antitumor efficacy, T-cell response durability upon rechallenge and overall prolongation of survival compared to conventional CAR T-cell treatment. There is considerable interest in modulating CAR T-cells to control tumors more successfully. Our identification of the reciprocal nature of BLIMP1 and NR4A3 activity as a distinct mechanism regulating the balance of T-cell differentiation and function represents an actionable and therapeutically effective strategy for achieving this desired outcome.
Results
TCF7- and HAVCR2-expressing CD8+ populations within infusion products are associated with favorable and poor CAR T-cell-mediated therapeutic potency, respectively.
We retrospectively performed scRNA-seq analysis on five autologous PSMA CAR T-cell infusion products administered to patients with mCRPC (fig. S1A). These products were selected based on sample availability and to represent a range of outcomes including in vivo CAR T-cell expansion and prostate-specific antigen (PSA) response (22). 20,702 cells were obtained after eliminating low-quality cells, consisting mostly of T-cells and rare B-cells (fig. S1B and C). To identify transcriptional features associated with CAR T-cell therapeutic potency, we focused on CD8+ T-cells. Clustering all CD8+ T-cells revealed four major cell states: early memory-like TCF7- and C-C Motif Chemokine Receptor 7 (CCR7)-expressing CD8+ clusters; effector-like Granzyme A (GZMA)- and HAVCR2-expressing CD8+ sub-populations (Fig. 1A, fig. S1D).
Figure 1. TCF7- and HAVCR2-expressing CD8+ populations within infusion products are associated with favorable and poor CAR T-cell therapeutic potency, respectively.
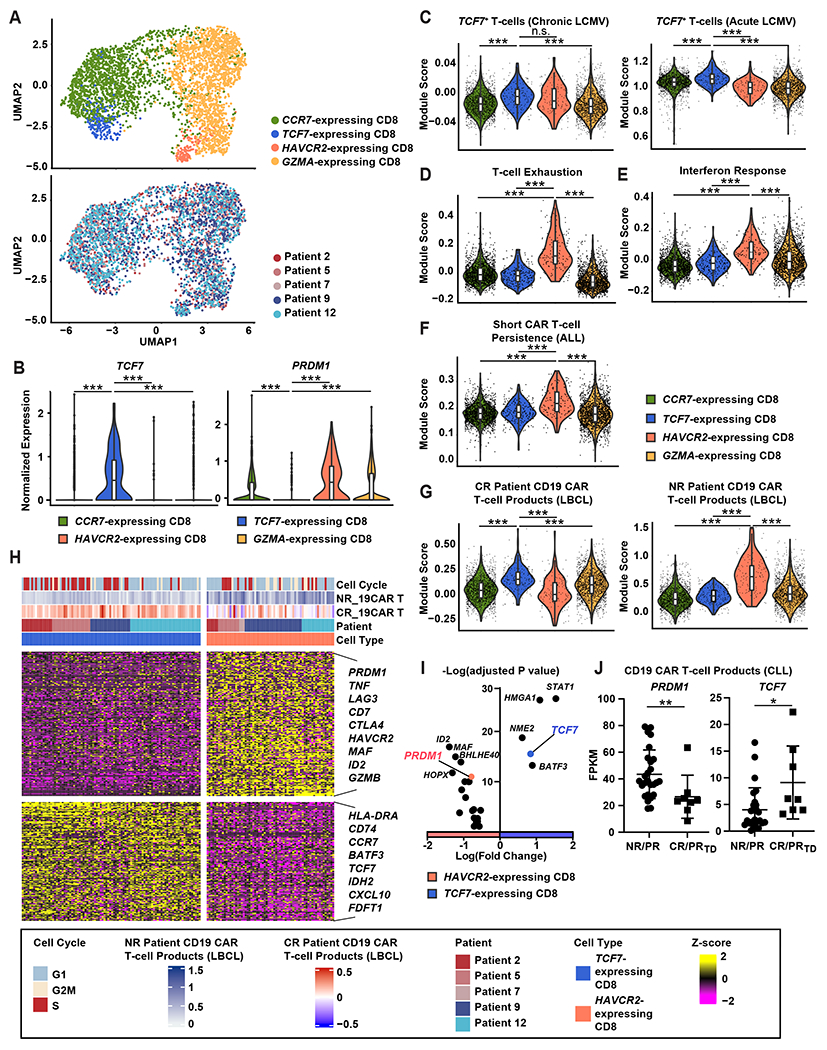
(A) Uniform manifold approximation and projection (UMAP) plots showing sub-clustering of CD8+ T-cells from CAR T-cell infusion products for patients with mCRPC. Cells are labeled according to marker gene expression and patient origin. (B) Expression of PRDM1 and TCF7 is shown for CD8+ T-cell subclusters. (C to E) Scores of gene signatures enriched in (C) TCF7-expressing T-cells in LCMV clone 13 (GSE83978; left) and LCMV Armstrong infection (GSE83978; right), (D) exhausted T-cells (GSE136796) and (E) the IFN response (dbGaP phs002323.v1.p1) are shown. (F and G) Shown are CD8+ T-cell subcluster-specific gene signature scores enriched in (F) pre-manufactured T-cells from patients with ALL with poor CD19 CAR T-cell persistence (dbGaP phs002323.v1.p1) and (G) anti-CD19 CAR T-cell infusion products of complete responder patients (CR) and non-responder (NR) patients with LBCL (GSE151511). (H) Differentially expressed genes were compared between TCF7- and HAVCR2-expressing CD8+ clusters. Top bars indicate cell clusters, patient origin, CD19 CAR T-cell response score (GSE151511), and cell cycle. (I) Differential expression of transcription factors between TCF7- and HAVCR2-expressing CD8+ clusters is shown. (J) PRDM1 and TCF7 expression was evaluated in CD19 CAR-T infusion products from patients with chronic lymphocytic leukemia (CLL; CR: complete response; PRTD: very good partial response; PR: partial response; NR: no response); FPKM: Fragments per kilo base of transcript per million mapped fragments. *P < 0.05, **P < 0.01, ***P < 0.001, n.s., not significant as measured by Kruskal-Wallis test with a post-hoc Dunn’s multiple comparison test. Violin plots in (B) to (G) indicate the distribution of data with rectangles in the middle of the density curves showing the ends of the first and third quartiles and central horizontal line depicting the median. Horizontal bars in (J) show the mean.
A TCF7-expressing CD8+ subset was the only cell population that expressed a high abundance of TCF7 (Fig. 1B, fig. S1D). In accordance with this TCF7 expression profile, these clusters were enriched in TCF7 stem cell-like T-cell signatures that are associated with robust antiviral responses in mouse models of acute and chronic lymphocytic choriomeningitis virus (LCMV) infection (Fig. 1C) (28–30). In contrast, a HAVCR2-expressing CD8+ cluster was markedly enriched in T-cell exhaustion signatures, whereas CCR7- and TCF7-expressing CD8+ populations possessed low exhaustion scores (Fig. 1D). Consistent with this analysis, the HAVCR2-expressing CD8+ T-cell population exhibited upregulation of multiple inhibitory molecule transcripts compared to TCF7-expressing CD8+ cells (fig. S1E).
A recent study that characterized leukapheresed T-cells of pediatric patients with acute lymphocytic leukemia (ALL) demonstrated that enrichment of an interferon (IFN) response signature is associated with poor CAR T-cell persistence (31). We thus queried the CD8+ T-cell clusters identified in our analysis for this battery of genes and found that HAVCR2-expressing CD8+ T-cells in mCRPC patient infusion products had significantly (P < 0.001) higher IFN response signature scores compared to the other populations (Fig. 1E). Accordingly, the HAVCR2-expressing CD8+ state is associated with short CD19 CAR T-cell persistence in ALL, which often prognosticates poor outcome (Fig. 1F).
To determine the generalizable nature of our initial findings, we next sought to investigate CD8+ clusters associated with response to CAR T-cell therapy in LBCL (21). Intriguingly, the TCF7-expressing CD8+ population highly expressed a gene set enriched in anti-CD19 CAR T-cell infusion products derived from complete responders (CR), whereas the HAVCR2-expressing CD8+ T-cell population exhibited the highest scores for a gene set that characterizes non-responder (NR) anti-CD19 CAR T-cells (Fig. 1G). These results were concordant with exhaustion signatures in other independent data sets (fig. S1F). We then scored PSMA CAR T-cell infusion products for the TCF7 and HAVCR2 profiles and analyzed how these signature scores correlate with PSA decline in patients with mCRPC following CAR T-cell transfer (fig. S1G and H). In accordance with our recently reported clinical findings (22), there is a clear dose- and lymphodepletion-dependent relationship with peripheral blood CAR T-cell expansion and early antitumor effects, as determined by serum PSA decrease (fig. S1H). Patients 2 and 5 were treated in the cohort that received the lowest dose of CAR T-cells in this trial, without lymphodepleting preconditioning prior to cell infusion. It is intriguing that, independently of conditioning and cell dose, Patient 2 pre-infusion CAR T-cells with a highly enriched TCF7 signature and a low HAVCR2 score exhibited superior in vivo expansion and PSA response induction, compared to the infusion product of Patient 5 with lower TCF7 and elevated HAVCR2 population scores (fig. S1G and H). These results suggest that increasing TCF7-expressing CD8+ populations and depleting HAVCR2-expressing populations in CAR T-cell infusion products may enhance CAR T-cell efficacy.
Among the transcription factors upregulated in the HAVCR2-expressing CD8+ population, we focused on BLIMP1, which is encoded by PRDM1 (Fig. 1H and I). PRDM1 is known to play a central role in driving T-cell exhaustion and terminal differentiation (27, 32, 33). In addition, high expression of PRDM1 is associated with loss of T-cell stemness and self-renewal capacity through repression of TCF7 (34). Consistent with these results, HAVCR2-expressing CD8+ cells displayed the highest expression of PRDM1, whereas TCF7-expressing CD8+ T-cells exhibited low expression of PRDM1 among the CD8+ clusters (Fig. 1B). To further validate these findings, we compared PRDM1 and TCF7 expression in cohorts of patients with chronic lymphocytic leukemia (CLL) treated with autologous CD19 CAR T-cells. As expected, pre-infusion CAR T-cells from CR and very good partially responding (PRTD) individuals exhibited significantly lower expression of PRDM1 (P < 0.01) and elevated TCF7 (P < 0.01), compared to poorly functional products from non-responders (NR) or conventional partial responders (PR) (Fig. 1J).
The frequencies of CD4+ and CD8+ T-cells within the clinical CAR infusion products profiled in this study are presented in fig. S2A. scRNA-seq analysis of the CD4+ compartment was also performed (fig. S2B to D). Clustering all CD4+ T-cells revealed four major cell states: TCF7-, CCR7-, Marker of Proliferation Ki-67 (MKI67) and Cytotoxic T-Lymphocyte Associated Protein 4 (CTLA4)-expressing CD4+ clusters. CTLA4-expressing CD4+ cells displayed the highest expression of PRDM1 whereas TCF7-expressing CD4+ T-cells exhibited low PRDM1 among the CD4+ clusters (fig. S2D). We therefore originally hypothesized that PRDM1 mediates CAR T-cell exhaustion and attrition of stemness. Thus, PRDM1 KO may mitigate T-cell exhaustion and improve CAR T-cell expansion, persistence and antitumor efficacy.
CRISPR/Cas9-mediated PRDM1 KO potentiates early memory differentiation of PSMA CAR T-cells.
To examine how BLIMP1 deficiency affects CAR T-cell fate, we knocked out PRDM1 in PSMA CAR T-cells using Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) technology (Fig. 2A to C). Because the dominant negative transforming growth factor (TGF)-β receptor component used in our recently published clinical study (22) can potentially confound findings, this was not included in the tested constructs. CAR T-cells with KO at the Adeno-associated virus integration site 1 (AAVS1) safe harbor locus were used as a negative control for gene editing in subsequent in vitro and in vivo functional studies (35). PSMA CAR expression was comparable between control AAVS1 KO and PRDM1 KO CAR T-cells (fig. S3). Next, we characterized the phenotype and function of PRDM1 KO CAR T-cells during an in vitro “stress test” involving repetitive PSMA stimulation through the CAR (Fig. 2D). PRDM1 KO CAR T-cells initially showed increased effector cytokine production compared to control CAR T-cells at 24 hours after the first restimulation (Fig. 2E). In addition, we found that, whereas the expansion capacity of AAVS1 KO CAR T-cells gradually declined following repetitive stimulation, PRDM1 KO CAR T-cells sustained high proliferative capacity even after multiple rounds of stimulation and concomitantly upregulated cell cycle-related gene signatures (Fig. 2F to H). This enhanced expansion capacity of PRDM1 KO CAR T-cells is likely attributed to increased memory formation. Accordingly, PRDM1 KO CAR T-cells displayed increased expression of CD62L, CCR7, MYB, ID3, and TCF7 as well as enriched transcriptomic signatures of memory precursor effector cells, fatty acid oxidation, and the tricarboxylic acid cycle, indicating that PRDM1 KO CAR T-cells are skewed toward an early memory fate (Fig. 2I to M).
Figure 2. CRISPR/Cas9-mediated PRDM1 KO potentiates early memory PSMA CAR T-cell differentiation.
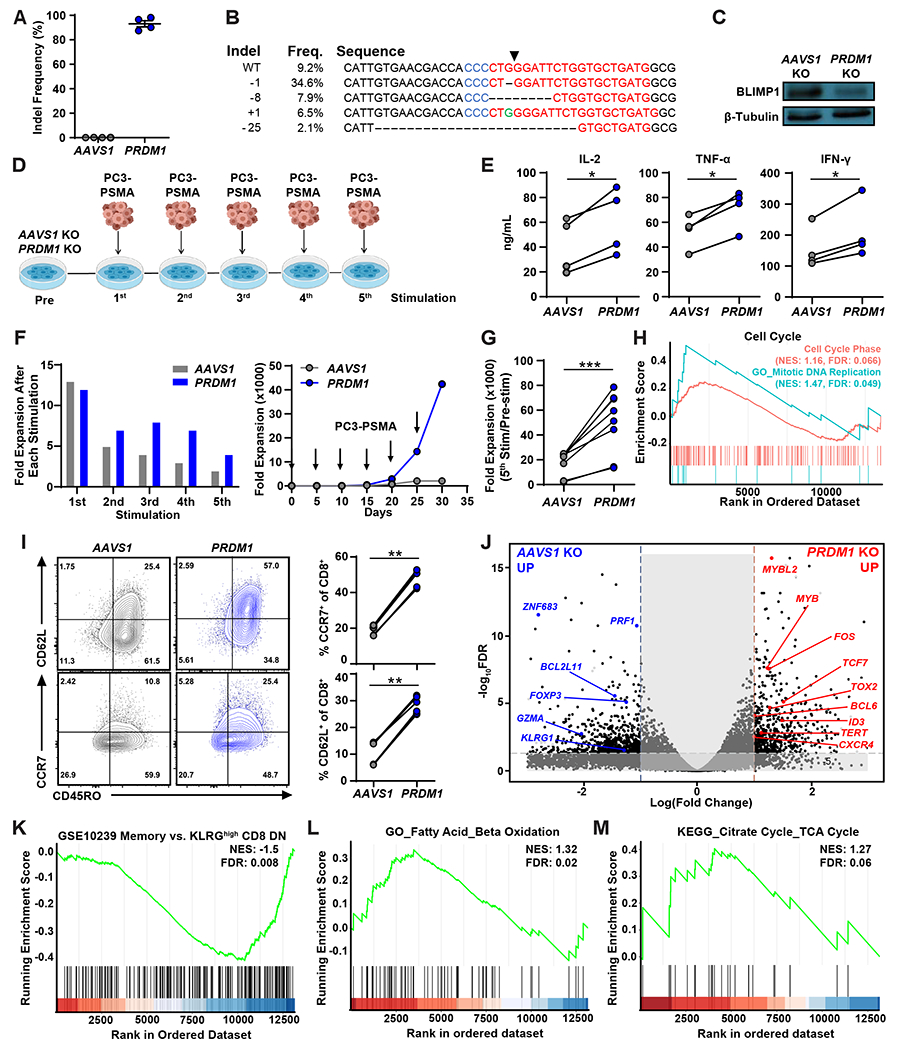
(A) PRDM1 editing efficiency was measured by Sanger sequencing and subsequent TIDE (Tracking of Indels by Decomposition) analysis. Data are presented as scatter points, where the mean and S.E.M. bars are included. (B) Amplicon sequencing of PRDM1 indel variants generated by CRISPR/Cas9-mediated gene editing is shown. Arrow indicates cleavage site. (C) Representative Western blot analysis for BLIMP1 expression is shown. (D) Schematic of the restimulation assay used to “stress test” PRDM1 KO PSMA CAR T-cells. CAR T-cells were challenged with PSMA-expressing PC3 prostate tumor targets every 4 to 5 days at an effector to target (E:T) ratio of 3:1. (E) Effector cytokines produced by CAR T-cells after the initial tumor cell challenge. (F) Representative CAR T-cell expansion kinetics during the restimulation assay for one donor are shown. Left: CAR T-cell expansion after each stimulation, Right: Cumulative CAR T-cell expansion. Arrows indicate the timing of PC3-PSMA tumor cell challenge. (G) Summary of the expansion capacity of AAVS1 and PRDM1 KO CAR T-cells during the restimulation assay with four different donors. (H) Gene set enrichment analysis (GSEA) of PRDM1 KO versus AAVS1 KO CAR T-cells comparing gene signatures related to the cell cycle and mitotic DNA replication. CAR-T samples were harvested on day 5 following the first tumor challenge. NES: normalized enrichment score, FDR: false discovery rate. (I) Early memory marker expression was measured by flow cytometry after two consecutive tumor cell stimulations. (J) Volcano plot displaying the results of differential gene expression analysis when comparing PRDM1 KO to control AAVS1 KO CAR T-cells. (K to M) GSEA of PRDM1 KO versus AAVS1 KO CAR T-cells comparing gene sets associated with (K) memory T-cells (GSE10239), (L) GO_Fatty acid_Beta oxidation, and (M) the KEGG_TCA cycle. All knockout and restimulation experiments were conducted with CAR T-cells manufactured from 4 different healthy donors. RNA-seq experiments were conducted with CAR T-cells manufactured from 2 different healthy individuals, each with replicates generated from two independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, as measured by paired t-tests.
PRDM1 KO increases TCF7 expression and enhances early memory CAR T-cell differentiation in a TCF7-dependent manner.
We next sought to investigate whether PRDM1 KO derepresses TCF7 expression. PRDM1 KO increased expression of TCF7 and genes encoding other transcription factors that are crucial for maintaining T-cell stemness, such as Myeloblastosis (MYB), B-cell lymphoma 6 (BCL6) and Inhibitor of DNA-binding 3 (ID3) (Fig. 3A and B) (36–38). Moreover, PRDM1 KO CAR T-cells were enriched in TCF7-expressing stem cell-like T-cell signatures, suggesting that BLIMP1 inhibits TCF7-mediated stemness (Fig. 3C). Consistent with our hypothesis, PRDM1 KO CAR T-cells exhibited depletion of HAVCR2-expressing CD8+ cell population gene signatures and transcriptionally resembled TCF7-expressing CD8+ T-cells observed in PSMA CAR T-cell infusion products (Fig. 3D). To examine whether TCF7 upregulation is required for the maintenance of early memory differentiation and robust CAR T-cell proliferative potential as observed in Fig. 2, we knocked out TCF7 (Fig. 3E) and subsequently assessed CAR T-cell expansion in a restimulation assay. We found that TCF7 depletion counteracted the effect of PRDM1 KO by reducing proliferative capacity, which was accompanied by decreased frequencies of CCR7 and CD62L expressing memory CAR T-cells (Fig. 3F to H). Because lack of polyfunctionality is a hallmark of terminal differentiation and T-cell dysfunction (38), we next stimulated PRDM1 and TCF7 single or double KO CAR T-cells and measured expression of interleukin (IL)-2, interferon (IFN)-γ and tumor necrosis factor (TNF)-α. PRDM1 ablation increased frequencies of polyfunctional CAR T-cells compared to AAVS1 KO control CAR T-cells (Fig. 3I), which coincides with the observed increased early memory differentiation of PRDM1 KO CAR T-cells. Conversely, TCF7 depletion decreased the frequency of polyfunctional PRDM1 KO CAR T-cell populations (Fig. 3I), suggesting that PRDM1 deficiency enhances CAR T-cell polyfunctionality, at least in part, by TCF7 upregulation. Taken together, these data imply that PRDM1 knockout enhances CAR T-cell early memory differentiation in a TCF7-dependent manner.
Figure 3. PRDM1 KO increases TCF7 expression and enhances early memory CAR T-cell differentiation in a TCF7-dependent manner.
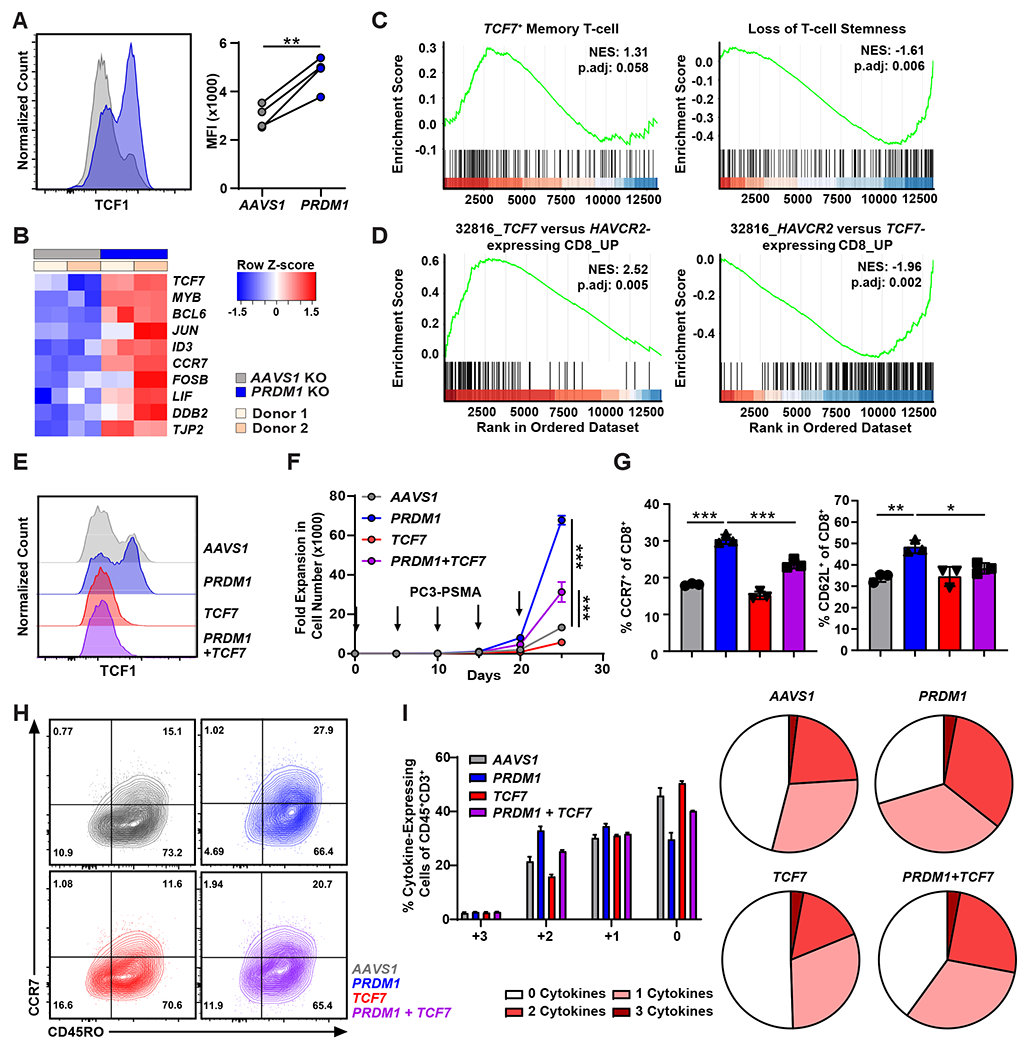
(A) TCF1 expression was measured by flow cytometric analysis of PRDM1 KO versus AAVS1 KO PSMA CAR T-cells. CAR T-cells derived from n = 4 different healthy individuals. MFI: mean fluorescence intensity. (B) Analysis of CAR T-cells for transcripts enriched in TCF7-expressing stem cell-like T-cells previously observed in LCMV mouse models (GSE83978). (C) GSEA of PRDM1 KO relative to AAVS1 KO CAR T-cells comparing gene sets associated with a TCF7+ T-cell memory state (GSE83978) and loss of stemness (GSE84105). p.adj, adjusted P value. (D) GSEA of PRDM1 KO versus AAVS1 KO CAR T-cells evaluating gene sets enriched in TCF7+ (left) and HAVCR2+ CD8+ clusters (right). Gene signatures were derived from CAR T-cell infusion products of patients with mCRPC shown in Figure 1. (E) Representative histogram shows TCF1 expression by flow cytometry in PRDM1 and TCF7 KO CAR T-cell variants. (F) Expansion kinetics are shown during a restimulation assay of gene-edited CAR T-cells. CAR T-cells were challenged with PC3-PSMA target cells every 5 days at an E:T of 3:1. Arrows indicate the timing of PC3-PSMA cell restimulation. Data represent the mean ± S.D. (n = 3 independent experiments). (G) Frequencies of CAR T-cell variants expressing CCR7 and CD62L and (H) representative flow cytometry plots showing CCR7 and CD45RO expression following the first in vitro tumor cell challenge. Data indicate mean ± S.D. from n = 3 independent experiments. (I) CAR T-cell polyfunctionality (IL-2, IFN-γ and TNF-α expression) was evaluated after a 15-hour co-culture with PC3-PSMA cells. Data show mean ± S.D. from n = 3 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001. Data in panel (A) and panels (F and G) were analyzed using a paired t-test and one-way ANOVA test with a post-hoc Tukey’s multiple comparison test, respectively.
PRDM1 KO hampers T-cell effector function and tumor control during chronic CAR stimulation despite increases in CAR T-cell proliferative capacity.
To evaluate the effector functions of PRDM1 KO CAR T-cells in the setting of repetitive antigen exposure, we first assessed cytokine concentrations 24 hours after the first and fifth rounds of in vitro PC3-PSMA tumor cell stimulation. PRDM1 KO initially increased effector cytokine production as shown in Fig. 3I and Fig. 4A. However, after multiple tumor cell stimulations, PRDM1 KO CAR T-cells exhibited dramatically reduced effector cytokine secretion (Fig. 4A). Further, after five consecutive tumor challenges, the cytolytic activity of PRDM1 KO CAR T-cells was impaired compared to that of control CAR T-cells (Fig. 4B). This result is consistent with a previous study in which PRDM1 deficiency profoundly compromised the cytotoxic activity of antigen-specific CD8+ T-cells during chronic viral infection (32).
Figure 4. PRDM1 KO marginally enhances solid tumor control, despite increases in CAR T-cell early memory phenotype and proliferative capacity.
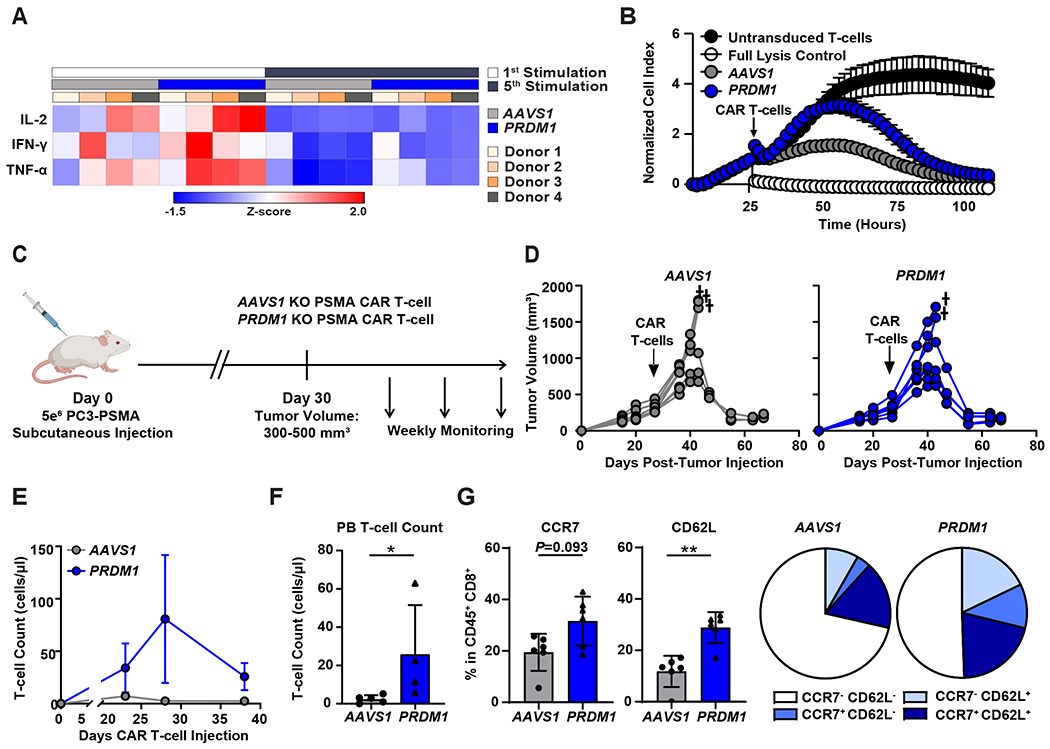
(A and B) CAR T-cells were restimulated five times with PC3-PSMA target cells every 4 to 5 days at an E:T ratio of 3:1. (A) The heat map shows the relative effector cytokine secretion by AAVS1 KO and PRDM1 KO CAR T-cells after first and fifth tumor cell restimulations. (B) Killing kinetics of AAVS1 KO and PRDM1 KO CAR T-cells are shown. CAR T-cells were isolated after the fifth restimulation timepoint and co-cultured with PC3-PSMA cells at a ratio of E:T = 3:1. Cytotoxicity was monitored by real-time cellular impedance monitoring technology (xCELLigence). T-cells without CAR transduction were used as a negative control and 20% Tween-20 treatment served as a full lysis control. Data are expressed as mean ± S.D. from n = 6 individual donors. (C) A schematic of the high tumor burden PC3-PSMA xenograft mouse model. Male NSG mice were subcutaneously transplanted with 5 × 106 PC3-PSMA cells, and 3.5 × 105 PSMA CAR T-cells were given intravenously when tumor volume reached about 500 mm3. (D) Tumor growth was monitored by caliper measurements. † = death. (E) CAR T-cell expansion kinetics in the peripheral blood are shown. (F) Absolute numbers of human T-cells in the peripheral blood on day 38 post-CAR T-cell injection are shown. (G) Frequencies of peripheral blood CAR T-cells expressing CCR7 and CD62L were measured by flow cytometry. Data are shown as mean ± S.D.; n = 6 for (D) and (G) and n = 4 to 5 for (E) and (F). *P < 0.05, **P < 0.01, as determined by Mann Whitney U tests.
Based on the above results, we next assessed the in vivo antitumor activity of PRDM1 KO CAR T-cells in xenogeneic mouse models. When tested against a relatively low burden of flank-engrafted PC3-PSMA prostate tumor cells, PRDM1 KO PSMA CAR T-cells exhibited a modest enhancement of tumor control compared to control CAR T-cells (fig. S4A and B). Similarly, and consistent with a recent report (23), PRDM1 KO anti-CD19 CAR T-cells better suppressed cancer growth compared to control CAR T-cells in a B-cell acute lymphoblastic leukemia (ALL) model (NALM-6), although these CAR T-cells eventually failed to eradicate tumors (fig. S4C to E). In an in vivo “stress test” in which tumor burden is escalated to reveal CAR T-cell functional limits, PRDM1 KO CAR T-cells showed comparable antitumor activity to AAVS1 KO CAR T-cells (Fig. 4C and D). Despite lack of improved tumor control over AAVS1 KO CAR T-cells, PRDM1 KO CAR T-cells exhibited enhanced in vivo expansion and persistence (Fig. 4E and F). Additionally, consistent with our in vitro studies, PRDM1 KO CAR T-cells maintained a higher fraction of central memory T-cells (Fig. 4G, fig. S4F), indicating that PRDM1 deficiency improves CAR T-cell expansion and persistence by preserving early memory pools. Together, these results suggest that, despite improvements in expansion and persistence, PRDM1 KO alone is not sufficient to potentiate robust and sustained CAR T-cell antitumor efficacy in aggressive tumor models.
PRDM1 KO CAR T-cells fail to maintain high effector function due to upregulation of exhaustion-related transcription factors.
To elucidate the mechanism by which PRDM1 KO CAR T-cells fail to maintain effector function following chronic antigen stimulation, we performed bulk RNA-seq on CAR T-cells harvested after several rounds of tumor challenge. We observed that PRDM1 KO increased the expression of early memory-related genes, including MYB, LEF, CCR7, IL7R, and CD28, even after multiple stimulations (Fig. 5A). Intriguingly, together with these early memory-related genes, PRDM1 KO resulted in upregulation of genes encoding multiple exhaustion-related transcription factors such as the Nuclear Receptor Subfamily 4A (NR4A) transcription factors as well as Thymocyte Selection-associated HMG BOX (TOX), TOX2, and Interferon Regulatory Factor 4 (IRF4; Fig. 5B, left). To rule out potential model-dependent effects, we took advantage of a publicly available RNA sequencing expression dataset of CD8+ TILs from B16F10 (melanoma) tumor-bearing PRDM1 conditional knockout (cKO) syngeneic mice. Concordant with our findings in CAR T-cells, PRDM1 cKO CD8+ TILs exhibited upregulated expression of NR4A3, NR4A1, and IRF4 compared to wild type counterparts (Fig. 5B, right).
Figure 5. PRDM1 KO CAR T-cells fail to sustain antitumor effector function due to upregulation of exhaustion-related transcription factors.
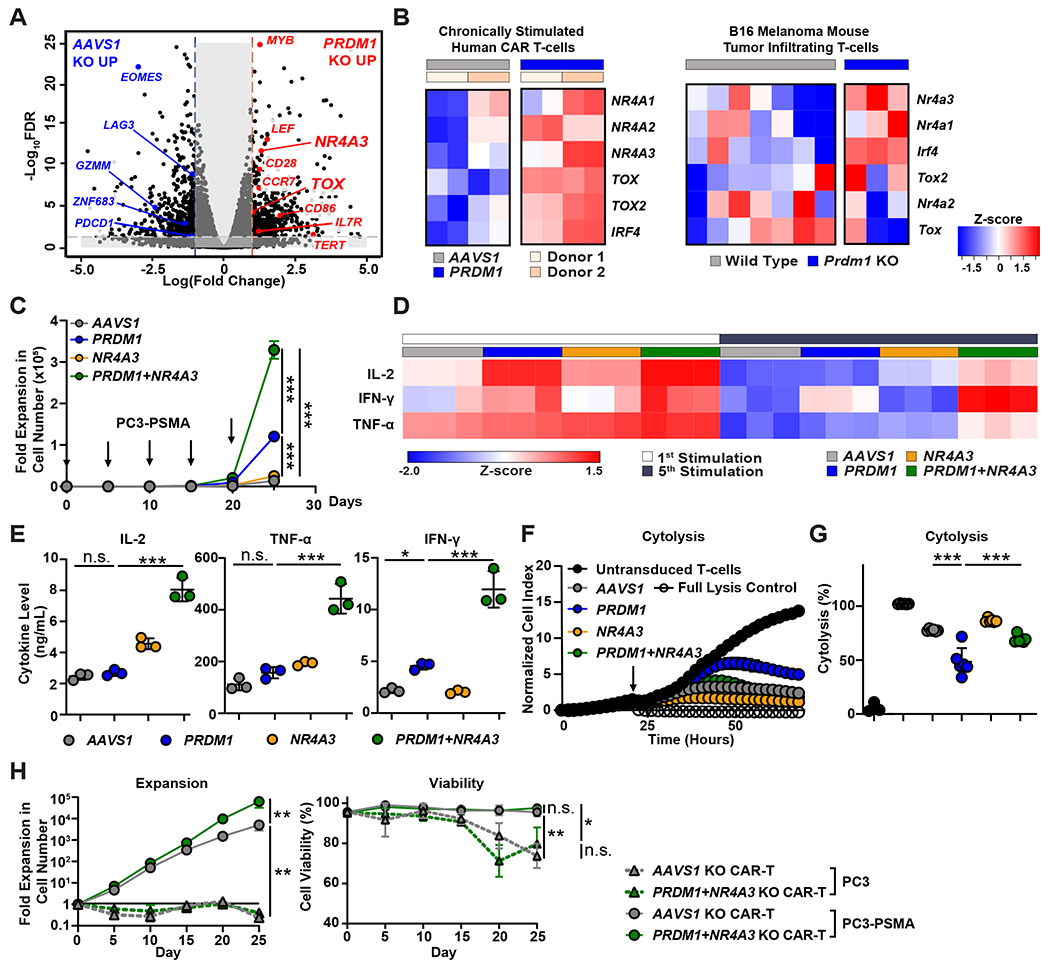
(A) CD8+ CAR T-cells were isolated after the fourth tumor restimulation for bulk RNA-seq. The volcano plot illustrates differential gene expression analysis in PRDM1 KO compared to control AAVS1 KO CAR T-cells after the fourth consecutive tumor cell challenge. (B) The heat map shows expression of transcription factor-encoding genes associated with T-cell exhaustion. RNA-seq experiments were conducted with CAR T-cells manufactured from 2 different individuals, each with replicates generated from two independent experiments (left). A similar profile of exhausted murine TILs (GSE113221) is shown in the right panel. (C) Expansion kinetics of PRDM1 and NR4A3 KO CAR T-cells during a restimulation assay. PSMA CAR T-cells were challenged with PC3-PSMA tumor cells every 5 days at an E:T ratio of 3:1. Arrows indicate the timing of PC3-PSMA challenge. Data are mean ± S.D. from n = 3 different donors. (D and E) Concentrations of effector cytokines produced by AAVS1 KO, PRDM1 KO, NR4A3 KO and PRDM1/NR4A3 dual KO CAR T-cells are shown. (D) The heat map shows effector cytokine secretion 24 hours following the first and fifth tumor cell challenges. (E) Graphical summaries are shown for effector cytokine production after the fifth CAR T-cell stimulation with tumor targets. Data are mean ± S.D. (n = 3). (F) Killing kinetics are shown for the indicated CAR T-cell/tumor cell co-cultures. (G) CAR T-cells were isolated after the fifth round of antigen stimulation and co-cultured with PC3-PSMA tumor cells at an E:T ratio of 3:1 for a “stressed” cytotoxicity assay. Data in (F and G) indicate mean ± S.D. (n = 6). For (C to G), data are representative of 3 independent experiments performed with engineered CAR T-cells manufactured from 3 different healthy individuals. (H) CAR T-cells were repetitively challenged with PC3 or PC3-PSMA tumor targets every 5 days at an E:T ratio of 3:1. The expansion capacity (left) and viability (right) of CAR T-cells were assessed over time. Data are depicted as the mean ± S.D. from n = 3 independent donors assayed in 2 independent experiments. For all panels, *P < 0.05, **P < 0.01, ***P < 0.001, n.s., not significant, as determined by a one-way ANOVA test with a post-hoc Tukey’s multiple comparison test.
Combinatorial PRDM1 and NR4A3 KO sustains the effector function of chronically-stimulated CAR T-cells.
We hypothesized that the observed compensatory upregulation of exhaustion-associated transcription factor genes can limit the effector function of PRDM1 KO CAR T-cells during chronic stimulation and that deletion of these exhaustion factors will render PRDM1 KO CAR T-cells capable of maintaining antitumor effector function. PRDM1 KO CAR T-cells exhibited reduced effector function, despite inhibitory receptor downregulation (fig. S5A). Because NR4A3 was the most significantly upregulated exhaustion-related transcription factor gene examined in PRDM1 KO CAR T-cells after multiple episodes of antigen stimulation (P < 0.001, Fig. 5B) and NR4A3 is significantly elevated in hypofunctional NR/PR CLL patient CD19 CAR T-cells (P < 0.01, fig. S5B), we next knocked out both NR4A3 and PRDM1 (fig. S5C) and functionally characterized the gene-edited CAR T-cells. We observed that, whereas NR4A3 single KO CAR T-cells exhibited a similar degree of expansion as control CAR T-cells, PRDM1/NR4A3 dual KO CAR T-cells exhibited the highest degree of antigen-induced proliferative capacity (Fig. 5C). We also measured the frequency of CAR T-cells expressing cytotoxic perforin and granzyme molecules. NR4A3/PRDM1 double KO partially restored cytotoxic function in the setting of perforin and granzyme expression in CD8+CAR+ T-cells (fig. S5D). PRDM1/NR4A3 KO CAR T-cells also exhibited increased frequencies of CCR7, CD62L, and TCF1 (encoded by TCF7) expressing cells compared to AAVS1 KO CAR T-cells (fig. S5E and F).
Because CD4+ CAR T-cells are important to antitumor efficacy (39, 40), we also determined the impact of the various KOs on CD4+ CAR T-cells with respect to differentiation phenotypes, as well as cytotoxic molecule expression during the aforementioned in vitro stress tests. Similar to CD8+ CAR T-cells, PRDM1 KO increased frequencies of CD4+ CAR T-cells expressing early memory markers such as CCR7 and TCF7 as compared to controls (fig. S5G). Further, combined KO of PRDM1 and NR4A3 had a strong effect on rescuing expression of cytotoxic perforin and granzyme molecules in CD4+ CAR T-cells (fig. S5H).
Next, we assessed effector functions in the context of PRDM1/NR4A3 KO and found that PRDM1/NR4A3 KO CAR T-cells maintained an elevated abundance of effector cytokine production after multiple tumor challenges, whereas PRDM1 and NR4A3 single KO CAR T-cells showed similar IL-2 and TNF-α secretion as control CAR T-cells (Fig. 5D and E). This potency enhancement conferred by PRDM1 and NR4A3 dual KO was consistent in the context of both high and low PSMA expression (fig. S5I and J). Furthermore, unlike PRDM1 single KO CAR T-cells which lost cytotoxic function following serial stimulation, PRDM1/NR4A3 KO CAR T-cells displayed sustained killing activity over time (Fig. 5F and G). To determine whether PRDM1/NR4A3 KO CAR T-cells exhibit aberrant growth patterns potentially indicative of transformation, we tested whether CAR T-cell proliferation is antigen-dependent using irradiated PC3 cells with and without PSMA expression as stimuli. We observed that PRDM1/NR4A3 KO CAR T-cells fail to expand, accompanied by a reduction in viability when co-cultured with PSMA-negative PC3 cells, suggesting that cell expansion and survival is antigen-dependent (Fig. 5H).
Upregulation of exhaustion-related transcription factors in PRDM1 KO CAR T-cells is attributed to increased chromatin accessibility and calcineurin-NFAT signaling.
We next sought to investigate the mechanism of exhaustion-associated transcription factor upregulation in PRDM1 KO CAR T-cells. Emerging evidence indicates that PRDM1 epigenetically regulates the transcription of memory-related genes by directly binding to promoter regions and recruiting histone modifiers (41). We therefore investigated whether PRDM1 KO affects the chromatin accessibility of exhaustion-related transcription factor genes by performing Assay for Transposase-Accessible Chromatin with sequencing (ATAC–seq) on control and PRDM1 KO CAR T-cells. PRDM1 KO increased global chromatin accessibility (Fig. 6A). Transcription motif enrichment analysis revealed that a BLIMP1 motif was one of the most enriched transcription factor binding sites in PRDM1 KO CAR T-cells (Fig. 6B and C), suggesting that BLIMP1 can act as an epigenetic repressor. Consistent with previous chromatin immunoprecipitation sequencing (ChIP-seq) results (41), PRDM1 KO increased chromatin accessibility at loci corresponding to early memory and stemness genes such as TCF7, CD28, CCR7, SELL, and MYB (fig. S6A). We found that, even before tumor challenge, PRDM1 KO increased chromatin accessibility at exhaustion-related transcription factor gene regions, including those of TOX, TOX2, and NR4A3, and that a subset of these open regions colocalized with the above BLIMP1 motif (Fig. 6D). These observations indicate that PRDM1 contributes to epigenetic repression of a battery of exhaustion-associated transcription factors, and that PRDM1 KO CAR T-cells are predisposed to upregulate exhaustion-associated transcription factors.
Figure 6. Upregulation of exhaustion-related transcription factors in PRDM1 KO CAR T-cells is attributed to increased chromatin accessibility and calcineurin-NFAT signaling.
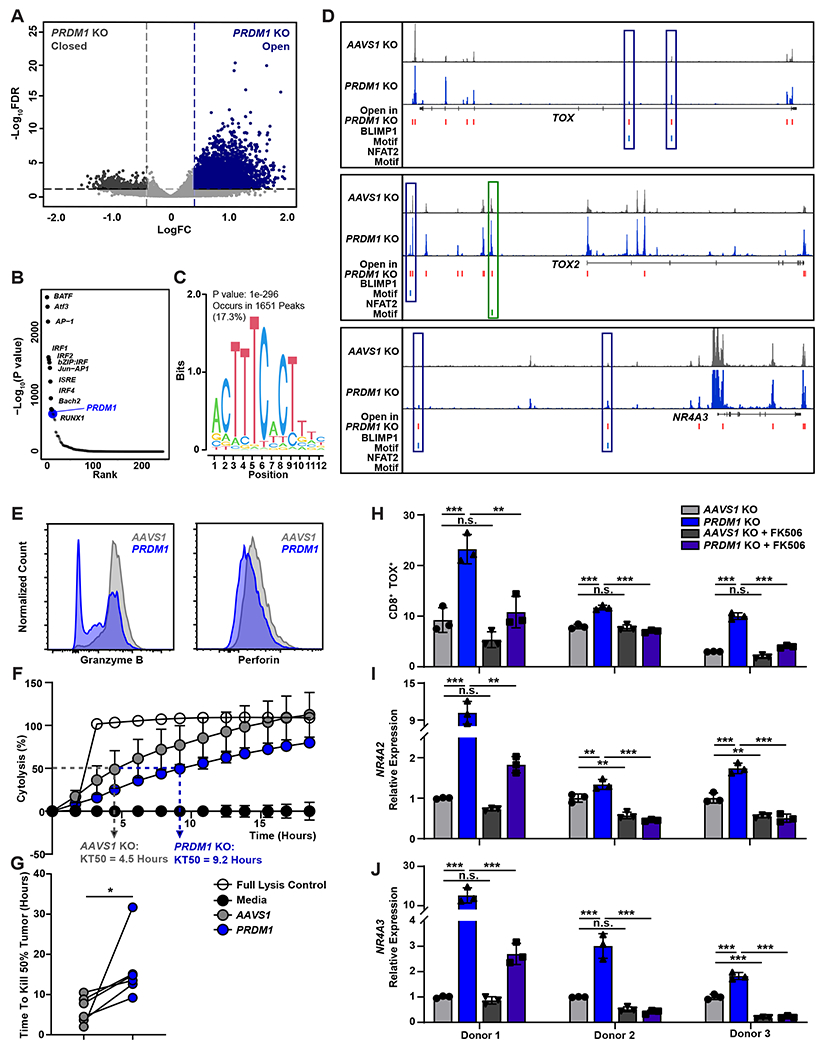
(A to D) ATAC-seq analysis of AAVS1 KO and PRDM1 KO CAR T-cells is shown. At the end of manufacturing, CAR+ T-cells were enriched and subjected to ATAC-seq analysis. (A) The volcano plot shows differentially accessible chromatin regions. (B) Top transcription factor motifs enriched in PRDM1 KO compared to AAVS1 KO CAR T-cells are shown. (C) A BLIMP1 binding motif enriched in open chromatin regions of PRDM1 KO CAR T-cells is shown. The P value was calculated by HOMER motif analysis. (D) ATAC–seq tracks of TOX, TOX2, and NR4A3 loci are shown. Open chromatin regions in PRDM1 KO CAR T-cells and the binding motifs of BLIMP1 and NFAT2 are indicated. ATAC-seq experiments were conducted with CAR T-cells manufactured from 2 different donors, each with replicates generated from two independent experiments. (E) Expression of granzyme B and perforin were measured by flow cytometry. (F and G) A cytotoxicity assay was used to determine the time needed to kill 50% of tumor target cells (KT50). (F) Representative killing kinetics of AAVS1 and PRDM1 KO CAR T-cells are shown. Data show mean ± S.D. (n = 3). (G) KT50 values were compared between AAVS1 and PRDM1 KO CAR T-cells. Data were generated from 6 independent experiments with CAR T-cells manufactured from 4 different donors. (H to J) Expression of exhaustion-related TOX, NR4A2 and NR4A3 following repetitive tumor cell challenges is shown. AAVS1 KO and PRDM1 KO CAR T-cells were challenged with PC3-PSMA cells every 2 to 4 days at an E:T ratio of 1:1 in the presence or absence of 100 nM FK506. Following two consecutive rounds of stimulation, CAR T-cells were isolated and expression of exhaustion-associated transcription factors or corresponding genes were measured. (H) TOX expression was measured by flow cytometry. (I) NR4A2 and (J) NR4A3 expression were measured by qRT-PCR. Data indicate mean ± S.D. (n = 3). *P < 0.05, *P < 0.01, ***P < 0.001, n.s., not significant. Data in (G) and (H to J) were analyzed using a paired t-test and one-way ANOVA test with a post-hoc Tukey’s multiple comparison test, respectively.
Given that CAR T-cell dysfunction can be induced by prolonged exposure to cancer cells due to low cytotoxic activity (42), we investigated how the impaired cytotoxicity of PRDM1 KO CAR T-cells affects regulation of exhaustion-associated transcription factors. PRDM1 KO CAR T-cells downregulated Granzyme B and Perforin expression (Fig. 6E, fig. S5D and H). This decrease in cytotoxic protein expression led to delayed tumor clearance, which caused PRDM1 KO CAR T-cells to be exposed to target cancer cells for twice as long as control CAR T-cells (Fig. 6F and G). Due to prolonged exposure to cancer cells, PRDM1 KO increased expression of NFAT2 (fig. S6B). As TOX and NR4A transcription factor families are induced by calcineurin-NFAT signaling, we used the FK506 calcineurin inhibitor to examine the involvement of NFAT in exhaustion-related transcription factor upregulation in PRDM1 KO CAR T-cells (43, 44). FK506 treatment either completely or partially counteracted PRDM1 KO-mediated upregulation of TOX protein expression, as well as NR4A2 and NR4A3 transcription, during restimulation, suggesting that PRDM1 KO may induce upregulation of exhaustion-associated transcription factors through increased NFAT signaling (Fig. 6H to J).
PRDM1/NR4A3 dual KO enhances in vivo CAR T-cell antitumor activity by preserving TCF1+ CD8 T-cells and increasing effector functions.
Based on the observation that PRDM1/NR4A3 dual KO sustains the proliferative ability, effector functions, and early memory phenotype of CAR T-cells, we next examined whether PRDM1/NR4A3 KO CAR T-cells would elicit enhanced antitumor activity in vivo. Whereas AAVS1, PRDM1, and NR4A3 single KO CAR T-cells controlled tumor growth in about 50% of mice in the high-burden PC3-PSMA cell xenograft model, PRDM1/NR4A3 double KO CAR T-cells successfully suppressed tumor growth in all treated mice, in association with overall prolongation of survival (Fig. 7A and B). Because advanced prostate cancer commonly progresses within two years following the initiation of androgen-ablative therapy often with osseous in addition to visceral metastases, we also tested our approach in an intraosseous PC3-PSMA prostate tumor model. Bioluminescent tumor burden was also significantly decreased by a single infusion of PRDM1/NR4A3 KO PSMA CAR T-cells compared to AAVS1 KO (P < 0.05) CAR T-cells in mice bearing intraosseous tumors generated by PC3-PSMA cell engraftment (Fig. 7C). We subsequently collected tumor and blood samples to characterize the immunophenotype of CAR T-cells. We found that PRDM1/NR4A3 double KO significantly increased the absolute numbers of CAR T-cells in the tumor (P < 0.05) and peripheral blood (P < 0.001) compared to AAVS1 KO CAR T-cells fig. S7A). This increased T-cell number may be due in part to the elevated frequencies of early memory T-cells with optimal proliferative potential observed with PRDM1 KO (fig. S7B), which is consistent with our previous results. To investigate how PRDM1 and NR4A3 depletion affect T-cell dysfunction, we first evaluated the frequencies of inhibitory receptor-expressing CAR T-cells. Although PRDM1 single KO CAR T-cells demonstrated reduced Programmed Death-1 (PD-1) and TIM-3 expression in the peripheral blood, their inhibitory receptor expression was comparable to that of AAVS1 KO CAR T-cells in the tumor where CAR T-cells receive persistent antigen stimulation (Fig. 7D and E, fig. S7C). In contrast, PRDM1/NR4A3 KO CAR T-cells demonstrated a significant reduction in the proportion of PD-1+TIM-3+ CD8 T-cells in both the tumor (P < 0.05) and peripheral blood as compared to AAVS1 KO CAR T-cells (P < 0.01, Fig. 7D and E). Also, as observed in vitro, PRDM1 KO led to a substantial increase in TCF1 (encoded by TCF7) expression in CAR T-cells in vivo. Both PRDM1 single KO and PRDM1/NR4A3 double KO CAR T-cells exhibited increased stem cell-like TIM-3−TCF1+ CD8 T-cells (45) in the tumor and peripheral blood, although only PRDM1/NR4A3 KO CAR T-cells showed a significant decrease in the frequencies of TIM-3+TCF1− exhausted T-cells in the tumor (P < 0.05, Fig. 7F and G, fig. S7D).
Figure 7. PRDM1/NR4A3 dual KO enhances in vivo CAR T-cell antitumor activity by preserving TCF1+CD8+ T-cells and increasing effector function.
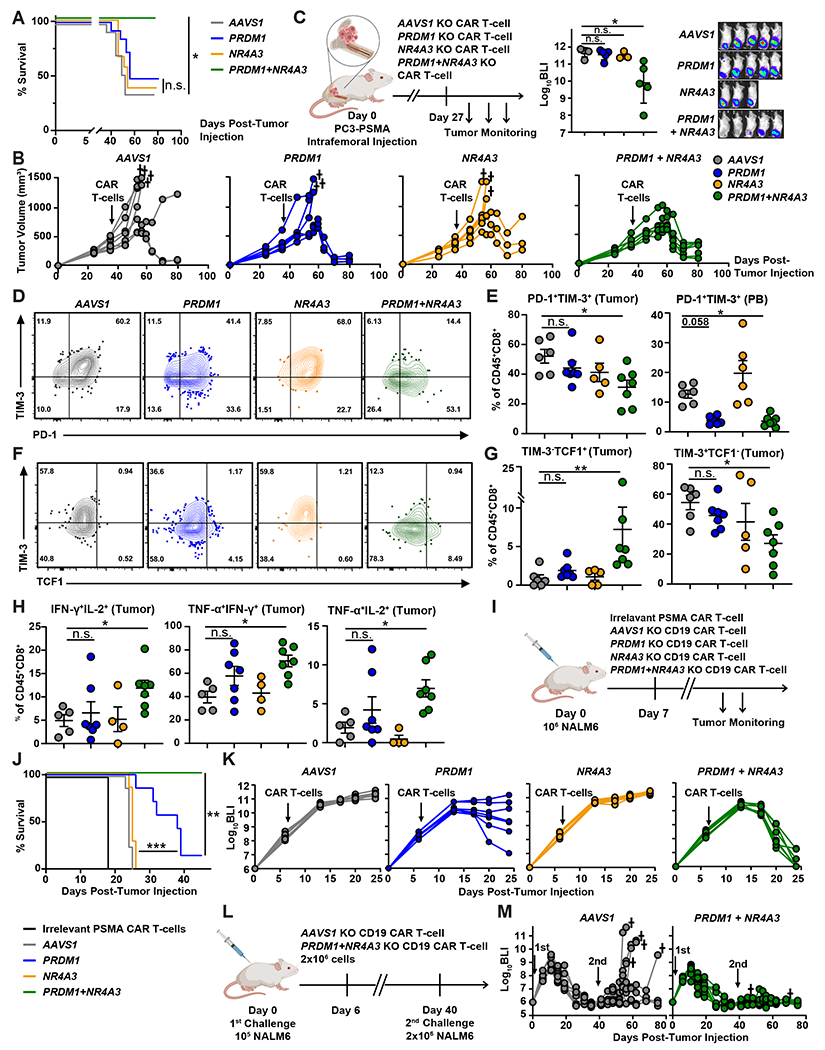
(A and B) Male NSG mice were subcutaneously engrafted with 5 × 106 PC3-PSMA tumor cells, and 3.5 × 105 PSMA CAR T-cells were given intravenously when tumor volume reached about 500 mm3. (A) Kaplan–Meier curves showing overall survival in each group. The Gehan-Breslow-Wilcoxon test was used for statistical analysis (n = 12 to 14 mice per group). (B) Tumor growth was monitored over time in a representative experiment. (C) Male NSG mice were intrafemorally injected with 2 × 105 PC3-PSMA tumor cells. On day 27, 1 to 2 × 105 PSMA CAR T-cells were injected intravenously, and tumor burden was measured by bioluminescent imaging (BLI, (p/sec/cm2/sr)). (D to H) Tumor and peripheral blood (PB) samples were harvested from mice injected subcutaneously with PC3-PSMA tumor cells on day 45 post-tumor implantation, when the tumor size was comparable between the groups. Samples were stained with hCD45, murine CD45 (mCD45), CD4, and CD8 antibodies and analyzed by flow cytometry. (D) Representative flow cytometry plots (E) and quantification show frequencies of cells expressing PD-1 and TIM-3 in CD45+CD8+ T-cells derived from PB and tumors. Representative flow cytometry plots in (D) show a tumor sample. (F) Representative flow cytometry plots and (G) quantification show frequencies of TIM-3- and TCF1-expressing CD45+CD8+ T-cells derived from tumors. (H) Effector cytokine expression was measured by flow cytometry following ex vivo stimulation of CAR TILs. TILs were activated with 50 ng/mL PMA and 1 μg/mL ionomycin in presence of 5 μg/mL Brefeldin A for 6-hours, followed by IFN-γ and TNF-α staining (n = 4 to 7). Statistical analysis in (C, E, G, and H) was conducted using a Kruskal-Wallis test with a post-hoc Dunn’s multiple comparisons test, *P < 0.05, **P < 0.01, n.s., not significant; mean ± S.E.M. shown. (I) A schematic of the NALM-6 xenograft model. Briefly, NSG mice were intravenously injected with 1 × 106 NALM6-CBG cells. On day 7 post tumor injection, 3 × 105 gene-edited anti-CD19 or control CAR T-cells were treated (n = 7 to 8 per group). (J) survival and (K) graphical summaries of longitudinal bioluminescent tumor burden are shown for NSG mice treated, as indicated. Data are representative of two independent experiments. The Gehan-Breslow-Wilcoxon test was used for survival analysis shown in (J). (L) A schematic of the NALM-6 rechallenge model is shown. Briefly, NSG mice were intravenously injected with 1 × 105 NALM6-CBG cells. On day 6 post-tumor injection, 2 × 106 anti-CD19 CAR T-cells were infused (n = 9 to 10 per group). Surviving mice were rechallenged with a second dose of NALM-6 cells on day 40. (M) Longitudinal tumor burden is shown.
To investigate whether PRDM1/NR4A3 dual KO enhances CAR TIL effector function, we reactivated these TILs ex vivo and assessed intracellular cytokine production. Consistent with our in vitro results (Fig. 5D and E), PRDM1 or NR4A3 single KO failed to improve effector cytokine production, whereas PRDM1/NR4A3 double KO CAR T-cells maintained higher polyfunctionality compared to control CAR T-cells (Fig. 7H, fig. S7E).
We also examined the antitumor activity of PRDM1/NR4A3 KO CAR T-cells in the NALM-6 B-ALL model. Similar to the results obtained from the PC3-PSMA model, NR4A3 single KO CD19 CAR T-cells failed to suppress tumor growth. PRDM1 single KO moderately enhanced tumor control and survival (Fig. 7I to K). However, when PRDM1 KO was combined with NR4A3 KO, CAR T-cells induced rapid tumor clearance and durable antitumor efficacy (Fig 7I to K). Further, we assessed the efficacy of PRDM1 and NR4A3 single KO or PRDM1/NR4A3 double KO mesothelin-directed CAR T-cells in an in vivo model of highly resistant pancreatic adenocarcinoma incorporating AsPC1 tumor cells expressing endogenous mesothelin (fig. S7F). In this study, only PRDM1/NR4A3 double knockout anti-mesothelin CAR T-cells mediated a significant reduction in tumor burden over time (P < 0.05, fig. S7G).
Additionally, in association with robust antitumor activity against NALM-6 B-ALL (fig. S7H), we found that PRDM1/NR4A3 KO enhanced CD19 CAR T-cell expansion compared to AAVS1 KO CAR T-cells (fig. S7I). We also observed increased central memory T-cell differentiation and reduced proportions of peripheral blood CAR T-cells co-expressing multiple inhibitory receptors as compared to control CAR T-cells (fig. S7J to L). To evaluate the durability of PRDM1/NR4A3 double KO CAR T-cell therapeutic efficacy, we conducted a study in which CAR T-cell-treated NSG mice were rechallenged with NALM-6 cells 40 days after the initial leukemia cell transfer. Because of the aggressive nature of this model, we administered a relatively high dose of CAR T-cells across all groups during the primary challenge to ensure initial tumor clearance. PRDM1/NR4A3 dual KO CAR T-cells demonstrated better control of tumor growth than control CAR T-cells following rechallenge (Fig. 7L and M). These collective findings suggest that PRDM1 depletion increases CAR T-cell expansion and mitigates dysfunction by increasing frequencies of TCF1+CD8+ T-cells and skewing fate away from the TIM-3+CD8+ state. NR4A3 KO rescues the in vivo potency-enhancing effect of PRDM1 KO on CAR T-cell function by reducing exhaustion and inducing durable effector activity.
Discussion
The full therapeutic potential of CAR T-cell therapies in cancer is often hindered by poor expansion, persistence, and lack of durable effector function of the transferred cellular product. Unfortunately, to date, efficacy in solid tumors has been limited in particular (6–8), and the underpinnings of CAR T-cell intrinsic antitumor potency in these indications have not been elucidated. Our results suggest that cellular and molecular diversity of infused CAR T-cells is a major factor contributing to the variability in proliferative capacity and antitumor activity among patients with metastatic solid tumors. Quantifiable phenotypes associated with pre-infusion PSMA CAR T-cells in mCRPC also shared with CD19 CAR T-cell products in leukemia and lymphoma are potentially actionable by enriching desirable, or eliminating undesirable, cellular populations or functional states during ex vivo manufacturing. Further, our study identifies the mechanisms by which distinct CAR T-cell populations lead to therapeutic potency and offers an engineering strategy for potentiating enrichment of highly functional products to improve efficacy after infusion.
A central finding from our study is that the presence of stem cell-like TCF7-expressing CD8+ and absence of HAVCR2-expressing CD8+ exhausted T-cells are indicative of CAR T-cell expansion and antitumor activity in both non-hematopoietic and hematopoietic cancers, suggesting that the intrinsic fitness of the engineered T-cells, in addition to absolute T-cell number and spatial distribution within a patient’s tumor, is crucial for induction of effective tumor immunity. TCF7-expressing CD8+ T-cells are a distinct CD8+ T-cell population that expands and generates cytotoxic progenitors which are critical for elicitation of robust antitumor function in response to checkpoint blockade therapies (15, 45, 46). Additionally, the TCF7 regulon not only associates with a favorable naïve T-cell state, but is also maintained in effectors among patients with long-term CAR T-cell persistence who experience durable remission (31). This is concordant with a previous report suggesting that TCF7 mediates the persistence of CD8+ effector T-cells as well as their differentiation toward a central memory phenotype (47). Similar cell populations were identified in the CD4+ T-cell compartment, and CD4+ CAR T-cells may be critical for maintaining long-term persistence and antitumor function (48). Examination of larger patient cohorts is needed to validate and build on these molecular determinants of CAR T-cell activity and outcome.
In contrast to TCF7-expressing CD8+ T-cells, we found poorly functional cells expressing exhausted or dysfunctional transcriptomic programs associated with lack of in vivo PSMA CAR T-cell proliferative potency and PSA responses in mCRPC. HAVCR2-expressing CD8+ T-cells upregulated pathways associated with exhaustion and IFN responses, exhibited a lack of persistence, and resulted in poor CAR T-cell clinical efficacy; these cells also expressed a low degree of TCF7, which is predictive of negative outcome in patients treated with checkpoint inhibitors (49). Notably, we discovered that unlike TCF7-expressing CD8+ T-cells, HAVCR2-expressing CD8+ T-cells express a high abundance of PRDM1 that has been reported to negatively regulate TCF7 and mediate terminal T-cell differentiation and exhaustion (32, 33). We demonstrate that CRISPR/Cas9-mediated knockout of PRDM1 not only successfully depletes HAVCR2-expressing CD8+ T-cell and increases TCF7-expressing CD8+ gene signatures during manufacturing, but also substantially enhances in vivo CAR T-cell proliferation and increased frequencies of TIM-3−TCF1+CD8+ peripheral blood and tumor-infiltrating CAR T-cells.
Although single deletion of PRDM1 favorably modulates CAR T-cell differentiation, antitumor effector activity is ultimately hampered by this modification in the setting of chronic antigen exposure. PRDM1 regulates CD8+ T-cell effector function and is required for granzyme B expression (32, 33). In acute LCMV infection, despite a reduction in cytotoxic molecule production, the cytolytic activity of virus-specific PRDM1 KO CD8+ T-cells was marginally affected and both wild type and PRDM1 KO CD8+ T-cells successfully cleared the infection (32). However, in chronic LCMV infection, the killing activity of LCMV-directed PRDM1 KO CD8+ T-cells is impaired, suggesting that loss of PRDM1 can profoundly compromise cytotoxicity in the context of exhaustion where the cytolytic potential of antigen-specific CD8+ T-cells is relatively low (33). Consistent with these previous studies, the cytotoxic capacity and effector cytokine production of PRDM1 KO CAR T-cells were substantially compromised compared to conventional CAR T-cells after multiple rounds of tumor antigen exposure. In accordance with these findings, despite a profound increase in expansion capacity, PRDM1 KO CAR T-cells minimally enhanced antitumor activity over control CAR T-cells in xenogeneic murine models of prostate cancer and aggressive B-cell leukemia, and ultimately became progressively dysfunctional due to exacerbated loss of effector activity. The marginally enhanced tumor control observed with this single modification may be attributed to the large increase in the number of PRDM1 KO CAR T-cells in vivo, despite upregulation of exhaustion pathways and concomitantly diminished effector function.
Until now, PRDM1 was presumed to induce exhaustion modules in antigen-specific T-cells (33). Detection of lower frequencies of peripheral blood PRDM1 KO PSMA CAR T-cells, but not CAR TILs co-expressing multiple inhibitory receptors, suggests that reduction of inhibitory receptor expression in PRDM1-deficient CAR T-cells was insufficient to promote effective antitumor immunity. We therefore examined whether other transcription factors may regulate the exhaustion module and compensate for the absence of PRDM1. We discovered that PRDM1 KO CAR T-cells chronically challenged with tumor targets display upregulation of a battery of exhaustion-associated transcription factors, including those belonging to the NR4A and TOX families which are reported to drive a cell-intrinsic program of T-cell hyporesponsiveness (50). These findings provide evidence that in the setting of PRDM1 deletion, expression of certain inhibitory receptors and genes encoding exhaustion-related transcription factors are decoupled and downregulation of specific inhibitory receptors may instead be reflective of changes to the activation or differentiation state of CAR T-cells.
A recent study also analyzed PRDM1 KO alone on antitumor T-cell responses (23). Interestingly, there are key differences between some of our findings and those presented in this report. While we build on the observation that TCF7 is a critical determinant of CAR T-cell memory differentiation in the context of PRDM1 deficiency using TCF7 KO experiments, the progressive suppression of robust antitumor effector function mediated by PRDM1 KO T-cells was not previously reported. Yoshikawa and colleagues (23) speculated that upregulation of TOX resulting from PRDM1 deletion may diminish the antitumor response through upregulation of inhibitory receptors. Notably, we provide a functionally validated mechanism by which PRDM1 KO regulates a battery of genes encoding exhaustion-related transcription factors, particularly NR4A3 not reported in (23), which together regulate CAR T-cell phenotype and effector activity. The decoupling of exhaustion-related transcription factor upregulation and inhibitory receptor expression observed in our study and not by Yoshikawa et al. may be attributed to several reasons. These discrepancies may include differences in the PRDM1 KO CAR- and TCR-transgenic T-cells used and the transfer of a fewer number of engineered T-cells in our study. Nevertheless, our findings underscore the need to balance a reciprocal upregulation of NR4A3 and NFAT signaling in the setting of PRDM1 deficiency to generate effective CAR T-cell-mediated responses in a variety of tumor types. Further, we provide a multiplex gene-editing strategy to potentiate the expansion of clinically relevant populations of stem-like CAR T-cells resistant to exhaustion.
T-cells in cancer and chronic viral infection differentiate into effector or exhausted T-cells, in association with global and site-specific changes to the epigenetic landscape, including transcription factor binding sites (51–53). Indeed, PRDM1 KO increased accessibility of many exhaustion-related chromatin regions (such as NR4A3, TOX, and TOX2) that normally become accessible in CD8+ TILs (54); a number of these open regions colocalized with a BLIMP1 binding motif, suggesting that BLIMP1 may be acting as a transcriptional repressor. Further study is required to investigate whether BLIMP1 directly regulates chromatin accessibility through recruitment of epigenetic regulators such as G9a and Histone Deacetylase 2 (HDAC2) to form a functional complex regulating exhaustion-related transcription factor gene expression, as has been previously described (41). In addition to epigenetic regulation, PRDM1 KO increased calcineurin-NFAT signaling, which is necessary and sufficient to induce exhaustion-related transcription factor expression (43, 44). This increased NFAT signaling may be attributed to prolonged exposure of PRDM1 KO CAR T-cells to cancer cells in association with the delayed killing kinetics we demonstrated. Thus, our findings suggest that PRDM1 can suppress expression of exhaustion-associated transcription factors by directly or indirectly regulating transcriptional and epigenetic reprogramming as well as the killing activity of CAR T-cells.
We recognize potential limitations of our present study. First, the tumors utilized in the mouse models are cell line-derived and may not behave exactly as primary tumors in humans. Further, use of immunocompromised mice engrafted with human cancer cell lines and T-cells falls short of recapitulating the interaction between gene-edited CAR T-cells and the native TME. Taking these limitations into account, the mouse models we used still offer good methods for assessment of CAR T-cell-augmenting regulators, though subsequent direct testing in human patients will be critical. Second, genotoxicity is a theoretical concern with dual KO of PRDM1 and NR4A3. However, we recently reported results from a clinical trial where we edited three genes at separate loci with no evidence of genotoxicity (55). Finally, for clinical applications, it is important to recognize that if elimination of the PRDM1/NR4A3 double-edited cells is compromised, a situation whereby a very large number of T-cells accumulate that are marginally less effective at tumor cell killing may considerably increase the risk of toxicity. This can be addressed in dose-escalation studies using a more conservative treatment regimen as compared to the initial doses evaluated in our prior phase I study (22). Given these uncertainties, initial clinical trials of this strategy should test PRDM1/NR4A3 dual KO CAR T-cell products at comparably lower concentrations, prior to incorporation of lymphodepletion, which we anticipated to enhance T-cell proliferation, antitumor activity and treatment-related toxicity.
In summary, this study provides a framework for developing the next-generation of CAR T-cells by identifying cellular and molecular characteristics associated with clinical response and engineering CAR T-cells to favor enrichment of desirable cell populations, as well as depletion of undesirable cell populations, in infusion products. Our work also highlights the fundamental role of PRDM1 in the regulation of human T-cell memory and provides a deeper understanding of how PRDM1 mediates T-cell exhaustion (fig. S8). Additional experiments will be required to completely reveal the transcriptional networks involved. From a therapeutic standpoint, however, combined disruption of PRDM1 and NR4A3 expression or activity has the direct potential to generate genetically reprogrammed T-cells capable of augmenting both immediate and long-term antitumor responses, since this strategy potentiates the formation of long-lived memory cells resistant to exhaustion imposed by tumors.
Materials and Methods
Study design
The objective of this study was to define the functional effects of PRDM1 and NR4A3 KO in CAR T-cells. The number of biological replicates (referring to T cells from different healthy donors), type of statistical used and P values are reported in the figure legends. No statistical methods were used to pre-determine sample size. These were estimated based on preliminary experiments. All in vitro functional experiments in the main text were carried out at least three times and no outliers or other data points were excluded from our analyses. For animal experiments, NSG mice were treated with CAR T cells from one to two different healthy donors. Cages of mice were randomly assigned to treatment groups. Investigators were not blinded to group allocation during experiments and assessment of outcome.
Infusion product samples were acquired from patients with mCRPC who enrolled in a completed clinical trial of single-agent PSMA CAR T-cell therapy, which was approved by the Institutional Review Board (IRB) of the University of Pennsylvania. All ethical regulations were followed. This study is registered at ClinicalTrials.gov (NCT03089203) (22). Correlative analyses presented in this report are exploratory, given the post-hoc nature of the investigation. Peripheral blood mononuclear cells (PBMCs) were collected for small-scale CAR T-cell production by leukapheresis from healthy individuals. Study participants provided written informed consent according to the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice. Correlative assays conducted on patient samples, such as quantitative polymerase chain reaction (qPCR) for CAR T-cell quantification, were carried out according to our previously published methods (20). Antitumor responses, as measured by maximum decline in PSA from baseline for the patients whose CAR T-cell infusion products were analyzed in this study, were previously reported (22).
Cell lines
PC3, PSMA-expressing PC3, and NALM-6 cells engineered to express click beetle green luciferase and green fluorescent protein (CBG-GFP), were kindly provided by Carl H. June and Marco Ruella (University of Pennsylvania), respectively. AsPC1 cells were obtained from the American Type Culture Collection (ATCC). PC3 and PC3-PSMA cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetus bovine serum (FBS) and streptomycin/penicillin. NALM-6 and AsPC1 cells were maintained in Roswell Park Memorial Institute (RPMI)-1640 media supplemented with 10% FBS and streptomycin/penicillin (R10 media). HEK 293T cells, used for lentivirus production, were obtained from ATCC and cultured in R10 media. Low-passage banks of cells were tested for mycoplasma with a MycoAlert kit (Lonza), according to the manufacturer’s instructions. Authentication of cell lines was carried out by the University of Arizona Genetics Core, based on criteria established by the International Cell Line Authentication Committee. Short-tandem-repeat profiling revealed that these cell lines were above the 80% match threshold. Mycoplasma testing and authentication were routinely performed before and after molecular engineering.
Lentivirus production
Vector construction and lentiviral production were conducted as previously described (56). In brief, CARs comprised of anti-PSMA (56), anti-CD19 (57), or anti-Mesothelin (58) single-chain variable fragments (scFv) fused to 4-1BB and CD3ζ stimulatory endodomains were subcloned into the pTRPE vector. Lentivirus supernatant was collected from 293T cells transfected with the pTRPE transfer vector and packaging plasmids using Lipofectamine 2000 (Thermo Fisher Scientific) and concentrated using ultracentrifugation.
Lentiviral transduction and T-cell culture
Healthy donor T-cells were isolated from PBMCs using the Pan T Cell Isolation Kit according to the manufacturer’s instructions (Miltenyi Biotec). T-cells were activated with anti-CD3/CD28 antibody coated Dynabeads (Thermo Fisher Scientific) at 3:1 bead:cell ratio in T-cell media (OpTmizer CTS SFM media (Gibco) supplemented with 5% human AB serum and 100u/mL human IL-2). Following a 24-hour incubation, lentivirus encoding the PSMA CAR was added to the culture at a multiplicity of infection (MOI) of 2.5. CAR T-cell expansion was carried out as previously described (56).
CRISPR/Cas9-mediated gene editing
Following T-cell activation as described above, beads were removed using a magnet on day 3 and electroporation was carried out with a P3 primary cell 4D-nucleofector kit (Lonza). 2 × 106 CAR T-cells were transfected with 12 μg TrueCut S. pyogenes Cas9 (Invitrogen) and 0.2nmol of chemically-modified tracrRNA and crRNA (Integrated DNA Technologies). Following electroporation, CAR T-cells were cultured in T-cell media. The crRNA sequences used in this study were: AAVS1: 5’-CCATCGTAAGCAAACCTTAG-3’, PRDM1: 5’-CATCAGCACCAGAATCCCAG-3’, TCF7: 5’-TCAGGGAGTAGAAGCCAGAG-3’, NR4A3: 5’-CCTTGGCAGCACTGAGATCA-3’. The frequency of targeted mutations generated by double strand break were determined by Sanger sequencing and subsequent TIDE (tracking of indels by decomposition) analysis. Primers used for targeted amplification were: PRDM1-F1: tctcagaaggagccacaggaacgg, PRDM1-R1: cacccaccctatgctgcaagttgc, NR4A3-F1: gaggagaggatgacacttcctctctgtttc, NR4A3-R1: ctgcccagcacctccatgtacttcaagcag. Western blot and flow cytometric analysis were conducted to confirm KO at the protein level.
Western blot analysis
T-cells (1 × 106 cells) were suspended in a low-salt lysis buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM PMSF, 2 μg/ml aprotinin, 2 μg/ml leupeptin) and allowed to swell on ice for 30 minutes. After centrifugation (1000 × g), supernatants obtained from cells lysates (30 μg) were analyzed by 10% SDS-polyacrylamide gel electrophoresis and electrophoretically transferred to PVDF membranes (Millipore). The membranes were washed with phosphate-buffered saline (PBS) containing 0.1% Tween 20 (PBST), and then blocked for 1-hour in 5% skim milk in PBST. After washing with PBST, membranes were incubated overnight with one of the following antibodies: Mouse monoclonal anti-β-Tubulin (Sigma-Aldrich #T8318; 1:100) or rabbit monoclonal anti-BLIMP1 (Cell Signaling #9115; 1:100) or mouse monoclonal anti-NR4A3 (Sigma-Aldrich #SAB1404566; 1:100). Membranes were washed with PBST and treated with 1:1000 diluted horseradish peroxidase-coupled goat anti-mouse or anti-rabbit secondary antibodies (Thermo Fisher Scientific) in PBST for 1-hour. After washing, the membranes were incubated in Pierce ECL western blotting substrate (Thermo Fisher Scientific) and visualized on X-ray film.
Flow cytometry
Cell surface anti-human antibodies were diluted in FACS buffer (PBS + 2% FBS). PSMA CAR expression was measured using an allophycocyanin (APC)-conjugated recombinant human PSMA protein (Sino Biological). T-cell immunophenotyping was carried out using the following antibodies: PD-1-Brilliant Violet (BV) 421 (BioLegend #329920; 1 μL/100 μL), CD45-BV570 (BioLegend #304226; 1 μL/100 μL), CD8-BV650 (BioLegend #301042; 1 μL/100 μL), CD8-APC-H7 (BD Biosciences #560179; 1 μL/100 μL), CD4-BV785 (BioLegend #317442; 1 μL/100 μL), TIM-3-phycoerythrin (PE) (BioLegend #345006; 1 μL/100 μL), CCR7-PE-CF594 (BD Biosciences #562381; 1 μL/100 μL), CD62L-PE-Cy5 (BioLegend #304808; 1 μL/100 μL), LAG3-PE-Cy7(eBioscience #25-2239-42; 1 μL/100 μL), hCD45-APC (BD Biosciences #340943; 1 μL/100 μL), murine CD45-Peridinin chlorophyll protein (PerCP)-Cy5.5 (BioLegend #103132; 1 μL/100 μL), CD127-BV570 (BioLegend #351307; 1 μL/100 μL), human leukocyte antigen (HLA)-DR-Alexa Fluor 700 (BioLegend #307625; 1 μL/100 μL), CD25-APC (eBioscience #17-0259-42; 1 μL/100 μL). For intracellular staining, cells were first permeabilized and washed using the FoxP3 Transcription Factor Staining Buffer Kit (eBioscience) and subsequently stained with following antibodies: IL2-PE-CF594 (BD Biosciences #562384; 3 μL/100 μL), IFN-γ-BV570 (BioLegend #502534; 3 μL/100 μL), TNF-α-Alexa Fluor700 (BioLegend #502928; 3 μL/100 μL), Perforin-BV421 (BioLegend # 353307; 2 μL/100 μL), Perforin-APC (BioLegend #308112; 2 μL/100 μL), Granzyme B-PE-Cy5.5 (Invitrogen #GRB18; 0.04 μL/100 μL), TCF1-Alexa Fluor 488 (Cell Signaling Technology #6444S; 1 μL/100 μL), TOX-APC (Miltenyi Biotech #130-107-785; 3 μL/100 μL), NFATC1-PE (BioLegend #649606; 3 μL/100 μL) according to our previously published methods (20). Samples were then analyzed using an LSRFortessa (BD Biosciences), FlowJo software (FlowJo, LLC) or FCS Express (De Novo Software).
CAR T-cell restimulation assay
CAR T-cell expansion capacity and effector function were assessed using a restimulation assay, as previously described (20, 56, 59). Briefly, AAVS1 KO, PRDM1 KO, TCF7 KO, NR4A3 KO, PRDM1 + TCF7 dKO, PRDM1 + NR4A3 dKO PSMA CAR-positive T-cells were isolated using a Biotin-goat anti-mouse IgG F(ab)2 fragment (Jackson ImmunoResearch #115-065-072) and anti-biotin kits (Miltenyi Biotech). Following isolation, CAR T-cell purity was confirmed by flow cytometric analysis and CAR T-cells were then serially exposed (every 2 to 5 days) to irradiated PC3-PSMA cells at an effector-to-target (E:T) ratio of 3:1 or 1:1. Restimulation assays were carried out for 25 to 30 days, since cell counts become largely unreliable beyond the fifth and sixth rounds of stimulation due to senescent proliferative arrest and a marked decrease in the viability of chronically stimulated CAR T-cells. Supernatants were collected 24-hours post-tumor challenge for cytokine analysis using the LEGENDplex human CD8 panel (BioLegend), and absolute numbers of T-cells in culture were monitored using a Luna automated cell counter (Logos Biosystems) during the assay.
Cytotoxicity assay
The longitudinal killing capacity of engineered CAR T-cells against PC3-PSMA cells was assessed using the xCELLigence system (ACEA Biosciences Inc.). CAR-expressing T-cells were magnetically enriched prior to the cytotoxicity assay. 2 × 104 PC3-PSMA cells were seeded in E-Plate VIEW 96 PET microwell plates. After 24-hours, PSMA CAR T-cells or control (untransduced) T-cells were added to wells containing tumor targets to achieve the desired E:T ratios. 20% Tween 20 was added to separate wells consisting of PC3-PSMA cells alone as a full lysis control. Electrical impedance was monitored in 20-minute intervals over 7 days and cytotoxicity was assessed by calculating the normalized cell index and % cytolysis.
Quantitative real-time PCR (qRT-PCR)
After 2 to 4 rounds of restimulation, CD8+ CAR T-cells were isolated from culture using CD8 microbeads (Miltenyi Biotec) for qRT-PCR. First, total RNA was extracted from CD8+ CAR T-cells using RNA Clean & Concentrator kits (Zymo Research). cDNA was then synthesized using the PrimeScript 1st strand cDNA Synthesis Kit (Takara Bio) per the manufacturer’s protocol and qRT-PCR was conducted using Applied Biosystems TaqMan Fast Advanced Master Mix (Thermo Fisher Scientific) on the QuantStudio3 (Applied Biosystems). The primer/probe sets used for these experiments were as follows: CD3E: Hs01062241_m1, NR4A2: Hs00428691_m1 and NR4A3: Hs00545009_g1 (Thermo Fisher Scientific).
Mouse xenograft studies
Mouse studies were performed with 6- to 8-week-old male NOD/SCID/IL-2Rγ-null (NSG) mice in compliance with a University of Pennsylvania Institutional Animal Care and Use Committee approved protocol. For the subcutaneous PC3-PSMA model, 106 or 5 × 106 PC3-PSMA-CBG-GFP tumor cells were premixed with Matrigel (Corning) and injected into the flanks of NSG mice. When the average tumor size reached 150 to 200 mm3 or 500 mm3, 3.5 × 105 PSMA CAR T-cells were injected intravenously. Tumor growth weekly monitored weekly by taking caliper measurements (tumor volume = (length × width2)/2). Animals were euthanized when tumor volume exceeded 1500 mm3 or 2 cm in diameter. For the intraosseous PC3-PSMA model, male NSG mice were intrafemorally transplanted with 2 × 105 PC3-PSMA cells. On day 27, 1 to 2 × 105 PSMA CAR T-cells were infused intravenously, and tumor burden was measured by injecting mice intraperitoneally with luciferin and quantifying bioluminescence using an IVIS Spectrum In Vivo Imaging System (PerkinElmer). To characterize CAR T-cell phenotype and function, peripheral blood and tumor tissues were isolated from mice. Blood samples were obtained at peak CAR T-cell expansion by cheek bleeding. Tumors isolated on day 13 post-CAR T-cell injection were minced with a scalpel and treated with 100 U/mL collagenase IV and 0.25mg/mL DNase I for 1 hour at 37°C. The absolute number of human T-cells in the peripheral blood and tumors was quantified using 123count eBeads (Thermo Fisher Scientific). To assess the effector function of TILs, these ex vivo isolated T-cells were stimulated with 50 ng/mL phorbol 12-myristate 13-acetate (PMA) and 1mg/mL ionomycin in the presence of 5 ug/mL Brefeldin A for 6-hours and expression of IL-2, IFN-γ and TNF-α were assessed by intracellular staining. We used PMA/ionomycin stimulation to assess TIL effector function to eliminate potential confounding variables associated with antigen receptor expression or signaling.
To examine the antitumor efficacy of CAR T-cells in pancreatic cancer, 4 × 106 AsPC1 cells were premixed with Matrigel and subcutaneously injected into NSG mice. On day 30, when tumor volume reached 300 to 400 mm3, mesothelin-directed CAR T-cells were intravenously injected, and tumor growth was monitored using caliper measurements. For the B-cell acute lymphoblastic leukemia model, NSG mice were intravenously injected with 1 × 106 NALM-6-CBG cells. On day 7 post-tumor injection, 3 × 105 CD19 CAR T-cells were intravenously infused, and tumor growth was assessed twice weekly using bioluminescent imaging (20). Peripheral blood was isolated on day 24 and flow cytometric T-cell immunophenotyping was carried out.
To assess the memory recall response of CAR T-cells, NSG mice were engrafted with 105 NALM-6-CBG cells. On day 6 post-tumor injection, 2 × 106 CD19 CAR T-cells were intravenously infused (n = 9 to 10 mice per group). The mice that had exhibited initial control of NALM-6 cells and survived were rechallenged with 2 × 106 NALM-6 cells on day 40. Tumor growth was monitored using bioluminescence imaging, as described above.
Single-cell RNA-seq
PSMA CAR T-cell infusion products from 5 different patients were thawed in a 37°C water bath and dead cells were eliminated using the Dead Cell Removal Kit (Miltenyi Biotec). PSMA CAR-positive T-cells were enriched by magnetic cell separation using a Biotin-goat anti-mouse IgG F(ab)2 fragment (Jackson ImmunoResearch #115-065-072) and anti-biotin kits (Miltenyi Biotech). scRNA-seq libraries were prepared using Chromium Single Cell 3’ v3.1 Reagent Kits (10X Genomics) following the manufacturer’s instructions. Isolated CAR T-cells were washed and resuspended in PBS containing 0.04% BSA and about 20,000 cells were loaded per reaction to capture about 10,000 cells. Sequencing was performed on a Novaseq 6000 (Illumina) at a depth of at least 20,000 reads per cell.
Reads were aligned to the human reference genome (GRCh38) using Cell Ranger version 6.0.0. Subsequent quality control and downstream analyses were performed using Seurat 4.0. Cells were filtered based on the following criteria to eliminate low-quality cells: 1) a minimum of 1000 genes and a maximum of 6000 genes detected per cell; and 2) less than 15% of mitochondrial gene counts. After quality control, 20,702 cells remained. Gene-cell matrices from five CAR T-cell products were then integrated using SelectIntegrationFeatures, PrepSCTIntegration, FindIntegrationAnchors, and IntegrateData functions in Seurat. CD4-positive and CD8-positive cells were clustered using FindNeighbors/FindClusters, followed by differential gene expression analysis using the FindClusters command. Module scores were then calculated using AddModuleScore to assess the enrichment of gene signatures in CD4 and CD8 subclusters.
Bulk RNA-seq
On days 0, 5 and 20 of the CAR T-cell restimulation assay, before stimulation and after the first and fourth tumor challenge respectively, CD8+ CAR T-cells were isolated using CD8 microbeads (Miltenyi Biotec) and total mRNA was extracted with RNA Clean & Concentrator kits (Zymo Research). Bulk RNA-seq was conducted by Novogene using the Novaseq6000 system (paired-end 150bp) at 40 × 106 reads per sample. Reads were pseudoaligned to the human genome (GRCh38) transcriptomes using kallisto v0.46.0.
Differential expression analyses between AAVS1 KO PSMA CAR T-cells and PRDM1 KO PSMA CAR T-cells were performed using the edgeR v3.34.0 and limma v3.48.0 packages. Briefly, expression data were normalized using a trimmed mean of log expression ratios method and transformed into log2(counts per million). Linear models were used to assess differential expression, and P values were adjusted using the Benjamini-Hochberg method. Gene set enrichment analysis was conducted using GSEABase v1.54.0, clusterProfiler v4.0.2, and msigdbr v7.4.1.
ATAC-seq analysis
After CAR T-cell manufacturing, dead cells were eliminated using the Dead Cell Removal Kit (Miltenyi Biotec) and CD8+ CAR T-cells were isolated using CD8 microbeads (Miltenyi Biotec). 100,000 cells per sample were cryopreserved. Nuclei were isolated from CD8+ T-cells for each replicate, followed by the transposition reaction in the presence of Tn5 transposase (Illumina, Inc.) for 45 minutes at 37°C. Purification of transposed DNA was then completed with the MinElute Kit (Qiagen) and fragments were barcoded with dual indexes (Illumina Nextra). Library preparation and sequencing were performed by Novogene using NovaSeq 6000 (paired-end 150bp reads) at a depth of 30 × 106 reads per sample.
FASTQ files for each sample were trimmed of adapter contamination using cutadapt (https://github.com/marcelm/cutadapt/). They were aligned to the hg19 reference genome using Bowtie2, restricting to properly aligned and properly paired reads between 10 and 1000 base pairs. Mitochondrial reads were removed (https://github.com/jsh58/harvard/blob/master/removeChrom.py). Files were sorted using samtools, and PCR duplicates were removed using Picard. BAM files were indexed using samtools in order to visualize tracks in IGV. Peak calling was performed with MACS2 with a false discovery rate (FDR) q-value of 0.01. The R package Diffbind was used to remove ENCODE blacklisted regions (https://sites.google.com/site/anshulkundaje/projects/blacklists), then to identify peaks differentially opened between the control and the PRDM1 knockout. The findMotifsGenome script from HOMER was used to map the hg19 genome for occurrences of the BLIMP1 motif (derived from ENCODE data accessible by GEO at GSE31477) and the NFAT2 motif (60).
Statistical analyses
Raw, individual-level data are presented in data file S1. Normality analyses were conducted using Shapiro-Wilk test and D’Agostino & Pearson tests. Pairwise comparisons were performed using the Mann Whitney U test and Student’s t-test as appropriate. For comparisons of three or more groups, one-way ANOVA with Tukey’s multiple comparisons test and Kruskal-Wallis test with a post-hoc Dunn’s multiple comparison test were used. Mouse survival was assessed using the Gehan-Breslow-Wilcoxon test. Statistical tests were performed in Prism 9 (GraphPad Software) and P values <0.05 were considered significant.
Supplementary Material
Acknowledgements
The authors thank Carl June and Steven Albelda (University of Pennsylvania) for critical review of the manuscript. We acknowledge the Human Immunology Core at the University of Pennsylvania for providing leukocytes and the Hospital of the University of Pennsylvania Apheresis Unit for peripheral blood mononuclear cell collections. We would also like to recognize the contributors who supported development and execution of the clinical trial from which clinical samples were obtained: Whitney Gladney, Anne Chew, Diane Frazee, Mary Truran, Elizabeth Veloso, Holly McConville, Jonathan Aguedelo, Samantha Hower, Sophia Ngo, Amy Marshall, Joan Gilmore, Lester Lledo, Karen Dengel, Vanessa Gonzalez, Irina Kulikovskaya, Fang Chen, Lifeng Tian, the Clinical Cell and Vaccine Production Facility (CVPF), the Translational and Correlative Studies Laboratory (TCSL), and the Product Development Lab (PDL) from the University of Pennsylvania Center for Cellular Immunotherapies (CCI) are thanked.
Funding
This work was supported by a Prostate Cancer Foundation Challenge Award (to N.H.), an Alliance for Cancer Gene Therapy Investigator Award in Cell and Gene Therapy for Cancer (to J.A.F. and N.H.), a Prostate Cancer Foundation Young Investigator Award (to V.N.), U54 CA244711 and P01 CA214278 (to M.M.D. and J.A.F.), U01 AG066100/Samuel Waxman Cancer Research Foundation Grant (to J.A.F.) and ACC P30 Core Grant P30 CA016520-42 (to S.F.L., N.H. and J.A.F.).
Competing Interests
I.Y.J., S.F.L., M.M.D, D.L.S. and J.A.F. have filed patent applications in the field of T-cell therapy for cancer and have received royalties. Patents related to this work are as follows: Biomarkers and CAR T-cell therapies with enhanced efficacy (US20200087376A1) and CAR T-cell therapies with enhanced efficacy (US20210047405A1). M.M.D. and J.A.F. have received research funding from Tmunity Therapeutics. M.M.D. serves on the Scientific Advisory Board for Cellares Corporation. S.F.L. has served as a consultant for Novartis Pharmaceuticals, Kite Pharma, and Wugen and receives clinical trial funding from Novartis Pharmaceuticals. J.A.F. is a member of the Scientific Advisory Boards of Cartography Bio. and Shennon Biotechnologies Inc.
Data and Materials Availability
All data associated with this study are in the paper or supplementary materials. Raw and analyzed sequencing datasets have been deposited to Gene Expression Omnibus (GEO) under the accession number GSE210265. Additional requests for raw and analyzed data or materials should be made to the PENN Center for Innovation (pciinfo@pci.upenn.edu) and will be promptly reviewed to determine whether the application is subject to any intellectual property or confidentiality requirements. Patient-related information not included in this report were collected as part of a clinical trial and might be subject to patient confidentiality. Any data and materials that can be shared will be released following execution of a material transfer agreement.
References
- 1.Porter DL, Hwang W-T, Frey NV, Lacey SF, Shaw PA, Loren AW, Bagg A, Marcucci KT, Shen A, Gonzalez V, Ambrose D, Grupp SA, Chew A, Zheng Z, Milone MC, Levine BL, Melenhorst JJ, June CH, Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Science Translational Medicine 7, 303ra139–303ra139 (2015); published online EpubSeptember 2, 2015 ( 10.1126/scitranslmed.aac5415). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, Timmerman JM, Stiff PJ, Friedberg JW, Flinn IW, Goy A, Hill BT, Smith MR, Deol A, Farooq U, McSweeney P, Munoz J, Avivi I, Castro JE, Westin JR, Chavez JC, Ghobadi A, Komanduri KV, Levy R, Jacobsen ED, Witzig TE, Reagan P, Bot A, Rossi J, Navale L, Jiang Y, Aycock J, Elias M, Chang D, Wiezorek J, Go WY, Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. The New England Journal of Medicine 377, 2531–2544 (2017); published online Epub2017-12-28 ( 10.1056/NEJMoa1707447). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, Qayed M, De Moerloose B, Hiramatsu H, Schlis K, Davis KL, Martin PL, Nemecek ER, Yanik GA, Peters C, Baruchel A, Boissel N, Mechinaud F, Balduzzi A, Krueger J, June CH, Levine BL, Wood P, Taran T, Leung M, Mueller KT, Zhang Y, Sen K, Lebwohl D, Pulsipher MA, Grupp SA, Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. The New England Journal of Medicine 378, 439–448 (2018); published online Epub2018-02-01 ( 10.1056/NEJMoa1709866). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, Liedtke M, Rosenblatt J, Maus MV, Turka A, Lam LP, Morgan RA, Friedman K, Massaro M, Wang J, Russotti G, Yang Z, Campbell T, Hege K, Petrocca F, Quigley MT, Munshi N, Kochenderfer JN, Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N Engl J Med 380, 1726–1737 (2019); published online EpubMay 2 ( 10.1056/NEJMoa1817226). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, Vogl DT, Weiss BM, Dengel K, Nelson A, Plesa G, Chen F, Davis MM, Hwang WT, Young RM, Brogdon JL, Isaacs R, Pruteanu-Malinici I, Siegel DL, Levine BL, June CH, Milone MC, B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest 129, 2210–2221 (2019); published online EpubMar 21 ( 10.1172/JCI126397). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Long KB, Young RM, Boesteanu AC, Davis MM, Melenhorst JJ, Lacey SF, DeGaramo DA, Levine BL, Fraietta JA, CAR T Cell Therapy of Non-hematopoietic Malignancies: Detours on the Road to Clinical Success. Front Immunol 9, 2740 (2018) 10.3389/fimmu.2018.02740). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yong CSM, Dardalhon V, Devaud C, Taylor N, Darcy PK, Kershaw MH, CAR T-cell therapy of solid tumors. Immunology and cell biology 95, 356–363 (2017); published online EpubApr ( 10.1038/icb.2016.128). [DOI] [PubMed] [Google Scholar]
- 8.Newick K, O’Brien S, Moon E, Albelda SM, CAR T Cell Therapy for Solid Tumors. Annual review of medicine 68, 139–152 (2017); published online EpubJan 14 ( 10.1146/annurev-med-062315-120245). [DOI] [PubMed] [Google Scholar]
- 9.Terakura S, Yamamoto TN, Gardner RA, Turtle CJ, Jensen MC, Riddell SR, Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood 119, 72–82 (2012); published online EpubJan 5 ( 10.1182/blood-2011-07-366419). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, Riddell SR, Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 30, 492–500 (2016); published online EpubFeb ( 10.1038/leu.2015.247). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, Wang E, Douek DC, Price DA, June CH, Marincola FM, Roederer M, Restifo NP, A human memory T cell subset with stem cell-like properties. Nat Med 17, 1290–1297 (2011); published online EpubSep 18 ( 10.1038/nm.2446). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, Boesteanu AC, Wang Y, O’Connor RS, Hwang WT, Pequignot E, Ambrose DE, Zhang C, Wilcox N, Bedoya F, Dorfmeier C, Chen F, Tian L, Parakandi H, Gupta M, Young RM, Johnson FB, Kulikovskaya I, Liu L, Xu J, Kassim SH, Davis MM, Levine BL, Frey NV, Siegel DL, Huang AC, Wherry EJ, Bitter H, Brogdon JL, Porter DL, June CH, Melenhorst JJ, Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 24, 563–571 (2018); published online EpubMay ( 10.1038/s41591-018-0010-1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh N, Perazzelli J, Grupp SA, Barrett DM, Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci Transl Med 8, 320ra323 (2016); published online EpubJan 6 ( 10.1126/scitranslmed.aad5222). [DOI] [PubMed] [Google Scholar]
- 14.Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, Johnson J, Staupe RP, Bengsch B, Xu C, Yu S, Kurachi M, Herati RS, Vella LA, Baxter AE, Wu JE, Khan O, Beltra J-C, Giles JR, Stelekati E, McLane LM, Lau CW, Yang X, Berger SL, Vahedi G, Ji H, Wherry EJ, TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity 51, 840–855.e845 (2019); published online Epub2019-11-19 ( 10.1016/j.immuni.2019.09.013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, Pawlak M, Dionne D, Xia J, Rozenblatt-Rosen O, Kuchroo VK, Regev A, Anderson AC, Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1-CD8+ Tumor-Infiltrating T Cells. Immunity 50, 181–194.e186 (2019); published online Epub2019-01-15 ( 10.1016/j.immuni.2018.11.014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R, Viral Persistence Alters CD8 T-Cell Immunodominance and Tissue Distribution and Results in Distinct Stages of Functional Impairment. Journal of Virology 77, 4911–4927 (2003); published online Epub2003-4 ( 10.1128/JVI.77.8.4911-4927.2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finney OC, Brakke HM, Rawlings-Rhea S, Hicks R, Doolittle D, Lopez M, Futrell RB, Orentas RJ, Li D, Gardner RA, Jensen MC, CD19 CAR T cell product and disease attributes predict leukemia remission durability. J Clin Invest 130, (2019); published online EpubMar 12 ( 10.1172/JCI125423). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blaeschke F, Stenger D, Kaeuferle T, Willier S, Lotfi R, Kaiser AD, Assenmacher M, Doring M, Feucht J, Feuchtinger T, Induction of a central memory and stem cell memory phenotype in functionally active CD4(+) and CD8(+) CAR T cells produced in an automated good manufacturing practice system for the treatment of CD19(+) acute lymphoblastic leukemia. Cancer immunology, immunotherapy : CII 67, 1053–1066 (2018); published online EpubJul ( 10.1007/s00262-018-2155-7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, Kaplan RN, Patterson GH, Fry TJ, Orentas RJ, Mackall CL, 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nature Medicine 21, 581–590 (2015); published online Epub2015/06/01 ( 10.1038/nm.3838). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, Boesteanu AC, Wang Y, O’Connor RS, Hwang W-T, Pequignot E, Ambrose DE, Zhang C, Wilcox N, Bedoya F, Dorfmeier C, Chen F, Tian L, Parakandi H, Gupta M, Young RM, Johnson FB, Kulikovskaya I, Liu L, Xu J, Kassim SH, Davis MM, Levine BL, Frey NV, Siegel DL, Huang AC, Wherry EJ, Bitter H, Brogdon JL, Porter DL, June CH, Melenhorst JJ, Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nature Medicine 24, 563–571 (2018); published online Epub2018/05/01 ( 10.1038/s41591-018-0010-1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng Q, Han G, Puebla-Osorio N, Ma MCJ, Strati P, Chasen B, Dai E, Dang M, Jain N, Yang H, Wang Y, Zhang S, Wang R, Chen R, Showell J, Ghosh S, Patchva S, Zhang Q, Sun R, Hagemeister F, Fayad L, Samaniego F, Lee HC, Nastoupil LJ, Fowler N, Eric Davis R, Westin J, Neelapu SS, Wang L, Green MR, Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nature Medicine 26, 1878–1887 (2020); published online Epub12/2020 ( 10.1038/s41591-020-1061-7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Narayan V, Barber-Rotenberg JS, Jung IY, Lacey SF, Rech AJ, Davis MM, Hwang WT, Lal P, Carpenter EL, Maude SL, Plesa G, Vapiwala N, Chew A, Moniak M, Sebro RA, Farwell MD, Marshall A, Gilmore J, Lledo L, Dengel K, Church SE, Hether TD, Xu J, Gohil M, Buckingham TH, Yee SS, Gonzalez VE, Kulikovskaya I, Chen F, Tian L, Tien K, Gladney W, Nobles CL, Raymond HE, Prostate I Cancer Cellular Therapy Program, Hexner EO, Siegel DL, Bushman FD, June CH, Fraietta JA, Haas NB, PSMA-targeting TGFbeta-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat Med 28, 724–734 (2022); published online EpubApr ( 10.1038/s41591-022-01726-1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshikawa T, Wu Z, Inoue S, Kasuya H, Matsushita H, Takahashi Y, Kuroda H, Hosoda W, Suzuki S, Kagoya Y, Genetic ablation of PRDM1 in antitumor T cells enhances therapeutic efficacy of adoptive immunotherapy. Blood, (2021); published online EpubDec 3 ( 10.1182/blood.2021012714). [DOI] [PubMed] [Google Scholar]
- 24.Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, Callahan DJ, Sun Z, Sun T, Tabib T, Pennathur A, Corry DB, Luketich JD, Lafyatis R, Chen W, Poholek AC, Bruno TC, Workman CJ, Vignali DAA, Adaptive plasticity of IL-10(+) and IL-35(+) Treg cells cooperatively promotes tumor T cell exhaustion. Nat Immunol 20, 724–735 (2019); published online EpubJun ( 10.1038/s41590-019-0346-9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, Jacob J, Calame K, Kaech SM, Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 31, 296–308 (2009); published online EpubAug 21 ( 10.1016/j.immuni.2009.05.014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin H, Blackburn SD, Intlekofer AM, Kao C, Angelosanto JM, Reiner SL, Wherry EJ, A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity 31, 309–320 (2009); published online EpubAug 21 ( 10.1016/j.immuni.2009.06.019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, Nyman J, Marjanovic ND, Kowalczyk MS, Wang C, Kurtulus S, Law T, Etminan Y, Nevin J, Buckley CD, Burkett PR, Buenrostro JD, Rozenblatt-Rosen O, Anderson AC, Regev A, Kuchroo VK, Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature 558, 454–459 (2018); published online Epub6/2018 ( 10.1038/s41586-018-0206-z). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pais Ferreira D, Silva JG, Wyss T, Fuertes Marraco SA, Scarpellino L, Charmoy M, Maas R, Siddiqui I, Tang L, Joyce JA, Delorenzi M, Luther SA, Speiser DE, Held W, Central memory CD8(+) T cells derive from stem-like Tcf7(hi) effector cells in the absence of cytotoxic differentiation. Immunity 53, 985–1000 e1011 (2020); published online EpubNov 17 ( 10.1016/j.immuni.2020.09.005). [DOI] [PubMed] [Google Scholar]
- 29.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, Sharpe AH, Freeman GJ, Germain RN, Nakaya HI, Xue HH, Ahmed R, Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016); published online EpubSep 15 ( 10.1038/nature19330). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, Pradervand S, Thimme R, Zehn D, Held W, T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 45, 415–427 (2016); published online EpubAug 16 ( 10.1016/j.immuni.2016.07.021). [DOI] [PubMed] [Google Scholar]
- 31.Chen GM, Chen C, Das RK, Gao P, Chen C-H, Bandyopadhyay S, Ding Y-Y, Uzun Y, Yu W, Zhu Q, Myers RM, Grupp SA, Barrett DM, Tan K, Integrative Bulk and Single-Cell Profiling of Premanufacture T-cell Populations Reveals Factors Mediating Long-Term Persistence of CAR T-cell Therapy. Cancer Discovery 11, 2186–2199 (2021) 10.1158/2159-8290.Cd-20-1677). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, Jacob J, Calame K, Kaech SM, Blimp-1 promotes terminal differentiation of virus-specific CD8 T cells and represses the acquisition of central memory T cell properties. Immunity 31, 296–308 (2009); published online Epub2009-8-21 ( 10.1016/j.immuni.2009.05.014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin H, Blackburn SD, Intlekofer AM, Kao C, Angelosanto JM, Reiner SL, Wherry EJ, A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity 31, 309–320 (2009); published online Epub2009-08-21 ( 10.1016/j.immuni.2009.06.019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu T, Shin HM, Moseman EA, Ji Y, Huang B, Harly C, Sen JM, Berg LJ, Gattinoni L, McGavern DB, Schwartzberg PL, TCF1 Is Required for the T Follicular Helper Cell Response to Viral Infection. Cell Reports 12, 2099–2110 (2015); published online Epub2015-09-29 ( 10.1016/j.celrep.2015.08.049). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Y, Cao J, Xiong M, Petersen AJ, Dong Y, Tao Y, Huang CT-L, Du Z, Zhang S-C, Engineering Human Stem Cell Lines with Inducible Gene Knockout using CRISPR/Cas9. Cell Stem Cell 17, 233–244 (2015); published online Epub2015-08-06 ( 10.1016/j.stem.2015.06.001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ji Y, Pos Z, Rao M, Klebanoff CA, Yu Z, Sukumar M, Reger RN, Palmer DC, Borman ZA, Muranski P, Wang E, Schrump DS, Marincola FM, Restifo NP, Gattinoni L, Repression of the DNA-binding inhibitor Id3 by Blimp-1 limits CD8+ T cell memory formation. Nature immunology 12, 1230–1237 (2011); published online Epub2011-11-06 ( 10.1038/ni.2153). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, Anderson SM, Handon R, Kenyon E, Elkahloun A, Wu W, Lang PA, Gattinoni L, McGavern DB, Schwartzberg PL, The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Science Immunology 1, eaai8593 (2016); published online Epub2016-12-23 ( 10.1126/sciimmunol.aai8593). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gautam S, Fioravanti J, Zhu W, Le Gall JB, Brohawn P, Lacey NE, Hu J, Hocker JD, Hawk NV, Kapoor V, Telford WG, Gurusamy D, Yu Z, Bhandoola A, Xue H-H, Roychoudhuri R, Higgs BW, Restifo NP, Bender TP, Ji Y, Gattinoni L, The transcription factor c-Myb regulates CD8+ T cell stemness and antitumor immunity. Nature Immunology 20, 337–349 (2019); published online Epub2019-03 ( 10.1038/s41590-018-0311-z). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miggelbrink AM, Jackson JD, Lorrey SJ, Srinivasan ES, Waibl-Polania J, Wilkinson DS, Fecci PE, CD4 T-Cell Exhaustion: Does It Exist and What Are Its Roles in Cancer? Clin Cancer Res 27, 5742–5752 (2021); published online EpubNov 1 ( 10.1158/1078-0432.CCR-21-0206). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tay RE, Richardson EK, Toh HC, Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther 28, 5–17 (2021); published online EpubFeb ( 10.1038/s41417-020-0183-x). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shin Hyun M., Kapoor Varun N., Guan T, Kaech Susan M., Welsh Raymond M., Berg Leslie J., Epigenetic Modifications Induced by Blimp-1 Regulate CD8+ T Cell Memory Progression during Acute Virus Infection. Immunity 39, 661–675 (2013); published online Epub10/2013 ( 10.1016/j.immuni.2013.08.032). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh N, Lee YG, Shestova O, Ravikumar P, Hayer KE, Hong SJ, Lu XM, Pajarillo R, Agarwal S, Kuramitsu S, Orlando EJ, Mueller KT, Good CR, Berger SL, Shalem O, Weitzman MD, Frey NV, Maude SL, Grupp SA, June CH, Gill S, Ruella M, Impaired Death Receptor Signaling in Leukemia Causes Antigen-Independent Resistance by Inducing CAR T-cell Dysfunction. Cancer Discovery 10, 552–567 (2020); published online Epub04/2020 ( 10.1158/2159-8290.CD-19-0813). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, Attanasio J, Yan P, George SM, Bengsch B, Staupe RP, Donahue G, Xu W, Amaravadi RK, Xu X, Karakousis GC, Mitchell TC, Schuchter LM, Kaye J, Berger SL, Wherry EJ, TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 571, 211–218 (2019); published online Epub2019-07 ( 10.1038/s41586-019-1325-x). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seo H, Chen J, González-Avalos E, Samaniego-Castruita D, Das A, Wang YH, López-Moyado IF, Georges RO, Zhang W, Onodera A, Wu C-J, Lu L-F, Hogan PG, Bhandoola A, Rao A, TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proceedings of the National Academy of Sciences of the United States of America 116, 12410–12415 (2019); published online Epub2019-6-18 ( 10.1073/pnas.1905675116). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, Luther SA, Speiser DE, Held W, Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211 e110 (2019); published online EpubJan 15 ( 10.1016/j.immuni.2018.12.021). [DOI] [PubMed] [Google Scholar]
- 46.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, Sharpe AH, Freeman GJ, Germain RN, Nakaya HI, Xue H-H, Ahmed R, Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016); published online Epub2016-9-15 ( 10.1038/nature19330). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Differentiation and Persistence of Memory CD8+ T Cells Depend on T Cell Factor 1. Immunity 33, 229–240 (2010); published online Epub2010/08/27 ( 10.1016/j.immuni.2010.08.002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, Gao P, Bandyopadhyay S, Sun H, Zhao Z, Lundh S, Pruteanu-Malinici I, Nobles CL, Maji S, Frey NV, Gill SI, Tian L, Kulikovskaya I, Gupta M, Ambrose DE, Davis MM, Fraietta JA, Brogdon JL, Young RM, Chew A, Levine BL, Siegel DL, Alanio C, Wherry EJ, Bushman FD, Lacey SF, Tan K, June CH, Decade-long leukaemia remissions with persistence of CD4(+) CAR T cells. Nature 602, 503–509 (2022); published online EpubFeb ( 10.1038/s41586-021-04390-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, Freeman SS, Reuben A, Hoover PJ, Villani A-C, Ivanova E, Portell A, Lizotte PH, Aref AR, Eliane J-P, Hammond MR, Vitzthum H, Blackmon SM, Li B, Gopalakrishnan V, Reddy SM, Cooper ZA, Paweletz CP, Barbie DA, Stemmer-Rachamimov A, Flaherty KT, Wargo JA, Boland GM, Sullivan RJ, Getz G, Hacohen N, Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175, 998–1013.e1020 (2018); published online Epub2018-11-01 ( 10.1016/j.cell.2018.10.038). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen J, Lopez-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, Yoshimura A, Scott-Browne JP, Rao A, NR4A transcription factors limit CAR T cell function in solid tumours. Nature 567, 530–534 (2019); published online EpubMar ( 10.1038/s41586-019-0985-x). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, Barnitz RA, Bartman C, Bengsch B, Huang AC, Schenkel JM, Vahedi G, Haining WN, Berger SL, Wherry EJ, Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165 (2016); published online EpubDec 2 ( 10.1126/science.aaf2807). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, Tsao HW, Godec J, LaFleur MW, Brown FD, Tonnerre P, Chung RT, Tully DC, Allen TM, Frahm N, Lauer GM, Wherry EJ, Yosef N, Haining WN, The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169 (2016); published online EpubDec 2 ( 10.1126/science.aae0491). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, Good Z, Belk JA, Daniel B, Klysz D, Malipatlolla M, Xu P, Bashti M, Heitzeneder S, Labanieh L, Vandris P, Majzner RG, Qi Y, Sandor K, Chen LC, Prabhu S, Gentles AJ, Wandless TJ, Satpathy AT, Chang HY, Mackall CL, Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 372, (2021); published online EpubApr 2 ( 10.1126/science.aba1786). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seo H, Gonzalez-Avalos E, Zhang W, Ramchandani P, Yang C, Lio CJ, Rao A, Hogan PG, BATF and IRF4 cooperate to counter exhaustion in tumor-infiltrating CAR T cells. Nat Immunol 22, 983–995 (2021); published online EpubAug ( 10.1038/s41590-021-00964-8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, Mangan PA, Kulikovskaya I, Gupta M, Chen F, Tian L, Gonzalez VE, Xu J, Jung IY, Melenhorst JJ, Plesa G, Shea J, Matlawski T, Cervini A, Gaymon AL, Desjardins S, Lamontagne A, Salas-Mckee J, Fesnak A, Siegel DL, Levine BL, Jadlowsky JK, Young RM, Chew A, Hwang WT, Hexner EO, Carreno BM, Nobles CL, Bushman FD, Parker KR, Qi Y, Satpathy AT, Chang HY, Zhao Y, Lacey SF, June CH, CRISPR-engineered T cells in patients with refractory cancer. Science 367, (2020); published online EpubFeb 28 ( 10.1126/science.aba7365). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, Maus MV, Fraietta JA, Zhao Y, June CH, Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Molecular Therapy 26, 1855–1866 (2018); published online Epub07/2018 ( 10.1016/j.ymthe.2018.05.003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, Campana D, Riley JL, Grupp SA, June CH, Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 17, 1453–1464 (2009); published online EpubAug ( 10.1038/mt.2009.83). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haas AR, Tanyi JL, O’Hara MH, Gladney WL, Lacey SF, Torigian DA, Soulen MC, Tian L, McGarvey M, Nelson AM, Farabaugh CS, Moon E, Levine BL, Melenhorst JJ, Plesa G, June CH, Albelda SM, Beatty GL, Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol Ther 27, 1919–1929 (2019); published online EpubNov 6 ( 10.1016/j.ymthe.2019.07.015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, Lacey SF, Melenhorst JJ, McGettigan SE, Cook DR, Zhang C, Xu J, Do P, Hulitt J, Kudchodkar SB, Cogdill AP, Gill S, Porter DL, Woyach JA, Long M, Johnson AJ, Maddocks K, Muthusamy N, Levine BL, June CH, Byrd JC, Maus MV, Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 127, 1117–1127 (2016); published online Epub2016-03-03 ( 10.1182/blood-2015-11-679134). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jolma A, Kivioja T, Toivonen J, Cheng L, Wei G, Enge M, Taipale M, Vaquerizas JM, Yan J, Sillanpää MJ, Bonke M, Palin K, Talukder S, Hughes TR, Luscombe NM, Ukkonen E, Taipale J, Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Research 20, 861–873 (2010); published online Epub06/01/2010 ( 10.1101/gr.100552.109). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are in the paper or supplementary materials. Raw and analyzed sequencing datasets have been deposited to Gene Expression Omnibus (GEO) under the accession number GSE210265. Additional requests for raw and analyzed data or materials should be made to the PENN Center for Innovation (pciinfo@pci.upenn.edu) and will be promptly reviewed to determine whether the application is subject to any intellectual property or confidentiality requirements. Patient-related information not included in this report were collected as part of a clinical trial and might be subject to patient confidentiality. Any data and materials that can be shared will be released following execution of a material transfer agreement.