


Abstract
Recently, we cloned two highly related human genes, hChlR1 (DDX11) and hChlR2 (DDX12), which appear to be homologs of the Saccharomyces cerevisiae CHL1 gene. Nucleotide sequence analysis suggests that these genes encode new members of the DEAH family of DNA helicases. While the enzymatic activity of CHL1 has not been characterized, the protein is required for the maintenance of high fidelity chromosome segregation in yeast. Here we report that the hChlR1 protein is a novel human DNA helicase. We have expressed and purified hChlR1 using a baculovirus system and analyzed its enzymatic activity. The recombinant hChlR1 protein possesses both ATPase and DNA helicase activities that are strictly dependent on DNA, divalent cations and ATP. These activities are abolished by a single amino acid substitution in the ATP-binding domain. The hChlR1 protein can unwind both DNA/DNA and RNA/DNA substrates. It has a preference for movement in the 5′→3′ direction on short single-stranded DNA templates. However, unlike other DNA helicases, the hChlR1 DNA helicase can translocate along single-stranded DNA in both directions when substrates have a very long single-stranded DNA region. The enzymatic activities of hChlR1 suggest that DNA helicases are required for maintaining the fidelity of chromosome segregation.
INTRODUCTION
Helicases catalyze the destabilization of hydrogen bonds between complementary nucleic acids (1). DNA duplexes, RNA duplexes and/or DNA–RNA hybrids must be transiently unwound during multiple cellular processes including replication, repair, recombination, transcription and splicing (2,3). Therefore these enzymatic activities are ubiquitous and essential to cells. Many helicases have been identified in both eukaryotes and prokaryotes (2–4). All known helicases contain seven conserved domains. The contributions of some of the domains to the enzymatic helicase activity have been elucidated. For instance, domains I and II are needed for ATP binding and ATP hydrolysis, respectively (5). Domain VI seems to be required for the binding of polynucleotides to the protein (5). Domain II has also been used to specify two major helicase subfamilies, which are called DEAD and DEAH based on the single letter amino acid sequence of this motif (6).
Recently, we cloned two genes, hChlR1 (DDX11) and hChlR2 (DDX12), that appear to be the human homologs of the yeast CHL1 gene (7,8). Analysis of the nucleotide sequence of the hChlR1 gene suggested that it encoded a DNA helicase, since it contained all seven conserved helicase domains. The yeast CHL1 gene also contains all seven conserved helicase domains. Although the enzymatic activity of CHL1 has not been characterized, a CHL1 gene containing a single amino acid substitution in the ATP-binding domain is unable to complement CHL1 null mutants, suggesting that enzymatic activity is required for CHL1 function (Holloway-Gerring and Hieter, unpublished results). The exact biological function of the yeast CHL1 gene is not known, however yeast strains lacking this gene show abnormal chromosome transmission (9). CHL1-negative mutants also show a G2/M cell cycle delay that is RAD9 independent. However, the recombination rate of CHL1 mutant yeast strains is similar to that of wild-type yeast. Moreover, CHL1 mutations have been shown to be synthetically lethal with either kar3 or mad2 (10,11; Holloway-Gerring et al., unpublished results). The kar3 and mad2 genes encode a kinesin-like protein involved in mitosis and a protein required for a mitotic checkpoint, respectively. These observations suggest that the Chl1 protein functions after DNA synthesis and before the completion of mitosis, possibly as part of a mitotic checkpoint. The requirement for CHL1 protein for the maintenance of high fidelity chromosome transmission supports the hypothesis that hChlR1 and hChlR2 are involved in maintaining faithful chromosome segregation in human cells.
Recently, two other human helicase genes, WRN and BLM, have been shown to participate in the maintenance of genome stability (12–18). Mutations in these two human RecQ-like DNA helicases have been shown to be responsible for Werner’s and Bloom’s syndromes, respectively (19,20). These two human DNA helicases are homologs of the yeast Sgs1 protein. Mutations in the SGS1 gene, which encodes DNA helicase, cause increased recombination of repeated sequences and genome instability in yeast (21–23). The nature of the symptoms associated with these human diseases suggests that while both helicases cause genome instability, each helicase also has a unique function(s) in the cell. Since the diseases are clearly distinct, clarification of the specific functions of these two helicases will assist in understanding these diseases and the basis of the genome instability. Here we show that the hChlR1 protein, which is encoded by the hChlR1 gene, is indeed a novel human DNA helicase. The homology of this gene to the CHL1 gene in yeast suggests that the hChlR1 protein, like WRN and BLM, may also be involved either directly or indirectly in the maintenance of genome stability in humans.
MATERIALS AND METHODS
Recombinant Werner syndrome gene product
The recombinant Werner gene product, the WRN DNA helicase, was a gracious gift from Dr Lawrence A. Loeb (University of Washington).
Construction of baculoviruses expressing hChlR1
The full-length hChlR1 cDNA was excised from pBluescript/hChlR1 using NcoI and HindIII and cloned into pFASTBac HTB digested with NcoI and HindIII (Life Science). The resulting construct was then transposed according to the manufacturer’s instructions. The viral DNA was transfected into Sf21 cells and high titer viral stocks were prepared. To generate enzymatically inactive protein the lysine residue at amino acid 50 was changed to an arginine by site-directed mutagenesis (24–27). This mutation alters the A box of the ATP-binding site consensus domain. A similar mutation in the RAD3 protein abolishes both ATPase and DNA helicase activity (27). Furthermore, mutant chl1 alleles containing substitutes in the corresponding residue in yeast CHL1 were unable to complement a chl1 deletion yeast strain (Holloway-Gerring and Hieter, unpublished results). Mutation of this residue was confirmed by manual DNA sequencing. The mutant hChlR1 cDNA was then cloned in pFASTBac HTb as described above for the wild-type protein.
Purification of hChlR1 protein
Recombinant hChlR1 containing a hexa-histidine tag at the N-terminus was expressed in High Five insect cells and purified by Ni2+ column chromatography as described below. A mutant protein, which should be enzymatically inactive, was made by replacement of the essential lysine with arginine in Domain I using site-directed mutagenesis and the protein was purified using the same protocol.
The cells (5 × 107) were collected 48 h after infection with the recombinant baculovirus at a m.o.i. of 3–6 and extracted with lysis buffer (5 ml) containing 10 mM HEPES pH 7.9, 1% NP-40, 1 mM MgCl2 and 0.5 mM CaCl2. Five milliliters of cell extract were incubated with 1 ml of Ni2+ resin. The resin was poured into a column and after washing with 50 mM HEPES pH 7.9, 300 mM NaCl, 10% glycerol and proteinase inhibitors (Complete™ EDTA-free; Boehringer) containing either 20 or 50 mM imidazole (10 ml each) was eluted with the same buffer containing 200 mM imidazole.
The purified proteins were electrophoresed on SDS–polyacrylamide gels and visualized with Coomassie brilliant blue. The concentrations of the purified proteins were estimated by comparison with known amounts of bovine serum albumin electrophoresed on the same gel. The protein preparations were 90–95% pure based on visual analysis of the stained gel.
Western blot analysis was performed using rabbit polyclonal Hel1 antisera to the hChlR1 protein as described previously (8). The Hel1 antisera was made to amino acids 2–130 of the hChlR1 protein and then purified by affinity chromatography (8). A peroxidase-labeled goat anti-rabbit IgG antibody (Kirkegaard & Perry Laboratories) was used as the secondary antibody. The signals were detected with an ECL Western Blotting Kit (Amersham Pharmacia Biotech).
ATPase assay
The ATPase activity of hChlR1 protein was detected by measuring the release of free phosphate during ATP hydrolysis (28,29). The reaction was carried out at 37°C in a volume of 100 µl. The assay mixture had the following components: 20 mM HEPES pH 7.4, 25 mM KCl, 2 mM MgCl2, 0.1 mM EDTA, 0.5 mM dithiothreitol, 1.0 mM [32P]ATP, calf thymus DNA and 10 µl of hChlR1 protein. The reaction was initiated by the addition of hChlR1 protein. At the indicated times, 20 µl aliquots were removed from the assay mixtures and 50 µl of the phosphate reagent (4 N H2SO4, 4% ammonium molybdate and 0.02 M silicotungstic acid) and 200 µl of xylene:isobutyl alcohol (65:35 v/v) were added. The samples were vigorously vortexed to extract the phosphomolybdate complex, formed from the free phosphate, into the organic phase. An aliquot (100 µl) was removed from the organic phase and the radioactivity was quantified using a liquid scintillation counter.
Preparation of helicase substrates
Ten different DNA and RNA oligonucleotides were used to construct the substrates for helicase assays. The sequences of these oligonucleotides were M13-18 (5′-CAGGGTTTTCCCAGTCAC-3′), M13-24 (5′-CGCCAGGGTTTTCCCAGTCACGAC-3′), M13-40 (5′-CGATTAAGTTGGGTAACGCC-AGGGTTTTCCCAGTCACGAC-3′), M13-18(RNA) (5′-CA-GGGUUUUCCCAGUCAC-3′), M13-35 (5′-AGCATGTCA-ATCATATGTACCCCGGTTGATAATCA-3′), M13-26 (5′-GG-TACCGAGCTCGAATTCGTAATCAT-3′), M13-19C (5′-GG-TGACTGGGAAAACCCTG-3′), DR-45 (5′-TCGCGGAAGCT-TGGCTGCAGAATATTGCTAGCGGGAATTCGGCGCG-3′), DR-1735 (5′-GCAGCCAAGCTTCCGCG-3′) and DR-1753 (5′-TCGCCCTTAAGCCGCGC-3′). These oligonucleotides are complementary to nucleotides 6313–6330 (M13-18), 6310–6333 (M13-24), 6310–6349 (M13-40), 6313–6330 [M13-18(RNA)], 6827–6861 (M13-35) and 6220–6247 (M13-26) of the M13mp18(+) DNA, respectively.
For the preparation of the 3′-end-labeled substrates, each of the oligonucleotides (1 pmol) was annealed with 2.5 µg of the single-strand M13mp18(+) DNA as described (14) and the free oligonucleotides removed by passage through a Sephacryl S-400 HR column (Pharmacia). After ethanol precipitation, the 3′-end of the annealed oligonucleotide was labeled by an extension reaction using the Klenow fragment (exo–) of Escherichia coli DNA polymerase I and [α-32P]dGTP (3000 Ci/mmol). The free nucleotides were removed by S-400HR column chromatography.
5′-End-labeled substrates were prepared by phosphorylating the oligonucleotide with T4 polynucleotide kinase and [γ-32P]ATP (3000 Ci/mmol) prior to annealing the oligonucleotide to M13mp18 DNA. Blunt-ended substrate was prepared by annealing M13-18 and M13-19C and then filling-in the end with Klenow fragment (exo–) and [α-32P]dCTP (3000 Ci/mmol). Two kinds of linear DNA substrates were used to determine the directionality of hChlR1 helicase translocation on a DNA substrate. For the 5′→3′ directionality experiments, M13-35 was annealed to M13mp18(+) DNA and the 3′-end was labeled by extension reaction with Klenow fragment (exo–) and [α-32P]dGTP (3000 Ci/mmol). The resulting partially duplexed DNA was digested with RsaI, which cuts the DNA in a blunt-ended manner leaving a 3′-end-labeled oligonucleotide. For 3′→5′ directionality experiments, M13-26 annealed to M13mp18(+) was digested with EcoRI and the cleavage sites were filled-in with Klenow fragment (exo–) in the presence of TTP and [α-32P]dATP (3000 Ci/mmol). We also prepared another pair of substrates to determine the directionality. These substrates consisted of an 18 base oligonucleotide annealed to the end of a 45 base oligonucleotide creating a substrate that was blunt at either the 5′- or 3′-end. The 18 base oligonucleotide was 32P-labeled by fill-in reaction with Klenow fragment (exo–) in the presence of [α-32P]dATP (3000 Ci/mmol) after annealing DR-45 with either DR-1735 or DR-1753.
Helicase assay
The helicase activity was measured by detecting the displacement of labeled oligonucleotide from the partially duplexed substrate. For these experiments, the labeled substrate was incubated with purified hChlR1 protein at 37°C for 10 min in 30 µl of the reaction mixture containing 50 mM Tris–HCl pH 7.4, 1 mM MgCl2, 2 mM 2-mercaptoethanol, 1 mM ATP and 0.5 mg/ml BSA. The reaction was terminated by adding 6 µl of stop solution containing 50 mM EDTA, 2% SDS, 40% glycerol, 0.3% bromophenol blue and 0.3% xylene cyanol. The reaction product was separated from the substrate by electrophoresis on a 12% polyacrylamide gel in 1× TBE buffer and visualized by autoradiography.
RESULTS
Purification of the recombinant hChlR1 protein from insect cells
In order to biochemically characterize the hChlR1 protein we used the baculovirus expression system to produce the recombinant protein with a hexa-histidine tag. High Five insect cells were infected with recombinant baculovirus expressing either the wild-type or mutant hChlR1 protein. The hChlR1 protein was purified from cellular extracts by Ni2+ column affinity chromatography. The purity of the hChlR1 proteins was assessed by SDS–PAGE and Coomassie brilliant blue staining (Fig. 1A). A major band accounting for 90–95% of the protein was visible at the predicted size (112 kDa) of hChlR1 for both wild-type and mutant protein preparations. For unknown reasons, cells expressing the mutant protein, which contains a single amino acid substitution, expressed ~20-fold more protein than cells infected with the wild-type virus under identical infection and purification conditions.
Figure 1.
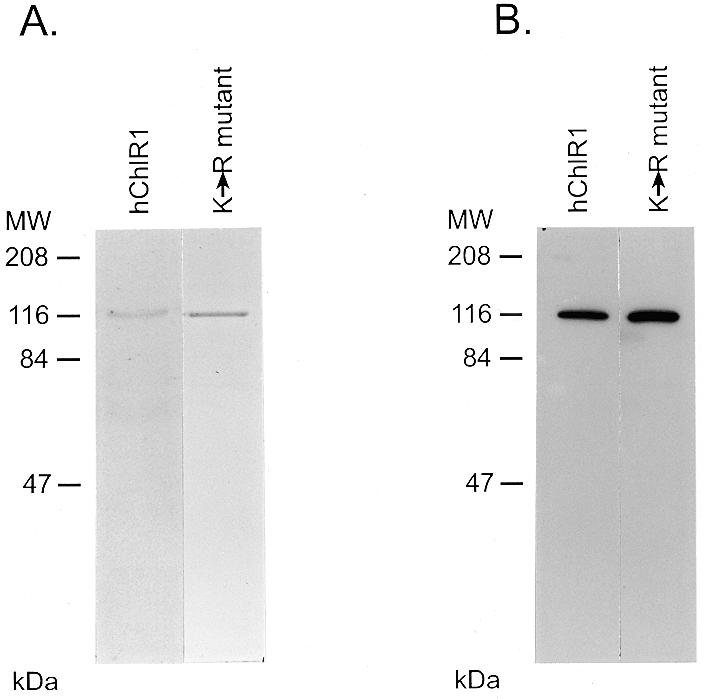
Purification of wild-type and mutant hChlR1 proteins by Ni2+ column chromatography. High Five insect cells were infected with baculoviruses carrying either the wild-type or a mutant hChlR1 cDNA with a single amino acid change in the ATP-binding site. Forty-eight hours after infection the cells were collected and lysed. Both the proteins were purified by Ni2+ affinity column chromatography. The purified proteins were electrophoresed through SDS–polyacrylamide gels. (A) The proteins were stained with Coomassie brilliant blue. The following concentrations of hChlR1 proteins were loaded on the gel: lane 1, 150 ng of hChlR1; lane 2, 300 ng of the K-R mutant. (B) Western blotting using affinity-purified Hel1 antisera which recognizes the N-terminus of the hChlR1 protein. Lane 1, 7.5 ng of hChlR1; lane 2, 15 ng of the K-R mutant.
To confirm that this 112 kDa protein was the hChlR1 gene product, we detected the protein by western blot analysis using an affinity-purified Hel1 antiserum which recognizes the N-terminus of the hChlR1 protein (Fig. 1B). The major Coomassie brilliant blue stained bands in both preparations were specifically recognized by this antiserum. Thus, the major Coomassie brilliant blue stained proteins are hChlR1 proteins.
ATPase activity of the purified hChlR1 protein
All helicases have DNA-dependent ATPase activity since the energy obtained from ATP hydrolysis is required for unwinding of nucleic acid duplexes (1–3). Therefore, we determined whether the hChlR1 protein functioned as an ATPase. We analyzed the ATPase activity using equal volumes of the mutant and wild-type enzymes in order to equalize the amount of contaminants in the reactions (Fig. 2A). It should be noted that samples from the mutant protein preparation contain ~20-fold more hChlR1 protein than the wild-type protein preparation (1500 and 75 ng of hChlR1 protein, respectively). The wild-type fraction hydrolyzed ATP in the presence of DNA in a time-dependent manner. This ATPase activity was strictly DNA-dependent since the ATPase activity was reduced >90% when DNA was not added to the ATPase reaction. The level of ATPase activity varied as a function of hChlR1 protein concentration, and this activity was abolished by boiling the protein prior to the reaction (data not shown). In contrast, the mutant hChlR1 protein exhibited only minimal background activity even after long incubations. In addition, this low level of ATPase activity was not influenced by DNA.
Figure 2.

Wild-type hChlR1 protein, but not mutant hChlR1 protein, hydrolyzes ATP in the presence of calf thymus DNA. ATPase activity was detected by measuring the release of inorganic phosphate from [γ-32P]ATP. (A) Release of free phosphate during a 150 min assay. The results are expressed as the means ± SD of the experiment performed in triplicate. Diamond, no hChlR1 protein; closed circle, wild-type hChlR1 protein with 100 µg/ml calf thymus DNA; open circle, wild-type hChlR1 protein without calf thymus DNA; closed square, mutant hChlR1 protein with 100 µg/ml calf thymus DNA; open square, mutant hChlR1 protein without calf thymus DNA. (B) Comparison of the specific activities of the wild-type and mutant hChlR1 proteins. The results were calculated using means of the triplicate samples from the experiments. Closed bar, in the presence of 100 µg/ml calf thymus DNA; open bar, in the absence of calf thymus DNA.
To compare the ATPase activities of the wild-type and mutant hChlR1 proteins, we calculated the specific activities of two preparations in the presence of DNA (Fig. 2B). There was an ~125-fold difference in the specific activity of the wild-type and mutant hChlR1 proteins. These data clearly indicate that the lysine in the ATP-binding domain is essential for ATPase activity. At the same time, it also confirms that hChlR1 protein functions as an ATPase.
We further characterized this ATPase activity in terms of pH, cation and nucleic acid dependency. Maximal ATP hydrolysis was observed at pH 7.4. This activity was drastically decreased at pH 5.0 (Fig. 3A). This ATPase activity also required an appropriate divalent cation. Mg2+ ions supported the activity most effectively. Ca2+ and Mn2+ ions partially substituted for Mg2+, but Zn2+ did not (Fig. 3B). As shown in Figure 2, this ATPase activity was stimulated by the addition of DNA to the reaction. Therefore, we examined the stimulatory effect of several nucleic acids on ATPase activity (Fig. 3C). Both calf thymus DNA and M13mp18 single-stranded DNA had the ability to stimulate ATPase activity, but circular double-stranded pcDNA3.0, blunt-ended pcDNA3.0 and two different types of RNAs (rRNA and tRNA) had no effect. The data examining the ATPase activity of the various plasmid forms suggest that hChlR1 is a single-stranded DNA-dependent ATPase. However, the observations that sheared calf thymus DNA stimulated ATPase activity while double-stranded pcDNA did not are somewhat contradictory, although it is possible that the calf thymus DNA preparation contained single-strand DNA. Nonetheless, this data indicates that the ATPase activity catalyzed by hChlR1 protein requires DNA as a cofactor. These data also verify that hChlR1 protein is a DNA-dependent ATPase.
Figure 3.

Characterization of hChlR1 ATPase activity. ATPase activity was detected in reaction buffer(s) at several different pH (A), containing different cations (B) or containing different types of nucleic acids (C). These experiments were performed in duplicate. The results are shown as the percentage of the value obtained under standard experimental conditions (pH 7.4, Mg2+ and 100 µg/ml calf thymus DNA).
DNA helicase activity of the purified hChlR1 protein
DNA helicase activity is often detected by displacement of a labeled oligonucleotide from a partially duplexed DNA substrate. To test whether hChlR1 protein exhibited helicase activity, we prepared a 3′-end-labeled substrate, which consisted of M13mp18 DNA and a complementary radioactively labeled oligonucleotide, allowing us to specifically detect the signal of the displaced oligonucleotides. 3′-End-labeled oligonucleotides were used for these studies since both wild-type and mutant hChlR1 protein preparations contained low levels of a co-purifying nuclease or phosphatase that removed the 5′-end label (see Fig. 5B). We cannot exclude the possibility that hChlR1 protein itself is responsible for this activity. Indeed, WRN helicase has recently been reported to contain 3′→5′ DNA exonuclease activity as well as DNA helicase activity (30–32). A similar contaminating activity has also been reported by others studying the WRN helicase (30).
Figure 5.

Substrate dependence of hChlR1 DNA helicase activity. Helicase activity was detected using different types of substrates. The structure of the substrate is shown at the left side of each panel. Asterisks denote the 32P-labeled end. The heat-denatured substrate was applied in lanes marked Denatured. (A) Three oligonucleotide substrates of different lengths were annealed to the M13mp18 DNA and used in the helicase assay. (B) RNA/DNA heteroduplexes, which consisted of an 18mer RNA oligonucleotide annealed to M13mp18 DNA, and DNA/DNA duplexes were used as helicase substrates. The sequence of the 18mer RNA oligonucleotide was identical to that of the 18mer DNA. (C) A 19mer blunt-ended DNA substrate prepared by fill-in reaction with Klenow fragment (exo–).
Using the substrates described above, we examined whether hChlR1 protein had the ability to unwind duplexed DNA. As shown in Figure 4, hChlR1 protein displaced the oligonucleotide from a partially duplexed DNA substrate in a concentration-dependent manner. This displacement activity required both Mg2+ and ATP. In contrast, the mutant protein did not unwind the substrate, demonstrating that ATP binding and hydrolysis is essential for helicase activity.
Figure 4.
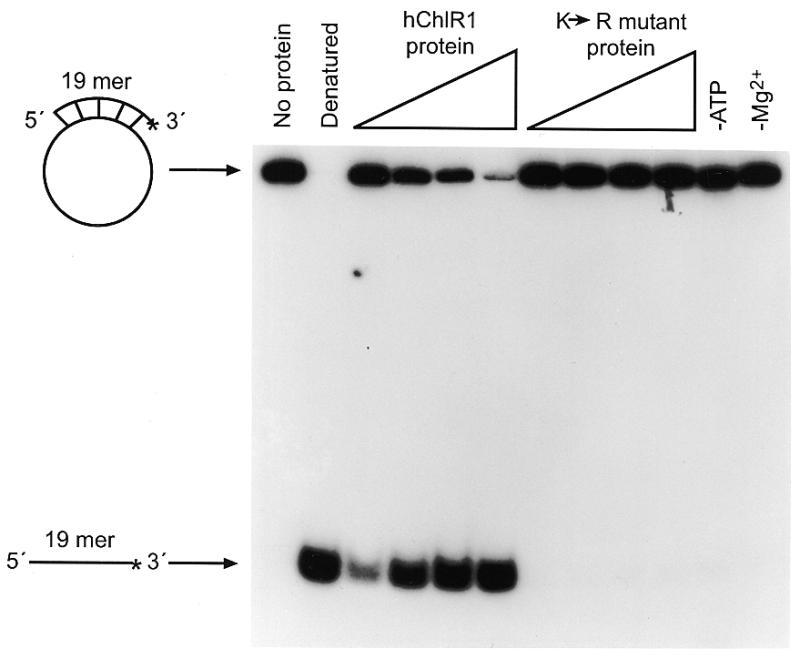
The wild-type hChlR1 protein, but not mutant hChlR1 protein, unwinds DNA/DNA duplexes. Helicase activity was detected by separating the labeled oligonucleotide displaced from a partial duplex substrate on a non-denaturing polyacrylamide gel. The structure of the substrate is shown on the left side. Asterisks denote the 32P-labeled end. The heat-denatured substrate was applied in the second lane shown as Denatured. –ATP and –Mg2+ indicate that ATP or Mg2+, respectively, was removed from the reaction (lane 6), indicating that the DNA helicase activity of hChlR1 protein requires ATP and Mg2+.
To better understand the enzymatic characteristics of this helicase, we examined the substrate, nucleotide and divalent cation requirements of hChlR1 helicase activity. First, the length of the oligonucleotide annealed to M13mp18 DNA was varied. Oligonucleotides of 18, 24 and 40 nt were designed to the same region on M13mp18 DNA and labeled as described previously. hChlR1 protein efficiently unwound the 19 nt substrate. It slightly displaced the 25 nt substrate, but not the 41 nt substrate (Fig. 5A), suggesting that either this enzyme has low processivity or that a high concentration of the protein is required to unwind longer substrates. Next, we used a RNA/DNA heteroduplex containing a 5′-end-labeled ribonucleotide as substrate (Fig. 5B). The RNA oligonucleotide was displaced from the substrate by hChlR1 protein. This data indicates that hChlR1 protein can unwind not only DNA/DNA substrates but also a RNA/DNA hybrid substrate. We also examined whether hChlR1 protein was capable of unwinding a blunt-ended substrate (Fig. 5C). hChlR1 protein could not unwind the blunt-ended substrate, which was consistent with the observation that blunt-ended DNA did not support ATPase activity.
The nucleotide dependence was examined using an unhydrolyzable ATP analog, ATPγS, in addition to eight different nucleotides (Fig. 6A). Although ATP could support the helicase activity, no other nucleotide, including ATPγS, could substitute for ATP. Therefore, this helicase appears to strictly require ATP hydrolysis for helicase activity. With respect to divalent cation dependence, the same divalent cations used for the ATPase assays were examined (Fig. 6B). Mg2+ and Mn2+ ions supported the helicase activity most effectively. Ca2+ ions were less effective, but still supported the unwinding activity. Zn2+ ions did not substitute for other ions, which, once again, was consistent with the results from the ATPase activity experiments. Thus, the cation preferences for the helicase and ATPase activities were similar.
Figure 6.

Nucleotide and cation dependence of hChlR1 DNA helicase activity. (A) All of the normal nucleoside 5′-triphosphates and an unhydrolyzable ATP analog, ATPγS, were used as cofactors of the hChlR1 DNA helicase. The concentration of nucleotide added to the reaction buffer was 1 mM. (B) Reaction buffers including different divalent cations at a concentration of 1 mM were used for helicase assay. The structure of the substrate is shown at the left side of each panel. Asterisks denote the 32P-labeled end. The heat-denatured substrate was applied in lanes marked Denatured.
Since most helicases translocate on single-stranded DNA in one direction (1–3), it was important to determine the directionality of hChlR1 protein for its classification and further functional studies. We prepared two different substrates for this purpose. These substrates consisted of a long linear M13 single-stranded DNA with two short oligonucleotides generated by restriction enzyme digestion of a large oligonucleotide that was annealed to circular single-stranded M13 DNA. The positions of 32P-labeled ends are shown in Figure 7A. Because hChlR1 protein cannot unwind a blunt-ended substrate, as shown in Figure 5C, the protein must begin to unwind from the single-stranded M13 DNA portion. Thus, the combination of substrates provided the necessary information on the ability of the protein to unwind DNA in a single direction.
Figure 7.

Directionality of hChlR1 DNA helicase. The linearized M13mp18 DNAs with blunt-ended partially duplexed termini were used as substrates (A). To distinguish either 3′→5′ or 5′→3′ directional movement, the oligonucleotides annealed to M13mp18 DNA were labeled as shown on the left side of each panel. WRN helicase, which is known to translocate only in a 3′→5′ direction, was used in order to confirm that the substrates were properly prepared. (B) Substrates with a short single-stranded DNA were used to examine the directionality of hChlR1 DNA helicase. The structures of two substrates used in this experiment are shown together on the left side of the panel. Asterisks denote the 32P-labeled end. The heat-denatured substrate was applied in lanes marked Denatured.
Surprisingly, hChlR1 protein was able to displace the labeled oligonucleotide from both substrates (Fig. 7A). In contrast, WRN helicase unwound the substrate only in the 3′→5′ direction, as reported previously. These data indicate that hChlR1 protein translocates on single-stranded DNA in both directions on this substrate. Furthermore, the WRN directionality data confirm that substrates were prepared correctly.
In order to further understand this directionality and determine whether hChlR1 helicase is bi-directional on all types of substrates, we examined hChlR1 helicase activity using a different type of substrate. These substrates consisted of a 32P-labeled 19 base oligonucleotide annealed to either the 5′- or 3′-end of a 45 base oligonucleotide. hChlR1 protein unwound only the 5′→3′ substrate (Fig. 7B), suggesting that this protein has a preference for the 5′→3′ direction and that the 3′→5′ unwinding activity requires very long regions of single-stranded DNA.
DISCUSSION
hChlR1 and hChlR2, two highly related human genes encoding putative helicases, were previously isolated in our laboratory (7,8). The proteins predicted by the open reading frames of these genes contain the seven domains conserved among known helicases (7,8). Here, we have demonstrated that hChlR1 protein is indeed an enzyme that possesses both ATPase and DNA helicase activities. Details concerning the divalent cation and substrate preferences, as well as the directionality of the helicase activity, have also been elucidated.
Our characterization of hChlR1 helicase distinguishes it from other known human DNA helicases. The divalent cation dependence and experiments using a blunt-ended substrate and/or unhydrolyzable ATP analog indicate that the ATPase activity of hChlR1 protein parallels the DNA helicase activity. This helicase activity efficiently unwound a 19 nt substrate, but exhibited only minimal activity towards a 25 nt substrate, suggesting that it has low processivity. However, we cannot exclude the possibility that another cofactor, such as a single-strand DNA-binding protein, is required for maximal enzyme activity. It is known that some DNA helicases are stimulated by the addition of single-strand DNA-binding proteins, such as RPA, E.coli SSB and the T4 gene 32 protein, and that these helicases unwind longer substrates (33,34). We examined this possibility in preliminary experiments using the T4 gene 32 protein and purified recombinant human RPA. We failed to detect any increase in hChlR1 helicase activity when either of these proteins was added to the reaction (data not shown). However, hChlR1 DNA helicase might require a different single-strand DNA-binding protein. Another potential explanation for the low processivity might be the presence of a hexa-histidine tag at the N-terminus of the hChlR1 protein we purified. However, several other laboratories have recently characterized hexa-histidine tagged WRN and BLM DNA helicases (13,14,35). These helicases have much higher processivity than hChlR1 helicase. Therefore we believe it is unlikely that the tag decreases the processivity of hChlR1 helicase. Instead the data suggests that the cofactor which is necessary for maximal processivity is not present in insect cells.
The most unique feature of hChlR1 helicase is its ability to translocate bi-directionally on the bound single-stranded DNA on long single-stranded substrates. Most helicases analyzed to date show specific directionality, either 3′→5′ or 5′→3′. Exceptionally, it has been reported that eukaryotic translation initiation factors 4A and 4F, in combination with eIF-4B, unwind RNA in a bi-directional manner (36). Thus, hChlR1 protein may be the first DNA helicase that can move on single-stranded DNA in both directions. However, hChlR1 is translocated only in the 5′→3′ direction when short single-stranded DNA substrates were used in the helicase assay. The 5′→3′ directionality of hChlR1 might be important for its cellular function, since long single-stranded DNA substrates similar to those used in this study are not generally formed in cells. Therefore in vivo hChlR1 helicase may translocate only in the 5′→3′ direction.
As we previously reported, all of the proliferating cell lines we analyzed express hChlR1 protein (8). In contrast, when human cells cease to proliferate, hChlR1 mRNA and protein expression is extinguished. In addition, when quiescent fibroblasts are stimulated to re-enter the cell cycle by serum addition, hChlR1 expression increases as the cell population enters S phase. It is therefore possible that hChlR1 might function in cellular proliferation, such as in DNA replication. However, based on the homology between the hChlR1 gene and the yeast CHL1 gene, we believe it is more likely that hChlR1 plays a more direct role in cell division. Yeast CHL1 null mutants show a substantial (200-fold) increase in the rate of missegregation of specific chromosomes, with chromosome loss and non-disjunction contributing equally to this phenotype (9). Therefore, hChlR1 may function to maintain the fidelity of chromosome transmission and the maintenance of genome stability. Interestingly, the genes responsible for two human disorders characterized by genome instability, Werner’s and Bloom’s syndromes, are RecQ-like DNA helicases, strongly suggesting that DNA helicases are involved in maintaining genome stability. If hChlR1 DNA helicase also plays a role in chromosome segregation, it would be the first DEAH/D family member outside the RecQ subclass with a role in genome stability. Further experiments will clarify how hChlR1 DNA helicase functions in human cells.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Mark Wold for purified human RPA and Drs L.A. Loeb and A.S. Kamath for providing WRN helicase and helpful comments. Their work is supported by NIH grant CA-77852. We acknowledge Dr V.J. Kidd for constructive discussions, Dr Joseph Amann for generation of the hChlR1 site mutant and Gail Richmond and Jose Grenet for excellent technical assistance. We also thank Dr C. Naeve and the Hartwell Center for Bio-informatics and Biotechnology for assistance with the synthesis of oligonucleotides. Support for these studies was provided by the American Lebanese Syrian Associated Charities (ALSAC) and Cancer Center Core Grant CA21765 from the NIH to St Jude Children’s Research Hospital and by Daiichi Pharmaceutical Corp. (Y.H.).
REFERENCES
- 1.Lohman T.M. and Bjornson,K.P. (1996) Annu. Rev. Biochem., 65, 169–214. [DOI] [PubMed] [Google Scholar]
- 2.Matson S.W. and Kaiser-Rogers,K.A. (1990) Annu. Rev. Biochem., 59, 289–329. [DOI] [PubMed] [Google Scholar]
- 3.Borowiec J.A. (1996) In DePamphilis,M.L. (ed.), DNA Replication in Eukaryotic Cells. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 545–574.
- 4.Ellis N.A. (1997) Curr. Opin. Genet. Dev., 7, 354–363. [DOI] [PubMed] [Google Scholar]
- 5.Hodgman T.C. (1988) Nature, 333, 22–23. [DOI] [PubMed] [Google Scholar]
- 6.Linder P., Lasko,P.F., Ashburner,M., Leroy,P., Nielsen,P., Nishi,K., Schnier,J. and Slonimski,P.P. (1999) Nature, 337, 121–122. [DOI] [PubMed] [Google Scholar]
- 7.Amann J., Valentine,M., Kidd,V.J. and Lahti,J.M. (1996) Genomics, 32, 260–265. [DOI] [PubMed] [Google Scholar]
- 8.Amann J., Kidd,V.J. and Lahti,J.M. (1997) J. Biol. Chem., 272, 3823–3832. [DOI] [PubMed] [Google Scholar]
- 9.Gerring S.L., Spencer,F. and Hieter,P. (1990) EMBO J., 9, 4347–4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meluh P.B. and Rose,M.D. (1990) Cell, 60, 1029–1041. [DOI] [PubMed] [Google Scholar]
- 11.Li R. and Murray,A.W. (1991) Cell, 66, 519–531. [DOI] [PubMed] [Google Scholar]
- 12.Lombard D.B. and Guarente,L. (1996) Trends Genet., 12, 283–286. [DOI] [PubMed] [Google Scholar]
- 13.Gray M.D., Shen,J.-C., Kamath-Loeb,A.S., Sopher,A.B.L., Martin,G.M., Oshima,J. and Loeb,L.A. (1997) Nature Genet., 17, 100–103. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki N., Shimamoto,A., Imamura,O., Kuromitsu,J., Kitao,S., Goto,M. and Furuichi,Y. (1997) Nucleic Acids Res., 25, 2973–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karow J.K., Chakraverty,R.K. and Hickson,I.D. (1997) J. Biol. Chem., 272, 30611–30614. [DOI] [PubMed] [Google Scholar]
- 16.Lebel M. and Leder,P. (1998) Proc. Natl Acad. Sci. USA, 95, 13097–13102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chester N., Kuo,F., Kozak,D., O’Hara,C.D. and Leder,P. (1998) Genes Dev., 12, 3382–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamagata K., Kato,J., Shimamoto,A., Goto,M., Furuichi,Y. and Ikeda,H. (1998) Proc. Natl Acad. Sci. USA, 95, 8733–8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu C.-E., Oshima,J., Fu,Y.-H., Wijsman,E.M., Hisama,F., Alisch,R., Matthews,S., Nakura,J., Miki,T., Ouais,S., Martin,G.M., Mulligan,J. and Schellenberg,G.D. (1996) Science, 272, 258–262. [DOI] [PubMed] [Google Scholar]
- 20.Ellis N.A., Groden,J., Ye,T.-Z., Straughen,J., Lennon,D.J., Ciocci,S., Proytcheva,M. and German,J. (1995) Cell, 83, 655–666. [DOI] [PubMed] [Google Scholar]
- 21.Lu J., Mullen,J.R., Brill,S.J., Kleff,S., Romeo,A.M. and Sternglanz,R. (1996) Nature, 383, 678–679. [DOI] [PubMed] [Google Scholar]
- 22.Morozov V., Mushegian,A.R., Koonin,E.V. and Bork,P. (1997) Trends Biochem. Sci., 22, 417–418. [DOI] [PubMed] [Google Scholar]
- 23.Watt P.M., Hickson,I.D., Borts,R.H. and Louis,E.J. (1996) Genetics, 144, 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Notarnicola S.M. and Richardson,C.C. (1993) J. Biol. Chem., 268, 27198–27207. [PubMed] [Google Scholar]
- 25.Korangy F. and Julin,D.A. (1992) J. Biol. Chem., 267, 1727–1732. [PubMed] [Google Scholar]
- 26.Patel S.S., Hingorani,M.M. and Ng,W.M. (1994) Biochemistry, 33, 7857–7868. [DOI] [PubMed] [Google Scholar]
- 27.Sung P., Higgins,D., Prakash,L. and Prakash,S. (1988) EMBO J., 7, 3263–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei J., Gaut,J.R. and Hendershot,L.M. (1995) J. Biol. Chem., 270, 26677–26682. [DOI] [PubMed] [Google Scholar]
- 29.Wei J. and Hendershot,L.M. (1995) J. Biol. Chem., 270, 26670–26676. [DOI] [PubMed] [Google Scholar]
- 30.Huang S., Li,B., Gray,M.D., Oshima,J., Mian,S. and Campisi,J. (1998) Nature Genet., 20, 114–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen J.-C., Gray,M.D., Oshima,J., Kamath-Loeb,A.S., Fry,M. and Loeb,L.A. (1998) J. Biol. Chem., 273, 34139–34144. [DOI] [PubMed] [Google Scholar]
- 32.Kamath-Loeb A.S., Shen,J.-C., Loeb,L.A. and Fry,M. (1998) J. Biol. Chem., 273, 34145–34150. [DOI] [PubMed] [Google Scholar]
- 33.Shen J.-C., Gray,M.D., Oshima,J. and Loeb,L.A. (1998) Nucleic Acids Res., 26, 2879–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hughes P. and Baldacci,G. (1997) Nucleic Acids Res., 25, 3881–3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bahr A., DeGraeve,F., Kedinger,C. and Chatton,B. (1998) Oncogene, 17, 2565–2571. [DOI] [PubMed] [Google Scholar]
- 36.Rozen F., Edery,I., Dever,T.E., Merrick,W.C. and Sonenberg,N. (1990) Mol. Cell. Biol., 10, 1134–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]