

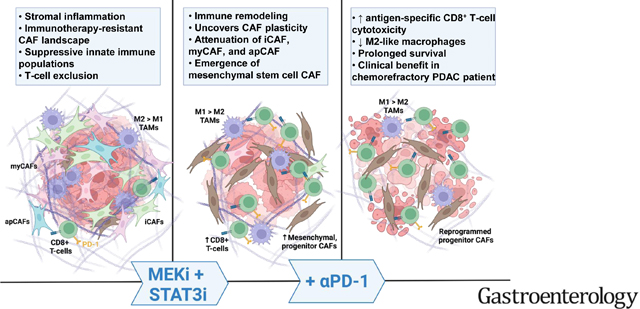
Abstract
BACKGROUND:
We have shown that reciprocally activated RAS/MEK and JAK/STAT3 pathways mediate therapeutic resistance in pancreatic ductal adenocarcinoma (PDAC), while combined MEK and STAT3 inhibition (MEKi+STAT3i) overcomes such resistance and alters stromal architecture. We now determine if MEKi+STAT3i reprograms the cancer-associated fibroblast (CAF) and immune microenvironment to overcome resistance to immune checkpoint inhibition in PDAC.
METHODS:
CAF and immune cell transcriptomes in MEKi (trametinib)+STAT3i(ruxolitinib)- vs. vehicle-treated Ptf1acre/+;LSL-KrasG12D/+;Tgfbr2flox/flox (PKT) tumors were examined via single-cell RNA sequencing (scRNAseq). CRISPR/Cas9-silencing of CAF-restricted Map2k1/Mek1 and/or Stat3 enabled interrogation of CAF-dependent effects on immunologic remodeling in orthotopic models. Tumor growth, survival, and immune profiling via time-of-flight mass cytometry were examined in PKT mice treated with vehicle, anti-PD1 monotherapy, and MEKi+STAT3i combined with anti-PD1.
RESULTS:
MEKi+STAT3i attenuates Il6/Cxcl1-expressing pro-inflammatory and Lrrc15-expressing myofibroblastic CAF phenotypes while enriching for Ly6a/Cd34-expressing CAFs exhibiting mesenchymal stem cell-like features via scRNAseq in PKT mice. This CAF plasticity is associated with M2-to-M1 reprogramming of tumor-associated macrophages, and enhanced trafficking of CD8+ T-cells which exhibit distinct effector transcriptional programs. These MEKi+STAT3i-induced effects appear CAF-dependent, since CAF-restricted Mek1/Stat3 silencing mitigates inflammatory-CAF polarization and myeloid infiltration in-vivo. Addition of MEKi+STAT3i to PD-1 blockade not only dramatically improves anti-tumor responses and survival in PKT mice, but also augments recruitment of activated/memory T-cells while improving their degranulating and cytotoxic capacity, compared with anti-PD-1 monotherapy. Importantly, treatment of a patient with chemotherapy-refractory metastatic PDAC with MEKi (Trametinib), STAT3i (Ruxolitinib), and PD-1 inhibitor (Nivolumab) yielded clinical benefit.
CONCLUSIONS:
Combined MEKi+STAT3i mitigates stromal inflammation and enriches for CAF phenotypes with mesenchymal stem cell-like properties to overcome immunotherapy resistance in PDAC.
Keywords: Pancreatic ductal adenocarcinoma, immunotherapy, immune checkpoint inhibition, stromal plasticity, mesenchymal stem cell
LAY SUMMARY
This study highlights the novel role of combination targeted therapies that reprograms tumor-permissive cancer-associated fibroblasts and suppressive immune microenvironments to invigorate anti-tumor immune responses, thereby sensitizing pancreatic cancers to immunotherapy.
Graphical Abstract
INTRODUCTION
The major contributors to therapeutic resistance that have been difficult to overcome in pancreatic ductal adenocarcinoma (PDAC) are mutations in the KRAS oncogene,1 presence of a dense desmoplastic stroma that acts as a barrier to drug delivery and effector immune cell infiltration,2 and an immunosuppressive tumor microenvironment (TME) that renders the tumor resistant to immune checkpoint inhibition (ICI) immunotherapy.3
The failure of ICI in PDAC4, 5 has been attributed not only to the fibroinflammatory desmoplastic stroma that promotes T-cell exclusion, but also to functional heterogeneity in the cancer-associated fibroblast (CAF) compartment as a key stromal-mediated mechanism of immunotherapy resistance.6, 7 In particular, IL1-mediated polarization of CAFs toward a secretory phenotype, characterized by elaboration of IL-6, CXCL1, and LIF,8 propagates pro-inflammatory tumor-stromal-immune crosstalk in the PDAC TME that drives therapeutic resistance.9 These tumor-permissive inflammatory CAFs (iCAF)—which co-exist in a dynamic equilibrium with other functionally divergent CAF sub-populations such as myofibroblastic CAFs (myCAFs) and antigen-presenting CAFs (apCAFs)10, 11—beckon immunosuppressive tumor-associated macrophages (TAM) and myeloid-derived suppressor cells (MDSC) to the TME, dampen anti-tumor adaptive immunity, and promote immune evasion in PDAC.12, 13 These underlying complexities in CAF immunobiology, coupled with the clinical failure of broad-based stromal-depleting strategies in PDAC patients,14, 15 warrant the development of more nuanced therapies to mitigate tumor-permissive inflammatory CAF programming while preserving its tumor-restraining counterparts.
KRAS mutations—another key contributor to therapeutic resistance—are not only the predominant oncogenic driver in over 90% of PDAC, but also promote an inflammatory program that establishes immune privilege in the TME.1, 16 Since targeting KRAS has remained elusive, our approach has been to target downstream effectors of RAS through MEK inhibition (MEKi). However, clinical trials of MEKi have been unsuccessful in PDAC17 due to the emergence of resistance mechanisms. We have shown that a key mechanism of MEKi resistance is reciprocal activation of STAT3 signaling. As such, JAK/STAT3 inhibition (STAT3i) combined with MEKi (MEKi+STAT3i) overcomes this parallel feedback loop activation, attenuates tumor growth in patient-derived xenograft models, and improves survival in the aggressive Ptf1acre/+;LSL-KrasG12D/+;Tgfbr2flox/flox (PKT) genetically engineered mouse model (GEMM).18, 19
In the present study, we show that combined MEKi+STAT3i uncovers stromal plasticity by attenuating CAFs from Il6/Cxcl1-expressing secretory phenotypes and enriching for Ly6a/Cd34-expressing CAF phenotypes with mesenchymal stem cell (MSC)-like features via single-cell RNA sequencing (scRNAseq) in PKT mice. This remodeling of CAF heterogeneity is associated with a striking attenuation in and reprogramming of TAMs, as well as enhanced trafficking of CD8+ T-cells which exhibit a distinct effector and anti-apoptotic transcriptional program. These immune repercussions are in part CAF-dependent, since CRISPR/Cas9 genetic silencing of CAF-restricted Mek1/Stat3 results in attenuation of iCAF polarization and MDSC/TAM infiltration in vivo. The addition of MEKi+STAT3i to PD-1 blockade overcomes ICI resistance by significantly enhancing the recruitment, degranulating capacity, and functional cytotoxicity of CD8+ T-cells, thereby augmenting anti-tumor responses and dramatically improving survival in PKT mice. Importantly, we demonstrate the clinical efficacy and tolerability of combined Trametinib (MEKi), Ruxolitinib (STAT3i), and Nivolumab (anti-PD1) treatment in a patient with chemotherapy-refractory PDAC, providing evidence of its translatability in PDAC patients.
RESULTS
Combined MEK and STAT3 inhibition remodels stromal fibrosis and attenuates inflammatory CAF phenotypes in a fibroblast-dependent manner
We extend our previous results18 to show that combined MEKi+STAT3i significantly remodels the tumor stroma in autochthonous PKT tumors (Fig. 1A) and orthotopically transplanted K-rasLSL.G12D/+;p53R172H/+;Pdx1Cre/+ (KPC) tumors (Fig. S1A), as well as reduces intratumoral mucin content and cellular proliferation, while increasing microvessel density compared with vehicle treatment in PKT tumors (Fig. S1B). To examine mechanistic underpinnings of MEKi+STAT3i-induced stromal remodeling in vivo, we performed scRNAseq in PKT mice—a GEMM characterized by dense stromatogenic response and immune exclusion20—treated with MEKi+STAT3i or vehicle. Using differentially expressed gene signatures, we attributed clusters to their putative identities (Fig. S2A). Specifically, CAF clusters were nominated by Col1a1, Col1a2, Pdpn, and Fap expression (Fig. S2B).21 Uniform Manifold Approximation and Projection (UMAP) was used to display discrete intratumoral cellular compartments in PKT mice (Fig. 1B).
Figure 1. Combined MEK and STAT3 inhibition remodels stromal fibrosis and attenuates inflammatory fibroblast phenotypes in the TME in a CAF-dependent manner.

(A) Trichrome blue, α-SMA, and Sirius Red staining in tumor sections from PKT mice treated with vehicle or MEKi+STAT3i for 4 weeks; relative areas of positive staining of respective markers from tissue sections in vehicle- and MEKi+STAT3i-treated mice are indicated in adjacent histograms (scale bar=50 μm). Data are shown as mean ± SEM; (B) Concatenated UMAP plot showing annotated clusters from 12,680 single cells undergoing RNA sequencing from vehicle- and MEKi+STAT3i-treated PKT mice (n=3 each); (C) Bubble plot representing pathway enrichment analysis performed on genes differentially downregulated in MEKi-STAT3i treated CAF cluster using fgsea (log fold change (FC)>1). Reactome, KEGG, and GO pathways with p-adjusted value<0.05 are displayed with normalized enrichment score (NES) indicated on x-axis; (D) Volcano plot of select differentially regulated genes related to innate immune response and stromal organization in MEKi+STAT3i-treated vs. vehicle-treated CAF single-cell transcriptomes. Vertical dashed line indicates an adjusted p-value=0.1, horizontal dashed lines indicate an absolute log2FC=0.5. Transcripts achieving non-significance (NS), p-value, log2FC, or p-value and log2FC significance threshold are indicated in adjoining legend; (E) CAF clusters in vehicle and MEKi+STAT3i-treated superimposed UMAP plot (left); violin plots depicting log expression level of Cxcl1 and Il6 genes in CAF single-cell transcriptomes comparing MEKi+STAT3i vs. vehicle-treated CAFs; (F) RT-qPCR analysis from whole tumor-derived RNA in MEKi+STAT3i-treated vs. vehicle-treated primary PKT tumors (n=4 mice/group) depicting relative fold change of Cxcl1, Il6, and Lif gene expression; (G) Venn diagram of differentially expressed genes in bulk RNA sequencing derived from KPC CAF cell lines that underwent CRISPR-Cas9 genetic silencing of either Mek1KO alone, Stat3KO alone or combined Mek1KO/Stat3KO. Numbers in each circle (intersection) represent the unique number of differentially regulated genes in respective comparisons; 1837 genes were differentially expressed in CAF-Mek1KOStat3KO, and were utilized for further analysis; (H) Bubble plot representing pathway enrichment analysis performed on genes differentially downregulated in CAF-Mek1KOStat3KO cells. Reactome, KEGG, and GO pathways with p-adjusted value<0.05 are displayed with normalized enrichment score (NES) indicated on x-axis; (I) Representative contour plots of CD45−PDPN+CD31− cells showing MHC-II and Ly6C gates to indicate iCAF (Ly6C+MHC-II−), myCAF (Ly6C−MHC-II−), and apCAF (Ly6C+MHC-II+) populations from experiments in which tumors were isolated from orthotopically injected C57BL/6 mice co-injected with KPC6694c2 tumor cells and either CAF-EV or CAF-Mek1KO/Stat3KO CAF (ratio 1:9 respectively) after 2 weeks (n=7–8 mice/group). Adjacent histogram shows quantification of iCAF:myCAF ratio at endpoint analysis. ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001.
Compared with vehicle treatment, combined MEKi+STAT3i resulted in substantial reprogramming of both the CAF and tumor-cell transcriptomes (Fig. S3A&B). Deeper investigation of the differentially expressed single-cell CAF transcriptomes revealed significant downregulation of pathways related to inflammatory cytokine/chemokine response, innate immune trafficking, NF-κB signaling, and extracellular matrix deposition (ECM) in MEKi+STAT3i-treated PKT tumors (Fig. 1C). Moreover, several genes implicated in promoting a pro-inflammatory secretome and/or innate immune cell recruitment (Cxcl2, Ccl6), peripheral immune tolerance (Tgfb1), and stromal organization/equilibrium (Mgp, Junb, Timp1) were differentially under-expressed in MEKi+STAT3i-treated CAF transcriptomes (Fig. 1D). In particular, expression of iCAF-defining genes Cxcl1 and Il610 was significantly reduced following MEKi+STAT3i treatment (Fig. 1E). These findings were validated by qPCR analysis of whole tumor-derived RNA showing significant diminution of Cxcl1 (P=0.002), Il6 (P=0.01), and Lif (P<0.001) expression in MEKi+STAT3i-treated PKT mice (Fig. 1F) and orthotopically injected KPC tumor-bearing mice (data not shown).
To determine if MEKi+STAT3i attenuates iCAF skewness in a fibroblast-dependent manner, we performed CRISPR/Cas9-enabled genetic editing of Mek1 alone (Mek1KO), Stat3 alone (Stat3KO), and dual Mek1/Stat3 (Mek1KOStat3KO) in PDPN+ KrasG12D/+;Trp53fl/+;Pdx-Cre (KPC) tumor-derived CAFs, and confirmed target silencing by western blotting (Fig. S4A). Bulk RNA sequencing of these cells revealed a distinct transcriptome in Mek1KOStat3KO CAFs comprising 1837 unique genes (Fig. 1G) that reflected downregulation of pathways related predominantly to focal adhesion/ECM deposition, collagen organization, and cytokine signaling (Fig. 1H; Fig. S4B). Compared to empty vector control-CAFs (CAF-EV), the secretome of CAF-Mek1KOStat3KO demonstrated reduction in several myeloid chemoattractants, such as IL-6, CXCL1, GM-CSF, and CCL2 in vitro (Fig. S4C)—confirmed by observation of largely similar secretory patterns (in particular, >50% reduction in CXCL1 and IL-6) when KPC CAFs were treated with Trametinib+Ruxolitinib in vitro compared with vehicle (Fig. S4D)—together recapitulating effects of MEKi+STAT3i on scRNAseq CAF transcriptomes in vivo.
Furthermore, orthotopic co-injection of KPC6694c2 tumor cells with CAF-Mek1KOStat3KO (1:9 ratio) in syngeneic C57Bl/6 mice not only resulted in significant reduction in pancreatic tumor weights (P=0.0009; Fig S4E) but also in iCAF (CD45−CD31−EpCAM−PDPN+Ly6C+MHC-II−) to myCAF (Ly6C−MHC-II−) cellular ratios by flow cytometry compared with CAF-EV tumors (P=0.009; Fig. 1I). Collectively, these data suggest that MEKi+STAT3i remodels stromal inflammation, attenuates iCAF phenotypes, and governs tumor growth in a fibroblast-dependent manner.
Combined MEK and STAT3 inhibition uncovers stromal plasticity and enriches for a CAF phenotype with MSC-like properties
To illustrate how MEKi+STAT3i shapes CAF heterogeneity, interrogation of CAF-designated barcoded events on scRNAseq revealed the presence of four discrete sub-clusters (Fig. 2A) harboring distinct gene expression profiles (Fig. S5A). While CAF2, CAF3, and CAF4 sub-populations dominated the CAF landscape in vehicle-treated mice, MEKi+STAT3i treatment revealed a significant enrichment in the CAF1 cluster while substantially attenuating CAF2, CAF3, and CAF4 populations (Fig. 2B). Given this striking MEKi+STAT3i-induced remodeling of the fibroblast microenvironment, we investigated the transcriptional and functional divergence between CAF1 and CAF2/CAF3/CAF4 subsets. Principal component analysis based on the most highly variable genes, as measured by variance-stabilizing transformation (Fig. S5B), and matrix plots of population concordance, as measured by Pearson’s correlation and Euclidean distances prior to hierarchical clustering of differentially expressed genes between CAF1–4 sub-clusters (Fig. 2C), revealed the largest degree of transcriptional separation between CAF1 and CAF2–4.
Figure 2. Combined MEK and STAT3 inhibition uncovers stromal plasticity and emergence of a CAF population with mesenchymal progenitor-like properties.

(A) Superimposed UMAP plot highlighting emergent CAF sub-clusters in vehicle- and MEKi+STAT3i treated PKT tumors subjected to single-cell RNA sequencing (scRNAseq); (B) Adjacent UMAP plots and circumplex plots showing absolute cell count of each CAF sub-cluster in vehicle- and MEKi+STAT3i-treated mice, respectively; (C) Pearson correlation analysis of transcriptional divergence between CAF sub-clusters in the combined vehicle- and MEKi+STAT3i CAF Seurat object. 1.00=complete correlation, 0.00=no correlation; (D) Bubble plot of pathway enrichment analysis highlighting the strongly differentially enriched pathways in each CAF cluster using fgsea (log(FC)>1. Reactome, KEGG, and GO pathways with adjusted p-value<0.05 were selected; (E) Stacked violin plots depicting expression level of select genes uniquely expressed in each CAF sub-cluster CAF1–4; (F) Dot plot depicting the expression of iCAF-defining genes Il6, Cxcl1, and Il33 across different CAF sub-subsets comparing vehicle- and MEKi+STAT3i-treated cohorts; (G) Single-cell lineage trajectory analysis performed using Monocle3 pipeline colored by Pseudotime timestamps (adjoining legend) in CAF-only cluster depicting 4 major putative CAF cellular sub-clusters in superimposed UMAP plot. The major trajectory is depicted by the continuous solid starting at (1), and two minor bifurcations indicated by branchpoints (2) and (3). CAF sub-clusters are nominated by gene expression identity and putative pathway enrichment into mesenchymal progenitor (CAF1), secretory/inflammatory (CAF2), antigen-presenting (CAF3), and myofibroblastic (CAF4); (H) Expression of lineage state-defining markers (Cd34, Il6, Cd74, Tgfb1) on Pseudotime scale across the CAF sub-clusters.
Closer inspection of differentially expressed single-cell transcriptomes in CAF1–4 sub-clusters revealed distinct transcriptional programs that reflected functional specialization. Differentially expressed genes were subjected to pathway enrichment analysis to determine functional signatures defining each CAF subset. CAF1—the most transcriptionally disparate from the other three sub-clusters—was strongly enriched in pathways reflecting fundamental processes related to fibroblast biology, such as matrix synthesis/organization, response to wounding, and elastic fiber formation (Fig. 2D). This distinct signature correlated with the concentration of markers Ly6a (Sca-1), an MSC marker detected early in pancreatic neoplasia but progressively lost during tumorigenesis21; Col14a1, a discriminatory marker for matrix fibroblasts22; and the fibrocytic marker Cd34, suggesting its putative origin from a bone marrow-derived hematopoietic/mesenchymal progenitor-like lineage (Fig. 2E). Based on these features, CAF1 was nominated as MSC-like mesenchymal progenitor CAF.
To validate these findings, histologic analysis in tumor sections from orthotopic KPC mice revealed disproportionate concentration of CD34 and PDPN co-expressing fibroblasts in MEKi+STAT3i-treated tumors which were not observed in vehicle-treated tumors. In addition, these MSC-like CD34+PDPN+ CAFs appear to preferentially encircle non-malignant ductal/acinar structures in MEKi+STAT3i-treated tumors (Fig. S5C). CAF1 also strongly express the MSC marker Meflin (lslr; Fig. S5D), recently proposed as a marker of tumor-restraining CAFs in PDAC,23 together raising the intriguing possibility that these MEKi+STAT3i-licensed CAF1 populations may have analogous properties.
The CAF2 sub-cluster was significantly enriched for pathways associated with pro-inflammatory/immunomodulatory function, including cytokine signaling involved in innate immune responses (Fig. 2D). The concentration of secretory markers Il6, Cxcl1, and Il33 in the CAF2 subset was consistent with its inflammatory transcriptional program (Fig. 2F). The global reduction in CAF-specific Il6/Cxcl1 expression following MEKi+STAT3i (Fig. 1E), therefore, was driven predominantly by attenuation of CAF2 (Fig. 2F). CAF2 was nominated as secretory/inflammatory CAF.
The CAF3 sub-cluster was enriched in genes related to antigen-presenting function Cd74, H2-ab1, H2-Eb1 (Fig. 2E) and MHC class II-related pathways (Fig. 2D); interestingly, Fcer1g—a classical macrophage marker—and FcγR-dependent phagocytosis pathways were also enriched in CAF3, suggestive of a shared lineage with myeloid-derived antigen-presenting cells. Thus, CAF3 was nominated as antigen-presenting CAF.
The CAF4 sub-cluster was enriched in Tgfb1, Acta2, and Ccn2 (Fig. 2E), indicative of canonical myofibroblastic functions such as smooth muscle contraction and focal adhesion, as well as enrichment of TGF-β signaling (Fig. 2D). To validate this attenuation of Tgfb1-enriched CAF4 subsets in MEKi+STAT3i-treated PKT tumors, we observed significant reduction in Tgfb1 expression (P<0.001) and TGF-β signaling (P-adj=0.036) in Mek1KOStat3KO compared with EV CAF transcriptomes in vitro (Fig. S5E). Notably, the myCAF marker Lrrc15—recently implicated as a master regulator of CAF-mediated resistance to immunotherapy6—was exclusively expressed in CAF4 (Fig. 2D), and its expression abrogated in MEKi+STAT3i-treated PKT tumors (Fig. S5F). CAF4 was therefore nominated as myofibroblastic CAF.
Pseudotime lineage reconstruction reveals differential developmental trajectories of CAF subsets following MEKi+STAT3i
We next investigated if the transcriptionally disparate CAF subsets uncovered by MEKi+STAT3i treatment had convergent or divergent developmental trajectories. Ordering of cells in pseudotime lineage reconstruction analysis24 arranged CAFs into a major trajectory commencing with mesenchymal progenitor CAFs—transcriptionally similar to Hosein et al. FB2, progressively lost during PDAC tumorigenesis21—with two minor bifurcations: secretory/inflammatory CAFs (i.e., CAF2) and myofibroblastic CAFs (i.e., CAF4) (Fig. 2G). CAFs expressing MSC markers Cd34 and Ly6a preferentially distributed at the beginning of all paths (Fig. 2H; Fig. S6), while lineage-determining genes Il6, Cd74, and Tgfb1 increased in density and expression levels progressively in CAFs nominated as secretory/inflammatory, antigen-presenting, and myofibroblastic, respectively (Fig. 2H; Fig. S6).
Interestingly, the enrichment of canonical mesothelial markers Myrf, Gpm6a, and Nkain4 in the predicted lineage paths of secretory/inflammatory as well as antigen-presenting CAFs (Fig. S6) suggest a co-evolution of these populations from a shared mesothelial ontology. Expression of these mesothelial markers dissipated in the lineage paths at the 2nd trajectory bifurcation toward myofibroblastic CAFs, revealing instead an enrichment in contractile genes Acta2 and Ccn. Taken together, these data suggest that MEKi+STAT3i treatment uncovers CAF plasticity by enriching for an MSC-like progenitor CAF subset while attenuating mesothelial- and myofibroblast-derived CAF populations in the TME.
MEKi+STAT3i reprograms the myeloid microenvironment and facilitates intratumoral T-cell trafficking via a CAF-dependent manner
Given the mitigation of IL-6/CXCL1-producing iCAF subsets following MEKi+STAT3i treatment, we examined if this reprogramming of stromal inflammation results in remodeling of the innate immune microenvironment in vivo.25 Indeed, flow cytometric analysis of PKT tumors (n=8–10 mice/arm) revealed a significant decrease in CD11b+ myeloid cells and F4/80+ macrophages (Fig. S7A).
To comprehensively characterize innate immune remodeling following MEKi+STAT3i at single-cell resolution in PKT mice, we performed multiparametric profiling of pooled tumor samples utilizing time-of-flight mass cytometry (CyTOF). Compared with vehicle treatment, MEKi+STAT3i, resulted in broad-based changes in the innate (CD11b+), adaptive (CD4+ and CD8+), and humoral (CD19+) immune compartments (Fig. 3A). Compared with vehicle treatment, MEKi+STAT3i treatment resulted in a dramatic decrease in CD11b+ myeloid cells, F4/80+ macrophages, M2-like macrophages (F4/80+CD206+), and MDSCs (CD11b+F4/80−Ly6G+/Ly6C+) (Fig. 3B). scRNAseq of PKT tumors analysis confirmed these broad-based immunologic changes following MEKi+STAT3i (Fig. 3C). Beyond decreased abundance of monocyte/macrophage and granulocytic MDSC identities, MEKi+STAT3i resulted in transcriptional reprogramming of tumor-infiltrating monocytic/macrophage sub-clusters from an alternatively activated M2-like phenotype—characterized by enrichment of Arg1, Thbs1, and Chil3—to a classically activated M1-like phenotype—characterized by enrichment of MHC-II genes H2-Eb1, H2-Ab1, and M1-master regulator Ciita26 (Fig. 3D).
Figure 3. Combined MEK and STAT3 inhibition reprograms the immunosuppressive myeloid microenvironment and facilitates intratumoral T-cell trafficking in part via a CAF-dependent manner.

(A) Mass cytometry time-of-flight (CyTOF) FlowSOM plots depicting changes in total myeloid (CD11b+), T-cell (CD4+ and CD8+) and B-cell (CD19+) populations in PKT mice treated with vehicle or MEKi+STAT3i (n=7–8 mice/arm); (B) Representative viSNE plots demonstrating changes in CD11b+ myeloid sub-populations F4/80+ macrophages, CD206+ M2-like macrophages, and Ly6GC myeloid-derived suppressor cells between vehicle- and MEKi+STAT3i-treated PKT mice as analyzed by CyTOF. Parent CD11b+ cell populations are denoted by dashed line; (C) Circumplex plot from single-cell RNA sequencing (scRNAseq) analysis in PKT mice treated with either vehicle (grey) or MEKi+STAT3i (green) showing relative abundance of T-cell, B-cell, granulocytes, and monocyte/macrophage immune cell clusters between treatment groups; (D) Volcano plot of transcripts associated with monocyte/macrophage polarization toward M1 (Ciita, H2-Ab1, H2-Eb1) or M2 (Chil3, Arg1, Thbs1) skewness that are significantly overexpressed (right) or underexpressed (left) in MEKi+STAT3i-treated vs. vehicle-treated scRNAseq monocyte/macrophage transcriptomes. FDR-corrected P-value and log2(fold change) thresholds were established at ≤0.05 and ≥0.5, respectively; (E) Flow cytometric analysis comparing global CD3+ T-cell populations, shown in representative contour plots (left), as well as CD4+ and CD8+ T-cell subsets, shown in histograms (right), between vehicle and MEKi+STAT3i-treated PKT mice (n=8–10 mice/arm); (F) Representative viSNE plots demonstrating changes in CD3+, CD4+, CD8+, and CD69+ T-cell populations between vehicle- and MEKi+STAT3i-treated PKT mice as analyzed by CyTOF. Parent CD3+, CD4+, or CD8+ T-cell populations are denoted by dashed line, where applicable; (G) Schematic showing generation of CRISPR/Cas9 genetic silencing of Mek1 and Stat3 in KPC cancer-associated fibroblasts, and orthotopic injection of KPC tumor cells with either empty vector (EV) CAFs or Mek1KOStat3KO CAFs in syngeneic C57B/l6 mice, followed by immunophenotyping of established tumors by flow cytometry. Histograms showing total numbers of (H) CD11b+, F4/80+, and Ly6GC+ myeloid cells, and (I) TCR-β, CD8+, and CD4+ T-cells in CAF-EV vs. CAF-Mek1KOStat3KO tumors (n=7–8 mice/group). Data are shown as mean ± SEM. ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001.
This innate immune remodeling following MEKi+STAT3i was associated with significant increases in proportions of total (CD3+), T-helper (CD4+), cytotoxic (CD8+), and activated (CD69+) T-cell populations by flow cytometry (Fig. 3E) and CyTOF immunoprofiling (Fig. 3F), as well as improved effector (CD44+) and central (CD62L+) memory T-cell markers, and expanded TCRβ expression via CyTOF profiling (Fig. S7B) in PKT mice. These broad-based changes in innate and adaptive immune populations following MEKi+STAT3i treatment were validated in orthotopic KPC tumor-bearing models via flow cytometry (Fig. S7C).
We next sought to determine if CAF-specific silencing of Mek1/Stat3 recapitulates the immunologic remodeling following systemic MEKi+STAT3i treatment. Flow cytometric immunophenotyping in orthotopic models using KPC6694c2 tumor cells co-injected with either CAF-EV or CAF-Mek1KOStat3KO (1:9 ratio) (Fig. 3G) revealed a significant reduction in CD11b+, F4/80+, and Ly6GC+ myeloid populations (Fig. 3H) while conversely demonstrating significantly increased TCR-β+, CD8+, and CD4+ T-cell populations in CAF-Mek1KOStat3KO compared with CAF-EV tumors (Fig. 3I). Taken together, these data suggest that combined MEKi+STAT3i reprograms stromal inflammation to promote an anti-tumor immune microenvironment by decreasing immunosuppressive myeloid cell populations and augmenting T-cell trafficking, in part, via a CAF-dependent mechanism.
Combined MEKi+STAT3i controls PDAC growth in a T-cell dependent manner in vivo
Given these immunologic changes, we sought to determine if the anti-tumor effects of MEKi+STAT3i are T-cell dependent by performing concurrent CD4+ and CD8+ T-cell depletion in this model (Fig. S8A–C). Non-T-cell depleted PKT mice treated with MEKi+STAT3i showed significant decreases in tumor weight compared with vehicle treatment and demonstrated 100% survival up to 80 days of treatment (Fig. 4A&B). In contrast, T-cell depleted mice treated with MEKi+STAT3i showed no decrease in tumor weight compared with vehicle-treated mice (Fig. 4A), despite confirmation of pERK and pSTAT3 inhibition in tumor lysates (Fig. 4C). Furthermore, median survival of T-cell depleted MEKi+STAT3i mice was significantly diminished compared with MEKi+STAT3i treatment without T-cell depletion (median 60.5 vs. not reached [NR] days, respectively; log-rank p=0.0001; Fig. 4B). These results show that MEKi+STAT3i-induced tumor control and survival improvements are T-cell dependent.
Figure 4. Combined MEK and STAT3 controls PDAC growth in a T-cell dependent manner and overcomes immunotherapy resistance in vivo.

PKT mice were treated with vehicle, MEKi+STAT3i, or MEKi+STAT3i following T-cell depletion with anti-CD4 and anti-CD8 antibodies (scheme in Fig. S8). Differences in (A) pancreas weight at sacrifice and (B) overall survival were compared among treatment arms. (C) Representative western blot demonstrating target inhibition of pERK1/2 and pSTAT3 in mice treated with MEKi+STAT3i ± T-cell depletion; (D) Metascape pathway enrichment analysis depicting top 17 signaling pathways from MSigDB compendium differentially upregulated in MEKi+STAT3i-treated compared with vehicle-treated T-cell single-cell transcriptomes from scRNAseq analysis in PKT tumors. Bolded pathways highlight those related to interferon signaling, T-cell activation, chaperone-mediated protein folding, and negative regulation of T-cell apoptosis; (E) Volcano plot of transcripts associated with aforementioned pathways that are significantly overexpressed (right) or underexpressed (left) in MEKi+STAT3i-treated vs. vehicle-treated single cell T-cell transcriptomes. FDR-corrected P-value and log2(fold change) thresholds were established at ≤0.05 and ≥0.5, respectively; (F) Mass cytometry time-of-flight (CyTOF) analysis of PKT mice treated with vehicle or MEKi+STAT3i showing an increase in PD-1+ tumor-infiltrating CD8+ T-cells, as visualized by representative viSNE plots (F, left). Flow cytometric analysis showing increase in degranulating CD8+PD-1+CD107a+ T-cells and non-degranulating CD8+PD-1+CD107a− T-cells in MEKi+STAT3i treated vs. vehicle-treated mice (n=8–10 mice/arm) (F, right); (G) Representative images from H&E sections from PKT mice treated with vehicle, αPD-1, MEKi+STAT3i, or MEKi+STAT3i plus αPD-1 for 4 weeks (G, left). Percent tumor area (G, right) at endpoint sacrifice were compared between treatment arms (n=5 mice/arm); (H) Kaplan-Meier survival plot depicting overall survival in PKT mice treated with vehicle, αPD-1, MEKi+STAT3i, or MEKi+STAT3i plus αPD-1 beginning at 4–4.5 weeks of age. Log-rank survival comparisons between individual treatment groups and overall cohort, as well as adjoining table with median survival times, are provided. Where applicable, data are shown as mean ± SEM. ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001.
MEKi+STAT3i treatment promotes a distinct transcriptional and functional reprogramming in tumor-infiltrating T-cells
We next sought to investigate if transcriptional and functional reprogramming of the T-cell compartment underlies the T-cell dependent effects of MEKi+STAT3i-induced tumor control. Interrogation of single-cell T-cell transcriptomes revealed global skewness towards effector versus exhausted programming in single-cell T-cell transcriptomes of MEKi/STAT3i-treated tumors (Fig. S9A), as well as enrichment of pathways related to interferon signaling, T-cell activation, chaperone-mediated protein folding, and negative regulation of T-cell apoptosis (Fig. 4D). In particular, we observed differential regulation of genes related to TCR activation (e.g., lymphocyte-specific protein tyrosine kinase Lck upregulation27), anti-exhaustion programming (e.g., Stat1 upregulation28, Apoe downregulation), effector differentiation/function (e.g., Hsp90aa1, Hsp90ab1, Hspd1, Hsph1 upregulation29) in MEKi+STAT3i-treated T-cell transcriptomes (Fig. 4E).
In further exploring the functional status of tumor-infiltrating CD8+ T-cells in MEKi+STAT3i-treated PKT mice, CyTOF profiling revealed a significant upregulation of antigen-experienced tumor-infiltrating PD1+CD8+ T-cells in PKT tumors treated with MEKi+STAT3i compared with vehicle treatment (Fig. 4F left). Despite a significant increase in antigen-experienced degranulating CD8+PD1+CD107a+ T-cells following MEKi+STAT3i, we also observed an increased proportion of PD1+CD107a− non-degranulating CD8+ T-cells (Fig. 4F right). Of note, we found no significant changes in PD-L1 expression in PKT tumors between vehicle treatment, MEKi or STAT3i monotherapy, and combined MEKi+STAT3i treatment (Fig. S9B&C). These data suggest that MEKi+STAT3i treatment—directly or indirectly—may uncouple effector/anti-apoptotic transcriptional programs from dysfunctional programs in tumorinfiltrating CD8+ T-cells to augment their degranulating capacity in vivo.
Treatment with MEKi+STAT3i overcomes resistance to PD-1 blockade in PKT mice
Viewed broadly, these data suggest that the addition of MEKi+STAT3i to ICI may overcome resistance to immunotherapy. Accordingly, PKT mice were treated with vehicle, αPD-1 monotherapy, MEKi+STAT3i, and MEKi+STAT3i combined with αPD-1 antibody (Fig. S10A). Consistent with our previous data18, MEKi+STAT3i significantly reduced tumor burden compared with vehicle treatment in PKT mice while αPD-1 monotherapy had no effect on tumor burden compared with vehicle treatment. Importantly, the addition of MEKi+STAT3i to αPD-1 blockade significantly reduced tumor burden (Fig. 4G) and pancreas weight (Fig. S10B) compared to αPD-1-treated as well as MEKi+STAT3i-treated mice. In fact, in this aggressive PDAC model, where the pancreata of vehicle- and αPD-1-treated mice are completely tumor-replaced by 6 weeks, MEKi+STAT3i-treated mice showed only 19% tumor area while the addition of MEKi+STAT3i to αPD-1 treatment maintained normal pancreas architecture with <6% tumor area after 4 weeks of treatment (~8 weeks of age; Fig. 4G left).
Addition of MEKi+STAT3i to PD-1 blockade significantly improves survival in PKT mice
Confirming our previous data,18 MEKi+STAT3i significantly increased survival compared with vehicle treatment (median 103 vs. 44 days; p=0.0013; treatment schema in Fig. S10C). Consistent with the lack of response to αPD-1 monotherapy in PDAC,30 we observed no difference in survival between αPD-1 monotherapy and vehicle treatment in PKT mice (median 57 vs. 44 days, p=0.34). The addition of MEKi+STAT3i to PD-1 blockade dramatically improved survival compared with αPD-1 monotherapy (median 181 vs. 57 days, p=0.0005), as well as combined MEKi+STAT3i (median 181 vs. 103 days, p=0.0064; Fig. 4H). There was no difference in body weight between vehicle and treatment cohorts, indicating that the combination of MEKi+STAT3i with αPD-1 is well tolerated with no additional toxicity compared with either αPD-1 monotherapy or MEKi+STAT3i in vivo (Fig. S10D). These results highlight the significant role of MEKi+STAT3i in overcoming resistance to ICI in an immunologically inert autochthonous PDAC model.
Addition of MEKi+STAT3i to PD-1 blockade improves recruitment, activation, and functional cytotoxicity of tumor-infiltrating T-cells
CyTOF-based immune profiling of PKT tumors revealed a significant increase in tumor-infiltrating CD4+ and CD8+ T-cells, as well as CD3+ T-cells demonstrating T-cell activation markers CD44 and CD69 and central memory marker CD62L with the addition of MEKi+STAT3i to PD-1 blockade compared with αPD-1 monotherapy and vehicle treatment (Fig. 5A). Corroboration of these broad-based changes in adaptive immune populations by flow cytometry in the CD4+ and CD8+ T-cell compartments separately showed that, compared with vehicle treatment and αPD-1 monotherapy, addition of MEKi+STAT3i to PD-1 blockade significantly augmented proportions of activated (CD69+), effector memory (CD44+CD62L−), central memory (CD44+CD62L+), tissue resident memory (CD44+CD62L−CD103+), and degranulating effector (CD44+CD62L−CD107a+) tumor-infiltrating CD4+ and CD8+ T-cells (Fig. 5B, Fig. S11).
Figure 5. Addition of MEKi+STAT3i to PD-1 blockade augments recruitment, activation, and functional cytotoxicity of tumor-infiltrating T-cells.

(A) viSNE plots demonstrating levels of tumor-infiltrating CD3+ cell subsets (CD4+, CD8+, CD69+, CD44+, and CD62L+) in vehicle, αPD-1, and combined MEKi+STAT3i/αPD-1 treated PKT mice as analyzed by CyTOF (n=7–8 mice/arm); (B) Analysis by flow cytometry showing changes in total CD3+, CD4+, and CD8+ tumor-infiltrating T-cells as well as levels of activated CD69+ and degranulating effector (CD4+CD62−CD107+) CD4+/CD8+ T-cells in PKT mice treated with vehicle, αPD-1, or MEKi+STAT3i/αPD-1 (n=8–10 mice/arm); (C) Schematic depicting ex vivo tumor cell:splenocyte co-culture experiment. Splenocytes were isolated from PKT mice following treatment with vehicle, αPD-1, and MEKi+STAT3i/αPD-1 and co-cultured with irradiated PKT tumor cells for 72 hours. IFN-γ release was determined by ELISA (right). (D) ELISA demonstrating granzyme B levels in total tumor lysate in vehicle, αPD1, and MEKi+STAT3i/αPD-1 treatment arms (n=3 mice/arm). Data are shown as mean±SEM. Scale bar = 50 μm. ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
We next investigated if the addition of MEKi+STAT3i to PD-1 blockade enhances functional cytotoxicity of tumor-infiltrating CD8+ T-cells. Addition of MEKi+STAT3i to PD-1 blockade significantly increased CD8+ T-cell-mediated cytolysis of PKT-derived EpCAM+ tumor cells in ex vivo CD8+ T-cell:tumor cell IFN-γ release assays (Fig. 5C) and increased tumor-specific granzyme-B levels in PKT tumor lysates (Fig. 5D), compared with αPD-1 monotherapy or vehicle treatment.
Together, these data show that the addition of MEKi+STAT3i to PD-1 blockade invigorates the adaptive immune compartment to overcome T-cell dysfunction and ICI resistance. Moreover, it is plausible that MEKi+STAT3i, which improves tumor-infiltrating CD8+ T-cell antigen experience but does not completely rescue their degranulating capacity (Fig. 4F), primes an activated CD8+ T-cell compartment for ICI to further augment the cytolytic function of PDAC-infiltrating CD8+ T-cells.
Dampening of suppressive innate immune microenvironment in PKT mice is driven by MEKi+STAT3i but not anti-PD-1 monotherapy
Given recent evidence that myeloid cell-specific PD-1 ablation—independent of T-cell-directed anti-PD-1 effects—may contribute to effector T-cell infiltration and antitumor immunity,31 we sought to investigate the differential effects of PD-1 blockade alone and combined MEKi+STAT3i/αPD-1 on the innate immune microenvironment in PKT tumors. CyTOF-based immune profiling revealed that αPD-1 monotherapy had no impact on innate immune infiltration in PKT tumors. The addition of MEKi+STAT3i to PD-1 blockade, however, significantly reduced the recruitment of tumor-infiltrating F4/80+ macrophages, CD206+ M2-like macrophages, and Ly6GC+ MDSCs compared with αPD-1 monotherapy or vehicle treatment (Fig. 6A). Flow cytometric analysis (Fig. 6B) and tissue-based immunofluorescence (Fig. 6C) confirmed a significant decrease in CD11b+ myeloid, F4/80+ macrophages, and Ly6G+ MDSCs following MEKi+STAT3i/αPD-1 compared with αPD-1 monotherapy or vehicle treatment. These data reinforce that the dampening of immunosuppressive innate populations in the PDAC TME is driven predominantly by MEKi+STAT3i without significant contributions from αPD-1 monotherapy.
Figure 6: Differential effects on innate immune remodeling are driven by MEKi/STAT3i with minimal contribution from anti-PD-1 monotherapy in PKT mice.

(A) viSNE plots depicting mass cytometry time-of-flight (CyTOF) analysis of CD11b+ myeloid cell subsets (F4/80+, CD206+, and Ly6G+C+) in PKT mice treated with vehicle, αPD-1, and MEKi+STAT3i plus αPD-1 for 4 weeks (n=7–8 mice/arm); (B) Levels of total myeloid (CD11b+), macrophage (CD11b+F4/80+), and PMN-MDSCs (Ly6G+Ly6CloF4/80−) cells were determined by flow cytometry in PKT tumors treated with vehicle, αPD1, and MEKi+STAT3i+αPD1 (n=7–8 mice/arm); (C) Immunofluorescent staining and quantification of CD11b+, F4/80+, and Ly6G+ levels in PKT mice among indicated treatment arms vehicle, αPD-1, and MEKi+STAT3i plus αPD-1 (n=3–4 mice/arm). Data are shown as mean ± SEM. Scale bar = 50 μm. ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
Combination MEKi+STAT3i and PD-1 blockade demonstrates efficacy in a metastatic PDAC patient with chemotherapy-refractory disease
A 69-year-old male presented with a resectable microsatellite stable, low-tumor mutation burden PDAC in the pancreatic body. He received neoadjuvant fluorouracil (5-FU), leucovorin, irinotecan, and oxaliplatin (FOLFIRINOX) for three months before undergoing curative-intent distal pancreatectomy/splenectomy. Pathology revealed a margin-negative resection, and a 2-cm focus of residual adenocarcinoma with 0/33 involved lymph nodes. He then completed three months of adjuvant FOLFIRINOX (Fig. 7A). Two months following completion of chemotherapy, he was diagnosed with a local recurrence in the pancreatic bed as well as a metastatic lesion in the liver (Fig. 7B). Second-line chemotherapy with gemcitabine/nab-paclitaxel was ineffective and he developed progression of disease with significant reduction in performance status.
Figure 7: Combination MEK, STAT3, and PD-1 inhibition demonstrates efficacy in a metastatic PDAC patient with chemotherapy-refractory disease, laying groundwork for clinical trial.

(A) Treatment timeline for patient with chemotherapy refractory PDAC prior to initiation of Trametinib, Ruxolitinib, and Nivolumab treatment. Pre-treatment (B) and post-treatment (C) PET/CT scan showing a significant reduction in size and FDG avidity of both locally recurrent tumor in pancreatic bed (white arrows) and in a segment VII liver metastasis (yellow arrows) on PET/CT imaging following 3 months of treatment with Trametinib, Ruxolitinib, and Nivolumab; (D) Design of Phase 1 clinical trial investigating Trametinib (MEKi), Ruxolitinib (STAT3i), and Retifanlimab (αPD-1) in advanced/metastatic pancreatic cancer patients. Proposed treatment schedules, as well as scheme for biopsies/blood collection for correlative endpoints, are shown.
After multidisciplinary discussion, he received off-label combination treatment with MEKi Trametinib (Mekinist/Novartis, 2mg PO QD), STAT3i Ruxolitinib (Jakafi/Incyte, 5mg PO BID), and αPD-1 Nivolumab (Opdivo/Bristol-Myers Squibb, 240mg IV Q2W) (Fig. 7A). Within one month of commencing treatment, the patient experienced a dramatic improvement in performance status. While CA19–9 biochemical response was not an actionable marker for his disease, PET/CT obtained after 3 months of treatment demonstrated reduction in size from 43 mm to 23 mm in the pancreatic bed mass, and metabolic response (SUVmax from 8.4 to 6.5) in the liver metastasis post-treatment (Fig. 7C). The regimen was well-tolerated with no major adverse events while on therapy.
Based on this encouraging signal of clinical efficacy and tolerability, and extensive preclinical rationale detailed herein, we are pursuing an investigator-initiated Phase I clinical trial (NCT05440942) investigating this treatment combination in advanced PDAC patients (Fig. 7D).
DISCUSSION
The present study uncovers a novel treatment strategy of CAF reprogramming that remodels stromal inflammation and myeloid-enriched immune microenvironments to activate functional T-cell anti-tumor immunity and overcome ICI resistance in PDAC. Resistance to ICI is a hallmark of PDAC,4, 5 defined by T-cell exclusion and tissue-level immune tolerance driven by KRAS-mediated tumor-intrinsic programs1, 16 as well as interwoven suppressive cellular networks comprising innate immune populations and tumor-permissive CAF signaling. Our therapeutic approach using MEKi+STAT3i not only disrupts oncogenic signaling via reciprocally activated RAS/MEK/ERK and JAK/STAT3 pathways in PDAC tumor cells18 but also promotes a fibroblast “switch” in the TME to overcome the critical barriers to ICI sensitivity in PDAC: stromal inflammation via IL-6/CXCL1-secreting iCAFs,8, 10, 11 Lrrc15+ myCAF-driven immunotherapy resistance,6 immunosuppressive myeloid cell-enriched tumor ecosystems, and T-cell exclusion and dysfunctional programming.3 We leverage these mechanistic underpinnings in highly aggressive and ICI-resistant preclinical models to demonstrate an impressive improvement in tumor burden and survival when combining MEKi+STAT3i with PD-1 inhibition, compared with either MEKi+STAT3i alone or anti-PD1 monotherapy. Importantly, the efficacy and tolerability of Trametinib (MEKi), Ruxolitinib (STAT3i), and Nivolumab (anti-PD1) in a patient with metastatic chemo-refractory PDAC offers encouraging signals of its clinical translatability, and is currently being pursued in an investigator-initiated Phase 1 trial (NCT05440942).
Signaling crosstalk between PDAC tumor cells and CAFs promote its aggressive biology. Consistent with our group’s previous discovery of reciprocally activated MEK/ERK (downstream of RAS) and STAT3 signaling in tumor cells promoting therapeutic resistance,18, 19 others have reported CAF-mediated factors such as TGF-β in further instigating tumor cell-specific MEK/ERK and STAT3 co-activation to perpetuate therapy-resistant signaling.32 It is plausible, therefore, that combined MEKi+STAT3i treatment restrains PDAC outgrowth by not only ameliorating reciprocal MEK/ERK and STAT3 signaling in tumor cells, but also dampening tumor-permissive tumor cell-stromal crosstalk via attenuation of TGF-β+ myofibroblastic CAF populations in the PDAC TME. The latter observation is supported by the fact that Mek1/Stat3-silenced CAFs, in which TGF-β signaling is attenuated, restrains tumor growth in orthotopic transplantation models in vivo.
In parallel, our scRNAseq analyses demonstrate emergence of an MSC-like CAF subset in MEKi+STAT3i-treated PKT tumors, defined by expression of Cd34, Ly6a, and Lslr (Meflin). A recent study revealed that Meflin+ MSC-CAFs were precursors to α-SMA+ myCAF populations— consistent with our pseudotime lineage reconstruction studies. Genetic ablation of these MSC-like CAFs resulted in significant tumor progression with poorly differentiated histology in a PDAC model,23 suggesting that MSC-like CAF subtypes may have tumor-restraining properties. To this end our data suggest that MEKi+STAT3i-licensed CD34+PDPN+ co-expressing CAFs—which transcriptionally resemble MSC-like fibroblasts—are spatially contiguous with normal-appearing, but not malignant, ductal structures33 in MEKi+STAT3i-treated tumors. As such, a provocative mechanism by which MEKi+STAT3i exerts tumor control may be by uncovering stromal plasticity and reprogramming the CAF compartment towards a putative tumor-restraining MSC-like fibroblast phenotype. It is also possible that these CD34+ MSC-like CAFs persist in the TME and reflect an alternative fate trajectory of mesenchymal progenitors due to the absence of TGFβ+ myCAF signaling following MEKi+STAT3i treatment. Ongoing work in our laboratory is focused on understanding the prognostic and predictive relevance of CD34+ MSC-like CAFs in PDAC patients, as has recently been explored in breast cancer.34
Growing evidence highlights a prominent role for CAFs in restraining efficacy of ICI in multiple solid tumors,35 and implicates subtype-specific heterogeneity in myCAF populations as a primary driver of immunotherapy resistance.6, 36 For instance, myCAF subpopulations in breast cancer patients nominated as ecm-myCAF (overexpressing LRRC15) and TGFβ-myCAF govern ICI sensitivity via a regulatory T-cell-dependent mechanism.36 In addition, an Lrrc15+ signature was shown to be a marker of TGF-β-driven myCAFs in the PDAC TME and predicted poor responses to immunotherapy in multiple solid cancers.6 As such, one of the key mechanisms by which MEKi+STAT3i overcomes immunotherapy resistance in the present study may be by attenuating immunoregulatory Lrrc15+ myCAF sub-phenotypes in the TME. In our upcoming phase 1 trial, we will interrogate the dynamics of stromally-expressed LRRC15 during treatment and its potential as a biomarker of response to MEKi+STAT3i/anti-PD1 therapy.
The present study adds to the growing taxonomy and ontogeny of CAF heterogeneity in the PDAC TME by describing the emergence of an MSC-like CAF following MEKi+STAT3i, which transcriptionally resembles the FB2-CAF detected in early KrasLSL-G12D;Cdkn2aflox/flox;Ptf1aCre/+ (KIC) tumors but progressively lost during PDAC progression.21 Intriguingly, FB2 CAFs (Ly6a+Ly6c1+) appeared to be an incipient progenitor-like state that converged towards Il6-expressing FB1 fibroblasts—representing secretory/inflammatory CAFs.21 Our pseudotime lineage reconstruction analysis strengthens this observation by suggesting that bone marrow-derived MSC-like fibroblast populations (Cd34+Ly6a+Ly6c1+) may be precursors for mesothelial-derived antigen-presenting,37 secretory/inflammatory, and/or myofibroblastic CAF populations that predominate and co-exist in a dynamic spatially regulated equilibrium in the “mature” PDAC TME.38 Notwithstanding the fact that CAF signatures in murine—from which our inferences are largely drawn in the present study—and human tumors may vary substantially, one might speculate that a mesenchymal-to-mesothelial transition is an incipient event in CAF lineage commitment, as has been described in developmental fibroblast biology.39 If this hypothesis is true, treatment with MEKi+STAT3i uncovers stromal plasticity and mitigates stromal inflammation by either: (1) arresting the chronology of this lineage commitment fate from mesenchymal progenitor CAFs to Il6+Cxcl1+Il33+ secretory/inflammatory CAFs; and/or (2) promoting dynamic transcriptional reprogramming of secretory/inflammatory CAFs towards its MSC progenitor-like state. Our data shed light on how CAF lineage fate determination and functional specialization in PDAC might converge.38 While ongoing work in our laboratory is interrogating CAF subtype-specific function—particularly of the putative bone marrow-derived MSC-like populations apparently driven by MEKi+STAT3i treatment—these data lend support to recent evidence from lineage tracing studies linking stromal evolution from distinct cells of origin to transcriptional heterogeneity among PDAC CAFs,40 as well as dynamic sculpting of CAF plasticity and/or functional specialization during therapeutic trajectories in PDAC.41
Mechanistically, we show that MEKi+STAT3i-induced remodeling of the TME is in part CAF-dependent, since genetic silencing of CAF-restricted Mek1/Stat3 not only dampens iCAF polarization but also significantly reduces intratumoral TAM/MDSC infiltration. Beyond these indirect CAF-mediated effects on the myeloid landscape, it is also possible that direct effects of MEKi+STAT3i treatment on myeloid cellular signaling and/or myelopoiesis contributes to the observed monocyte/macrophage reprogramming from an M2- to M1-like fate. While the tolerogenic role of myeloid-specific STAT3 signaling has been well established in pancreatic and other solid tumors,42 the role of myeloid-specific MEK/ERK signaling43 and its immunologic repercussions is incompletely understood. The findings herein warrant further investigation into the differential contributions of MEK/ERK and STAT3 signaling in myelopoiesis, myeloid-specific lineage commitment, and multi-directional crosstalk with disparate immune constituents of the PDAC TME.
In summary, our data present novel insight into a combinatorial targeted therapy that overcomes ICI resistance in PDAC by unmasking CAF plasticity and mitigating stromal inflammation, reprogramming the immunosuppressive TME,3 and establishing an immunotherapy-permissive T-cell landscape. These data provide timely insight into the concept that context-dependent targeting of inflammatory and/or myofibroblastic CAFs with preservation (or selection) of their tumor-restraining CAF counterparts—and not indiscriminate stromal depletion44—will be more effective in overcoming immune exclusion and therapeutic resistance in PDAC. Furthermore, the immunologic changes associated with stromal remodeling following MEKi+STAT3i treatment and the T-cell dependence of MEKi+STAT3i-mediated tumor control underscores a complex relationship between diverging signaling cues from heterogenous CAF subtypes, tolerogenic myeloid cell signaling, and effector immune infiltration and programming.45 Our long-standing efforts in understanding the multi-faceted role of MEK/ERK and JAK/STAT3 signaling in PDAC culminate with clinical evidence of exceptional response to combination MEKi+STAT3i plus PD1 inhibition in a chemorefractory patient with metastatic PDAC. While such dramatic responses are not uniformly expected in all patients enrolled in our upcoming Phase 1 trial investigating this combination regimen in advanced PDAC (NCT05440942), the biologic consequences of MEKi+STAT3i treatment described here will be incorporated into a predictive biomarker discovery platform utilizing pre- and on-treatment tumor biopsies that may inform strategic selection of future patients for these immunotherapeutic combinations.
MATERIALS AND METHODS
In vivo studies
Tumor-bearing PKT mice were generated as previously described.19 For endpoint analyses (i.e., flow cytometry, CyTOF, histology), PKT mice were treated with vehicle (0.5% HPMC+0.1% Tween80), trametinib (MEKi, Novartis, 2.5 mg/kg, oral gavage three times weekly), ruxolitinib (STAT3i, TargetMol, 15 mg/kg, oral gavage three times weekly), αPD-1 antibody (BioXCell, Clone #BE0273, 200 μg/mouse, intraperitoneal injection twice weekly), or combined MEKi+STAT3i or MEKi+STAT3i with αPD1 beginning at 4–4.5 weeks of age. Mice were sacrificed after four weeks of treatment or when moribund. For survival studies, MEKi (2.5 mg/kg) and STAT3i (15 mg/kg) were administered by oral gavage 5 days/week starting at 4.5 weeks of age; αPD-1 antibody dosing remained intraperitoneal injection twice weekly. Six weeks after treatment initiation, frequencies of MEKi+STAT3i and αPD1 antibody dosing were reduced to three times weekly and once weekly, respectively, and continued until mice were moribund (Fig. S10). T-cell depletion experiments in PKT mice and generation of orthotopic KPC tumor-bearing transplantation models are described in Supplementary Methods.
Single cell RNA sequencing and cluster annotation
For details, refer to Supplementary Methods.
Differential gene expression, pathway analysis, and pseudotime analysis in scRNAseq dataset
For details, refer to Supplementary Methods.
CRISPR/Cas9 gene editing in KPC CAFs
To genetically ablate Mek1 and Stat3 in PDPN+ CAFs isolated from KPC mice (see Supplementary Methods), transEDIT™ Lentiviral gRNA target gene sets specific for mouse Map2k1 and Stat3 (Cat. #CCMV1101) were used (Table S3). CAFs were transduced with these lentiviral particles in conjugation with a Cas9-expressing (pCLIP-Cas9-nuclease) vector. Cells were kept in appropriate antibiotic selection (puromycin or blasticidin) for the enrichment of transduced cells. To further increase genome editing efficiency, cells transduced with CRISPR/Cas9 vectors expressing a fluorescent protein were subjected to FACS to seed single cells into a 96 well plate at a density of 1–2.0 cells in 100μL/well. Adherent cells grown out from one colony (single clonal population) were identified using microscopy and expanded. Ensuing CAF-EV, CAF-Mek1KO, CAF-Stat3KO, CAF-Mek1KOStat3KO constructs were validated for knockout of respective genes by western blot analysis using antibodies specific for total MEK1 (CST#12671S) and STAT3 protein (CST#9139S) (Fig. S4).
Bulk RNA sequencing and analysis of CRISPR/Cas9-edited CAF constructs
For details, refer to Supplementary Methods.
Flow cytometry
Preparation of single cell suspensions from pancreatic tumors or spleen samples harvested from PKT or orthotopically injected mice were performed as described previously.9 Prior to flow cytometry staining, samples were thawed, washed prior to incubation with FcR blocking reagent (Miltenyi Biotec), and subsequently stained with fluorescently conjugated antibodies listed in Table S1. For details of flow cytometry procedures, refer to Supplementary Methods. Gating strategies are depicted in Fig. S12.
CyTOF immunoprofiling
Single-cell suspensions from PKT tumors were flow sorted using FACS Aria III (BD Life Sciences) to remove non-cellular contaminants. Samples were resuspended and viability staining performed using 2.5 μM cisplatin (Fluidigm) working solution following by FcR blocking and surface staining for 30 min at room temperature using tagged antibodies as shown in Table S2. Fixation was then performed using 1.6% formaldehyde solution (ThermoFisher). Cells were suspended in 125 nM Cell-ID Intercalator-Ir solution (Fluidigm) overnight at 4°C. The following day, cells were washed and resuspended in 0.1X EQ Four Element Calibration Beads (Fluidigm) before acquisition. Data were acquired by Helios Mass Cytometer (Fluidigm) and analyzed using FlowJo v10 and Cytobank (Beckman Coulter). To generate viSNE plots, individual files from each treatment group were concatenated into a single file and then a subset of equal events were selected at random through FlowJo DownSample plugin. Total number of CD45+ cells from each group were used to create viSNE through Cytobank with 2000 iterations, perplexity of 30, and theta of 0.5.
Histologic Analysis, Western Blotting, qPCR
Histologic analysis, western blotting, and qPCR analysis was performed as described previously.18, 19, 46 For details, refer to Supplementary Methods.
T-cell Functional Cytotoxicity Assays
PKT mice were treated with vehicle, αPD-1, or MEKi+STAT3i with αPD-1 for 4 weeks before sacrifice and processing of tumor samples for flow cytometry. FACS-sorted viable EpCAM+ tumor cells and total splenocytes were isolated from each mouse. Tumor cells were irradiated (4 Gy for 7 minutes) and plated in a direct co-culture system with isolated splenocytes. Condition media was collected after 72 hours of co-culture and protein content determined by BCA method. Secreted IFN-γ was measured by ELISA as per manufacturer’s instruction (R&D Systems).
For measurement of intratumoral granzyme-B levels, pancreatic tumor homogenates were prepared from PKT mice treated with vehicle, αPD-1, or MEKi+STAT3i with αPD-1 for 4 weeks. Tumor lysates were quantified and 40 μg protein loaded for analysis of granzyme-B levels by ELISA (R&D Systems).
Statistical analysis
Descriptive statistics were calculated using Prism 9.0 (GraphPad, La Jolla, CA). Results are shown as mean±SEM. Multiple comparisons were performed using ANOVA followed by Tukey’s multiple comparisons test. The paired two-sided Student’s t-test was used for two-group comparison. Survival curves were estimated using the Kaplan-Meier method and differences between groups were assessed using the log-rank test. An α-cutoff ≤0.05 was used to define statistical significance.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT:
The hallmarks of immunotherapy resistance in pancreatic ductal adenocarcinoma (PDAC) are stromal inflammation and tolerogenic immune ecosystems. Novel therapeutic strategies to overcome these barriers to immunotherapy sensitivity are urgently needed.
NEW FINDINGS:
Combinatorial MEK and STAT3 inhibition mitigates stromal inflammation, enriches for mesenchymal stem cell (MSC)-like CAFs exhibiting a tumor-restraining phenotype, and reprograms the immune microenvironment to overcome immunotherapy resistance in PDAC.
LIMITATIONS:
Lineage tracing studies are needed to characterize the tumor-restraining function and cell-of-origin of MSC-like CAFs. Future work will identify biomarkers of response to this combination therapy in PDAC patients.
IMPACT:
These preclinical studies lay the foundation for an upcoming Phase I clinical trial investigating the tolerability and efficacy of Trametinib, Ruxolitinib, and Retifanlimab (anti-PD1) in advanced PDAC patients (NCT05440942).
Grant Support:
This work was supported by the National Institutes of Health grant 2R01 CA161976 (to N.B.M.), American Association for Cancer Research Translational Research Grant 15-65-25-MERC (to N.B.M.), National Institutes of Health T32 CA211034 (to N.B.M.), University of Miami Sylvester Comprehensive Cancer Center P30 CCSG under National Institutes of Health Award P30CA240139 (to N.B.M., J.D., and N.S.N.), KL2 career development grant by the Miami Clinical and Translational Science Institute under National Institutes of Health Award UL1TR002736 (to J.D.), University of Miami Stanley Glaser Foundation (to J.D.), American College of Surgeons Franklin Martin Career Development Award (to J.D.), Association for Academic Surgery Joel J. Roslyn Faculty Award (to J.D.), National Institutes of Health R21 CA209536 (to N.S.N.), and American Cancer Society IRG 98-277-13 (to N.S.N.)
Footnotes
Disclosures: The authors declare no potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Availability:
Single cell RNA sequencing matrix files will be deposited in NIH GEO database upon acceptance of the manuscript. All other data are available in the main text or the supplementary materials.
REFERENCES
- 1.Collins MA, Bednar F, Zhang Y, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 2012;122:639–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kerr EM, Gaude E, Turrell FK, et al. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 2016;531:110–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beatty GL, Eghbali S, Kim R. Deploying Immunotherapy in Pancreatic Cancer: Defining Mechanisms of Response and Resistance. Am Soc Clin Oncol Educ Book 2017;37:267–278. [DOI] [PubMed] [Google Scholar]
- 4.O’Reilly EM, Oh DY, Dhani N, et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marabelle A, Le DT, Ascierto PA, et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol 2020;38:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dominguez CX, Muller S, Keerthivasan S, et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov 2020;10:232–253. [DOI] [PubMed] [Google Scholar]
- 7.Hosein AN, Brekken RA, Maitra A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol 2020;17:487–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biffi G, Oni TE, Spielman B, et al. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov 2019;9:282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dosch AR, Singh S, Dai X, et al. Targeting Tumor-Stromal IL6/STAT3 Signaling through IL1 Receptor Inhibition in Pancreatic Cancer. Mol Cancer Ther 2021;20:2280–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohlund D, Handly-Santana A, Biffi G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 2017;214:579–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elyada E, Bolisetty M, Laise P, et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov 2019;9:1102–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang H, Zhang Y, Gallegos V, et al. Targeting TGFbetaR2-mutant tumors exposes vulnerabilities to stromal TGFbeta blockade in pancreatic cancer. EMBO Mol Med 2019;11:e10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi Y, Gao W, Lytle NK, et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019;569:131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catenacci DV, Junttila MR, Karrison T, et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J Clin Oncol 2015;33:4284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ko AH, LoConte N, Tempero MA, et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016;45:370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.di Magliano MP, Logsdon CD. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 2013;144:1220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Infante JR, Somer BG, Park JO, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer 2014;50:2072–81. [DOI] [PubMed] [Google Scholar]
- 18.Nagathihalli NS, Castellanos J, Lamichhane P, et al. Inverse Correlation of STAT3 and MEK Signaling Mediates Resistance to RAS Pathway Inhibition in Pancreatic Cancer. Cancer Res 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagathihalli NS, Castellanos JA, Shi C, et al. Signal Transducer and Activator of Transcription 3, Mediated Remodeling of the Tumor Microenvironment Results in Enhanced Tumor Drug Delivery in a Mouse Model of Pancreatic Cancer. Gastroenterology 2015;149:1932–1943 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ijichi H, Chytil A, Gorska AE, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev 2006;20:3147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hosein AN, Huang H, Wang Z, et al. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie T, Wang Y, Deng N, et al. Single-Cell Deconvolution of Fibroblast Heterogeneity in Mouse Pulmonary Fibrosis. Cell Rep 2018;22:3625–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mizutani Y, Kobayashi H, Iida T, et al. Meflin-Positive Cancer-Associated Fibroblasts Inhibit Pancreatic Carcinogenesis. Cancer Res 2019;79:5367–5381. [DOI] [PubMed] [Google Scholar]
- 24.Qiu X, Hill A, Packer J, et al. Single-cell mRNA quantification and differential analysis with Census. Nat Methods 2017;14:309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Somerville TD, Biffi G, Dassler-Plenker J, et al. Squamous trans-differentiation of pancreatic cancer cells promotes stromal inflammation. Elife 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buxade M, Huerga Encabo H, Riera-Borrull M, et al. Macrophage-specific MHCII expression is regulated by a remote Ciita enhancer controlled by NFAT5. J Exp Med 2018;215:2901–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li L, Guo X, Shi X, et al. Ionic CD3-Lck interaction regulates the initiation of T-cell receptor signaling. Proc Natl Acad Sci U S A 2017;114:E5891–E5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryan N, Anderson K, Volpedo G, et al. STAT1 inhibits T-cell exhaustion and myeloid derived suppressor cell accumulation to promote antitumor immune responses in head and neck squamous cell carcinoma. Int J Cancer 2020;146:1717–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bae J, Munshi A, Li C, et al. Heat shock protein 90 is critical for regulation of phenotype and functional activity of human T lymphocytes and NK cells. J Immunol 2013;190:1360–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steele CW, Karim SA, Leach JDG, et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016;29:832–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strauss L, Mahmoud MAA, Weaver JD, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ligorio M, Sil S, Malagon-Lopez J, et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019;178:160–175 e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rhim AD, Oberstein PE, Thomas DH, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014;25:735–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Westhoff CC, Jank P, Jacke CO, et al. Prognostic relevance of the loss of stromal CD34 positive fibroblasts in invasive lobular carcinoma of the breast. Virchows Arch 2020;477:717–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riaz N, Havel JJ, Makarov V, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017;171:934–949 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kieffer Y, Hocine HR, Gentric G, et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov 2020;10:1330–1351. [DOI] [PubMed] [Google Scholar]
- 37.Huang H, Wang Z, Zhang Y, et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell 2022;40:656–673 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sahai E, Astsaturov I, Cukierman E, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer 2020;20:174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheng G The developmental basis of mesenchymal stem/stromal cells (MSCs). BMC Dev Biol 2015;15:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Helms EJ, Berry MW, Chaw RC, et al. Mesenchymal Lineage Heterogeneity Underlies Nonredundant Functions of Pancreatic Cancer-Associated Fibroblasts. Cancer Discov 2022;12:484–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steele NG, Biffi G, Kemp SB, et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin Cancer Res 2021;27:2023–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kortylewski M, Kujawski M, Wang T, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med 2005;11:1314–21. [DOI] [PubMed] [Google Scholar]
- 43.Allegrezza MJ, Rutkowski MR, Stephen TL, et al. Trametinib Drives T-cell-Dependent Control of KRAS-Mutated Tumors by Inhibiting Pathological Myelopoiesis. Cancer Res 2016;76:6253–6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Cutsem E, Tempero MA, Sigal D, et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa With Nab-Paclitaxel Plus Gemcitabine for Patients With Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J Clin Oncol 2020;38:3185–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carstens JL, Correa de Sampaio P, Yang D, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun 2017;8:15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagathihalli NS, Castellanos JA, VanSaun MN, et al. Pancreatic stellate cell secreted IL-6 stimulates STAT3 dependent invasiveness of pancreatic intraepithelial neoplasia and cancer cells. Oncotarget 2016;7:65982–65992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single cell RNA sequencing matrix files will be deposited in NIH GEO database upon acceptance of the manuscript. All other data are available in the main text or the supplementary materials.