

Abstract
Nuclear DEAD box protein p68 is immunologically related to SV40 large tumour antigen and both proteins possess RNA helicase activity. In this report, we describe the structural organisation of the human p68 gene and aspects of the regulation of its expression. Northern blot and primer extension analyses indicate that, although its level is variable, the p68 RNA helicase appears to be expressed from a single transcription start site in all tissues tested. Sequence analysis revealed that the p68 promoter harbours a ‘TATA’, a ‘CAAT’ and an initiator element and contains high affinity binding sites for Sp1, AP-2, CRE and Myc. This and functional promoter analyses in transient expression assays suggest that transcriptional regulation of the p68 gene is complex. Furthermore, there are indications that p68 expression is also regulated post-transcriptionally. Steady-state pools of poly(A)+ RNA from human cells contain completely spliced p68 mRNA and alternatively spliced forms that contain introns 8–11 or 8–12 (from a total of 12 introns) and are not translated. Analysis of a conditionally p68-overproducing HeLa cell line points to negative autoregulation at the level of splicing, which is confirmed by a recently reported association of p68 with spliceosomes in human cells.
INTRODUCTION
Nuclear protein p68 has been detected because of its immunological relationship with the simian virus large tumour antigen (1) and biochemical studies revealed that both proteins express a NTP-dependent RNA helicase activity (2,3). p68 belongs to the ‘DEAD/H box’ protein family (4), members of which share a core region of nine conserved amino acid motifs (including the DEAD box) thought to function in RNA binding and unwinding (5–7). The N- and C-terminal extensions from the core region of individual DEAD box proteins show little sequence homology to each other and seem to bring about the helicase activity in specific processes like RNA splicing, nuclear export, ribosome assembly or translation (for reviews see 8–11). Above all, DEAD box proteins have drawn our attention to the importance of RNA secondary and tertiary structure and their dynamic alterations in RNA-dependent processes whereby their biochemical functions are far from being understood.
A gene closely related to p68 has been found in Saccharomyces cerevisiae (DBP2) and also in Schizosaccharomyces pombe (dbp2) (12). Deletion of the DBP2 gene, like that of several other DEAD box genes, renders yeast cells cold-sensitive for growth, most probably reflecting the greater stability of RNA duplexes at low temperature (13 and references therein). The p68 gene of man and mouse as well as the yeast homologues each contain a large intron, which has been conserved during evolution with respect to size and relative position (12–14). A more extending analysis of the genomic DNA encoding human or mouse p68 is missing. In S.cerevisiae, the intron spans 1001 nt, which is twice the size of other known introns there, and is involved in a post-transcriptional autoregulatory feedback loop of DBP2 expression (13). Whether a similar p68 expression control also exists in higher cells is unknown, though recently p68 has been shown to be associated with spliceosomes (15).
Immunohistochemical studies revealed a weak granular staining pattern within the nucleus of interphase cells that spares the nucleoli, and during telophase p68 transiently enters pre-nucleolar bodies (16). In vitro studies suggest that the RNA unwinding activity of p68 is controlled by protein kinase C phosphorylation and calmodulin binding (17), though phosphorylation cannot be detected in vivo (H.Stahl, unpublished results). Recently, a 72 kDa nuclear DEAD box protein (p72) that shows striking homology to p68 has been identified in human cells and thus may have similar biological functions (18).
p68 is found in a wide range of vertebrate species (1,19). Very recently its chicken homologue has been found to be tightly associated with the 5-methylcytosine-DNA glycosylase complex of developing embryos (20). The glycosylase complex is required for active (methylcytosine) demethylation of DNA and contains RNA rich in CpG pairs that serves as a guide for the enzyme to its DNA sites. There is strong evidence that the pattern of DNA methylation is vital for normal development of vertebrates and, interestingly, a high abundance of p68 transcripts has been detected in differentiating embryonic tissues from chickens (20) and rodents (13). In adult rat tissues, p68 mRNA and an alternatively spliced RNA form of unknown structure have been found in the steady-state RNA pool and p68 mRNA and protein seem to be differentially expressed (14).
To gain more insight into the biological role of p68 in mammalian cells we determined the genomic organisation of its human gene, including the promoter region and upstream sequences. In order to understand its post-transcriptional regulation, we determined the structure of the alternatively spliced p68 transcripts and analysed the influence of p68 overproduced from a conditional allele on endogeneous p68 mRNA production in HeLa cells.
MATERIALS AND METHODS
Isolation of genomic clones and DNA sequencing
A human genomic DNA library (generated by insertion of partial Sau3A-digested human lymphocyte DNA in λL.47.1 and kindly given to us by Dr B. Horsthemke, Essen, Germany) was screened under conditions of high stringency (21). We used a 150 bp AhaII restriction fragment from the 5′-non-coding region of the human p68 cDNA (22) that was labelled by random priming using the non-radioactive DIG system (Boehringer Mannheim, Mannheim, Germany) as a probe. Six positive clones (out of 500 000) were identified; one clone (λp68cG) contained the entire p68 gene including 5′- and 3′-flanking sequences. Fragments of the insert of clone λp68cG were subcloned into M13mp18/mp19 vectors by standard techniques (18) and sequenced by the chain termination method (23). Sequences were compiled using the McVector™ and DNA Strider™ programs.
Primer extension analysis
Cytoplasmic and nuclear poly(A)+ RNA was purified from growing HeLa-S3 cells using the Oligotex™ procedure (Qiagen) according to the manufacturer’s protocols. A synthetic oligonucleotide (3 ng), corresponding to nucleotides 41–66 (5′-GGAAGGACACCGATGACACCAGCCG-3′) of the human p68 cDNA (22), was 5′-end-labelled with [γ-32P]dATP by T4 polynucleotide kinase (sp. act. 5 × 105 c.p.m./ng), annealed to the cytoplasmic poly(A)+ RNA (5 µg) and incubated in the presence of 4 U Tth (Thermus thermophilus) DNA polymerase (Boehringer Mannheim) at 68°C for 20 min and at 95°C for 5 min in reverse transcription buffer (90 mM KCl, 10 mM Tris–HCl pH 8.9, 0.9 mM MnCl2, 200 mM dNTPs). The reaction was stopped by incubation at 37°C with 0.25 µg DNase-free RNase (Boehringer Mannheim) for 30 min. Samples were analysed by PAGE (6% polyacrylamide, 7 M urea). The primer extension oligonucleotide was also used to generate a reference sequencing ladder. Results obtained by primer extension were confirmed by RNase A protection analysis of the 5′-end of p68 poly(A)+ RNA according to Zinn (24).
Promoter analysis
DNA fragments were analysed for their ability to direct the transient expression of chloramphenicol acetyltransferase (CAT) when cloned upstream of a promoterless CAT reporter gene (pBLCAT3; 25). Deletions of the p68 promoter region were constructed by exonuclease III trimming (21) of appropriately linearised plasmids. The exact lengths of derivative clones were determined by nucleotide sequencing.
Plasmid DNA (10 µg/90 mm dish, purified using a Qiagen™ plasmid kit) was calcium phosphate co-precipitated and added to 293 cells (seeded on 90 mm diameter dishes to give 50% confluency at the time of transfection; 26). HeLa-S3 cells were transfected by the DEAE–dextran method (27) with 1.0 µg plasmid DNA/90 mm dish, and the medium was renewed after 20 h. Cells were harvested after an additional incubation period of 24 h and extracts were assayed for CAT (28) and β-galactosidase (29) activity.
Northern blot analysis
Poly(A)+ RNA (prepared from cultured cells by use of the Oligotex Direct mRNA Kit; Qiagen) was loaded (100 ng/lane) onto a denaturing agarose (1.6%) gel containing 1.85% formaldehyde. After electrophoresis, the gel was blotted onto a positively charged nylon membrane and hybridized by standard procedures as described in the DIG System User’s Guide for Filter Hybridization (Boehringer Mannheim). Detection was performed using CDP-Star chemiluminescent substrate (Boehringer Mannheim) and blots were exposed to Fuji RX films for 1 min. Equal loading was confirmed by reprobing the blot with a DIG-labelled antisense β-actin RNA probe (Boehringer Mannheim). A Human Multiple Tissue Northern Blot (Clontech no. 7760-1) with poly(A)+ RNA from various human tissues was hybridized at 43°C with random primed 32P-labelled DNA in 5× SSPE buffer supplemented with 10× Denhardt’s solution, 100 µg/ml salmon sperm DNA, 50% formamide and 2% SDS. The blot was washed under high stringency conditions and analysed by autoradiography.
Western blot analysis
Cell extracts from adult mouse tissues or cultured cells were prepared by homogenisation in lysis buffer (10 mM Tris–HCl, pH 7.5, 100 mM NaCl, 1 mM EDTA, 1% SDS) and analysed by SDS–PAGE (30) and immunoblotting (31) with monoclonal antibody C10 (raised against a peptide representing the C-terminal 15 amino acids of p68; 32) or Pab 204 (1) as previously described (33). Secondary antibody incubation was done with an anti-mouse antibody conjugated to horseradish peroxidase (1:5000 dilution) and the membrane was treated with ECL reagents and subjected to autoradiography. p68 signals were quantitated with a Molecular Dynamics densitometer.
Isolation of cDNAs from alternatively spliced p68 RNAs
A HeLa-S3 cDNA library [oligo(dT) primed; Clontech] was screened under high stringency conditions according to standard protocols (21) with a 1017 bp MunI–StuI restriction fragment representing part of the intron 11 sequence of the human p68 gene as a hybridization probe (labelled by random priming using the non-radioactive DIG system; Boehringer Mannheim). Two clones out of 600 000 plaques were isolated with cDNA inserts of ~5 kb. DNA inserts were recovered by EcoRI restriction, the resulting fragments subcloned into the plasmid pMOS (Amersham) and sequenced with an ALFexpress™ and the AutoRead™ sequencing system (Pharmacia).
Analysis of p68 RNA by RT–PCR
First strand synthesis on poly(A)+ RNA (0.5 µg, prepared as described above and pretreated with DNase I) was primed with a p68-specific downstream primer (rev pr; see Fig. 6B) and performed by Moloney murine leukemia virus (M-MuLV) reverse transcriptase (15 min at 42°C, 5 min at 99°C, 5 min at 5°C). Taq polymerase was used for subsequent PCR amplification (System 9600 protocol, GeneAmp™ RNA PCR kit; Perkin-Elmer). PCR was set to 35 cycles (15 s at 95°C, 30 s at 60°C) after an initial step (105 s at 95°C) and finished by a final step (7 min at 72°C) using a MJ PTC-100 MaxiCycler™. The sequences of used p68-specific synthetic oligonucleotide primers and their location according to the p68 genomic sequence (see Fig. 3) were as follows: pr 1, 5′-ATGCTTTGCATGACTGCC-3′ (4607–4624); pr 2, 5′-ACTACCCTAACTCCTCAGAG-3′ (5537–5556); rev pr, 5′-CCCATTGGTGTAATTTGC-3′ (6115–6132). Recombinant plasmids with p68 genomic or cDNA sequences (1.0 fmol) were used as templates for control reactions.
Figure 6.
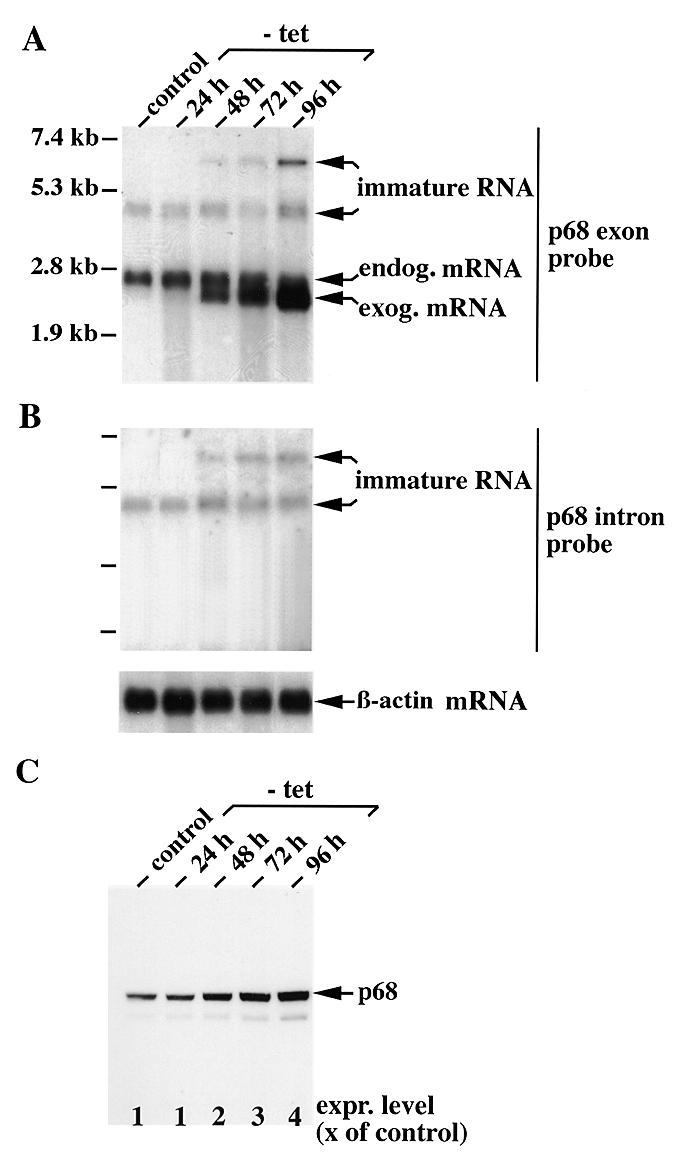
High level expression of exogenous p68 suppresses maturation of endogenous p68 mRNA. (A and B) Analysis of conditionally p68-overproducing HeLa cells for p68-specific transcripts at different times after induction. Poly(A)+ RNA was prepared from a conditionally p68-overproducing HeLa cell clone at indicated times after withdrawal (–tet) from or in the presence (control) of tetracycline (2 µg/ml) in the medium and analysed by northern blotting with DIG-labelled antisence RNA probes representing either (A) nucleotides 1–1300 (p68 exon probe) or (B) nucleotides 5336–6352 (p68 intron probe) of the human p68 cDNA or the human β-actin coding sequence (β-actin mRNA, bottom). Sizes of RNA markers are indicated on the left. (C) Western blot analysis of the same cell clone. Each lane was loaded with 20 µg protein extract from the respective cells and separated by 10% SDS–PAGE. After transfer to nitrocellulose, the blots were probed with the p68-specific monoclonal antibody Pab 204 (1). p68 signals were quantitated with a Molecular Dynamics densitometer and expressed as a multiple of the control.
Figure 3.

Analysis of the p68 gene promoter. (A) Sequence of the upstream p68 genomic DNA. The complete nucleotide sequence of the human p68 gene was determined, and the sequence of the 5′-flanking region is depicted (EMBL accession no. AF015812). The transcription initiation site is marked +1, and nucleotide numbers relative to the transcription start site are given on the left. Consensus sequences for transcription factor binding sites are underlined. (B) Activities of p68 promoter–CAT fusion genes in HeLa cell transfection assays. p68 promoter–CAT gene constructs are schematically shown on the left side of the figure. Numbers refer to p68 coordinates in (A). CAT activity was normalised for variations in transfection efficiency by dividing it by β-galactosidase activity and expressed in relation to the Herpes simplex virus thymidine kinase promoter activity obtained in parallel experiments. All results are mean values (±SD) from four independent transfection experiments.
Construction of conditionally p68-overproducing HeLa cells
For transfection of tTA-expressing HeLa cells (34) with a p68 expression vector controlled by a tTA-dependent promotor, plasmid pUHD-p68 was constructed. Briefly, a chimeric intron (nt 857–989 of pCIneo; Promega) was placed upstream of the p68 cDNA (nt 171–2018; 22) and an oligonucleotide encoding the KT3 epitope (ProProGluProGluThr; 42) was fused to its 3′-end and the whole construct was cloned into the SacII–BamHI side of pUHD10-3 (34). Transfection was performed with a mixture of 6 µg each of pUHD-p68 and pTKPURO DNA (which imparts puromycin resistance) and superfect lipid transfection reagent (Qiagen) according to the manufacturer’s instructions. Cells were selected in the presence of tetracycline (0.5 µg/ml) and puromycin (1 µg/ml) and the resulting colonies were cloned and further selected for induced p68 overproduction. Plasmid pUHD10-3 and tTA-expressing HeLa S3 cells were a generous gift from H. Bujard.
RESULTS
Structure of the human p68 gene
By sequencing we found that the genomic insert of clone λp68cG, isolated with a p68 cDNA probe (22), encodes the complete human p68 gene. The gene consists of 13 exons and 12 introns (Fig. 1A). Exon length varies between 60 and 706 bp, while the introns were between 81 and 1243 bp in size (Fig. 1B). Exon I contains the 5′-untranslated region (UTR), an AUG initiation codon and encodes the N-terminal 15 amino acids. Exons IV, VI, VII, VIII, IX, X, XI and XII each encode one or two of the nine conserved amino acid motifs characteristic of DEAD box proteins, with one motif (RGXD) being interrupted by an intron. As shown in Figure 1B, all intron–exon boundaries match with published consensus sequences (35) and each intron contains a potential lariat acceptor site upstream of its 3′ splice junction (36). The intron splice phase is type 0 (introns occuring between codons) for introns 2, 4 and 5, type I (between the first and second bases of a codon) for introns 3, 6, 10, 11 and 12 and type II (between the second and third bases of a codon) for introns 1, 8 and 9, respectively. As is typical for mammalian genes (37), the last exon of the p68 gene was found to be the largest one. It encodes the 134 C-terminal amino acids followed by a stop codon for translation (TAA) and a relatively long (301 bases) 3′-UTR with two potential polyadenylation consensus signals. A previous cDNA analysis has revealed that one of two adenosine residues positioned 16 nt downstream of the second signal functions as the poly(A) attachment site (22). The 3′-non-coding part of human p68 mRNA contains potential U-rich regulatory elements that are known to direct rapid mRNA turnover (for a review see 38).
Figure 1.

Structure of the human p68 gene (EMBL accession no. AF015812). (A) Relative distribution of exons and introns with an allocation of the conserved DEAD box protein motifs of p68 among exons. Filled boxes represent exons. Introns and flanking regions are depicted by thin lines (bar = 500 nt). Restriction sites used for subcloning are indicated. Amino acid consensus sequences of DEAD box proteins are given in single letter code. (B) Detailed structure of exon–intron boundaries. Exons are indicated by Roman numbers on the right and left side, with their sizes in parentheses. Nucleotide sequences at the exon and intron boundaries and around the putative branch sites are shown in comparison to the respective consensus sequences (M = A or C; R = A or G; Y = C or T; N = any nucleotide). The size of exon I is numbered from the transcription start site (see below). The size of exon XIII is 706 bp up to the polyadenylation site. Underneath the donor and acceptor sites, the amino acids are given in the three letter amino acid code. Note that some codons are split by intron sequences.
Localization of the transcription initiation site
Two p68-specific hybridization signals at ~2.3 and 4.4 kb can be detected in poly(A)+ RNA from rat (14) and human tissues (see below) by northern blot analysis. To examine whether p68 transcripts were synthesised from the same transcription initiation site in different tissues, primer extension analysis was performed using a primer complementary to the N-terminal sequence of p68 cDNA and poly(A)+ RNA from human liver, thymus, testis and HeLa cells. As shown in Figure 2, one major extended product of 63 bases was specifically generated in each case that ended with a T residue 171 bases upstream of the translational initiation codon. The same results were obtained by RNase A protection experiments using a complementary RNA probe that contained the p68 gene sequence from –433 to +170 (data not shown). Taken together, these experiments exclude that multiple transcriptional start sites are utilised within the 5′-UTR, including proximal upstream sequences (up to ~0.5 kb; see also Discussion). In comparison to the initiation site identified here, the human p68 cDNA contains three additional nucleotides (GGC) at the 5′-end (22). These nucleotides are also present in the genomic sequence but nevertheless may have been artificially introduced into the cDNA during library construction, since no signal was obtained at the respective position in the primer extension experiment.
Figure 2.
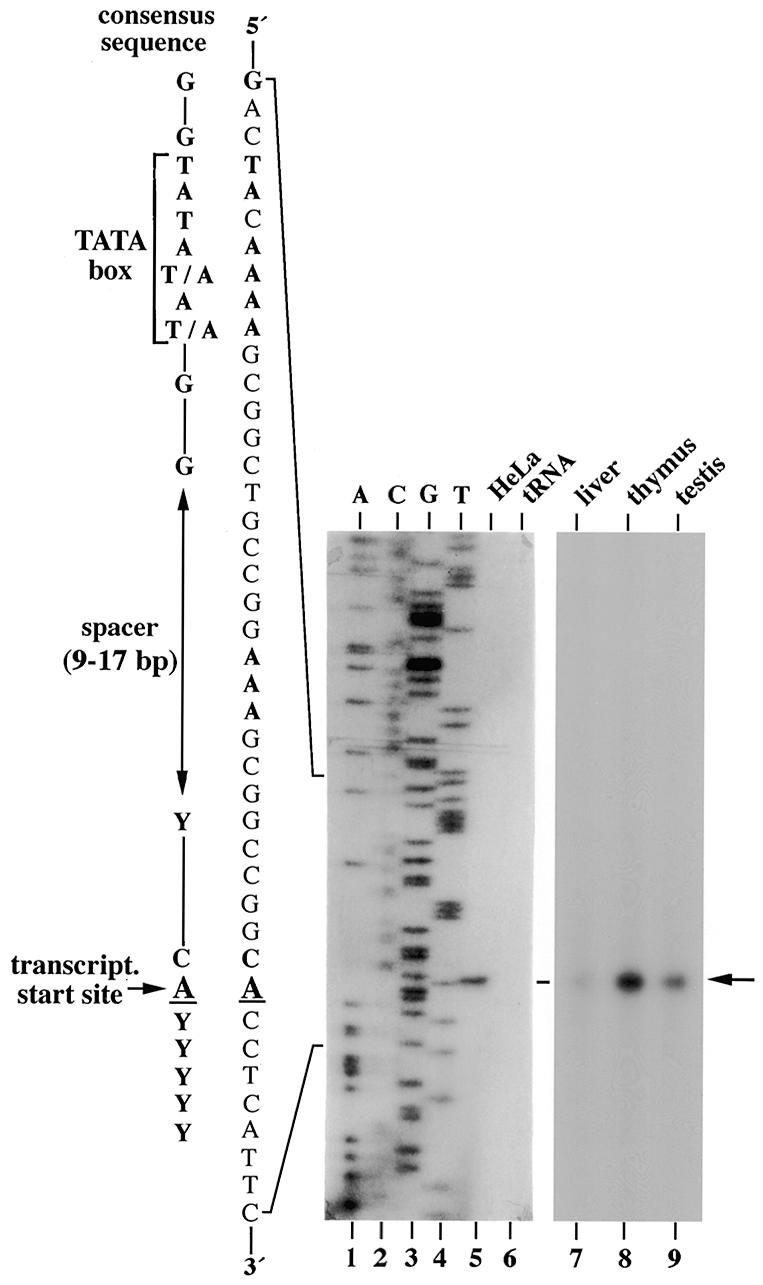
Identification of the transcription initiation site in the p68 gene. Primer extension reactions were performed with a p68-specific 32P-labelled primer (see Materials and Methods) and 5 µg of poly(A)+ RNA from HeLa-S3 cells or the indicated human tissues. tRNA was used in a control reaction. Products were analysed on a sequencing gel side-by-side with DNA sequencing reactions (lanes 1–4). The nucleotide sequence around the p68 transcription start site (indicated by an arrow) is shown on the left in comparison to the consensus sequence of TATA box-containing promoters (31).
Sequence analysis and promoter activity of the upstream genomic region
The sequence of the transcrition start site (C–1ACCTCA+5) resembles that of the consensus initiator element (Inr; CAPyPyPyPyPy) defined in eukaryotes (39), and an increased G and C content (63%) within 1 kb upstream of the start site of transcription is observed (Fig. 3A). The promoter region has a TATA (TACAAA, consensus TATAAA) and a CCAAT box (CCTAT) at positions –30 and –107, respectively (40). A search for additional promoter elements points, among others, to five Sp1-binding sites (consensus GGGCGG), a cyclic AMP response element (CRE; G/TA/TCGTCA), four AP-2 sites (GC/GC/GA/TGC/GCC), and one non-canonical (CACGG) and one canonical (CACGTG) E box motif (Fig. 3A).
We have analysed the promoter activity of the 5′-flanking region by transient transfection assays. The plasmids used contain 432 nt of the 5′-flanking sequence or shortened versions of it and the transcription initiation site, as well as 168 nt of the 5′-UTR of the human p68 gene fused to a promoterless CAT gene. They were transiently transfected into 293 (Fig. 3B) or HeLa cells (data not shown) together with plasmid pSVβ (29) to correct for variations in transfection efficiencies. When compared to the relatively weak HSV TK promoter, the p68 promoter was found to be much stronger in 293 cells and, to a lesser extent, also in HeLa cells. No activity was detected upon transfection of empty control vectors or when the –432/+168 p68 fragment was used in an antisense orientation (Fig. 3B). Removing nucleotides –432 to –382 of the p68 promoter had only a modest effect (<20% reduction in 293 cells), whereas a deletion of the next 5 bp, which destroys the canonical E box motif, cut the promoter activity in half. Deletions at nucleotides –177, –80 or –3 further reduced the promoter activity to ~10 and 5% of the original level or completely abolished it, respectively.
The p68 promoter region also contains a putative p53 binding site (TGGGAAGTTTAGAGACCATTC, consensus PuPuPuCA/TA/TGPyPyPy; 41) and a double-stranded oligonucleotide comprising this sequence was specifically bound by isolated p53 in band shift experiments, although with reduced affinity as compared with the established p53 consensus sequence. In preliminary transfection assays the same sequence, however, showed no effect on promoter activity independent of p53 and, therefore, it remains uncertain whether p68 is a p53-responsive gene (data not shown).
Cell type-specific expression of p68
We analysed the distribution of p68 mRNA in various human tissues by northern blot analysis. Two classes of p68 transcripts, ~2.3 and 4.4 kb in size, were detected with a cDNA probe in the poly(A)+ RNA from each tissue analysed (Fig. 4A, left). Their absolute level, as well as the ratio of the 2.3 to the 4.4 kb RNA, appeared different in each organ. Size comparison by gel electrophoresis with an in vitro transcript of full-length p68 cDNA identified the 2.3 kb product as mature p68 mRNA (data not shown) and, therefore, the 4.4 kb RNA was regarded as representing alternatively (or incompletely) spliced p68 RNA as has previously been found in rat (14). The blot was stripped and successively hybridized with probes specific for intron 1 or 11, which are the largest among the p68 gene introns. Now, only the 4.4 and not the 2.3 kb RNA was detected and only with the intron 11-specific probe (Fig. 4A, right, and data not shown). The 4.4 kb p68 RNA species was also found in the steady-state poly(A)+ RNA pool from HEF, 293 and HeLa cells in addition to mature mRNA and, again, the proportions of the two RNA classes to each other differed from cell line to cell line (Fig. 4B). To compare p68 RNA with protein levels, protein extracts of the cultured cells were analysed by western blotting with the p68-specific monoclonal antibody C10. This antibody binds strongly to native and denatured p68 from several species (32). The p68 content of the individual cell lines appeared to inversely correlate with the concentration of unspliced p68 RNA but not at all with that of mature mRNA (Fig. 4B). When different mouse organs were subjected to western blot analysis, p68 contents also did not correspond to their respective mRNA levels. The testes showed the highest level of p68 while all other tissues contained relatively low amounts, which nevertheless differed from each other (data not shown, but see also 14).
Figure 4.

Expression of the p68 gene in human cells. (A) Northern blot analysis of different human tissues. A Human Multiple Tissue Northern Blot [with 2 µg poly(A)+ RNA/lane; Clontech no. 7760-1] was hybridized with a p68 full-length 32P-labelled cDNA probe and analysed by autoradiography (left, exon DNA probe). The stripped membrane was rehybridized with an intron 11-specific (StuI–MunI 1017 bp fragment) 32P-labelled DNA probe (right, intron DNA probe). RNA markers, run in parallel, are indicated on the right, and estimated sizes of p68 RNAs are shown on the left. (B) p68 RNA (upper) and protein (lower) analysis of different human culture cells. Northern blot analysis with 2 µg poly(A)+ RNA from the indicated cell lines was performed as in (A) except that the blot was hybridized wih the p68 exon- and intron-specific and a β-actin probe at the same time. For western blot analysis, each lane was loaded with 20 µg protein extract from the respective cells and separated by 10% SDS–PAGE. After transfer to nitrocellulose, the blot was probed with the p68-specific monoclonal antibody C10. Only the p68 area of the membrane is shown.
Structures of incompletely spliced p68 transcripts
The 4.4 kb RNA apparently originates from alternative splicing of p68 pre-mRNA whereby the unspliced intron 11 sequence by itself cannot account for its full length. To learn more about this apparently regulated splicing process we determined the exact primary structure of this p68 RNA species. A HeLa cDNA library was screened with an intron 11-derived p68 DNA probe and two positive clones of ~4.4 kb (in addition to some smaller ones) were isolated. Sequence analysis revealed that the two cDNA clones indeed represent partially spliced p68 RNA still harbouring either introns 8–11 or 8–12 (Fig. 5A). An analysis of HeLa cell RNA by RT–PCR confirmed the existence of the two alternatively spliced p68 RNA forms in HeLa cells (Fig. 5B) while no other splice variants could be detected with respective primer combinations (data not shown). The occurrence of the alternatively spliced RNAs was, however, restricted to the nucleus, in contrast to mature p68 mRNA, which was found in both cellular compartments, with accumulation in the cytoplasm (Fig. 5B). Intron 8 contains several stop codons at its 5′-end and thus the 4.4 kb RNA encodes a truncated p68 polypeptide of 328 amino acids that should, however, not be expressed in light of its nuclear restriction. Accordingly, we were unable to identify truncated p68 in HeLa cells or human tissues by western blot analysis using polyclonal antibodies specific for an N-terminal p68 peptide (amino acids 45–59; data not shown).
Figure 5.
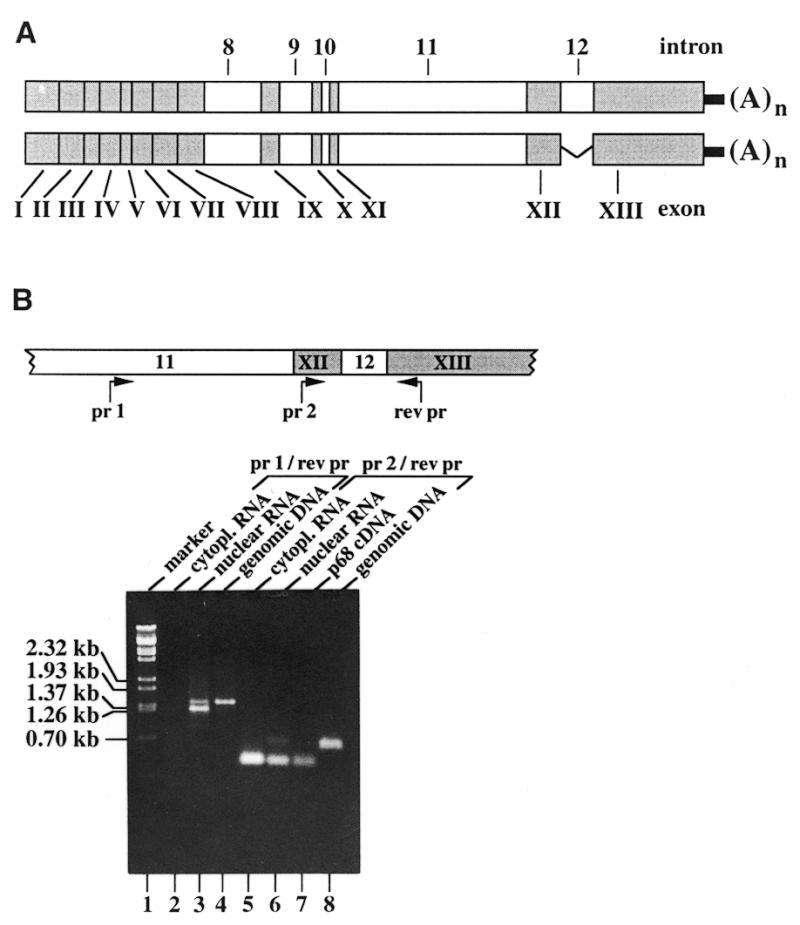
Structure and cellular localisation of the alternatively spliced human p68 RNA. (A) The structure of two alternatively spliced p68 cDNAs. Two p68-specific clones with inserts of ~4.4 kb were isolated by screening of a λgt11 HeLa cDNA library with a StuI–MunI p68 intron 11-specific probe. Determination of their sequence revealed that they are copies of alternatively spliced p68 poly(A)+ RNAs, the structures of which are schematically shown with open and filled boxes representing introns and exons, respectively. (A)n is the poly(A) tail. (B) RT–PCR analysis of HeLa-S3 poly(A)+ RNA. Cytoplasmic (lanes 2 and 5) and nuclear (lanes 3 and 6) poly(A)+ RNAs were analysed with the indicated probes. Their positions in the genomic DNA are shown in the scheme on top of this part (pr 1, intron 11-specific primer; pr 2, exon 12-specific primer; rev pr, reverse primer; arrows indicate direction of DNA synthesis). PCR reactions were performed with the same primers but with p68 cDNA (lane 7) or cloned genomic p68 DNA (lanes 4 and 8) used as controls. RT–PCR, with PCR products analysed by agarose gel electrophoresis and ethidium bromide staining. Sizes of λ DNA markers, run in parallel (lane 1), are indicated on the left side.
Analysis of conditionally p68-overproducing HeLa cells
We took advantage of HeLa cells that constitutively express a tetracycline-controlled transactivator (tTA) and integrated a p68 expression vector controlled by a tTA-dependent promotor (33). For a higher expression efficiency, the 5′- and 3′-UTRs were removed and the p68 cDNA was tagged with a chimeric intron at the 5′-end (42) and at the 3′-end with an oligonucleotide encoding the epitope of monoclonal antibody KT3 (43). Several clones were obtained that overexpressed p68 from the exogenous cDNA construct after withdrawal of tetracycline from the medium (see below). The KT3-tagged p68 was localised in the nucleus and showed RNA-dependent ATPase activity (data not shown). Furthermore, identically modified Dbp2p has been shown to rescue the cold-sensitive growth defect of yeast strain DBP2Δ (13). Exogenous p68 mRNA could first be detected by northern blot analysis 24 h after gene induction and its concentration increased strongly with time (up to 96 h after induction; Fig. 6A). Contemporaneously the cellular concentration of p68 increased more than 4-fold (Fig. 6C). p68-overproducing cells showed a slightly reduced growth rate but otherwise looked normal (data not shown). Interestingly, after induction the cells showed a decrease in endogenous p68 mRNA over time whereas the concentration of the alternatively spliced 4.4 kb RNA increased slightly. Moreover, we observed the simultaneous appearance of an even larger, most probably unspliced, p68 transcript of ~6.5 kb in the poly(A)+ RNA pool (Fig. 6A and B). The 4.4 and 6.5 kb p68 RNA species could also be detected with an intron 11-specific probe (Fig. 6B), and similar data were obtained with a second p68-overproducing HeLa cell clone. Taken together, these results indicate that the maturation of p68 primary transcripts is most probably inhibited at increasing cellular p68 concentrations at the level of splicing. The chimeric intron of the integrated p68 cDNA, in contrast, was quantitatively spliced out and unspliced p68 RNA of this size could not be detected in the steady-state poly(A)+ RNA pool (see Fig. 6A). Furthermore, maturation of other pre-mRNAs, like that of the highly p68-related DEAD box protein p72 (18), was not affected under the same conditions (data not shown). The inhibition of splicing by overproduced p68, therefore, seems to specifically affect endogenous p68 gene transcripts and apparently represents a negative autoregulatory control of p68 gene expression in human cells.
DISCUSSION
p68, a member of the DEAD box protein family, is highly conserved in higher eukaryotes (1,4). In humans it is encoded by a single gene locus (44) and its genomic organisation is shown here to be consistent with other eukaryotic genes. The transcribed part of the human p68 gene is ~6.7 kb in size and split into 13 exons. In parallel with mouse eIF-4A (45), the prototype of DEAD box proteins, the conserved RNA helicase domains, with the exception of the RGXD motif, are not interrupted by introns. Primer extension and RNase A protection experiments have identified only one start site within the 5′-UTR of the cDNA and further upstream up to ~0.5 kb in all human tissues tested. This is in contrast to the Ddx5 gene (the mouse homologue of human p68), where alternative start points seem to be used within this area in a tissue-specific manner (46). Our experiments do not exclude, however, that additional start sites for human p68 transcription may exist further upstream. In light of their structures, the resulting transcripts should, however, be larger than those identified here (see Fig. 5), except for the case when most of the 5′-transcribed sequence is spliced out. An Inr element (39) encompasses the transcriptional start site of the human p68 gene, and a suboptimal TATA box, TACAAA, is located 30 nt upstream. The TATA box binds TFIID and, along with Inr, helps assemble RNA polymerase II and other basal initiation factors into a stable preinitiation complex (for a review see 47). Point mutations in the consensus TATA box lower the rate of reinitiation several-fold (48,49). According to our transient expression studies, efficient initiation of transcription of the p68 gene seems to depend on the presence of binding sites for Sp1 (see Fig. 3B), a ubiquitous transcription factor that has been shown to specifically tether the basal transcription machinery to TATA-less promoters via interaction with the TFIID complex (50).
The p68 promoter contains a CRE, two E boxes and four binding sites for the retinoic acid-inducible transcription factor AP-2, which plays an essential role during murine development (51,52). According to our preliminary expression data, the canonical E box at least seems to be critical for p68 promoter activity (see Fig. 3). E box motifs can be recognized by Myc–Max heterodimers that are known to function in the regulation of many growth regulating genes (53). As in the human p68 gene, E boxes have also been found in association with CRE and AP-2 sites in other gene promoters resulting in negative or positive cooperativity (54–57). Furthermore, a canonical E box is also present in the 5′-region of the mouse Ddx5 gene (46) and in two other DEAD box protein genes, the Drosophila pitchoune (58) and the mouse MrDb (Myc-regulated DEAD box protein) (59) genes; in the latter case it was demonstrated to be a direct target for Myc. p68 has just been identified as functioning as a transcriptional co-activator specific for the activation function of human oestrogen receptor α and to bind to CBP, the CREB-binding protein (60). CBP bridges the CRE/CREB complex to components of the basal transcriptional apparatus, and it is possible that p68 directly influences its own transcription. Taken together, transcription of the p68 gene seems to be highly complex and its promoter structure suggests that p68 is developmentally regulated, as has been confirmed by respective analyses of mouse embryos (20).
In adult rats (14) and humans (60; this report) the p68 gene appears to be ubiquitously expressed, at least at the level of transcription, though to different extents. There seems, however, to be additional post-transcriptional control, at both the levels of splicing and translation. This is suggested by the existence of an alternatively spliced p68 poly(A)+ RNA, 4.4 kb in size (Fig. 4A), and the lack of a correlation between mRNA and protein content in tissues and cultured cells (Fig. 4B and data not shown; see also 14). We have shown here by cloning and sequence analysis that the alternatively spliced p68 poly(A)+ RNA contains either introns 8–11 or 8–12 in addition to the complete coding sequence (Fig. 5). The fact that intron 12 is spliced out from part of the larger RNA confirms the observation that splicing of the last intron seems to stimulate polyadenylation and vice versa (61). The 4.4 kb RNA seems not to be expressed. Its abundance in the nuclear steady-state pool, therefore, rather hints at a regulatory process involved in the maturation of p68 primary transcripts. A function of intron 11 in this process is suggested by the fact that it is conserved in size and relative position from yeast to man. Furthermore, it is involved in autoregulatory post-transcriptional expression control in S.cerevisiae that also requires intact Dbp2p (13). We have extended these findings to human cells by demonstrating that conditional overexpression of p68 from a cDNA causes a reduction in endogenous p68 mRNA, combined with an enrichment of unspliced and partially spliced RNA (Fig. 6). RNA transcription by polymerase II, polyadenylation, splicing and export from the nucleus are intimately linked in vivo (62), and this chain of reactions may be interrupted by a p68-induced secondary or tertiary RNA structure inaccessible to the progressing splicing machinery. Interestingly, accumulation of unspliced RNA was not observed in S.cerevisiae under the same conditions, which may hint at different handling or degradation pathways of immature mRNAs in the two species or, alternatively, at a function of the immature p68 RNA in higher cells. RNA analysis of the highly p68-related DEAD box protein p72 also revealed the existence of alternatively spliced RNA in human cells (18). In our inducible HeLa cell line, overexpression of p68 did not influence the steady-state pool of p72 mRNA, which points to a highly specific regulatory role of p68 in the processing of its primary transcript. How this role of p68 as a negative regulator of splicing can be reconciled with its function as a transcriptional co-activator is unclear. The two functions may come into effect at different cellular p68 concentrations, with co-activation at very low protein levels and splicing inhibition at higher ones. We note, however, that in light of the complexity of p68 gene expression, overexpression by nearly a factor of five is apparently not harmful to HeLa cells.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank R. Nagel for technical assistance and K. Roemer, G. Thiel and H. Uhlmann-Schiffler for helpful discussions and critical reading of the manuscript. This work was supported by a grant from the Deutsche Forschungsgemeinschaft (SFB 399) to H.S.
DDBJ/EMBL/GenBank accession no. AF015812
REFERENCES
- 1.Lane D.P. and Hoeffler,W.K. (1980) Nature, 288, 167–170. [DOI] [PubMed] [Google Scholar]
- 2.Hirling H., Scheffner,M., Restle,T. and Stahl,H. (1989) Nature, 339, 562–564. [DOI] [PubMed] [Google Scholar]
- 3.Scheffner M., Knippers,R. and Stahl,H. (1989) Cell, 57, 955–963. [DOI] [PubMed] [Google Scholar]
- 4.Ford M.J., Anton,I.A. and Lane,D.P. (1988) Nature, 322, 736–738. [Google Scholar]
- 5.Hodgman T.C. (1988) Nature, 333, 22–23. [DOI] [PubMed] [Google Scholar]
- 6.Linder P., Lasko,P.F., Ashburner,M., Leroy,P., Nielsen,P.J., Nishi,K., Schnier,J. and Slonimsky,P.P. (1989) Nature, 337, 121–122. [DOI] [PubMed] [Google Scholar]
- 7.Gorbalenya A.E. and Koonin,E.V. (1993) Curr. Opin. Struct. Biol., 3, 419–429. [Google Scholar]
- 8.Wassarman D.A. and Steitz,J.A. (1991) Nature, 349, 463–464. [DOI] [PubMed] [Google Scholar]
- 9.Schmid S.R. and Linder,P. (1992) Mol. Microbiol., 6, 238–292. [DOI] [PubMed] [Google Scholar]
- 10.Fuller-Pace F. (1994) Trends Cell Biol., 4, 271–274. [DOI] [PubMed] [Google Scholar]
- 11.de la Cruz J., Kressler,D. and Linder,P. (1999) Trends Biochem. Sci., 24, 192–198. [DOI] [PubMed] [Google Scholar]
- 12.Iggo R.D., Jamieson,D.J., MacNeill,S.A., Southgate,J., McPheat,J. and Lane,D.P. (1991) Mol. Cell. Biol., 11, 1326–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barta I. and Iggo,R. (1995) EMBO J., 14, 3800–3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stevenson R.J., Hamilton,S.J., MacCallum,D.E., Hall,P. and Fuller-Pace,F.V. (1998) J. Pathol., 184, 351–359. [DOI] [PubMed] [Google Scholar]
- 15.Neubauer G., King,A., Rappsilber,J., Calvio,C., Watson,M., Ajuh,P., Sleeman,J., Lamond,A. and Mann,M. (1998) Nature Genet., 20, 46–50. [DOI] [PubMed] [Google Scholar]
- 16.Iggo R.D. and Lane,D.P. (1989) EMBO J., 8, 1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buelt M.K., Glidden,B.J. and Storm,D.R. (1994) J. Biol. Chem., 269, 28367–28370. [Google Scholar]
- 18.Lamm G.M., Samantha,M.N., Fuller-Pace,F.V. and Lamond,A.I. (1996) Nucleic Acids Res., 24, 3739–3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lemaire L. and Heinlein,U.A.O. (1993) Life Sci., 52, 917–926. [DOI] [PubMed] [Google Scholar]
- 20.Jost J.P., Schwarz,S., Hess,D., Angliker,H., Fuller-Pace,F.V., Stahl,H., Thiry,S. and Siegmann,M. (1999) Nucleic Acids Res., 27, 3245–3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 22.Hloch P., Schiedner,G. and Stahl,H. (1990) Nucleic Acids Res., 18, 3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanger F., Nicklen,S. and Coulson,A.R. (1977) Proc. Natl Acad. Sci. USA, 74, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zinn K., DiMaio,D. and Maniatis,T. (1983) Cell, 34, 865. [DOI] [PubMed] [Google Scholar]
- 25.Luckow B. and Schütz,G. (1987) Nucleic Acids Res., 15, 5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graham F.L., Smiley,J., Russel,W.C. and Nairn,R. (1977) J. Gen. Virol., 36, 59–72. [DOI] [PubMed] [Google Scholar]
- 27.Banerji J., Olson,L. and Schaffner,W. (1983) Cell, 33, 729–739. [DOI] [PubMed] [Google Scholar]
- 28.Nielson D.A., Chang,T.-C. and Shapiro,D. (1989) Anal. Biochem., 179, 19–23. [DOI] [PubMed] [Google Scholar]
- 29.MacGregor G.R. and Nolan,G.P. (1989) In Murray,E.B. and Walker,J.M. (eds), Methods in Molecular Biology. Humana Press, Clifton, NJ, Vol. 7.
- 30.Laemmli U.K. (1970) Nature, 227, 680. [DOI] [PubMed] [Google Scholar]
- 31.Towbin H., Staehelin,T. and Gordon,J. (1979) Proc. Natl Acad. Sci. USA, 76, 4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oberosler P., Hloch,P., Ramsperger,U. and Stahl,H. (1993) EMBO J., 12, 2389–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramsperger U. and Stahl,H. (1995) EMBO J., 14, 3215–3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gossen M. and Bujard,H. (1992) Proc. Natl Acad. Sci. USA, 89, 5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Breathnach R. and Chambon,P. (1981) Annu. Rev. Biochem., 50, 349–383. [DOI] [PubMed] [Google Scholar]
- 36.Keller E.B. and Noon,W.A. (1984) Proc. Natl Acad. Sci. USA, 81, 7417–7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hawkins J.D. (1988) Nucleic Acids Res., 16, 9893–9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen C.Y.A. and Shyu,A.B. (1995) Trends Biochem. Sci., 20, 465–470. [DOI] [PubMed] [Google Scholar]
- 39.Smale S.T. and Baltimore,D. (1989) Cell, 57, 103–113. [DOI] [PubMed] [Google Scholar]
- 40.Weis L. and Reinberg,D. (1992) FASEB J., 6, 3300–3309. [DOI] [PubMed] [Google Scholar]
- 41.El-Deiry W.S., Kern,S.E., Pietenpol,J.A., Kinzler,K.W. and Vogelstein,W. (1992) Nature Genet., 1, 45–49. [DOI] [PubMed] [Google Scholar]
- 42.Matsumoto K., Wassarman,K.M. and Wolffe,A.P. (1998) EMBO J., 17, 2107–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MacArthur H. and Walter,G. (1984) J. Virol., 52, 483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iggo R.D., Gough,A., Xu,W., Lane,D.P. and Spurr,N.K. (1989) Proc. Natl Acad. Sci. USA, 86, 6211–6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nielsen P.J. and Trachsel,H. (1988) EMBO J., 7, 2097–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heinlein U.A.O. (1998) J. Pathol., 184, 345–347. [DOI] [PubMed] [Google Scholar]
- 47.Koleske A.J. and Young,R.A. (1995) Trends Biochem. Sci., 20, 113–116. [DOI] [PubMed] [Google Scholar]
- 48.Wobbe C.R. and Struhl,K. (1990) Mol. Cell. Biol., 10, 3859–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yean D. and Gralla,J. (1997) Mol. Cell. Biol., 17, 3809–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pugh B.F. and Tjian,R. (1991) Genes Dev., 5, 1935–1945. [DOI] [PubMed] [Google Scholar]
- 51.Shorle H., Meier,P., Buchert,M., Jaenisch,R. and Mitchell,P.J. (1996) Nature, 381, 235–238. [DOI] [PubMed] [Google Scholar]
- 52.Zhang J., Hagopian-Donaldson,S., Serbedzija,G., Elsemore,J., Plehn-Dujowich,D., McMahon,A.P., Flavell,R.A. and Williams,T. (1996) Nature, 381, 238–241. [DOI] [PubMed] [Google Scholar]
- 53.Schmidt E.V. (1996) Nature Genet., 14, 8–10. [DOI] [PubMed] [Google Scholar]
- 54.Rourke I.J., Hansen,T.V., Nerlov,C., Rehfeld,J.F. and Nielson,F.C. (1999) FEBS Lett., 448, 15–18. [DOI] [PubMed] [Google Scholar]
- 55.Chaudhary J. and Skinner,M.K. (1999) Endocrinology, 140, 1262–1271. [DOI] [PubMed] [Google Scholar]
- 56.Gaubatz S., Imhof,A., Dosch,R., Werner,O., Mitchell,P., Buettner,R. and Eilers,M. (1995) EMBO J., 14, 1508–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Batsche E., Muchardt,C., Behrens,J., Hurst,H.C. and Cremisi,C. (1998) Mol. Cell. Biol., 18, 3647–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zaffran S., Chartier,A., Gallant,P., Astier,M., Arquier,N., Doherty,D., Gratecos,D. and Semeriva,M. (1998) Development, 125, 3571–3584. [DOI] [PubMed] [Google Scholar]
- 59.Grandori C., Mac,J., Siebelt,F., Ayer,D.E. and Eisenman,R.N. (1996) EMBO J., 15, 4344–4357. [PMC free article] [PubMed] [Google Scholar]
- 60.Endoh H., Marujama,K., Masuhiro,Y., Kobayashi,Y., Goto,M., Tai,H., Yanagisawa,J., Metzger,D., Hashimoto,S. and Kato,S. (1999) Mol. Cell. Biol., 19, 5363–5372. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Cooke C., Hans,H. and Alwine,J.C. (1999) Mol. Cell. Biol., 19, 4971–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McCracken S., Fong,N., Yankulov,K., Ballantyne,S., Pan,G., Greenblatt,J., Patterson,S.D., Wickens,M. and Bentley,D.L. (1997) Nature, 385, 357–361. [DOI] [PubMed] [Google Scholar]