

Abstract
Epigenetic regulation of gene expression using histone deacetylase (HDAC) inhibitors is a promising strategy for developing new anticancer agents. The most common HDAC inhibitors are hydroxamates, which, though highly potent, have limitations due to their poor pharmacokinetic properties and lack of isoform selectivity. Trifluoromethylketones (TFMK) developed as alternatives to hydroxamates are rapidly metabolized to inactive trifluoromethyl alcohols in vivo, which prevented their further development as potential drug candidates. In order to overcome this limitation, we designed trifluoropyruvamides (TFPAs) as TFMK surrogates. The presence of an additional electron withdrawing group next to the ketone carbonyl group made the hydrate form of the ketone more stable, thus preventing its metabolic reduction to alcohol in vivo. In addition, this structural modification reduces the potential of the TFMK group to act as a covalent warhead to eliminate off-target effects. Additional structural changes in the cap group of the inhibitors gave analogues with IC50 values ranging from upper nanomolar to low micromolar in the cytotoxicity assay, and they were more selective for cancer cells over normal cells. Some of the most active analogues inhibited HDAC enzymes with low nanomolar IC50 values and were found to be more selective for HDAC8 over other isoforms. These molecules provide a new class of HDAC inhibitors with a metabolically stable metal-binding group that could be used to develop selective HDAC inhibitors by further structural modification.
1. Introduction
Cancer is still the second largest cause of death in the United States [1]. Despite improvement in treatment regimens, problems such as lack of target specificity and off–target toxicity persist. Targeted drug therapy is a possible way to counter these problems. Enzymes responsible for epigenetic regulation of gene expression provide several targets for rational design of anti-cancer agents. Epigenetic regulation includes DNA methylation and histone acetylation/deacetylation, which do not change the genetic sequence, but alter the rate of transcription of genes affected by these modifications [2]. Histone acetylation and deacetylation are regulated by two families of enzymes, histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively. Deacetylation of histones can reduce the expression of tumor suppressor genes, and their hyperacetylation can lead to their induction, leading to apoptosis [3]. HDACs also deacetylate non–histone proteins such as α–tubulin, which are involved in cancer cell migration and proliferation [2]. Hence, selective targeting of HDACs has been studied extensively. HDACs consist of zinc–dependent (Class I, II and IV) and NAD+dependent (Class III) enzymes [4]. Class I and II HDACs can be inhibited by small molecules containing Zinc-Binding Groups (ZBGs), which bind to a Zn2+ ion in the enzyme active site via mono or bidentate complexation, which is reversible in nature. These ZBGs include groups such as carboxylates, hydroxamates, and thiols. Of the four HDAC inhibitors approved by the US FDA for clinical use, vorinostat (SAHA) and belinostat (Beleodaq) approved for T–cell lymphoma and panobinostat (Farydak) approved for multiple myeloma (Fig. 1A) [5] are hydroxamates, while romidepsin (Istodax) approved for T–cell lymphoma has a thiol ZBG. Chidamide (Epidaza, Fig. 1A) is another hydroxamate that is in clinical trials in USA and in clinical use in China [6]. Typical Class I and II HDAC inhibitors consist of a ZBG, a linker, and a cap group (Fig. 1A) and are designed to mimic the endogenous ligand of the enzyme acetyl lysine (Fig. 1A). The cap group interacts with the outer rim of the HDAC catalytic groove, which varies among different HDAC isoforms, while the enzyme active site is relatively conserved [7]. Compared to the linear HDAC inhibitors, romidepsin has a depsipeptide cap group and it undergoes in situ activation by reduction of the disulfide bridge to release a thiol ZBG.
Fig. 1.

(A) HDAC inhibitors are designed to mimic acetyl–lysine, as shown by a ZBG (red), linker (green) and a cap group (blue). HDAC inhibitors vorinostat (SAHA), belinostat (Beleodaq), panabinostat (Farydak), romidepsin (Istodax) in clinical use and chidamide (Epidaza) in clinical trials.
The most common HDAC inhibitors comprise hydroxamates, which are very potent, and chelate very tightly to Zn2+ ion in a bidentate manner. However, hydroxamates have poor pharmacokinetic properties and their promiscuous metal-binding ability lead to HDAC-independent adverse effects, including cardiotoxicity, pulmonary embolism, thrombocytopenia with nausea and diarrhea [8,9]. In addition, the hydroxamates have the potential to undergo zinc–catalyzed Lossen-rearrangement to isocyanates, which are highly reactive electrophiles that can be targeted by numerous nucleophiles in the cells to cause a plethora of biological problems such as mutagenesis through reaction with nitrogen bases of DNA [9]. Hydroxamates are also rapidly cleared metabolically [10]. Hence, there is a need for alternate ZBGs. Even if the alternates are less potent than hydroxamates, modulation of the cap group can be used to strike a balance between potency and selectivity, improving their pharmacologic properties and reducing possible drug side effects. Alternates to hydroxamates include molecules such as trifluoromethylketones [11], thiols [12], α-ketoamides [13] and HDAC 6-selective mercaptoacetamides [14–16]. These inhibitors exhibited not only potent in vitro HDAC inhibitory activity, but significant anti-tumor activity; for example, α-ketoamides in an in-vivo tumor model [13]. Unfortunately, there is no established set of guidelines for designing ZBG, and a direct correlation between zinc-binding ability and biological activity has not been fully established. The existence of some HDAC inhibitors without ZBGs [17] add to the complexity. Our goal was to develop HDAC inhibitors with ZBGs of moderate Zn2+ binding ability and higher selectivity. Selection of appropriate metal-binding groups was based on the hypothesis that bidentate chelation leading to five-membered metallocycle intermediates/transition states is mostly preferred due to lack of strain. We designed HDAC inhibitors with alternative ZBGs (Fig. 2A). Among the alternates to SAHA, trifluoromethyl ketones (TFMKs) have been shown to be potent HDAC inhibitors, but they suffer from rapid reductive metabolism of the ketone carbonyl group in vivo, limiting their use as anticancer agents. To overcome this metabolic impediment, we designed and synthesized trifluoropyruvamides (TFPAs) as metabolically more stable variants of TFMK HDAC inhibitors.
Fig. 2.

(A) potential zinc binding groups for HDAC inhibition [18]. B) ZBGs designed as potential HDAC inhibitors. C) SAHA-like pharmacophore model used for HDAC inhibitor design.
2. Results and discussion
2.1. Design and molecular modeling
Computational analysis by Chen et al. revealed several possible bidentate ZBGs (Fig. 2A) that could be explored to design effective HDAC inhibitors [18]. We selected groups from this study as well as additional fragments we designed for computational screening (Fig. 2B). For preliminary investigation of the efficacy of these ZBGs, we designed HDAC inhibitors using a “SAHA–like model” (Fig. 2C) and compared their antiproliferative activity with that of SAHA. This model utilized anilides as the cap group and an alkyl linker. To determine the optimal length of the linker, alkyl chains of different lengths were used. During preliminary biological screening (Table 1), it was observed that among the several designed new ZBGs, only the TFPA 10b demonstrated antiproliferative activity. TFPAs are structural variants of TMFKs, which were originally developed around the same time as SAHA [11]. These molecules bind to HDACs using a four membered gem-diol-based metallocycle transition state using their hydrate form. However, TFMKs, especially the linear analogues, have exhibited poor half-lives of under 15 min as they are prone to reductive metabolism by carbonyl reductases (CBRs) [19] in vivo [11,20,21]. Since then, several variants have been synthesized, but without any reported improvements of the stability of the ZBG [22–25]. TFPAs resulted from the incorporation of an additional electron-withdrawing carbonyl functional group to increase the metabolic stability of the trifluoromethyl ketone via stabilization of its hydrate.
Table 1.
Evaluation of effect of different zinc-binding groups on HDAC inhibitory activity by growth inhibition assay using HCT116 cells at a maximum concentration of 100 μM.
| Compound | HCT116 IC50 | Compound | HCT116 IC50 |
|---|---|---|---|
|
| |||

|
>100 |

|
>100 |

|
>100 |

|
>100 |
|
|
>100 |

|
>100 |

|
>100 |

|
>100 |

|
>100 |
|
>100 |

|
>100 |
|
>100 |

|
>100 |

|
>100 |
|
|
>100 |

|
54.39 ± 6.02 |
|
|
>100 | ||
We hypothesized that HDAC–TFMK interactions via the hydrate form inside the active site are induced due to enhancement of electrophilicity of the carbonyl group by zinc-chelation, facilitating nucleophilic addition of a water molecule. The keto-form is predominant elsewhere as indicted by the appearance of the carbonyl carbon as a quartet at 191.5 ppm in the 13C NMR spectrum of the TFMK analogue (Fig. 3B). It is susceptible to reductive metabolism by carbonyl reductases (CBRs) in the human body [26]. We hypothesized that incorporation of an additional electron withdrawing group would buttress the effect of the trifluoromethyl group and stabilize the hydrate form, making it resistant to reductive metabolism. [27]. To explore this hypothesis, two different approaches were used. The first approach led to a perfluoro aryl-TFMK (compound 89, Table 1), which was inactive, presumably due to the steric nature of the aromatic ring. The second design that led to TFPAs incorporated an amide carbonyl as an extra electron withdrawing group to TFMK, while retaining its small size. This modification does not sacrifice the linearity of the molecule. It stabilizes the hydrate form sufficiently to enable its isolation as evident from the appearance of the carbonyl carbon as a quartet at 94.3 ppm in the 13C NMR spectrum of the TFPA analogue 10c (Fig. 3C). In contrast, the pyruvic amide 5c without the electron withdrawing TFM group was isolated in its keto-form with the carbonyl carbon appearing as a singlet at 197.2 ppm (Fig. 3A). TFMKs, due to their high electrophilicity, can act as highly reactive covalent warheads and lead to off-target effects, thus limiting their application in drug design. An added advantage of the conversion of TFMKs to TFPAs is that stabilization of the hydrate form reduces the potential of the TFMK group to act as a covalent warhead thereby minimize their potential off-target effects and making these molecules less likely to act as pan-assay interference compounds (PAINS). To compare the susceptibility of the two groups to nucleophilic addition, we studied the reactivity of the two groups towards addition of the methyl ester of N-Boc cysteine as a thiol-based nucleophile by 19F-NMR-based kinetic study (Supporting Information, Fig. S1). The TFMK showed the appearance of a new fluorine resonance corresponding to the thiol addition product, in addition to the signals corresponding to the ketone and the hydrate forms. In contrast, only a signal corresponding to the hydrate form was observed with the PFPA without any sign of thiol addition product. While this work was in progress, a somewhat similar, but conceptually different, design using trifluorolactic amides as HDAC 6-selective inhibitors was published [28]. However, their mechanism of zinc-binding is based on increasing the acidity of the alcohol by the trifluoromethyl group, facilitating its deprotonation, and creating a hydroxamate surrogate that binds to HDAC6 via a 5-membered metallocycle, unlike the 4-membered binding mode of TFPAs involving the gem diol group. However, the mode of binding of the trifluorolactic amides suggests the possibility of an additional binding mode of TFPA ZBG via a less-strained 5-membered metallocycle involving one of the hydrate-alcohol groups and the adjacent amide-carbonyl, in addition to the 4–membered metallocycle involving the two gem-diol hydroxyl groups. (Fig. 4).
Fig. 3.

A) A pyruvic amide (here compound 5c typically shows a13C NMR singlet at 197.2 ppm, which shows complete retention of the keto-form, despite the electron withdrawing ability of the adjacent amide group. B) The 13C NMR of the TFMK 91 shows the presence of a quartet at 191.5 ppm, despite the electron withdrawing ability of the trifluoromethyl group. C) Combining the amide and trifluoromethyl ketone groups resulted in a quartet at 94.3 ppm for compound 10c, which is characteristic of a hydrate.
Fig. 4.

Optimizing of the electrophilicity of carbonyl results in stabilized hydrates that can withstand Carbonyl Reductase reduction before reaching their targets inside cancer cells. Included is an alternate Zinc-binding mode.
To compare binding modes of the designed SAHA analogues with alternative ZBGs (Fig. 2B), they were docked on several HDACs (Supporting Information, Fig. S2). They all contained an anilide cap group and 6 methylene carbons as in SAHA. Also included were the SAHA–like TFMK analogue synthesized by Frey et al. [11] as a reference, along with SAHA for comparison of docking scores and binding poses. It was observed that all molecules bind to only HDAC4, HDAC6 and HDAC7 in the correct orientation. These three HDACs were used to compare the docking scores (Fig. 6B). The surface representation of each molecule aligned with SAHA inside the HDAC6 binding site is shown in Fig. 5.
Fig. 6.

A) Alignment of SAHA with TFMK and TFPA shows similar binding modes on HDAC6, even though the linkers and cap groups of the TFPA are not as well aligned as the TFMK. B) Docking scores of analogues against several HDACs showed TFPA, Diamide (Oxamide) and the Pyruvamide to have similar values against several HDACs. C) Docking scores of compounds 14 and 10a-d against several HDACs revealed 10b to be the best candidate. D) and E) Examination of the active site of HDAC6 with 10b and 10c revealed that the inhibitors were bound to Tyr745, a residue that is essential for enzymatic activity.
Fig. 5.

Alignment of binding poses of each compound with SAHA inside the HDAC 6 catalytic site.
All the molecules aligned with SAHA when docked against the designated HDACs with similar binding modes (Figs. 5 and 6A). The docking scores did not reveal any clear preferred candidate for the ZBGs (Fig. 6B). Hence, an in vitro screening of antiproliferative activity was carried out resulting in the identification of 10b as a potential lead molecule for structure optimization (Table 1). To determine the optimal linker length of 10b, analogues 14 and 10a-d (Scheme 2) of varying linker length were designed. It was observed that all TFPAs preferred to bind via the gem diol using a four-membered metallocyle transition state, like TFMKs. Docking data revealed that TFPAs bound only to HDAC2, HDAC6, HDAC7 and HDAC8 with minimum error. Results indicated that analogue 10b with a six-carbon linker had the best scores (Fig. 6C). A close examination of the interactions between compounds 10b, 10c and the HDAC6 active site residues (Fig. 6D and E) reveals Tyr745 as a common denominator. Tyr745 is essential for acetyl–lysine cleavage by stabilizing the tetrahedral intermediate formed when a molecule of water, acting as a nucleophile, attacks the acetyl lysine amide bond [29]. Hence, Tyr745 interactions with inhibitors can disrupt this process and inactivate the enzyme. Having identified 10b as a potential lead molecule, an SAR study around the cap group was undertaken. Docking scores of all cap group variants synthesized for SAR study (Supporting Information, Fig. S3, Table S1) revealed m–bromoanilide TFPA 76 to have better scores than SAHA against HDAC6 and HDAC8, and that it targets Tyr745 like 10b.
Scheme 2.

A) Synthesis of aminoalkanilides 4a-d. n = number of methylene groups B) An alternative synthetic route using Boc-protected amino acids was used to synthesize 14 and the SAR variants 53–87. n = number of methylene groups. C) Synthesis of compounds 51 and 52 from 50.
2.2. Chemistry
2.2.1. Synthesis
The final products 5a-c, 7a,b,d, 8b, 9a-d and 10a-d were synthesized by amide coupling of aminoalkanilides 4a-d with a ZBG appended to a carboxylic acid group as shown in Scheme 1. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCl) and dimethylaminopyridine (DMAP) in dichloromethane were used for amide coupling. However, due to solubility issues, some amines required other coupling agents such as (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b] pyridinium-3-oxide hexafluorophosphate (HATU) and 1-hydroxybenzotriazole (HOBt) along with N–methylmorpholine (NMM) in N,N–dimethylformamide (DMF). In the case of kojic anilides 9a-d (Scheme 2A), a third method using oxalyl chloride and catalytic DMF was used to generate the final products. Oximes 6a-c were synthesized from 5a-c using hydroxylamine hydrochloride in methanol at room temperature.
Scheme 1.

Synthesis of final products from aminoalkanilides 4a-d a) EDCl/DMAP, dichloromethane, r.t, overnight. b) HATU/HOBt/NMM, DMF, 4 h r.t. c) Oxalyl chloride/DMF (catalytic), DCM, r.t., overnight. d) NaOMe/MeOH, r. t., overnight.
The only reliable synthetic approach reported for preparation of TFPAs was by an Ugi–like mechanistic pathway using isocyanides and trifluoroacetic anhydride [30]. Considering the high cost of isocyanides needed to make a diverse library of analogues of varying cap groups, we instead focused on simple peptide couplings using amino-alkanamides resembling SAHA fragments (4a-d). Peptide coupling using EDCl/DMAP, and EDCl/HOBt failed to give any product. Using HATU, HOBt and NMM in the presence of molecular sieves under anhydrous conditions at room temperature for 4 h, we were able to synthesize TFPAs 10a-d (Scheme 2A), albeit in low yields (12–37%).
The aminoalkanilides 4a-d required for above syntheses were made by an approach starting from bromoalkanoic acids 1a-d (Scheme 2A). This involved peptide coupling to generate bromoanilides 2a-d, an SN2 reaction using sodium azide in DMF overnight at 80 °C to generate azidoanilides 3a-d, followed by reduction by catalytic hydrogenation using palladium on carbon to obtain aminoanilides 4a-d. The spectral data of all these intermediates are consistent with those previously reported [12,31–34].
For synthesis of the TFPA analogue 14 with a short three-methylene linker (Scheme 2B), more commonly available γ-aminobutyric acid (GABA) 11 was used instead of 4-bromobutanoic acid. Boc-protection of GABA (11) gave compound 12. This was followed by peptide coupling with aniline using EDCl and DMAP in dichloromethane overnight to give compound 13. Finally, a tandem Boc–deprotection/peptide coupling of the γ–aminobutyranilide 13 with trifluoropyruvic acid was used to obtain the TFPA 14 (50% yield). This approach was subsequently used for efficient synthesis of Boc-aminoarenes 17–50. Tandem Boc-deprotection/peptide coupling with trifluoropyruvic acid gave TFPAs 53–85 (4–42% yield) without the need to isolate and characterize the intermediate amines. Compounds 51–52 required for the synthesis of TFPAs 86 and 87, respectively were prepared from 50 by removal of 2,2,2-trichloroethoxycarbonyl (Troc) protecting group (Scheme 2C). Compound 51 underwent peptide coupling with trifluoropyruvic acid to yield 86, while compound 52 was first ethylated by refluxing with ethyl iodide and potassium carbonate in acetone for 6 h, followed by concentration in vacuo and peptide coupling like 51 to give 87. Some of the aromatic amines used as cap groups included 2-aminothiazoles. Compounds containing these fragments have shown to possess antibacterial, antimicrobial and other properties [35] and one such fragment has been used to synthesize a TFMK derivative before [11]. Thus, we used thiazole pharmacophores to generate potent TFPAs. They were either commercially available, or made using known synthetic methods (see Supporting Information for synthesis of 2-aminothiazoles). The spectral data of the functional group of all TFPAs is in excellent agreement with those previously reported by El Kaïm et al. in which they showed a13C quartet at 90 ppm (in DMSO–d6), with a J-value of 32 Hz, corresponding to the hydrate of –COCF3 carbonyl, with no signals corresponding to the keto-form [30]. Our molecules displayed similar hydrate quartet at 90–91 ppm (acetone-d6) or 94–95 ppm (MeOH-d4) (Fig. 3C and Supporting Information, NMR Spectra). The mass spectral data too corresponds to the hydrate forms. For use as a reference in biological assays, we synthesized TFMK 91 as described in literature [11].
2.3. Screening for cytotoxicity of TFPA-based analogues
Inhibition of growth of HCT116 colon cancer cells at a maximum concentration of 100 μM was used for the initial screening of the ZBGs for HDAC inhibitory potential (Table 1). Included in the assay are the perfluoroaromatic trifluoromethyl ketone 89 and the corresponding trifluoromethyl alcohol 88 (Synthesized according to Supporting Information, Scheme S1). All analogues except TFPA 10b were inactive. The lack of activity of compound 89 may possibly be due to the increased steric bulk of the perfluorophenyl group preventing the ZBG from entering the HDAC active site, even though it exists as a stable hydrate. TFPA 10b displayed an IC50 of 54.39 μM and was selected for lead optimization.
2.3.1. Lead optimization
To understand the effect of varying the linker length and to determine the optimal linker length, compounds 10a-d and 14 were tested against the HCT116 cell line (Fig. 7, Table 2). The results were contrary to the results of the docking studies; compound 10b (n = 5) which had the best docking score displayed the lowest activity with an IC50 value of 54.39 μM. Compound 10c (n = 6) was the most active with an IC50 value of 9.14 μM against the HCT116 cell line.
Fig. 7.

Chain elongation study to find the optimal linker length (n = 3–7).
Table 2.
Effect of linker length on growth inhibition of HCT116 cell line.
| IC50 (μM) | IC50 (μM) | IC50 (μM) | IC50 (μM) | IC50 (μM) | |
|---|---|---|---|---|---|
|
| |||||
| Number of CH2 (n) | 3 | 4 | 5 | 6 | 7 |
| Compound number | 14 | 10a | 10b | 10c | 10d |

|

|

|

|

|
|
| HCT116 | 34.83 ± 5.65 | 20.26 ± 1.23 | 54.39 ± 6.02 | 9.14 ± 2.25 | 41.67 ± 5.60 |
Having established the validity of the ZBG and the optimal length of the linker, we next focused on the SAR of the cap group of the most active analogue 10c (n = 6) to further improve its biological activity. This involved the synthesis of a library of analogues 53–87 (Table 3) using cap groups shown in Fig. 8, focusing on variance in the stereoelectronic effects. Their growth inhibitory activity on HCT116 cell line was measured at a maximum concentration of 40 μM (Table 3). SAHA and TFMK analogue 91 used as reference compounds had IC50 values of 1.18 μM and 10.07 μM, respectively (Table 3).
Table 3.
IC50 ± SD (μM) (n = 3) values of compounds 53–87 on HCT116 cell line. Included are compounds 90–92.
| Cap Group | IC50 | Cap Group | IC50 | Cap Group | IC50 |
|---|---|---|---|---|---|
|
| |||||

|
12.11 ± 1.82 |

|
18.53 ± 1.29 |

|
14.07 ± 0.83 |

|
9.11 ± 1.32 |

|
22.15 ± 1.97 |

|
6.48 ± 0.75 |

|
4.52 ± 0.63 |
|
4.23 ± 0.25 |

|
>40 |

|
7.57 ± 0.37 |

|
3.13 ± 0.17 |

|
17.20 ± 1.69 |

|
6.67 ± 1.02 |

|
10.07 ± 0.71 |

|
14.5 ± 1.02 |
|
|
2.66 ± 0.32 |

|
10.13 ± 1.2 |

|
3.06 ± 0.63 |
|
6.11 ± 0.72 |

|
29.88 ± 5.63 |

|
6.47 ± 1.18 |

|
10.82 ± 1.28 |

|
4.73 ± 0.87 |

|
10.99 ± 1.29 |

|
2.37 ± 0.23 |

|
3.87 ± 0.15 |

|
10.29 ± 1.36 |

|
12.05 ± 1.47 |

|
3.45 ± 0.36 |

|
7.56 ± 0.73 |

|
2.18 ± 0.32 |
|
0.93 ± 0.14 |
|
2.46 ± 0.25 |
|
|
1.02 ± 0.07 |
|
4.05 ± 0.87 |
|
>4011 |

|
10.07 ± 1.22 |

|
1.18 ± 0.07 |
|
>40 |
Fig. 8.

Cap groups selected for SAR studies of compound 10c.
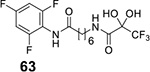
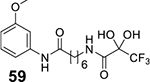
Of the analogues substituted at the para position of aniline, p–bromoanilide 65 (3.13 μM) and p-methoxyanilide 55 (4.52 μM) had the lowest IC50 values. Hence, we also investigated the effect of positional substitutions with bromine and methoxy at other positions. There was hardly any difference in the IC50 values of meta-bromo (3.06 μM) and para-bromo (3.13 μM) analogues, but ortho–bromo substitution 75 increased the IC50 value to 14.5 μM. A direct decreasing trend in activity was observed with the methoxy group going from 4.52 μM for para (55) to 6.11 μM for meta (59) and 10.07 μM for ortho (66). Among the bulkier aromatic groups, the best results were obtained for the 5-phenylthiazole variant 58 (2.66 μM). This is consistent with phenylthiazole TFMKs being among the most active variants reported d by Frey et al. [11]. We therefore decided to expand the investigation of SAR of thiazoles in much greater detail. We synthesized 4 and 5–positional substitution variants of thiazoles, starting with phenyl, methyl phenyl and diphenyl substituents. The 4, 5-diphenyl thiazole was of particular interest as a similarly substituted oxazole fragment existed as a part of the HDAC6-selective inhibitor Tubacin cap group [36]. However, we found out that 4–position substitution was detrimental for activity, as the 4, 5-diphenylthiazole 68 had drastically reduced activity (29.88 μM).
We next investigated the effect of fusing the thiazole ring with a phenyl group he as in benzothiazole vs the phenylthiazole. The results were similar with IC50 of 2.37 μM for benzothiazole 61 vs. 2.66 μM for 5-phenylthiazole 58. We then investigated the effect of substitution at the para–position of phenylthiazoles, and substitutions at 5 and 6–positions of benzothiazoles. Thus, we synthesized p-bromo (85), p-hydroxy (86), p-methoxy (84) and p-ethoxy (87) phenylthiazoles. Of these the p-methoxyphenylthiazole 84 produced an IC50 of 0.93 μM, making it more potent than SAHA (IC50 1.18 μM). Replacing the methoxy with an ethoxy in 87 reduced the activity to IC50 4.05 μM, while the corresponding free phenol analogue 86 had an IC50 of 1.02 μM, almost as active as the corresponding methoxy analogue 84. For the benzothiazole series, we synthesized 6-fluoro (77), 6-chloro (78) and 6-bromo (79), 5-methoxy (83) and 6-methoxy (82) benzothiazoles. Of these, only the 5–methoxybenzothiazole variant 83 (2.18 μM) produced better results than the unsubstituted parent molecule.
As metabolic reduction of TFMKs has been shown to abolish their ability to kill cancer cells [11], we wanted to test if reduction of TFPA would produce the same effect. We reduced TFPA 60 to the corresponding trifluoromethyl-α-hydroxyamide (TFMHA) 92. As expected, it was inactive on the HCT116 cell line (Table 3).
After screening the activity of the compounds against the HCT116 cell line, the five compounds with the lowest IC50 values (Compounds 61, 83, 84, 85 and 86) were tested on additional cell lines; NCI–H522 lung cancer cell line, HT1080 fibrosarcoma cell line, HeLa cervical cancer cell line, U2OS osteosarcoma cell line, MDA-MB-231 and MDA-MB-468 breast cancer cell line. SAHA was used as a positive control for comparison. All compounds showed comparable or better growth inhibitory activity than SAHA on some cell lines, while compounds 84 and 85 displayed better activity than SAHA against most of the cell lines (Fig. 9, Table 4).
Fig. 9.

Cell growth inhibition assay of selected cell lines found compounds 84 and 85 to be more active than SAHA in almost all.
Table 4.
IC50 ± SD (μM) (n = 3) values of five compounds against multiple cell lines, as compared to SAHA. Compound 45 is highlighted in red.
| Cell Line | Compound 84 | Compound 86 | Compound 83 | Compound 61 | Compound 85 | SAHA |
|---|---|---|---|---|---|---|
|
| ||||||
| H522 | 3.24 ± 0.56 | 1.90 ± 0.21 | 3.23 ± 0.41 | 2.86 ± 0.36 | 2.70 ± 0.34 | 2.38 ± 0.28 |
| HT1080 | 1.96 ± 0.21 | 1.42 ± 0.29 | 2.28 ± 0.47 | 2.65 ± 0.21 | 1.76 ± 0.22 | 3.56 ± 0.30 |
| HeLa | 1.60 ± 0.12 | 6.38 ± 0.92 | 1.86 ± 0.31 | 1.52 ± 0.23 | 1.22 ± 0.09 | 2.04 ± 0.31 |
| U2OS | 1.31 ± 0.18 | 0.74 ± 0.04 | 3.29 ± 0.46 | 1.41 ± 0.18 | 1.09 ± 0.09 | 2.23 ± 0.15 |
| MDA-MB-468 | 1.16 ± 0.26 | 1.10 ± 0.10 | 3.65 ± 0.11 | 1.31 ± 0.18 | 0.76 ± 0.08 | 2.85 ± 0.37 |
| MDA-MB-231 | 1.02 ± 0.09 | 1.46 ± 0.15 | 0.81 ± 0.10 | 1.83 ± 0.35 | 0.79 ± 0.10 | 2.23 ± 0.15 |
Ten selected compounds were then tested in NCI 60 cell one-dose assay, at a single concentration of 10 μM (Supporting Information, Figs. S4–S13), followed by a five-dose assay at five different concentrations for eight of the ten compounds that displayed sufficient cytostatic/cytotoxic activity at 10 μM (Supporting Information, Figs. S14–S21). The one-dose assay displays mean growth percentages, while the five-dose assay shows GI50, TGI and LC50 values (Table 5). Most of the compounds demonstrated potent cytostatic activity, with several of them (58, 84, 85) showing cytotoxic effects as well (Table 5). Interestingly, compound 85 exhibited the least mean growth percentage and GI50 value, concurring with our previous assay results (Table 4). Even though compound 84 was the most active on the HCT116 cell line, the results on both six additional cell lines (Fig. 9, Table 4) and the NCI–60 cell one-dose and five-dose assays (Table 5) showed that compound 85 had a better activity profile against multiple cell lines, making it more consistent. Hence, we picked compound 85 as the prototype inhibitor in our study.
Table 5.
NCI data of ten compounds against sixty cell lines displaying mean cell growth percentage (one-dose assay), GI50, TGI and LC50 values (five-dose assay). Positive growth percentages indicate cytostatic activity, while negative percentages imply cytotoxicity. The best results are highlighted in red. ND – Not Determined. GI50 values correspond to the drug concentration needed to prevent 50% cell growth. TGI (Total Growth Inhibition) values correspond to the concentrations needed to prevent complete cell growth. LC50 values are a measure of cytotoxicity, corresponding to the minimum concentrations needed to kill 50% of the target cells.
| Compound | 84 | 85 | 83 | 70 | 61 | 64 | 65 | 69 | 58 | 55 |
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Mean Growth % | −10.17 | −11.47 | −4.91 | 35 | −2.45 | 15.99 | 2.58 | 2.1 | −10.06 | 37.09 |
| GI50 (μM) | 1 | 0.93 | 1.23 | ND | 3.39 | 5.13 | 3.16 | 3.09 | 3.02 | ND |
| TGI (μM) | 8.51 | 11.4 | 12.3 | ND | 21.37 | 26.92 | 20.89 | 15.14 | 16.98 | ND |
| LC50 (μM) | 26.3 | 26.3 | 28.18 | ND | 28.84 | 35.48 | 31.62 | 29.51 | 22.91 | ND |
2.4. Isoform selectivity testing
The inhibitory potencies of the compounds were tested against HDAC1, HDAC2, HDAC3, HDAC4, HDAC6, and HDAC8. The overall results indicated that all the inhibitors possess an affinity for class 1 HDACs over class II HDACs (Supporting Information, Figs. S22–S27, Table S2–S7). The dose dependent curve data showed that the compounds displayed higher potency against HDAC8, with IC50 values in the low nanomolar range (Table 6). The most potent compound was 69 with an IC50 of 4.1 ± 0.6 nM against HDAC8. The most selective compounds were 65, 83, and 58 (Table 6). However, compound 83 is the standout inhibitor, exhibiting between 11-to-460-fold selectivity for HDAC8, when all three were compared using heat maps of the ratios of IC50 values against different HDACs (Supporting Information, Table S8 – Table S10). Hence, a case can be proposed for 83 being HDAC8 selective. However, we do not observe a direct correlation between HDAC8 selectivity and cell growth inhibitory activity. The results of the assay may not be enough to justify HDAC isoform selectivity in a cellular context, as HDACs exist as multiprotein complexes at a cellular level. In this context, it is important to note that selective HDAC6 inhibitors have been shown to display anticancer properties only at high concentrations resulting in low selectivity and off-target effects [37].
Table 6.
Inhibitory potencies against HDAC1, HDAC2, HDAC3, HDAC4, HDAC6 and HDAC8. Highlighted in red are the most selective inhibitors.
| Compound | IC50 (nM) | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| HDAC1 | HDAC2 | HDAC3 | HDAC4 | HDAC6 | HDAC8 | |
|
| ||||||
| 65 | 120 ± 20 | 3200 ± 300 | 230 ± 20 | 2600 ± 200 | 100 ± 10 | 11 ± 2 |
| 61 | 56 ± 3 | 580 ± 20 | 210 ± 10 | 430 ± 30 | 82 ± 7 | 5.3 ± 1.6 |
| 85 | 120 ± 10 | 2400 ± 100 | 59 ± 1 | 1700 ± 200 | 35 ± 4 | 10 ± 1 |
| 69 | 61 ± 4 | 1800 ± 100 | 72 ± 2 | 1100 ± 100 | 38 ± 3 | 4.1 ± 0.6 |
| 55 | 100 ± 10 | 1400 ± 100 | 180 ± 10 | 1600 ± 200 | 440 ± 20 | 51 ± 5 |
| 86 | 110 ± 10 | 3600 ± 300 | 99 ± 9 | 2300 ± 300 | 210 ± 20 | 30 ± 3 |
| 64 | 160 ± 10 | 4900 ± 200 | 270 ± 20 | 2700 ± 300 | 260 ± 50 | 52 ± 7 |
| 83 | 4100 ± 100 | 2500 ± 400 | 120 ± 10 | 1900 ± 400 | 100 ± 10 | 8.9 ± 0.7 |
| 58 | 180 ± 10 | 4800 ± 300 | 620 ± 50 | 2500 ± 300 | 350 ± 20 | 21 ± 3 |
| 84 | 97 ± 2 | 360 ± 40 | 100 ± 10 | 2500 ± 100 | 37 ± 10 | 13 ± 1 |
| SAHA | 33 ± 1 | 96 ± 10 | 20 ± 1 | 12000 ± 800 | 33 ± 3 | 540 ± 10 |
2.4.1. Hyperacetylation assay
Hallmark of HDAC inhibition is the hyperacetylation of several proteins which are substrates for HDACs. Thus, histone H3 hyper-acetylation assay of some active compounds was carried out by Western blot using antibodies for Histone H3 and pan–acetyl lysine. When tested at concentrations of 2.5, 5 and 10 μM, both acetyl–H3 and “pan” acetyl–lysine (Fig. 10) demonstrated elevated levels in a similar way to SAHA, confirming that these molecules target HDACs.
Fig. 10.

Western blot of compound 84 and SAHA revealed increased levels of acetylated H3 and “pan”-acetyl – lysine in a dose dependent manner.
2.5. Evidence of apoptosis
Pan HDACi SAHA has been reported to induce apoptosis in a caspase 8 dependent manner in HCT116 cells [39]. To determine whether our compounds induced apoptosis, we analyzed the enzyme Poly (ADP ribose) polymerase 1 (PARP1) by western blotting. Caspase-dependent proteolytic cleavage of PARP1 is one of the hall marks of apoptosis [40] and antibodies that specifically recognize the cleavage products are available. Western blot analysis indicated that our novel HDACi induced PARP1 cleavage suggesting induction of apoptosis (Fig. 11). HCT116 cells were treated with compounds 83, 84 and 85 for 24 h. Dose-dependent elevation of cleaved PARP1 was detectable after exposure to 83 and 85, but not compound 84. Absence of PARP1 cleavage in cells exposed to 84 may be a result of insufficient dose or time of treatment. The fact that 83 and 85 did induce PARP1 cleavage suggest that these analogues kill cells through an apoptotic mechanism.
Fig. 11.

Western blot analysis of PARP1 cleavage. HCT116 cells were treated with compounds 83, 84 and 85 at the indicated concentrations for 24 h. Western blots were probed with an antibody that recognizes a cleaved form of PARP (Cell Signaling Technology 5626) as a measure of caspase dependent cell death.
2.5.1. Selectivity for cancer vs normal cells
To determine the selectively of the inhibitors for cancer cells over normal cells, compound 55 was tested on the mesenchymal cancer cell line NCI–H522 and the mesenchymal normal cell line WI38, as well as the epithelial cancer cell line HCT116 and the epithelial normal cell line RPE (Fig. 12). The cancer cell lines were found to be more sensitive to the compound compared to normal cells. Moreover, at a maximum concentration of 30 μM, the NCI–H522 cells displayed above 20% survival while the HCT116 cells showed close to 10% cell survival. In comparison, the WI38 cells displayed greater than 80% survival while the RPE cells show more than 50% survival (Fig. 12B). Also, the IC50 values of 55 against NCI–H522 and HCT116 cell lines were 8.05 μM and 3.13 μM, but against RPE and WI38 they were inactive (>30 μM, Fig. 12A). This reaffirms the selectivity of TFPAs for cancer cells over normal cells.
Fig. 12.

A) Plot of Cell survival percentage against the concentration of 55 (shown as RB-5–55b) showed low IC50 values against NCI–H522 and HCT116 cell lines, while RPE and WI38 cell lines displayed high IC50 values. B) Cell survival assay using compound 55 shows that HCT116 and H522 cancer cell lines have low survival rates (<20%) while normal cell lines WI38 and RPE have survival rates of >50%.
2.5.2. Metabolism studies
Previous studies established that trifluoromethyl ketones are metabolically unstable and reduced in vivo to trifluoromethyl alcohols (TFMAs) [11,21]. This metabolic reduction caused a major loss in activity. The TFPAs possess a TFMK moiety, albeit with significantly enhanced electrophilicity. Our hypothesis was that in TFPAs, the hydrate form of the ketone is stabilized compared to TFMKs, and is therefore less susceptible to reductive metabolism. To prove the superior metabolic stability of the TFPAs over TFMKs, a metabolic stability assay was carried out. Metabolic stability studies are primarily conducted using liver microsomal assays, as the predominant cytochrome P450 enzymes present in them are responsible for the metabolism of most xenobiotics in the human body. However, P450 transformations are mostly oxidative in nature precluding their use to assess the stability of ketone drugs against reductive metabolism. A comprehensive review of ketone drug metabolism showed the presence of carbonyl reductases and additional reductases mainly in the liver cytosol, and a minor amount in microsomes [19]. Thus, using a liver extract containing both cytosol and microsomal enzymes would be more appropriate for this assay. This was accomplished by using the S9 fraction which is basically the supernatant obtained from centrifugation of hepatocytes at 9000 g and contains both liver cytosol and microsomal enzymes. We used LC-ESI-MS to monitor the reduction of TFPA 60 over time and compared it with TFMK 91.
2.6. Metabolic stability of TFPAs vs TFMKs
To test our hypothesis, we first tracked the disappearance of TFMK 91 m/z 302 (Fig. 13C) at 15-min intervals (Fig. 13A) and TFPA 60 m/z 370 M + 1 peak was monitored over a period of 2 h (Fig. 14A).
Fig. 13.

(A) The disappearance of the TFMK 91 302 M + 1 peak in 15 min, followed by (B) appearance of the TFMA 90 304 M + 1 peak in 15 min. Highlighted next are the mass spectra of the TFMK 91 (C) and the TFMA 90 (D). Note: The LC and the mass spectrometer were operated using different computers. The sample run was started from the autosampler using a method in one computer that causes the compound to flow through the column into the mass spectrometer, following which the mass spectra were recorded using a different program in another computer. This lack of synchronicity caused a slight difference in the retention time for the same compound in different runs.
Fig. 14.

LC-MS studies of the S9 fraction assay of compound 60 show no disappearance of the 370 M + 1 peak over time. LC-MS studies revealed that its 354 M + 1 peak did not form over time. Also included are the M+1 peaks of compounds 60 and 92 (B, C).
To test the rapid kinetics of TFMK metabolism, we analyzed the aliquots obtained from the S9 fraction assay for TFMK 91. The M+1 peak of m/z 302 completely disappeared in 15 min (Fig. 13A). Simultaneously, the formation of the TFMA 90 M + 1 peak of m/z 304 (Fig. 13D) was monitored over 30 min (Fig. 13B). It was formed in 15 min, just around the same time as the disappearance of compound 91. Hence, the LC-MS results show a quantitative biological reduction of TFMK 91 by metabolizing enzymes in 15 min, similar to data obtained by Frey et al. [11].
In contrast, we observed that TFPA 60 was resistant to reducing conditions imposed by the S9 fraction; the M+1 of m/z 370 for compound 60 (Fig. 14B) did not disappear over 2 h (Fig. 14A). Similarly, there were no traces of the M+1 peak of TFMHA 92 at m/z 354 (Fig. 14C) around a retention time of 8.13 min, over 2 h. This proves that there is no reduction to the alcohol taking place. Metabolism is a process executed by the liver to transform non–polar substances into polar products that can be excreted easily. Ketone drugs are generally hydrophobic in nature and metabolic reduction serves to introduce polarity into the molecule for excretion. Since TFPA 60 is more polar than TFMHA 92, it does not require metabolic reduction to increase polarity. This is a stark contrast to the quantitative biological reduction of compound 91 in 15 min.
3. Conclusion
The metabolic reduction of linear TFMKs has been a significant barrier that has prevented them from being considered as successful SAHA-alternatives for HDAC inhibition for 20 years. This was probably due to the inability of the ketone function to sustain the hydrate-form outside the active site of HDACs, making them vulnerable to reductive metabolism by carbonyl reductases in the liver. We hypothesized that this vulnerability may be overcome by structural modification that would stabilize the hydrate form while maintaining the linearity of the functional group such that it is small enough to enter the active site. This modification would entail enhancing the electrophilicity of the ketone group to favor its hydrate form and thereby prevent its reductive metabolism. This was successfully achieved by incorporating an additional electron-withdrawing amide group adjacent to the trifluoromethyl carbonyl group, resulting in TFPAs. An additional advantage of this structural modification is that it reduces the potential of the TFMK group to act as a covalent warhead and produce off-target effects. Molecular docking confirmed that the designed molecules can bind to several HDAC proteins by chelation of the hydrated pyruvamide group with the zinc ion in the HDAC active site and positioning the anilide cap group at the active site outer rim area. This mode of binding agrees with the binding of other well-known inhibitors such as SAHA. Biochemical studies showed that these compounds were potent cytotoxic agents. SAHA-like linkers containing six methylene groups were found to be optimal for antiproliferative activity of TFPAs. SAR studies of the cap-group of thiazole-based TFPAs identified several potent variants such as compound 85, which consistently outperformed SAHA. Additional cell survival assays demonstrated that TFPAs were more selective for cancer cells over normal cells. In HDAC isoform selectivity studies utilizing purified HDAC proteins, the compounds inhibited HDAC enzymes with low nanomolar IC50 values, while displaying some selectivity for HDAC 8. Compound 83 in particular displayed between 11 and 460-fold selectivity for HDAC8 over other isoforms. Western blot analysis revealed the ability of the compounds to inhibit HDAC proteins in a cellular environment as well. Metabolism studies using the S9-fraction assay showed that TFMK 91 undergoes complete metabolic reduction in 15 min, whereas TFPA 60 is completely unaffected over 2 h by the reducing enzymes present in the S9-fraction. Thus, the conversion of TFMK group to TFPA group has resulted in TFMK surrogates as metabolically stable potent HDAC inhibitors which maintain a stable keto-hydrate form resistant to reduction by carbonyl reductases. These compounds constitute a new class of HDAC inhibitors with a metabolically stable metal-binding group that may be used as a template for designing selective HDAC inhibitors by varying the nature of the linker and the cap group.
4. Experimental section
4.1. Materials and methods
All chemicals and solvents were purchased from commercial sources and used without further purification, unless stated otherwise. 1H and 13C NMR spectra were recorded on Brucker Avance 600 MHz, INOVA 600 MHz and Varian VXRS 400 MHz NMR spectrometers in deuterated solvents using residual undeuterated solvents as internal standard. High-resolution mass spectra (HRMS) were recorded on a Waters Synapt high-definition mass spectrometer (HDMS) equipped with nano-ESI source. Melting points were determined using a Fisher-Johns melting point apparatus. Purification of crude products was performed by either flash chromatography on silica gel (40–63 μm) from Sorbent Technologies or on a Teledyne ISCO CombiFlash Companion chromatography system on RediSep prepacked silica cartridges. Thin layer chromatography (TLC) plates (20 cm × 20 cm) were purchased from Sorbent Technologies (catalog #4115126) and were viewed under Model UVG-54 mineral light lamp UV-254 nm. A Shimadzu Prominence HPLC with an LC20AT solvent delivery system coupled to a Shimadzu Prominence SPD 20AV Dual wavelength UV/Vis absorbance Detector, a Shimadzu C18 column (1.9 m, 2.1 mm × 50 mm) and HPLC grade solvents (MeOH, H2O with 0.1% formic acid) were used to determine the purity of compounds by HPLC. All compounds are >95% pure by HPLC analysis. Details on materials and methods used for LC-ESI-MS analysis are provided in supporting information.
General Procedure 1.
Preparation of bromoanilides: A mixture of n–bromoalkanoic acid 1a-d (40 mmol), aniline (4 mL, 44 mmol, 1.1 equiv), EDCl (7.67 g, 40 mmol, 1 equiv) and 4–dimethylaminopyridine (10 mol%, 488 mg) was stirred in anhydrous dichloromethane (35 mL) at room temperature overnight. The mixture was washed with 1 M HCl (25 mL), saturated aqueous sodium bicarbonate (25 mL) and brine (25 mL). The organic layer was dried (Na2SO4), and the mixture was concentrated in vacuo to give 2a-d, which was used in the next reaction without further purification.
5-Bromo-N-phenylpentanamide (2a):
1H NMR (600 MHz, CDCl3) δ 7.52 (d, J = 7.8 Hz, 2H), 7.33 (t, J = 7.9 Hz, 2H), 7.12 (t, J = 7.4 Hz, 1H), 3.46 (t, J = 6.5 Hz, 2H), 2.42 (t, J = 7.2 Hz, 2H), 2.01–1.95 (m, 2H), 1.94–1.87 (m, 2H).
6-Bromo-N-phenylhexanamide (2b)
1H NMR (600 MHz, CDCl3) δ 7.48 (d, J = 7.9 Hz, 2H), 7.30 (t, J = 7.9 Hz, 2H), 7.08 (t, J = 7.4 Hz, 1H), 3.40 (t, J = 6.7 Hz, 2H), 2.36 (t, J = 7.5 Hz, 2H), 1.92–1.85 (m, 2H), 1.78–1.70 (m, 2H), 1.55–1.48 (m, 2H).
7-Bromo-N-phenylheptanamide (2c):
1H NMR (600 MHz, CDCl3) δ 7.52 (d, J = 7.9 Hz, 2H), 7.32 (t, J = 7.9 Hz, 2H), 7.11 (t, J = 7.4 Hz, 1H), 3.41 (t, J = 6.7 Hz, 2H), 2.37 (t, J = 7.5 Hz, 2H), 1.91–1.83 (m, 2H), 1.79–1.71 (m, 2H), 1.52–1.45 (m, 2H), 1.44–1.37 (m, 2H).
8-Bromo-N-phenyloctanamide (2d):
1H NMR (600 MHz, CDCl3) δ 7.52 (d, J = 7.9 Hz, 2H), 7.33 (t, J = 7.9 Hz, 2H), 7.11 (t, J = 7.5 Hz, 1H), 3.42 (t, J = 6.8 Hz, 2H), 2.37 (t, J = 7.5 Hz, 2H), 1.80–1.69 (m, 2H), 1.49–1.43 (m, 2H), 1.43–1.30 (m, 6H).
General Procedure 2.
Preparation of azidoanilides: A mixture of 2a-d (5 mmol) and sodium azide (260 mg, 20 mmol, 4 equiv) was heated in N, N-dimethylformamide (5 mL) at 80 °C overnight. The reaction mixture was diluted with ethyl acetate (30 mL) and washed with ice-water (5 × 25 mL). The organic layer was dried (Na2SO4) and concentrated in vacuo and the product was used in the next reaction without purification.
5-Azido-N-phenylpentanamide (3a):
1H NMR (600 MHz, CDCl3) δ 7.52 (d, J = 7.9 Hz, 2H), 7.32 (t, J = 7.8 Hz, 2H), 7.11 (t, J = 7.4 Hz, 1H), 3.32 (t, J = 6.7 Hz, 2H), 2.39 (t, J = 7.3 Hz, 2H), 1.85–1.77 (m, 2H), 1.71–1.63 (m, 2H).
6-Azido-N-phenylhexanamide (3b):
1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 7.8 Hz, 2H), 7.32 (t, J = 7.9 Hz, 2H), 7.11 (t, J = 7.4 Hz, 1H), 3.29 (t, J = 6.8 Hz, 2H), 2.38 (t, J = 7.4 Hz, 2H), 1.82–1.72 (m, 2H), 1.71–1.59 (m, 2H), 1.53–1.42 (m, 2H).
7-Azido-N-phenylheptanamide (3c):
1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 7.7 Hz, 2H), 7.33 (t, J = 7.9 Hz, 2H), 7.12 (t, J = 7.3 Hz, 1H), 3.28 (t, J = 6.8 Hz, 2H), 2.38 (t, J = 7.4 Hz, 2H), 1.81–1.72 (m, 2H), 1.67–1.61 (m, 2H), 1.49–1.38 (m, 4H).
8-Azido-N-phenyloctanamide (3d):
1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 7.8 Hz, 2H), 7.33 (t, J = 7.8 Hz, 2H), 7.11 (t, J = 7.3 Hz, 1H), 3.27 (t, J = 6.9 Hz, 2H), 2.37 (t, J = 7.5 Hz, 2H), 1.78–1.69 (d, J = 6.8 Hz, 2H), 1.67–1.52 (m, 2H), 1.47–1.31 (m, 6H).
General Procedure 3.
To a solution of 3a-d (4 mmol) in methanol (15 mL) was added 10% palladium on carbon. The suspension was stirred under an atmosphere of H2 (3 atm) overnight. The mixture was filtered through Celite and concentrated in vacuo to give 4a-d as a crude product, which was used in the next reaction without purification.
5-Amino-N-phenylpentanamide (4a):
1H NMR (600 MHz, CDCl3) δ 7.53 (d, J = 8.0 Hz, 2H), 7.33 (t, J = 7.9 Hz, 2H), 7.11 (t, J = 7.5 Hz, 1H), 2.77 (t, J = 6.8 Hz, 2H), 2.41 (t, J = 7.4 Hz, 2H), 1.84–2.77 (m, 2H), 1.59–1.52 (m, 2H).
6-Amino-N-phenylhexanamide (4b)
1H NMR (600 MHz, CDCl3) δ 7.52 (d, J = 7.9 Hz, 2H), 7.31 (t, J = 7.7 Hz, 2H), 7.10 (t, J = 7.3 Hz, 1H), 2.71 (t, J = 6.8 Hz, 2H), 2.37 (t, J = 7.4 Hz, 2H), 1.79–1.71 (m, 2H), 1.54–1.46 (m, 2H), 1.46–1.39 (m, 2H).
7-Amino-N-phenylheptanamide (5c):
1H NMR (600 MHz, CDCl3) δ 7.52 (d, J = 7.9 Hz, 2H), 7.31 (t, J = 7.7 Hz, 2H), 7.10 (t, J = 7.3 Hz, 1H), 2.71 (t, J = 6.8 Hz, 2H), 2.37 (t, J = 7.4 Hz, 2H), 1.79–1.71 (m, 2H), 1.54–1.46 (m, 2H), 1.46–1.39 (m, 2H).
8-Amino-N-phenyloctanamide (5d):
1H NMR (600 MHz, CDCl3) δ 7.49 (d, J = 7.9 Hz, 2H), 7.29 (t, J = 7.9 Hz, 2H), 7.07 (t, J = 7.4 Hz, 1H), 2.65 (t, J = 7.0 Hz, 2H), 2.33 (t, J = 7.5 Hz, 2H), 1.75–1.67 (m, 2H), 1.44–1.38 (m, 2H), 1.38–1.28 (m, 6H).
4.2. General procedure 4. synthesis of pyruvate amides
To a stirred solution of pyruvic acid (1.2 equiv.) in DCM (1 mL/mmol), oxalyl chloride (1.2 equiv.) was added dropwise at 0 °C, followed by the addition of catalytic amount of DMF (few drops). Then it was let to warm at room temperature and stirred for 3 h. The resulting mixture was transferred with a glass syringe to a suspension of the n-amino-anilide (1 equiv.) and triethylamine (1.6 equiv.) in DCM (1 mL/ mmol) at 0 °C, under nitrogen balloon. The reaction was left again to warm at room temperature and stirred overnight. Brine was added to the resulting mixture and was extracted with ethyl acetate. The combined organic layers were dried over anhydrous Na2SO4, filtered, and condensed under reduced pressure. The crude mixture was purified by on silica gel chromatography in Petroleum Ether/Ethyl acetate (0->70% ethyl acetate) yielding pure products.
4.3. General-procedure 5. synthesis of pyruvate oximes
To a stirred solution of the corresponding pyruvate amides in Ethanol (0.04 M) an aqueous solution of the hydroxylamine (2 equiv., 0.4 M concentration) was added, and the reaction was left to stir for 4 h at room temperature. The resulting mixture was worked up using brine and extracted with ethyl acetate. The combined organic layers were dried using sodium sulfate, filtered, and condensed under reduced pressure resulting in pure oximes. The isomeric mixture of oximes was used without further purification.
4.4. General-procedure 6. synthesis of oxamates
To a stirred solution of oxamic acid (1.2 equiv.) in DCM (1 mL/mmol), oxalyl chloride (1.2 equiv.) was added dropwise at 0 °C followed by the addition of catalytic amount of DMF (few drops). Then it was let to warm at room temperature and stirred for 3 h. The resulting mixture was transferred with a glass syringe to a suspension of the n-amino-anilide (1 equiv.) and triethylamine (1.6 equiv.) in DCM (1 mL/mmol) at 0 °C, under nitrogen balloon. The reaction was left again to warm at room temperature and stirred overnight. Brine was added to the resulting mixture and was extracted with ethyl acetate. The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to yield a white solid which was recrystallized using Ethyl acetate/acetone with few drops of MeOH to yield pure compounds.
4.5. General-procedure 7. synthesis of kojic anilides
A mixture of 5-acetoxy-4-oxo-4H-pyran-2-carboxylic acid 94 (Supporting Information) (0.25 mmol, 1 equiv), dimethylformamide (10 μL) and oxalyl chloride (150 μL of 2.0 M in dichloromethane, 0.3 mmol, 1.2 equiv) in anhydrous dichloromethane (1 mL) under an atmosphere of N2 was stirred for 4 h at room temperature. A solution of aminoalkanamide (0.26 mmol, 1.1 equiv) in anhydrous dichloromethane (500 μL) was added over 10 min and the mixture was stirred overnight at room temperature. Solvent was removed under reduced pressure and the residue was dissolved in ethyl acetate (5 mL), washed with brine (3 × 1 mL) and the solvent was removed under reduced pressure. The crude solid product was dissolved in anhydrous methanol, sodium methoxide (16 mg, 0.3 mmol, 1.2 equiv) was added, and the reaction was continued until completion as monitored by TLC. The solvent was removed under reduced pressure and the residue was dissolved in water (0.5 mL) and washed with ethyl acetate (0.5 mL) three times. The aqueous layer was acidified with 1 M HCl (1 mL), extracted with ethyl acetate (3 × 1 mL). The organic extract was dried over Na2SO4, concentrated in vacuo, and purified by flash chromatography (methanol/DCM 2–4%) to give compounds 9a-d.
5-(2-oxopropanamido)-N-phenylpentanamide (5a).
Synthesized according to general procedure 4. Yield 33%. 1H NMR (600 MHz, CDCl3) δ 7.54 (d, J = 7.9 Hz, 2H), 7.43 (s, 1H(NH)), 7.33 (t, J = 7.9 Hz, 2H), 7.11 (d, 1H- s,1H(NH) overlapping), 3.37 (q, J = 6.7 Hz, 2H), 2.49 (s, 3H), 2.42 (t, J = 7.4 Hz, 2H), 1.81–1.74 (m, 2H), 1.66 (dt, J = 14.0, 6.9 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 197, 170.8, 160.4, 129, 124.2, 119.6, 38.5, 36.7, 28.7, 24.5, 22.5.
6-(2-oxopropanamido)-N-phenylhexanamide (5b).
Synthesized according to general procedure 4. Yield 36%. 1H NMR (600 MHz, CDCl3) δ 7.53 (t, J = 9.4 Hz, 2H), 7.40 (s, 1H), 7.32 (t, J = 7.8 Hz, 2H), 7.10 (t, J = 7.4 Hz, 1H), 7.03 (s, 1H), 3.31 (dd, J = 13.6, 6.8 Hz, 2H), 2.47 (s, 3H), 2.37 (t, J = 7.4 Hz, 2H), 1.76 (dt, J = 15.1, 7.5 Hz, 2H), 1.63–1.55 (m, 2H), 1.46–1.37 (m, 2H). 13C NMR (151 MHz, MeOD) δ 196.3, 173, 172.3, 161.4, 138.4, 128.5, 123.9, 119.8, 38.5, 36.7, 29.2, 28.2, 26.5, 24.9, 24.2, 23.
7-(2-oxopropanamido)-N-phenylheptanamide (5c).
Synthesized according to general procedure 4. Yield 26%. 1H NMR (600 MHz, CDCl3) δ 7.53 (d, J = 8.0 Hz, 2H), 7.32 (dd, J = 15.7, 7.8 Hz, 2H), 7.24 (s, 1H), 7.10 (dd, J = 18.9, 11.5 Hz, 1H), 6.97 (s, 1H), 3.30 (dd, J = 13.4, 6.8 Hz, 2H), 2.49 (s, 3H), 2.36 (t, J = 7.4 Hz, 2H), 1.79–1.71 (m, 2H), 1.57 (dt, J = 14.0, 6.9 Hz, 2H), 1.41 (dd, J = 26.1, 12.5 Hz, 4H). 13C NMR (151 MHz, CDCl3) δ 197.34, 171.1, 160.1, 138.0, 128.9, 124.1, 119.6, 39.5, 37.7, 29.03, 28.6, 26.5, 25.3, 24.2. HRMS calcd for C16H23N2O3 (M+H) 291.1709 found 291.1693.
5-(2-(hydroxyimino)propanamido)-N-phenylpentanamide (6a).
Synthesized according to general procedure 5. Yield 90%, 1H NMR (600 MHz, CD3OD) δ 7.55 (d, J = 7.6 Hz, 2H), 7.31 (t, J = 8.0 Hz, 2H), 7.10 (t, J = 7.4 Hz, 1H), 2.42 (t, J = 7.4 Hz, 2H), 2.05 (s, 2H), 1.98 (s, 3H), 1.77–1.71 (m, 2H), 1.66–1.59 (m, 2H). 13C NMR (151 MHz, MeOD) δ 173.1, 165.3, 150.3, 138.6, 128.2, 123.6, 119.6, 38.5, 35.8, 29.1, 22.6, 8.3. HRMS calcd for C14H20N3O3 (M+H) 278.1505 found 278.1517.
6-(2-(hydroxyimino)propanamido)-N-phenylhexanamide (6b).
Synthesized according to general procedure 5. Yield 91%, 1H NMR (600 MHz, CD3OD) δ 7.50 (d, J = 5.3, 3.3 Hz, 2H), 7.26 (t, J = 10.7, 5.2 Hz, 2H), 7.04 (t, J = 7.4 Hz, 1H), 3.23 (t, J = 7.1 Hz, 2H), 2.34 (t, J = 7.5 Hz, 2H), 1.92 (s, 3H), 1.75–1.62 (m, 2H), 1.60–1.48 (m, 2H), 1.43–1.32 (m, 2H). 13C NMR (151 MHz, MeOD) δ 173.1, 165.1, 150.1, 138.5, 128.5, 124.2, 120.3, 38.9, 36.7, 28.9, 26.2, 25.1, 7.7.
7-(2-(hydroxyimino)propanamido)-N-phenylheptanamide (6c).
Synthesized according to general procedure 5. Yield 94%, 1H NMR (600 MHz, CD3OD) δ 7.51 (d, 2H), 7.26 (t, J = 7.9 Hz, 2H), 7.04 (t, J =7.4 Hz, 1H), 3.21 (t, J = 7.1 Hz, 2H), 2.33 (t, J = 7.5 Hz, 2H), 1.93 (s, 3H), 1.70–1.63 (m, 2H), 1.55–1.48 (m, 2H), 1.42–1.31 (m, 4H). 13C NMR (151 MHz, CD3OD) δ 173.1, 164.9, 150.1, 138.6, 128.2, 123.5, 120.1, 38.6, 36.5, 29, 28.4, 26.3, 25.4, 7.9.
N1-(5-oxo-5-(phenylamino)pentyl)oxalamide (7a).
Synthesized according to general procedure 6. yield 24%. 1H NMR (600 MHz, DMSO) δ 9.87 (s, 1H), 8.74 (t, J = 5.9 Hz, 1H), 8.04 (s, 1H), 7.77 (s, 1H), 7.58 (d, J = 7.6 Hz, 2H), 7.28 (t, J = 7.9 Hz, 2H), 7.02 (t, J = 7.4 Hz, 1H), 3.14 (dd, J = 13.3, 6.7 Hz, 2H), 2.31 (t, J = 7.3 Hz, 2H), 1.60–1.45 (m, 4H). 13C NMR (151 MHz, DMSO) δ 171.5, 162.7, 160.7, 139.7, 129.2, 123.3, 119.4, 39, 36.5, 28.9, 22.9.
N1-(6-oxo-6-(phenylamino)hexyl)oxalamide (7b).
Synthesized according to general procedure 6. yield 21%. 1H NMR (600 MHz, DMSO) δ 9.86 (s, 1H), 8.70 (t, J = 6.0 Hz, 1H), 8.04 (s, 1H), 7.76 (s, 1H), 7.59 (d, J = 7.6 Hz, 2H), 7.28 (t, J = 7.9 Hz, 2H), 7.02 (t, J = 7.4 Hz, 1H), 3.11 (dd, J = 13.5, 6.8 Hz, 2H), 2.29 (t, J = 7.5 Hz, 2H), 1.64–1.54 (m, 2H), 1.53–1.44 (m, 2H), 1.32–1.23 (m, 2H). 13C NMR (151 MHz, DMSO) δ 171.6, 162.8, 160.6, 139.8, 129.1, 123.4, 119.5, 39.2, 36.8, 29, 26.5, 25.3.
N1-(8-oxo-8-(phenylamino)octyl)oxalamide (7d).
Synthesized according to general procedure 6. yield 35%. 1H NMR (600 MHz, DMSO) δ 9.85 (s, 1H), 8.68 (t, J = 6.0 Hz, 1H), 8.03 (s, 1H), 7.76 (s, 1H), 7.59 (d, J = 7.7 Hz, 2H), 7.28 (t, J = 7.9 Hz, 2H), 7.02 (t, J = 7.4 Hz, 1H), 3.10 (dd, J = 13.5, 6.8 Hz, 2H), 2.29 (t, J = 7.5 Hz, 2H), 1.60–1.54 (m, 2H), 1.49–1.40 (m, 2H), 1.33–1.19 (m, 6H). 13C NMR (151 MHz, DMSO) δ 171.7, 162.9, 160.6, 139.8, 129, 123.6, 119.1, 39.2, 36.9, 29.2, 29.1, 29, 26.7, 25.6.
N-(6-oxo-6-(phenylamino)hexyl)-1H-pyrrole-2-carboxamide (8b).
To a stirred suspension of 1H-pyrrole-2-carboxylic acid (230 mg, 2.1 mmol) in DCM (8 mL), were added, EDC-HCl (600 mg, 3.1 mmol, 1.5 equiv.), cat. amount of DMAP (approximately 10 mol %) and 6-amino-N-phenylhexanamide 4b (428 mg, 2.1 mmol, 1 equiv.). The resulting mixture was stirred at room temperature overnight, after which it was diluted with brine and ethyl acetate. The organic layer was separated, dried over Na2SO4, filtered, and concentrated in vacuo. The crude mixture was separated on silica gel chromatography in ethyl acetate/Hexanes to provide N-(6-oxo-6-(phenylamino) hexyl)-1H-pyrrole-2-carboxamide (8b) (403 mg, 64% yield). 1H NMR (600 MHz, CD3CN) δ 9.93 (s, 1H), 8.32 (s, 1H), 7.57 (d, J = 8.0 Hz, 2H), 7.32 (t, J = 7.9 Hz, 2H), 7.09 (t, J = 7.4 Hz, 1H), 6.90 (d, J = 1.1 Hz, 1H), 6.79 (s, 1H), 6.65 (s, 1H), 6.17 (dt, J = 5.3, 1.6 Hz, 1H), 3.33 (dd, J = 13.2, 6.7 Hz, 2H), 2.34 (t, J = 7.4 Hz, 2H), 1.97 (dt, J = 4.8, 2.4 Hz, 2H), 1.74–1.66 (m, 2H), 1.63–1.56 (m, 2H), 1.45–1.38 (m, 2H). 13C NMR (151 MHz, CD3CN) δ 171.6, 160.8, 139.1, 128.8, 126.5, 123.4, 120.9, 119.5, 109.2, 108.81, 38.5, 36.6, 29.2, 26.2, 24.9. HRMS calcd for C17H22N3O2 (M+H) 264.1348 found 264.1354.
5-Hydroxy-4-oxo-N-(5-oxo-5-(phenylamino)pentyl)-4H-pyran-2-carboxamide (9a):
Synthesized according to General Procedure 7. 12.75 mg (16%). 1H NMR (400 MHz, CD3OD) δ 8.00 (s, 1H), 7.53 (d, J = 7.7 Hz, 2H), 7.29 (t, J = 7.9 Hz, 2H), 7.08 (d, J = 7.3 Hz, 1H), 7.05 (s, 1H), 3.43–3.38 (m, 2H), 2.42 (t, J = 7.2 Hz, 2H), 1.80–1.64 (m, 4H).13C NMR (151 MHz, CD3OD) δ 175, 172.8, 159.4, 155.8, 147.7, 139.2, 138.5, 128.4, 123.7, 119.8, 112.8, 39.2, 39.1, 36, 28.4, 22.7. HRMS-ESI M/z: [M+H]+calcd. 331.1293 for C17H18N2O5, found 331.1310.
5-Hydroxy-4-oxo-N-(6-oxo-6-(phenylamino)hexyl)-4H-pyran-2-carboxamide (9b):
Synthesized according to General Procedure 7. 14 mg (17.6%). 1H NMR (400 MHz, CD3OD) δ 7.99 (s, 1H), 7.51 (d, J = 7.6 Hz, 2H), 7.28 (d, J = 7.5 Hz, 2H), 7.06 (t, J = 7.4 Hz, 1H), 7.02 (s, 1H), 3.38 (q, J = 6.8 Hz, 2H), 2.38 (t, J = 7.4 Hz, 2H), 1.79–1.70 (m, 2H), 1.70–1.59 (m, 2H), 1.50–1.35 (m, 2H). 13C NMR (151 MHz, CD3OD) δ 175, 173.1, 159.4, 155.8, 147.7, 139.2, 138.5, 128.4, 123.7, 119.8, 112.7, 39.4, 39.2, 36.3, 28.6, 26, 25.1. HRMS-ESI M/z: [M+H]+calcd. 345.145 for C18H20N2O5, found 345.1437.
5-Hydroxy-4-oxo-N-(7-oxo-7-(phenylamino)heptyl)-4H-pyran-2-carboxamide (9c):
Synthesized according to General Procedure 7. 12.4 mg (14%). 1H NMR (600 MHz, CD3OD) δ 8.05 (s, 1H), 7.55 (d, J = 7.9 Hz, 2H), 7.30 (t, J = 7.8 Hz, 2H), 7.09 (t, J = 7.4 Hz, 1H), 7.07 (s, 1H), 3.41–3.38 (m, 2H), 2.40 (t, J = 7.4 Hz, 2H), 1.78–1.69 (m, 2H), 1.68–1.61 (m, 2H), 1.49–1.40 (m, 4H). 13C NMR (151 MHz, CD3OD): δ 175, 173.2, 159.4, 159.3, 155.9, 155.9, 147.6, 139.3, 138.5, 128.4, 123.7, 119.8, 112.8, 39.5, 39.4, 36.4, 28.7, 28.5, 26.3, 25.4. HRMS-ESI M/z: [M+H]+calcd. 359.1606 for C19H22N2O5, found 359.1618.
5-Hydroxy-4-oxo-N-(8-oxo-8-(phenylamino)octyl)-4H-pyran-2-carboxamide (9d):
Synthesized according to General Procedure 7. 20 mg (6%). 1H NMR (600 MHz, CD3OD) δ 8.01 (s, 1H), 7.55 (dd, J = 8.5, 0.9 Hz, 2H), 7.31 (t, J = 7.2 Hz, 2H), 7.09 (t, J = 7.4 Hz, 1H), 7.05 (s, 1H), 3.37 (t, J = 7.2 Hz, 2H), 2.38 (t, J = 7.5 Hz, 2H), 1.76–1.69 (m, 2H), 1.66–1.59 (m, 2H), 1.4–1.36 (m, 6H). 13C NMR (151 MHz, CD3OD): δ 175.4, 159.3, 155.7, 139.2, 138.5, 128.4, 123.7, 119.8, 112.7, 39.4, 36.5, 28.8, 28.6, 26.4, 25.4. HRMS-ESI M/z: [M+H]+calcd. 373.1763 for C20H24N2O5, found 373.1767.
4.6. Synthesis of 4-((tert-butoxycarbonyl)amino)butanoic acid (12)
In a 250 mL round–bottom flask equipped with a stir–bar, 4-aminobutanoic acid (11) (5 g, 48.5 mmol, 1 equiv) was first dissolved in a mixture of acetone (50 mL) and water (50 mL). Triethylamine (13.6 mL, 97 mmol, 2 equiv) was added to the reaction mixture, followed by di-tert-butyl dicarbonate (10.92 g, 75 mmol, 1.03 equiv). The mixture was stirred at room temperature for 4 h. Acetone was removed under reduced pressure and the residual water was acidified with 1 M HCl (200 mL) and extracted with ethyl acetate (3 × 100 mL). The organic extract was dried (Na2SO4), concentrated in vacuo and triturated in hexanes to yield 4-((tert-butoxycarbonyl) amino) butanoic acid (12) as a white solid (9.23 g, 94%).
4.7. Synthesis of tert-butyl(4-oxo-4-(phenylamino)butyl)carbamate (13)
In a 10 mL round – bottomed flask, a mixture of 4-((tert-butoxycarbonyl) amino) butanoic acid (2 mmol, 490 mg), aniline (2 mmol, 182 μL), EDCl (2 mmol, 384 mg, 1 equiv), DMAP (10 mol %, 24 mg) was stirred in dichloromethane (4 mL) under an atmosphere of nitrogen (balloon) overnight. Solvent was removed under reduced pressure, and the reaction mixture was diluted in ethyl acetate (10 mL) and washed with 1 M HCl (10 mL), saturated NaHCO3 (10 mL) (aq.) and brine (10 mL) The organic layer was dried (Na2SO4), concentrated in vacuo and purified using flash chromatography (ethyl acetate/hexanes 10–70%) to yield tert-butyl (4-oxo-4-(phenylamino) butyl) carbamate as a white solid, 450 mg, 81%. 1H NMR (600 MHz, CDCl3) δ 7.62 (d, J = 7.7 Hz, 2H), 7.33 (t, J = 7.9 Hz, 2H), 7.10 (t, J = 7.3 Hz, 1H), 3.30–3.22 (m, 2H), 2.46–2.34 (m, 2H), 1.95–1.84 (m, 2H), 1.47 (s, 9H).
4.8. Synthesis of 7-((tert-butoxycarbonyl)amino)heptanoic acid (16)
In a 250 mL round – bottom flask equipped with a stir–bar, 7-aminoheptanoic acid (15) (12.72 g, 70 mmol, 1 equiv) was first dissolved in a mixture of acetone (50 mL) and water (50 mL). Triethylamine (19.53 mL, 140 mmol, 2 equiv) was added to the reaction mixture, followed by di-tert-butyl dicarbonate (16.37 g, 75 mmol, 1.07 equiv). The mixture was stirred at room temperature for 4 h. Acetone was first removed under reduced pressure and the residual water was acidified with 1 M HCl (200 mL) and extracted with ethyl acetate (100 mL) three times. The organic extract was dried (Na2SO4), concentrated in vacuo and triturated in hexanes to yield 7-((tert-butoxycarbonyl) amino) heptanoic acid (16) as a white solid (15.7 g, 91%).
4.9. Synthesis of boc-arenes
In a 10 mL round–bottomed flask, a mixture of 7-((tert-butoxycarbonyl) amino) heptanoic acid (2 mmol, 490 mg), the respective aminoarene (2 mmol), EDCl (2 mmol, 384 mg, 1 equiv), DMAP (10 mol %, 24 mg) was stirred in dichloromethane (4 mL) under an atmosphere of nitrogen (balloon) overnight. Solvent was removed under reduced pressure, and the reaction mixture was diluted in ethyl acetate (10 mL) and washed with 1 M HCl (10 mL), saturated NaHCO3 (10 mL) (aq.) and brine (10 mL). The organic layer was dried (Na2SO4), concentrated in vacuo and purified using flash chromatography (ethyl acetate/hexanes 10–70%) to yield Boc – arenes 17–50.
An alternative procedure involved using a mixture of 7-((tert-butoxycarbonyl) amino) heptanoic acid (2 mmol, 490 mg), the aminoarene (2 mmol), HATU (2 mmol, 760 mg, 1 equiv), HOBt (1 mmol, 135 mg, 1 equiv) and NMM (660 μl, 6 mmol, 3 equiv) in dimethylformamide (4 mL) under an atmosphere of nitrogen (balloon) overnight. The reaction mixture was diluted in ethyl acetate (10 mL), washed with ice–water (5 × 10 mL) and dried (Na2SO4). The organic layer was concentrated in vacuo and purified in the same manner as above.
tert-butyl(7-((4-acetylphenyl)amino)-7-oxoheptyl)carbamate (17):
1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 8.7 Hz, 2H), 7.69 (d, J = 8.3 Hz, 2H), 3.14 (q, J = 6.1 Hz, 2H), 2.39 (t, 2H), 1.80–1.71 (m, 2H), 1.54–1.48 (m, 2H), 1.46 (s, 9H), 1.43–1.34 (m, 4H).Obtained a partially purified product that was used in the next step.
tert-butyl(7-((4-methoxyphenyl) amino)-7-oxoheptyl)carba mate (18):
White solid, 200 mg (29%) mp. 120–125 °C. 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 3.80 (s, 3H), 3.13 (q, J = 5.1 Hz, 2H), 2.33 (t, J = 7.3 Hz, 2H), 1.80–1.70 (m, 2H), 1.54–1.47 (m, 2H), 1.45 (s, 9H), 1.41–1.31 (m, 4H). [1].3CNMR (151 MHz, CDCl3): 171.3, 156.3, 156.2, 131.2, 121.7, 114.1, 79.2, 55.5, 40.2, 37.3, 29.9, 28.5, 28.5, 26.1, 25.5.
tert-butyl(7-((4-fluorophenyl) amino)-7-oxoheptyl)carbamate (19):
White solid, 145 mg (21.4%) mp. 90–95 °C. 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 8.6 Hz, 2H), 6.86 (d, J = 8.9 Hz, 2H), 3.17–3.06 (m, 2H), 2.33 (t, J = 7.3 Hz, 2H), 1.79–1.68 (m, 2H), 1.54–1.48 (m, 2H), 1.45 (s, 9H), 1.41–1.31 (m, 2H). [1].3CNMR (151 MHz, CDCl3): 171.6, 160, 158.4, 156.3, 134.2, 121.6, 121.5, 115.6, 115.5, 79.3, 40.1, 37.2, 29.9, 28.5, 28.3, 26, 25.4.
tert-butyl (7-oxo-7-(pyridin-4-ylamino) heptyl)carbamate (20):
1H NMR (400 MHz, CDCl3) δ 8.48 (d, J = 5.4 Hz, 2H), 7.61 (d, J = 4.6 Hz, 2H), 3.15 (q, 6.5 Hz, 2H), 2.40 (t, J = 6.9 Hz, 2H), 1.80–1.70 (m, 2H), 1.53–1.49 (m, 2H), 1.47 (s, 9H), 1.43–1.34 (m, 4H). Obtained a partially purified product, which was used in the next step. Also, it was slow to purification until 4–7% Methanol/DCM was used.
tert-butyl (7-oxo-7-((4-(pentafluoro-λ [6]-sulfaneyl)phenyl) amino)heptyl)carbamate (21):
1H NMR (400 MHz, CDCl3) δ 7.69 (s, 4H), 3.13 (q, J = 5.7 Hz, 2H), 2.37 (t, J = 7.1 Hz, 2H), 1.79–1.70 (m, 2H), 1.53–1.48 (m, 2H), 1.45 (s, 9H), 1.41–1.35 (m, 4H). Obtained a partially purified product, which was used in the next step.
tert-butyl (7-((2,4-difluorophenyl) amino)-7-oxoheptyl) carbamate (22):
White solid, 300 mg (42%) mp. 75–80 °C. 1H NMR (400 MHz, CDCl3) δ 8.26 (dd, J = 15.3, 9.0 Hz, 1H), 6.93–6.79 (m, 2H), 3.12 (q, J = 5.8 Hz, 2H), 2.40 (t, J = 7.5 Hz, 2H), 1.80–1.68 (m, 2H), 1.54–1.48 (m, 2H), 1.45 (s, 9H), 1.42–1.32 (m, 4H). [1].3CNMR (151 MHz, CDCl3): 171.6, 160, 158.4, 156.3, 134.2, 121.6 (d, J = 7.8 Hz), 115.6 (d, J = 22.4 Hz), 79.3, 40.1, 37.2, 29.9, 28.3, 26, 25.4.
tert-butyl (7-oxo-7-((2,4,6-trifluorophenyl) amino) heptyl) carbamate (23):
1H NMR (400 MHz, CDCl3) δ 6.73 (t, J = 8.1 Hz, 2H), 3.12 (q, J = 7.0 Hz, 2H), 2.34 (t, J = 7.5 Hz, 2H), 1.92–1.81 (m, 2H), 1.57–1.48 (m, 2H), 1.44 (s, 9H), 1.40–1.30 (m, 4H). Obtained a crude product which was used in the next step without purification.
tert-butyl (7-oxo-7-((5-phenylthiazol-2-yl) amino) heptyl) carbamate (24):
1H NMR (600 MHz, CDCl3) δ 7.79 (d, J = 7.2 Hz, 2H), 7.44 (t, J = 7.6 Hz, 2H), 7.37 (t, J = 7.4 Hz, 1H), 7.13 (s, 1H), 3.16–3.07 (m, 2H), 2.28 (t, J = 7.5 Hz, 2H), 1.71–1.59 (m, 2H), 1.47 (s, 9H), 1.43–1.30 (m, 4H), Obtained a partially purified product that was used in the next step.
tert-butyl (7-oxo-7-((3,4,5-trimethoxyphenyl) amino) heptyl) carbamate (25):):
[1]HNMR (400 MHz, CDCl3): δ 6.91 (s, 2H), 3.15 (d, J = 6 Hz, 2H), 2.36 (t, J = 7.2 Hz, 2H), 1.77–1.69 (m, 2H), 1.51–1.45 (m, 2H), 1.44 (s, 9H), 1.4–1.35 (m, 4H). [1].3CNMR (151 MHz, CDCl3): δ 171.6, 153.3, 134.4, 134.3, 97.3, 79.3, 61, 56.1, 40.12, 37.34, 30, 28.5, 28.3, 29.6, 25.4.
tert-butyl (7-(naphthalen-1-ylamino)-7-oxoheptyl)carbamate (26):
Pink solid, 778 mg, quantitative yield, mp. 90–95 °C. 1H NMR (600 MHz, CDCl3) δ 7.92–7.86 (m, 3H), 7.71 (d, J = 8.0 Hz, 1H), 7.54–7.50 (m, 2H), 7.47 (t, J = 7.7 Hz, 1H), 3.14 (d, J = 5.4 Hz, 2H), 2.51 (t, J = 7.2 Hz, 2H), 1.85–1.77 (m, 2H), 1.55–1.49 (m, 2H), 1.46 (s, 9H), 1.41–1.31 (m, 4H). [1].3C NMR (151 MHz, CDCl3): 172, 156.1, 134.1, 132.4, 128.7, 128.5, 127.3, 126.2, 126, 125.8, 125.7, 121.3, 120.9, 79.1, 40.4, 37.4, 30, 28.8, 28.5, 26.4, 25.7.
tert-butyl (7-((4-benzoylphenyl)amino)-7-oxoheptyl)carbamate (27):
1H NMR (600 MHz, CDCl3) δ 7.81 (d, J = 8.6 Hz, 2H), 7.77 (t, J = 8.3 Hz, 4H), 7.59 (t, J = 7.4 Hz, 1H), 7.48 (t, J = 7.7 Hz, 2H), 3.11 (d, J = 6.0 Hz, 2H), 2.38 (t, J = 7.2 Hz, 2H), 1.75–1.68 (m, 2H), 1.50–1.47 (m, 2H), 1.46 (s, 9H), 1.38–1.29 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 196, 172.3, 156.3, 142.7, 137.9, 132.5, 132.3, 131.6, 130.1, 129.9, 128.3, 128.1, 118.8, 79.3, 40.2, 37.3, 30, 28.5, 28.4, 26.1, 25.4.
tert-butyl (7-((2,5-dimethoxyphenyl amino)-7-oxoheptyl) carbamate (28):
Brown solid, 500 mg (65.7%) mp. 80–85 °C. 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 2.5 Hz, 1H), 6.79 (d, J = 8.9 Hz, 1H), 6.57 (dd, J = 8.9, 2.9 Hz, 1H), 3.85 (s, 3H), 3.79 (s, 3H), 3.12 (d, J = 6.3 Hz, 2H), 2.40 (t, J = 7.4 Hz, 2H), 1.78–1.70 (m, 2H), 1.54–1.47 (m, 2H), 1.45 (s, 9H), 1.42–1.32 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 171.2, 153.9, 141.8, 128.4, 110.6, 108.5, 105.7, 56.2, 55.8, 40.5, 37.9, 29.9, 28.9, 28.4, 26.6, 25.4.
tert-butyl (7-((4-((4-methylphenyl)sulfonamido)phenyl) ami no)-7-oxoheptyl)carbamate (29):
White solid, 588 mg (60.1%) mp. 140–145 °C. 1H NMR (600 MHz, CDCl3) δ 7.62 (d, J = 7.9 Hz, 2H), 7.46 (d, J = 8.4 Hz, 2H), 7.24 (d, J = 8.0 Hz, 2H), 7.02 (d, J = 8.4 Hz, 2H), 3.13 (d, J = 5.8 Hz, 2H), 2.40 (s, 3H), 2.34 (t, J = 7.2 Hz, 2H), 1.76–1.69 (m, 2H), 1.52–1.48 (m, 2H), 1.47 (s, 9H), 1.43–1.33 (m, 4H). [1].3C NMR (151 MHz, CDCl3): δ 171.5, 156.2, 143.9, 135.9, 132, 129.7, 127.3, 123.5, 120.5, 40.1, 37.3, 30, 28.5, 28.3, 25.9, 25.4, 21.6.
tert-butyl (7-((2-methoxyphenyl)amino)-7-oxoheptyl)carbamate (30):
1H NMR (600 MHz, CDCl3) δ 8.41 (dd, J = 8.0, 1.3 Hz, 1H), 7.05 (td, J = 7.9, 1.5 Hz, 1H), 6.98 (td, J = 7.8, 1.2 Hz, 1H), 6.90 (dd, J = 8.1, 1.1 Hz, 1H), 3.91 (s, 3H), 3.14 (q, J = 6.2 Hz, 2H), 2.41 (t, J = 7.5 Hz, 2H), 1.80–1.72 (m, 2H), 1.55–1.48 (m, 2H), 1.46 (s, 9H), 1.44–1.36 (m, 4H). [1].3C NMR (151 MHz, CDCl3): δ 171.1, 156, 147.7, 127.7, 123.5, 121.1, 119.7, 109.8, 79, 55.64, 40.5, 37.9, 29.9, 28.9, 28.4, 26.5, 25.5. tert-butyl (7-((4-chlorophenyl) amino)-7-oxoheptyl)carbamate (31): White solid, 597 mg (8%) mp. 105–110 °C. 1H NMR (600 MHz, CDCl3) δ 7.54 (d, J = 8.4 Hz, 2H), 7.30 (d, J = 8.9 Hz, 2H), 3.15 (q, J = 6.3 Hz, 2H), 2.36 (t, J = 7.3 Hz, 2H), 1.79–1.72 (m, 2H), 1.54–1.49 (m, 2H), 1.47 (s, 9H), 1.44–1.34 (m, 4H). [1].3C NMR (151 MHz, CDCl3): 171.6, 156.3, 136.8, 129, 121, 79.3, 40, 37.3, 30, 28.5, 28.5, 28.22, 25.9, 25.4.
tert-butyl (7-((4-bromophenyl)amino)-7-oxoheptyl)carbamate (32):
White solid, 552 mg (69%) mp. 115–120 °C. 1H NMR (600 MHz, CDCl3) δ 7.49 (d, J = 8.4 Hz, 2H), 7.44 (d, J = 8.8 Hz, 2H), 3.15 (q, J = 6.1 Hz, 2H), 2.36 (t, J = 7.3 Hz, 2H), 1.80–1.73 (m, 2H), 1.53–1.48 (m, 2H), 1.47 (s, 9H), 1.45–1.35 (m, 4H). [1].3C NMR (151 MHz, CDCl3): δ 171.59, 156.2, 137.3, 131.9, 121.3, 116.5, 79.3, 40.1, 37.3, 30, 28.5, 28.3, 25.9, 25.4.
tert-butyl (7-([1,1′-biphenyl]-4-ylamino)-7-oxoheptyl)carbamate (33):
White solid, 500 mg, (63%) mp. 150–155 °C. 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 8.4 Hz, 2H), 7.60–7.54 (m, 4H), 7.43 (t, J = 7.6 Hz, 2H), 7.33 (t, J = 7.3 Hz, 1H), 3.14 (q, J = 6.4 Hz, 2H), 2.38 (t, J = 7.3 Hz, 2H), 1.82–1.72 (m, 2H), 1.54–1.48 (m, 2H), 1.46 (s, 9H), 1.43–1.35 (m, 4H). [1].3C NMR (151 MHz, CDCl3): 171.5, 156.2, 140.6, 137.5, 136.9, 128.8, 127.6, 127.1, 126.9, 120, 79.3, 40.2, 37.4, 30, 28.6, 28.6, 26.13, 25.5.
tert-butyl (7-oxo-7-(thiazol-2-ylamino) heptyl)carbamate (34):
White solid, 512 mg (78%) mp. 85–90 °C. 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 3.6 Hz, 1H), 7.01 (d, J = 3.6 Hz, 1H), 3.11 (d, J = 6.2 Hz, 2H), 2.55 (t, J = 7.4 Hz, 2H), 1.84–1.73 (m, 2H), 1.54–1.47 (m, 2H), 1.44 (s, 9H), 1.40–1.31 (m, 4H). [1].3C NMR (151 MHz, CDCl3): δ 171.2, 160, 156, 136, 113.5, 79.1, 40.4, 36.1, 29.9, 28.9, 28.5, 26.5, 25.
tert-butyl (7-(benzo[d]thiazol-2-ylamino)-7-oxoheptyl)carbamate (35)
1H NMR (400 MHz, CDCl3) δ 7.82 (d, J = 7.8 Hz, 1H), 7.71 (d, J = 8.1 Hz, 1H), 7.44 (t, J = 7.7 Hz, 1H), 7.32 (t, J = 7.5 Hz, 1H), 3.17–3.04 (m, 2H), 2.52 (t, J = 7.4 Hz, 2H), 1.79–1.67 (m, 2H), 1.56–1.46 (m, 2H), 1.44 (s, 9H), 1.41–1.26 (m, 4H). Obtained a partially purified product that was used in the next step.
tert-butyl (7-((3-methoxyphenyl) amino)-7-oxoheptyl) carbamate (36):
White solid, 520 mg (74%) mp. 80–85 °C. [1].H NMR (400 MHz, CDCl3): δ 7.35 (s, 1H), 7.16 (t, J = 7.8 Hz, 1H), 7.03 (t, J = 7.8 Hz, 1H), 6.62 (d, J = 8.1 Hz, 1H), 3.74 (s, 3H), 3.06 (q, J = 6.3 Hz, 2H), 2.3 (t, J = 7.2 Hz, 2H), 1.78–1.68 (m, 2H), 1.43 (s, 9H), 1.4–1.26 (m, 4H). [1].3C NMR (151 MHz, CDCl3): δ 171.6. 160.1, 156.2, 139.4, 129.6, 11.8, 110, 105.3, 79.2, 40.2, 37.5, 30, 28.5, 26.2, 25.4.
tert-butyl (7-((4-methyl-5-phenylthiazol-2-yl)amino)-7-oxoheptyl)carbamate (37):
1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 7.3 Hz, 2H), 7.43 (t, J = 7.5 Hz, 2H), 7.35 (t, J = 7.3 Hz, 1H), 3.09 (q, J = 5.6 Hz, 2H), 2.49 (s, CH3), 2.15 (t, J = 6.7 Hz, 2H), 1.66–1.53 (m, 2H), 1.44 (s, 9H), 1.31–1.19 (m, 4H). [1].3C NMR (151 MHz, CDCl3): δ 171.6, 156.4, 144.2, 135, 129.6, 128.5, 128.4, 128.38, 127.7, 121.8, 79.1, 40.5, 35, 29.8, 28.6, 28.5, 26.4, 24.5, 12.1.
tert-butyl (7-((4,5-diphenylthiazol-2-yl) amino)-7-oxoheptyl) carbamate (38):
1H NMR (400 MHz, CDCl3) δ 7.42 (dd, J = 6.6, 3.0 Hz, 1H), 7.31 (m, 4H), 3.12 (d, J = 6.0 Hz, 2H), 2.41–2.30 (m, 2H), 1.76–1.61 (m, 4H), 1.45 (s, 9H), 1.40–1.22 (m, 4H). [1].3C NMR (151 MHz, CDCl3): δ 171.8, 158, 156, 143.7, 131.9, 129.5, 129.4, 129.1, 128.8, 128.7, 128.6, 128, 127.8, 126.7, 79.1, 40.5, 35, 29.8, 28.6, 28.5, 26.4, 24.5.
tert-butyl (7-((2-bromophenyl)amino)-7-oxoheptyl)carbamate (39):
1H NMR (600 MHz, CDCl3) δ 8.36 (d, J = 7.0 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.32 (t, J = 7.7 Hz, 1H), 6.98 (t, J = 7.5 Hz, 1H), 3.13 (d, J = 5.2 Hz, 2H), 2.44 (t, J = 7.4 Hz, 2H), 1.80–1.73 (m, 2H), 1.54–1.47 (m, 2H), 1.45 (s, 9H), 1.42–1.34 (m, 4H). Obtained a partially purified product that was used in the next step.
tert-butyl (7-((3-bromophenyl) amino)-7-oxoheptyl)carbamate (40):
White solid, 420 mg (52.6%) mp. 90–95 °C. 1H NMR (600 MHz, CDCl3) δ 7.83 (s, 1H), 7.47 (d, J = 7.0 Hz, 1H), 7.23 (d, J = 7.8 Hz, 1H), 7.17 (t, J = 8.0 Hz, 1H), 3.13 (d, J = 5.5 Hz, 2H), 2.35 (t, J = 7.3 Hz, 2H), 1.77–1.70 (m, 2H), 1.53–1.47 (m, 2H), 1.46 (s, 9H), 1.42–1.33 (m, 4H). [1].3C NMR (151 MHz, CDCl3): δ 171.9, 156.3, 139.6, 130.2, 126.9, 122.7, 122.5, 118.2, 79.3, 40.2, 37.2, 30, 28.5, 28.3, 26, 25.4.
tert-butyl (7-((6-chlorobenzo[d]thiazol-2-yl)amino)-7-oxoheptyl)carbamate (41):
1H NMR (400 MHz, CDCl3) δ 7.80 (s, 1H), 7.65 (d, J = 8.6 Hz, 1H), 7.40 (d, J = 8.6 Hz, 1H), 3.13 (q, J = 5.3 Hz, 2H), 2.52 (t, J = 7.3 Hz, 2H), 1.83–1.73 (m, 2H), 1.55–1.49 (m, 2H), 1.45 (s, 9H), 1.43–1.33 (m, 4H). Obtained a partially purified product that was used in the next step.
tert-butyl (7-((6-fluorobenzo[d]thiazol-2-yl) amino)-7-oxoheptyl)carbamate (42):
White solid, 400 mg (50.6%) mp. 150–155 °C. 1H NMR (400 MHz, CDCl3) δ 7.69 (dd, J = 8.8, 4.7 Hz, 1H), 7.52 (dd, J = 8.1, 2.5 Hz, 1H), 7.17 (td, J = 8.9, 2.6 Hz, 1H), 3.12 (q, J = 6.3 Hz, 2H), 2.49 (t, J = 7.3 Hz, 2H), 1.83–1.70 (m, 2H), 1.53–1.47 (m, 2H), 1.45 (s, 9H), 1.41–1.30 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 171.8, 160.4, 158.8, 156, 144.2, 133 (d, J = 10.5 Hz), 121.3 (d, J = 8.6 Hz), 114.7 (d, J = 24.6 Hz), 108 (d, J = 26.6 Hz), 79.2, 40.4, 36.3, 29.9, 28.6, 28.5, 26.3, 24.8.
tert-butyl (7-((6-bromobenzo[d]thiazol-2-yl) amino)-7-oxoheptyl)carbamate (43):
Yellow solid, 160 mg (17.%) mp. 180–185 °C. 1H NMR (600 MHz, DMSO) δ 8.25 (s, 1H), 7.67 (d, J = 8.5 Hz, 1H), 7.57 (d, J = 8.2 Hz, 1H), 6.79 (s, 1H), 2.89 (d, J = 5.8 Hz, 2H), 2.48 (t, J = 7.1 Hz, 2H), 1.64–1.57 (m, 2H), 1.37 (s, 9H), 1.32–1.20 (m, 4H). 13C NMR (151 MHz, DMSO) δ 173, 159.2, 156., 148.2, 134.1, 129.6, 124.7, 122.5, 115.8, 77.8, 35.5, 29.8, 28.7, 26.5, 24.9.
tert-butyl (7-((5-bromobenzo[d]oxazol-2-yl) amino)-7-oxoheptyl)carbamate (44):
Brown solid, 437 mg, (49.6%) mp. 140–145 °C. 1H NMR (400 MHz, CDCl3) δ 7.73 (s, 1H), 7.39–7.32 (m, 2H), 3.13 (q, J = 6.3 Hz, 2H), 2.75–2.61 (m, 2H), 1.83–1.72 (m, 2H), 1.55–1.47 (m, 2H), 1.45 (s, 9H), 1.41–1.25 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 171.3, 156.4, 156.1, 147.2, 126.7, 121.4, 117.5, 111.5, 79.2, 40.4, 36.7, 29.9, 28.6, 28.5, 26.3, 24.9, 24.6.
tert-butyl (7-(benzo[d]oxazol-2-ylamino)-7-oxoheptyl) carbamate (45): 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 6.8 Hz, 1H), 7.47 (d, J = 8.2 Hz, 1H), 7.32 (t, J = 7.0 Hz, 1H), 7.23 (t, J = 9.5 Hz, 1H), 3.14 (q, J = 6.6 Hz, 2H), 2.78–2.63 (m, 2H), 1.84–1.73 (m, 2H), 1.57–1.48 (m, 2H), 1.46 (s, 9H), 1.43–1.30 (m, 4H). Obtained a partially purified product that was used in the next step.
tert-butyl(7-((6-methoxybenzo[d]thiazol-2-yl) amino)-7-oxoheptyl)carbamate (46):
White solid, 446 mg (54. %) mp. 105–110 °C. [1].H NMR (400 MHz, CDCl3): 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 8.9 Hz, 1H), 7.30 (d, J = 2.4 Hz, 1H), 7.03 (dd, J = 8.9, 2.5 Hz, 1H), 3.86 (s, 3H), 3.05 (q, J = 5.7 Hz, 2H), 1.71–1.61 (m, 2H), 1.52–1.46 (m, 2H), 1.43 (s, 9H), 1.32–1.17 (m, 4H). [1].3C NMR (151 MHz, CDCl3): δ 171.6, 157.3, 156.9, 156, 142, 133.2, 121, 115.3, 104.4, 79.1, 55.9, 40.4, 36.3, 29.9, 28.7, 28.5, 26.4, 24.9.
tert-butyl(7-((5-methoxybenzo[d]thiazol-2-yl) amino)-7-oxoheptyl)carbamate (47):
White solid, 571 mg (70%) mp. 85–90 °C. [1].H NMR (400 MHz, CDCl3): δ 7.69 (d, J = 8.6 Hz, 2H), 7.24 (s, 1H), 6.97 (d, J = 8.7 Hz, 2H), 3.86 (s, 3H), 3.06 (d, J = 6.4 Hz, 2H), 2.46 (t, J = 6.5 Hz, 2H), 1.75–1.63 (m, 2H), 1.54–1.47 (m, 2H), 1.44 (s, 9H), 1.35–1.19 (m, 4H). [1].3C NMR (151 MHz, CDCl3): δ 171.8, 160.7, 159.1, 156, 149.1, 123.8, 122, 113.1, 104.4, 79.1, 55.7, 40.4, 36.3, 29.9, 28.8, 28.5, 26.4, 24.9.
tert-butyl(7-((5-(4-methoxyphenyl) thiazol-2-yl)amino)-7-oxoheptyl)carbamate (48):
1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 8.9 Hz, 2H), 7.00 (s, 1H), 6.95 (d, J = 8.8 Hz, 2H), 3.85 (s, 3H), 3.18–3.06 (m, 2H), 2.33 (t, J = 7.5 Hz, 2H), 1.79–1.60 (m, 2H), 1.45 (s, 9H), 1.34–1.21 (m, 4H). Obtained a partially purified product that was used in the next step.
tert-butyl(7-((5-(4-bromophenyl) thiazol-2-yl) amino)-7-oxo-heptyl)carbamate (49):
White solid, 374 mg (38.7%) mp. 140–145 °C. 1H NMR (600 MHz, CDCl3) δ 7.64 (d, J = 7.5 Hz, 2H), 7.51 (d, J = 7.5 Hz, 2H), 7.12 (s, 1H), 3.14–3.04 (m, 2H), 2.23–2.12 (m, 2H), 1.70–1.48 (m, 2H), 1.39 (s, 9H), 1.35–1.03 (m, 4H). [1].3C NMR (151 MHz, CDCl3): δ 171.3, 159, 156.1, 148.3, 133.2, 132, 127.7, 122, 108.3, 79.2, 40.4, 35.9, 29.9, 28.6, 28.5, 26.3, 24.8.
tert-butyl(7-oxo-7-((5-(4-(((2,2,2-trichloroethoxy)carbonyl) oxy)phenyl)thiazol-2-yl)amino)heptyl)carbamate (50):
White solid, 520 mg (44%) mp. 140–145 °C. 1H NMR (600 MHz, CDCl3) δ 7.88 (d, J = 8.8 Hz, 2H), 7.30 (d, J = 8.7 Hz, 2H), 7.16 (s, 1H), 4.91 (s, 2H), 3.13 (d, J = 5.2 Hz, 2H), 2.37 (t, J = 7.2 Hz, 2H), 1.79–1.63 (m, 2H), 1.47 (s, 9H), 1.38–1.23 (m, 4H). [1].3C NMR (151 MHz, CDCl3): δ 170.9, 158.3, 152.5, 150.6, 148.3, 132.6, 127.3, 121.2, 108.1, 94.1, 79.2, 40.3, 36, 29.9, 28.6, 28.5, 26.3, 24.8.
4.9.1. Synthesis of 51
A mixture of tert-butyl(7-oxo-7-((5-(4-(((2,2,2-trichloroethoxy) carbonyl) oxy)phenyl) thiazol-2-yl)amino) heptyl)carbamate (500 mg, 0.84 mmol) was dissolved in 19 ml of methanol, followed by addition of 1 ml 2 M NaOH (aq.) (2 mmol, 2.38 equiv) and the reaction was continued until completion, monitored by TLC. The solvent was removed under reduced pressure and the water layer was first cooled using an ice – bath, followed by slow addition of conc. HCl until a white solid was obtained. The solid was filtered and dried under vacuum to yield 7-((5-(4-hydroxyphenyl) thiazol-2-yl) amino) −7-oxoheptan-1-aminium chloride as a crude product that was used in the next reaction without further purification. [1].H NMR (600 MHz, CD3OD): δ 7.70 (d, J = 8.6 Hz, 2H), 6.86 (d, J = 8.6 Hz, 2H), 2.95 (t, J = 7.1 Hz, 2H), 2.61 (t, J = 7.3 Hz, 2H), 1.84–1.75 (m, 2H), 1.73–1.68 (m, 2H), 1.51–1.45 (m, 4H).
4.9.2. Synthesis of 52
A mixture of tert-butyl(7-oxo-7-((5-(4-(((2,2,2-trichloroethoxy) carbonyl) oxy)phenyl) thiazol-2-yl) amino) heptyl) carbamate (440 mg, 0.742 mmol) and sodium hydroxide (31 mg, 0.78 mmol, 1.05 equiv, 0.02 M) was stirred in methanol (39 ml) at room temperature for 3 h, after which the solvent was removed under reduced pressure, water was added to the reaction mixture (5 mL) and the reaction mixture was washed with ethyl acetate (5 mL). The aqueous layer was slowly acidified and extracted with ethyl acetate (5 mL) three times. The organic extract was dried (Na2SO4) concentrated and purified using flash chromatography (ethyl acetate/hexanes 30–70%) to give tert-butyl (7-((5-(4-hydroxyphenyl) thiazol-2-yl) amino)-7-oxoheptyl) carbamate (183 mg, 0.44 mmol) as a partially purified product that was used in the next step. 1H NMR (600 MHz, CD3OD) δ 7.71 (d, J = 8.7 Hz, 2H), 7.20 (s, 1H), 6.84 (d, J = 8.7 Hz, 2H), 3.05 (t, J = 7.0 Hz, 2H), 2.54 (t, J = 7.5 Hz, 2H), 1.79–1.71 (m, 2H), 1.54–1.48 (m, 2H), 1.45 (s, 9H), 1.44–1.35 (m, 4H).
5. Synthesis of TFPAs
5.1. General Procedure for preparation of linker variants of TFPAs
To a 10 mL round bottom flask was added N,N-dimethyl formamide (1 mL), trifluoro pyruvic acid (160 mg, 1 mmol, 1 equiv), HATU (380 mg, 1 mmol, eq,), HOBt (135 mg, 1 mmol, 1 equiv), NMM (330 μl, 3 mmol, 3 equiv), followed by 4a-d (1 mmol, 1 equiv) and molecular beads. The mixture was stirred for 4 h, ethyl acetate was added to the mixture (5 mL), and the mixture was washed several times with ice–water (3 mL). The organic layer was dried (Na2SO4) and the mixture was concentrated in vacuo and purified by flash chromatography (MeOH/DCM 1.5–2%) to yield the respective TFPAs.
N-phenyl-5-(3,3,3-trifluoro-2,2-dihydroxypropanamidopentanamide (10a):
white solid, 64 mg (19.1%) mp. 108–113 °C. 1H NMR (600 MHz, CD3OD): δ 7.56 (d, J = 7.6 Hz, 2H), 7.31 (t, J = 7.5 Hz, 2H), 7.10 (t, J = 7.4 Hz, 1H), 3.34–3.32 (m, 2H), 2.42 (t, J = 7.4 Hz, 2H), 1.79–1.71 (m, 2H), 1.68–1.57 (m, 2H). [13].CNMR (151 MHz, CD3OD): δ 172.9, 165.3, 138.5, 128.4, 123.7, 121.8 (q, J = 287.3 Hz), 119.80, 94.3 (q, J = 32 Hz), 38.8, 35.9, 28.4, 22.6. HRMS-ESI M/z: [M+H]+calcd. 335.1218 for C14H17F3N2O4, found 335.1218, 99% purity.
N-phenyl-6-(3,3,3-trifluoro-2,2-dihydroxypropanamido)hexanamide (10b):
white solid, 41 mg (11.8%) mp. 103–108 °C. 1H NMR (400 MHz, CD3OD) δ 7.53 (d, J = 7.6 Hz, 2H), 7.28 (t, J = 8.0 Hz, 2H), 7.07 (t, J = 7.4 Hz, 1H), 3.26 (q, J = 7.0 Hz, 2H), 2.36 (t, J = 7.4 Hz, 2H), 1.85–1.64 (m, 2H), 1.64–1.52 (m, 2H), 1.52–1.33 (m, 2H). [13].C NMR (151 MHz, CD3OD): δ 173.1, 165.2, 138.5, 128.4, 123.7, 121.8 (q, J = 287.2 Hz), 119.8, 94.3, (q, J = 32 Hz), 39, 36.4, 28.6, 26.1, 25.1. HRMS-ESI M/z: [M+H]+calcd. 349.1375 for C15H19F3N2O4, found 349.136, 99% purity.
N-phenyl-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (10c):
white solid, 72 mg (26.4%) mp. 123–128 °C. 1H NMR (600 MHz, CD3OD) δ 7.55 (d, J = 7.5 Hz, 2H), 7.31 (t, J = 8.4 Hz, 2H), 7.09 (t, J = 7.4 Hz, 1H), 3.28 (q, J = 7.0 Hz, 2H), 2.38 (t, J = 7.5 Hz, 2H), 1.77–1.68 (m, 2H), 1.64–1.53 (m, 2H), 1.47–1.32 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 172.2, 165.2, 138.5, 128.4, 123.7, 121.8, (q, J = 287.3 Hz), 119.9, 94.3 (q, J = 32 Hz), 39.1, 36.5, 28.7, 28.5, 26.2, 25.42. HRMS-ESI M/z: [M+H]+calcd. 363.1531 for C16H21F3N2O4, found 363.1531, 96% purity.
N-phenyl-8-(3,3,3-trifluoro-2,2-dihydroxypropanamido)octanamide (10d):
white solid, 139 mg (37%) mp. 103–108 °C. 1H NMR (600 MHz, CD3OD) δ 7.56 (d, J = 7.7 Hz, 2H), 7.31 (t, J = 8.0 Hz, 2H), 7.09 (t, J = 7.4 Hz, 1H), 3.27 (q, J = 6.1 Hz, 2H), 2.38 (t, J = 7.5 Hz, 2H), 1.74–1.67 (m, 2H), 1.59–1.52 (m, 2H), 1.44–1.33 (m, 6H). [13].C NMR (151 MHz, CD3OD): δ 173.3, 165.1, 138.5, 128.4, 123.7, 121.82 (q, J = 287.2 Hz), 119.9, 94.4 (q, J = 32 Hz), 39.2, 36.5, 28.8, 28.77, 28.6, 26.3, 25.4. HRMS-ESI M/z: [M+H]+calcd. 377.1688 for C17H23F3N2O4, found 377.1688, 99% purity.
5.2. General Procedure for preparation of SAR variants of TFPAs (53–85)
To a 10 mL round bottom flask was added the respective boc–arene (0.4 mmol), followed by 20% trifluoroacetic acid in dichloromethane (1 mL: 4 mL), and the mixture was stirred at room temperature for 4 h. The solvent was removed under reduced pressure, and the mixture was dried under vacuum, followed by addition of N,N-dimethyl formamide (1 mL), trifluoropyruvic acid (64 mg, 0.4 mmol, 1 equiv), HATU (152 mg, 0.4 mmol, eq,), HOBt (54 mg, 0.4 mmol, 1 equiv), NMM (132 μl, 1.2 mmol, 3 equiv). The mixture was stirred for 4 h, ethyl acetate was added to the mixture (5 mL), and the mixture was washed several times with ice–water (3 mL). The organic layer was dried (Na2SO4), the solvent was removed under reduced pressure, and the mixture was dissolved in silica and purified by flash chromatography (MeOH/DCM 1.5–2%) to yield the respective TFPAs (4–42%, 2 steps).
N-phenyl-4-(3,3,3-trifluoro-2,2-dihydroxypropanamido)butanamide (14):
White solid, 64 mg, (50%) mp. 105–110 °C. [1].HNMR (400 MHz, CD3OD): δ δ 7.56 (dd, J = 8.5, 0.9 Hz, 2H), 7.31 (t, J = 8.0 Hz, 2H), 7.09 (t, J = 7.4 Hz, 1H), 3.33 (p, J = 3.2 Hz, 2H), 2.42 (t, J = 7.5 Hz, 2H), 1.93 (p, J = 7.2 Hz, 2H). [13].C NMR (151 MHz, CD3OD): δ 172.3, 165.5, 138.4, 128.4, 123.8, 121.8 (q, J = 287.4 Hz), 119.9, 94.3 (q, J = 32 Hz), 38.8, 33.7, 24.9. HRMS-ESI M/z: [M+H]+calcd. 321.1062 for C13H15F3N2O4, found 321.1069, 99% purity.
N-(4-acetylphenyl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (53):
White solid, 26 mg, (16.19%) mp. 133–138 °C. 1H NMR (400 MHz, CD3OD) δ 7.95 (d, J = 8.8 Hz, 2H), 7.71 (d, J = 8.8 Hz, 2H), 3.26 (t, J = 6.7 Hz, 2H), 2.56 (s, 3H), 2.40 (t, J = 7.5 Hz, 2H), 1.75–1.65 (m, 2H), 1.60–1.50 (m, 2H), 1.45–1.32 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 173.5, 165.2, 143.4, 132.2, 129.3, 121.8 (q, J = 287.4 Hz), 118.7, 94.3 (q, J = 32 Hz), 39.1, 36.6, 28.7, 28.5, 26.2, 25.2, 25.1. HRMS-ESI M/z: [M+H]+calcd. 405.1637 for C18H23F3N2O5, found 405.1637, 98.9% purity.
N-(4-(pentafluoro-λ [6]-sulfaneyl)phenyl)-7-(3,3,3-trifluoro-2, 2dihydroxypropanamido)heptanamide (54):
White solid, 52 mg (26.6%) mp. 105–110 °C. 1H NMR (600 MHz, CD3OD) δ 7.76 (s, 4H), 3.28 (td, J = 7.0, 2.5 Hz, 2H), 2.42 (t, J = 7.5 Hz, 2H), 1.77–1.67 (m, 2H), 1.62–1.54 (m, 2H), 1.46–1.35 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 173.5, 165.2 (d, J = 4 Hz), 148.4 (m, J = 17 Hz), 141.9, 126.5 (t, J = 4 Hz), 121.8 (q, J = 287.3 Hz), 118.7, 94.4 (q, J = 32 Hz), 39.1, 36.5, 28.7, 28.5, 26.1, 25.1. HRMS-ESI M/z: [M+H]+calcd. 489.1094 for C16H20F8N2O4S, found 489.1093, 99% purity.
N-(4-methoxyphenyl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (55):
white solid, 49 mg (31%) mp. 105–110 °C. 1H NMR (400 MHz, CD3OD) δ 7.42 (d, J = 9.0 Hz, 2H), 6.86 (d, J = 9.0 Hz, 2H), 3.76 (s, 3H), 3.25 (t, J = 6.9 Hz, 2H), 2.33 (t, J = 7.5 Hz, 2H), 1.77–1.62 (m, 2H), 1.58–1.48 (m, 2H), 1.46–1.28 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 173, 165.2, 156.5, 132.4, 121.8 (q, J = 287.2 Hz), 121.7, 113.5, 94.3 (q, J = 32 Hz), 54.4, 39.1, 36.3, 28.7, 28.5, 26.2, 25.5. HRMS-ESI M/z: [M+H]+calcd. 393.1637 for C17H23F3N2O5, found 393.1634, 99% purity.
N-(4-fluorophenyl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (56):
White solid, 64 mg (42%) mp. 97–103 °C. [1].H NMR (400 MHz, CD3OD): δ 7.56 (d, J = 4.9 Hz, 2H), 7.05 (d, J = 8.3 Hz, 2H), 3.31–3.24 (m, 2H), 2.37 (t, J = 6.2 Hz, 2H), 1.75–1.67 (m, 2H), 1.61–1.53 (m, 2H), 1.47–1.35 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 173.1, 165.2 (d, J = 4 Hz), 160, 158.4, 134.7 (d, J = 2 Hz), 121.8 (q, J = 287.2 Hz), 121.6, 114.9, 114.7, 94.4 (q, J = 32 Hz), 54.4, 39.1, 36.3, 28.7, 28.5, 26.2, 25.4. HRMS-ESI M/z: [M+H]+ calcd. 381.1437 for C16H20F4N2O4, found 381.1432, 96% purity.
N-(pyridin-4-yl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido) heptanamide (57):
White solid, 19 mg (13.2%) mp. 150–155 °C. 1H NMR (400 MHz, CD3OD) δ 8.37 (d, J = 5.5 Hz, 2H), 7.64 (d, J = 6.3 Hz, 2H), 3.25 (t, J = 7.0 Hz, 2H), 2.41 (t, J = 7.5 Hz, 2H), 1.76–1.63 (m, 2H), 1.63–1.48 (m, 2H), 1.45–1.31 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 174, 165.2, 149.3, 146.8, 121.8 (q, J = 287.3 Hz), 113.6, 94.3 (q, J = 32 Hz), 39.1, 36.6, 28.7, 28.4, 26.1, 25. HRMS-ESI M/z: [M+H]+calcd. 364.1844 for C15H20F3N3O4, found 364.1484, 99% purity.
N-(5-phenylthiazol-2-yl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (58):
White solid, 18 mg (9.9%) mp. 143–148 °C. [1].H NMR (400 MHz, CD3OD): δ 7.91 (d, J = 8.2 Hz, 2H), 7.40 (t, J = 7.7 Hz, 2H), 7.37 (s, 1H), 7.31 (t, J = 7.4 Hz, 1H), 3.28 (q, J = 6.4 Hz, 2H), 2.51 (t, 2H), 1.77–1.72 (m, 2H), 1.62–1.55 (m, 2H), 1.47–1.38 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 172.4, 165.2, 158, 149.8, 134.6, 128.2, 128.1, 127.4, 127.2, 125.6, 125,56, 121.8 (q, J = 287.4 Hz), 107.1, 101.4, 94.3 (q, J = 32 Hz), 39.1, 35.1, 28.7, 28.4, 26.1, 24.9. HRMS-ESI M/z: [M+H]+calcd. 446.1361 for C19H22F3N3O4S, found 446.1376, 95% purity.
N-(3-methoxyphenyl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (59):
White solid, 40 mg (26%) mp. 120–125 °C. [1].H NMR (400 MHz, CD3OD): δ 7.91 (s, 1H), 7.47 (d, J = 7.1 Hz, 1H), 7.25–7.19 (m, 2H), 3.30–3.24 (m, 2H), 2.40–2.35 (m, 2H), 1.73–1.67 (m, 2H), 1.60–1.53 (m, 2H), 1.44–1.35 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 173.2, 165.2, 160.1, 139.7, 129.1, 121.8 (q, J = 286.9 Hz), 111.9, 109.2, 105.6, 94.3 (q, J = 32 Hz), 54.2, 39.1, 36.5, 28.7, 28.5, 26.2, 25.4. HRMS-ESI M/z: [M+H]+calcd. 393.1637 for C17H23F3N2O5, found 393.1644, 98% purity.
N-(thiazol-2-yl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido) heptanamide (60):
White solid, 55 mg (37%) mp. 137–142 °C. [1].H NMR (600 MHz, CD3OD): δ 7.43 (d, J = 3.6 Hz, 2H), 7.11 (d, J = 3.6 Hz, 2H), 3.31–3.24 (m, 2H), 2.50 (t, J = 7.5 Hz, 2H), 1.76–1.69 (m, 2H), 1.61–1.54 (m, 2H), 1.45–1.36 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 172.2, 165.2, 158.7, 136.9, 121.8 (q, J = 287.2 Hz), 113, 94.3 (q, J = 32 Hz), 39.1, 36.5, 35, 28.7, 28.4, 26.1, 24.9. HRMS-ESI M/z: [M+H]+ calcd. 370.1048 for C13H18F3N3O4S, found 370.1057, 99% purity.
N-(benzo[d]thiazol-2-yl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (61):
White solid, 20 mg (12%) mp. 150–155 °C. 1H NMR (600 MHz, Acetone-d6) δ 7.94 (d, J = 7.9 Hz, 1H), 7.71 (d, J = 8.1 Hz, 1H), 7.43 (t, J = 8.3 Hz, 1H), 7.31 (t, J = 8.1 Hz, 1H), 3.33 (q, J = 6.9 Hz, 2H), 2.63 (t, J = 7.5 Hz, 2H), 1.80–1.73 (m, 2H), 1.64–1.57 (m, 2H), 1.48–1.38 (m, 4H). 13C NMR (151 MHz, Acetone-d6) δ 171.9, 166.5, 157.9, 149, 132.1, 125.9, 123.5, 122.58 (q, J = 287.7 Hz), 121.3, 120.70, 90.8 (q, J = 32.4 Hz), 39.6, 35.5, 26.1, 24.8. HRMS-ESI M/z: [M+H]+calcd. 420.1204 for C17H20F3N3O4S, found 420.1208, 98% purity.
N-(2,4-difluorophenyl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (62):
White solid, 51 mg (32%) mp. 100–105 °C. 1H NMR (600 MHz, CD3OD) δ 7.77 (td, J = 8.9, 6.1 Hz, 1H), 7.07–7.00 (m, 1H), 6.99–6.93 (m, 1H), 3.29 (q, J = 6.9 Hz, 2H), 2.43 (t, J = 7.5 Hz, 2H), 1.75–1.68 (m, 2H), 1.62–1.55 (m, 2H), 1.47–1.36 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 173.7, 165.2, 160.6 (d, J = 11.4 Hz), 159.01 (d, J = 11.4 Hz), 155.88 (d, J = 12.2 Hz), 154.23 (d, J = 12.2 Hz), 126.21 (dd, J = 9.5, 2.5 Hz), 121.98 (dd, J = 12.2, 3.8 Hz), 121.80 (q, J = 287.3 Hz), 110.50 (dd, J = 22.2, 3.7 Hz), 103.44 (dd, J = 26.7, 24.3 Hz), 94.34 (d, J = 32.1 Hz), 39.1, 35.7, 28.69, 28.4, 26.1, 25.3. HRMS-ESI M/z: [M+H]+calcd. 399.1343 for C16H19F5N2O4, found 399.1327, 98% purity.
7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)-N-(2,4,6-trifluorophenyl)heptanamide (63):
White solid, 23 mg (13.8%) mp. 95–100 °C. 1H NMR (600 MHz, CD3OD) δ 6.96 (t, J = 8.3 Hz, 1H), 3.32–3.25 (m, 2H), 2.44 (t, J = 7.4 Hz, 2H), 1.76–1.69 (m, 2H), 1.61–1.55 (m, 2H), 1.49–1.37 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 25.34, 26.13, 28.31, 28.72, 35.13, 39.28, 174.1, 165.3 (d, J = 4.2 Hz), 161.88 (t, J = 15 Hz), 160.23 (t, J = 15 Hz), 159.5 (dd, J = 15.4, 7.4 Hz), 157.8 (dd, J = 15.4, 7.4 Hz), 121.8 (q, J = 287.2 Hz), 110.7 (td, J = 17.3. 5.1 Hz), 101.1–99.2 (m), 94.4 (m, J = 32 Hz), 39.3, 35.1, 28.7, 28.3, 26.1, 25.3. HRMS-ESI M/z: [M+H]+calcd. 417.1249 for C16H18F6N2O4, found 417.1256, 95% purity.
N-(4-chlorophenyl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (64):
White solid, 27 mg (17%) mp. 103–108 °C. 1H NMR (400 MHz, CD3OD) δ 7.55 (d, J = 8.8 Hz, 2H), 7.28 (d, J = 8.8 Hz, 2H), 3.26 (t, J = 7.2 Hz, 2H), 2.35 (t, J = 7.5 Hz, 2H), 1.74–1.63 (m, 2H), 1.61–1.50 (m, 2H), 1.44–1.30 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 173.2, 165.2, 137.4, 128.5, 128.3, 121.8 (q, J = 287.2 Hz), 121.1, 94.3 (q, J = 32 Hz), 39.1, 36.4, 28.7, 28.5, 26.1, 25.3. HRMS-ESI M/z: [M+H]+calcd. 397.1141 for C16H20ClF3N2O4, found 397.1169, 95% purity.
N-(4-bromophenyl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (65):
White solid, 19 mg (10.8%) mp. 105–110 °C. 1H NMR (400 MHz, CD3OD) δ 7.50 (d, J = 8.8 Hz, 2H), 7.42 (d, J = 8.9 Hz, 2H), 3.25 (t, J = 7.0 Hz, 2H), 2.35 (t, J = 7.5 Hz, 2H), 1.73–1.63 (m, 2H), 1.60–1.51 (m, 2H), 1.44–1.32 (m, 4H). 13C NMR (151 MHz, CD3OD) δ 173.2, 165.2, 137.9, 131.3, 121.8 (q, J = 287.2 Hz), 121.37, 115.9, 94.3 (q, J = 32 Hz), 39.1, 36.4, 28.7, 28.5, 26.1, 25.3. HRMS-ESI M/z: [M+H]+calcd. 441.0636 for C16H20BrF3N2O4, found 441.0641, 99% purity.
N-(2-methoxyphenyl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (66):
White solid, 18 mg (11.7%) mp. 105–110 °C. 1H NMR (400 MHz, CD3OD) δ 7.90 (d, J = 7.9 Hz, 1H), 7.08 (t, J = 8.6 Hz, 1H), 6.98 (d, J = 7.4 Hz, 1H), 6.89 (t, J = 8.2 Hz, 1H), 3.24 (q, J = 7.9 Hz, 2H), 2.41 (t, J = 7.5 Hz, 2H), 1.74–1.63 (m, 2H), 1.59–1.50 (m, 2H), 1.44–1.32 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 173.3, 165.2, 150.3, 126.7, 124.8, 122.3, 121.8 (q, J = 287.3), 120, 110.4, 94.3 (q, J = 32 Hz), 54.81, 54.80, 39.2, 36.3, 28.7, 28.4, 26.2, 25.5. HRMS-ESI M/z: [M+H]+calcd. 393.1637 for C17H23F3N2O5, found 393.1636, 99% purity.
N-(4-methyl-5-phenylthiazol-2-yl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (67):
White solid, 36 g (20%) mp. 143–148 °C. 1H NMR (400 MHz, Acetone-d6) δ 7.66 (d, J = 8.4 Hz, 2H), 7.42 (t, J = 7.7 Hz, 2H), 7.31 (t, J = 7.4 Hz, 1H), 3.31 (q, J = 6.5 Hz, 2H), 2.55 (t, J = 7.4 Hz, 2H), 2.51 (s, 3H), 1.77–1.68 (m, 3H), 1.63–1.54 (m, 2H), 1.46–1.35 (m, 4H). [13].C NMR (151 MHz, Acetone-d6): δ170.9, 166.4, 154, 135.6, 128.2, 128.1, 127, 122.6 (q, J = 287.7 Hz), 121.1, 90.7 (q, J = 32.5 Hz), 39.6, 35.3, 26.2, 24.9, 11.3. HRMS-ESI M/z: [M+H]+calcd. 460.1517 for C20H24F3N3O4S, found 460.1506, 99% purity.
N-(4,5-diphenylthiazol-2-yl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (68):
White solid, 22 mg (10.6%) mp. 143–148 °C. [1].H NMR (400 MHz, CD3OD): δ 1H NMR (600 MHz, CD3OD) δ 7.49–7.45 (m, 1H), 7.36–7.31 (m, 3H), 7.27 (dd, J = 3.7, 2.5 Hz, 1H), 3.28 (dt, J = 12.0, 6.9 Hz, 2H), 2.52 (t, J = 7.4 Hz, 2H), 1.79–1.71 (m, 2H), 1.62–1.56 (m, 2H), 1.48–1.38 (m, 4H). 13C NMR (151 MHz, CD3OD) δ 172.5, 165.2, 156.1, 144.3, 135, 132.29, 129.3, 128.6, 128.5, 127.8, 127.6, 127.4, 126.3, 121.8 (q, J = 287.2 Hz), 94.3 (q, J = 32 Hz), 39.1, 35.1, 28.7, 28.4, 26.1, 24.9. HRMS-ESI M/z: [M+H]+calcd. 522.1674 for C25H26F3N3O4S, found 522.1673, 99% purity.
N-([1,1′-biphenyl]-4-yl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (69):
White solid, 32 mg (18.2%) mp. 123–12 8 °C. [1].H NMR (600 MHz, Acetone-d6): δ 7.77 (d, J = 8.5 Hz, 2H), 7.68–7.57 (m, 4H), 7.43 (t, J = 7.7 Hz, 2H), 7.32 (t, J = 7.4 Hz, 1H), 3.31 (q, J = 6.7 Hz, 2H), 2.38 (t, J = 7.4 Hz, 2H), 1.75–1.65 (m, 2H), 1.65–1.52 (m, 2H), 1.46–1.34 (m, 4H). [13].C NMR (151 MHz, Acetone-d6): δ 171.1, 166.4, 140.6, 139.1, 135.6, 128.8, 127, 126.9, 126.4, 122.6 (q, J = 287.9 Hz), 119.4, 90.8 (q, J = 32.5 Hz), 39.6, 36.8, 26.2, 25.3. HRMS-ESI M/z: [M+H]+calcd. 439.1844 for C22H25F3N2O4, found 439.1844, 99% purity.
N-(2,5-dimethoxyphenyl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (70):
White solid, 17 mg (10%) mp. 108–113 °C. 1H NMR (400 MHz, Acetone) δ 8.1 (d, J = 2.5 Hz, 1H), 6.89 (d, J = 8.9 Hz, 1H), 6.67 (s, 1H), 6.56 (dd, J = 8.9, 3.0 Hz, 1H), 3.80 (s, 3H), 3.72 (s, 3H), 3.31 (q, J = 6.8 Hz, 2H), 2.44 (t, J = 7.4 Hz, 2H), 1.72–1.63 (m, 2H), 1.62–1.52 (m, 2H), 1.44–1.32 (m, 4H). [13].C NMR (151 MHz, Acetone-d6): δ 171.1, 166.4, 153.8, 142.6129.1, 122.6 (q, J = 285.87 Hz), 111, 107, 106.8, 90.8 (q, J = 32.5 Hz), 55.7, 55, 39.6, 36.9, 26.2, 25.3. HRMS-ESI M/z: [M+H]+calcd. 423.1742 for C18H25F3N2O6, found 423.1745, 99% purity.
7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)-N-(3,4,5-trimethoxyphenyl)heptanamide (71):
White solid, 29 mg (15.9%) mp. 98–103 °C. 1H NMR (400 MHz, CD3OD) δ 6.95 (s, 2H), 3.82 (s, 3H), 3.72 (s, 3H), 3.27 (t, J = 7.2 Hz, 2H), 2.35 (t, J = 7.5 Hz, 2H), 1.74–1.64 (m, 2H), 1.61–1.52 (m, 2H), 1.45–1.33 (m, 4H). [13].C NMR (151 MHz, CD3OD): 13C NMR (151 MHz, CD3OD) δ 173.1, 165.2, 153, 135, 134, 121.8 (q, J = 287.2 Hz), 94.3 (q, J = 33 Hz), 97.4, 59.8, 55.1, 39.11, 36.5, 28.7, 28.5, 26.2, 25.4. HRMS-ESI M/z: [M+H]+calcd. 453.1848 for C19H27F3N2O7, found 453.1866, 99% purity.
N-(naphthalen-1-yl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (72):
White solid, 48 mg (29%) mp. 123–128 °C. 1H NMR (400 MHz, CD3OD) δ 7.97 (d, J = 9.0 Hz, 1H), 7.90 (d, J = 9.2 Hz, 1H), 7.78 (d, J = 8.2 Hz, 1H), 7.59–7.45 (m, 4H), 3.29–3.22 (m, 2H), 2.55 (t, J = 7.5 Hz, 2H), 1.86–1.75 (m, 2H), 1.65–1.56 (m, 2H), 1.55–1.39 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 174.4, 165.22, 165.19, 134.3, 132.8, 128.8, 128, 126.2, 125.9, 125.7, 125.1, 122.9, 122, 121.8 (q, J = 287.4 Hz), 94.3 (q, J = 32 Hz), 39.2, 35.6, 28.7, 28.6, 26.2, 25.6. HRMS-ESI M/z: [M+H]+calcd. 413.163 for C20H23F3N2O4, found 413.1428, 99% purity.
N-(4-benzoylphenyl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (73):
Yellow solid, 24 mg (12.9%) mp. 113–118 °C. [1].H NMR (600 MHz, CD3OD): δ 7.80–7.73 (m, 6H), 7.64 (t, J = 7.4 Hz, 1H), 7.54 (t, J = 7.7 Hz, 2H), 3.30 (t, J = 7.2 Hz, 2H), 2.44 (t, J = 7.5 Hz, 2H), 1.77–1.70 (m, 2H), 1.64–1.57 (m, 2H), 1.48–1.37 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 196.2, 173.5, 157.5 (q, J = 36.5 Hz), 143.1, 137.8, 132.2, 132.1, 131.1, 129.4, 128.1, 118.6, 116.9 (q, J = 286.6 Hz), 39.3, 36.6, 28.5, 28.3, 26.1, 25.2. HRMS-ESI M/z: [M+Na]+calcd. 489.1613 for C23H24F3N2NaO5, found 489.1623, 96% purity.
N-(4-((4-methylphenyl)sulfonamido)phenyl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido) heptanamide (74):
White solid, 24 mg (11. %) mp. 115–120 °C. 1H NMR (400 MHz, CD3OD) δ 7.58 (d, J = 8.3 Hz, 2H), 7.38 (d, J = 8.9 Hz, 2H), 7.26 (d, J = 7.3 Hz, 2H), 6.98 (d, J = 8.9 Hz, 2H), 3.24 (t, J = 6.8 Hz, 2H), 2.36 (s, 3H), 2.31 (t, J = 7.4 Hz, 2H), 1.72–1.60 (m, 2H), 1.59–1.48 (m, 2H), 1.43–1.26 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 173.1, 165.2, 143.6, 136.6, 135.6, 133.4, 129.1, 126.9, 122.1, 121.8 (q, J = 287.2 Hz), 120.38, 94.4 (q, J = 32.5 Hz), 39.1, 36.4, 28.7, 28.5, 26.1, 25.4, 20. HRMS-ESI M/z: [M+H]+ calcd. 532.167 for C23H28F3N3O6S, found 532.1673, 98% purity.
N-(2-bromophenyl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (75):
White solid, 8 mg (4.5%) mp. 108–113 °C. 1H NMR (400 MHz, CD3OD) δ 7.62 (t, J = 7.4 Hz, 2H), 7.35 (t, J = 7.7 Hz, 1H), 7.12 (t, J = 7.7 Hz, 1H), 3.27 (t, J = 7.0 Hz, 2H), 2.44 (t, 2H), 1.79–1.67 (m, 2H), 1.62–1.51 (m, 2H), 1.50–1.33 (m, 4H). [13]. C NMR (151 MHz, CD3OD): δ 173.6, 165.2, 135.8, 132.5, 127.6, 127, 126.9, 121.8 (q, J = 287.2 Hz), 118.1, 94.3 (q, J = 32.9 Hz), 39.2, 39.1, 35.9, 28.7, 28.5, 28.47, 26.2, 26.1, 25.4. HRMS-ESI M/z: [M+H]+calcd. 441.0636 for C16H20BrF3N2O4, found 441.0651, 99% purity.
N-(3-bromophenyl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (76):
27 mg (15.3%). [1].H NMR (600 MHz, CD3OD): δ 7.91 (s, 1H), 7.47 (d, J = 7.1 Hz, 1H), 7.25–7.19 (m, 2H), 3.30–3.24 (m, 2H), 2.38 (t, J = 7.5 Hz, 2H), 1.73–1.67 (m, 2H), 1.60–1.53 (m, J = 9.1, 4.8 Hz, 2H), 1.44–1.35 (m, J = 2.6 Hz, 5H). [13]. C NMR (151 MHz, CD3OD): δ 173.4, 173.3, 165.2, 140.1, 129.9, 126.4, 122.4, 121.9, 121.8 (q, J = 287 Hz), 118.1, 94.3 (q, J = 32 Hz), 39.1, 36.4, 28.7, 28.5, 26.1, 25.3. HRMS-ESI M/z: [M+H]+calcd. 441.0636 for C16H20BrF3N2O4, found 441.063, 95% purity.
N-(6-fluorobenzo[d]thiazol-2-yl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (77):
White solid, 20 mg (11.4%) mp. 146–151 °C. [1].H NMR (600 MHz, CD3OD): δ 1.42 (m, 4H, CH2, J = 5.64 Hz), 1.58 (m, 2H, CH2, J = 7.14 Hz), 1.75 (m, 2H, CH2, J = 7.5 Hz), 2.54 (t, 2H, CH2, J = 7.5 Hz), 3.29 (q, 2H, CH2, J = 4 Hz), 7.21 (td, 1H, CH, J = 2.58, 9 Hz), 7.65 (dd, 1H, CH, J = 2.58, 8.4 Hz), 7.72 (dd, 1H, CH, J = 4.2, 4.68 Hz). [13].C NMR (151 MHz, CD3OD): δ 173, 165.2, 160.4, 158.8, 158.2, 145.2, 133.1 (d, J = 10.8 Hz), 121.8 (q, J = 287.2 Hz), 121.4 (d, J = 9.1 Hz), 113.8 (d, J = 24.8 Hz), 107.1 (d, J = 27 Hz), 39.1, 35.2, 28.7, 28.4, 26.1, 24.7. HRMS-ESI M/z: [M+H]+calcd. 438.1110 for C17H19F4N3O4S, found 438.1111, 98% purity.
N-(6-chlorobenzo[d]thiazol-2-yl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (78):
White solid, 13 mg (7.2%) mp. 165–170 °C. 1H NMR (400 MHz, CD3OD) δ 7.89 (d, J = 2.1 Hz, 1H), 7.68 (d, J = 8.6 Hz, 1H), 7.40 (dd, J = 8.7, 2.1 Hz, 1H), 3.26 (t, J = 8.0 Hz, 2H), 2.52 (t, J = 7.5 Hz, 2H), 1.77–1.67 (m, 2H), 1.61–1.50 (m, 2H), 1.47–1.34 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 173.1, 165.2, 159, 147.4, 133.5, 128.7, 126.3, 121.8 (q, J = 287.2 Hz), 121.4, 120.6, 39.1, 35.2, 28.7, 28.4, 26.1, 24.7. HRMS-ESI M/z: [M+H]+calcd. 454.0815 for C17H19ClF3N3O4S, found 454.0817, 98% purity.
N-(6-bromobenzo[d]thiazol-2-yl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (79):
White solid, 11 mg (5.5%) mp. 163–168 °C. [1].H NMR (600 MHz, Acetone-d6): δ 8.16 (d, J = 2.0 Hz, 1H), 7.64 (d, J = 8.6 Hz, 1H), 7.57 (dd, J = 8.6, 2.0 Hz, 1H), 3.35–3.31 (m, 2H), 2.64 (t, J = 7.5 Hz, 2H), 1.80–1.73 (m, 2H), 1.66–1.57 (m, 2H), 1.49–1.35 (m, 4H). [13].C NMR (151 MHz, Acetone-d6): δ 172.1, 166.4, 158.6, 148.2, 134.3, 129.1, 123.9, 122.6 (q, J = 287.2 Hz), 121.2, 115.7, 90.8 (q, J = 32 Hz), 39.6, 35.5, 28.7, 28.5, 26.1, 24.7. HRMS-ESI M/z: [M+H]+calcd. 498.0309 for C17H19BrF3N3O4S, found 498.0313, 98% purity.
N-(6-bromobenzo[d]oxazol-2-yl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (80):
White solid, 15 mg (7.8%) mp. 163–168 °C. 1H NMR (400 MHz, CD3OD) δ 7.69 (s, 1H), 7.43 (d, J = 8.4 Hz, 2H), 3.26 (t, J = 7.0 Hz, 2H), 2.52 (t, J = 7.2 Hz, 2H), 1.85–1.66 (m, 2H), 1.66–1.51 (m, 2H), 1.5–1.34 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 171.8, 165.2, 156.2, 146.5, 142.6, 126.4, 121.8 (q, J = 287.3 Hz), 120.8, 116.9, 111, 94.4 (q, J = 32.3 Hz), 39.1, 36, 28.7, 28.3, 26.1, 24.5. HRMS-ESI M/z: [M+H]+calcd. 482.0538 for C17H19BrF3N3O5, found 482.0528, 98% purity.
N-(benzo[d]oxazol-2-yl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (81):
White solid, 9 mg (5.5%) mp. 118–123 °C. [1].H NMR (400 MHz, Acetone-d6): δ 7.67–7.46 (m, 2H), 7.42–7.19 (m, 2H), 3.40–3.32 (m, 2H), 2.69 (q, J = 8.3 Hz, 2H), 1.93–1.71 (m, 2H), 1.67–1.55 (m, 2H), 1.57–1.35 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 171.8, 165.2, 155, 147.4, 140.7, 124.2, 123.7, 121.8 (q, J = 287.3 Hz), 119, 117.9, 109.5, 94.4 (q, J = 32 Hz), 39.2, 36, 28.7, 28.3, 26.1, 24.5. HRMS-ESI M/z: [M+H]+calcd. 404.1433 for C17H20F3N3O5, found 404.1439, 98% purity.
N-(6-methoxybenzo[d]thiazol-2-yl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (82):
White solid, 17 mg (9.4%) mp. 143–148 °C. [1].HNMR (600 MHz, CD3OD): δ 7.63 (d, J = 8.9 Hz, 1H), 7.43 (d, J = 2.5 Hz, 1H), 7.04 (dd, J = 8.8, 2.5 Hz, 1H), 3.87 (s, 3H), 3.29 (td, J = 6.9, 4.2 Hz, 2H), 2.53 (t, J = 7.5 Hz, 2H), 1.78–1.71 (m, 2H), 1.62–1.54 (m, 2H), 1.49–1.36 (m, 4H). [13].C NMR (151 MHz, CD3OD): 24.8, 26.1, 28.4, 28.7, 35.2, 39.1, 54.8, 94.3 (J = 32 Hz), 103.7, 114.8, 120.9, 121.8 (q, J = 286 Hz), 133.1, 142.6, 156.5, 157, 165.2, 172.8. HRMS-ESI M/z: [M+H]+calcd. 450.1310 for C18H22F3N3O5S, found 450.1306, 99% purity.
N-(5-methoxybenzo[d]thiazol-2-yl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (83):
White solid, 14 mg (7.8%) mp. 160–165 °C. [1].HNMR (400 MHz, CD3OD): δ 7.72 (d, J = 8.7 Hz, 1H), 7.29 (d, J = 2.4 Hz, 1H), 6.96 (dd, J = 8.7, 2.4 Hz, 1H), 3.87 (s, 3H), 3.29 (td, J = 7.0, 4.1 Hz, 2H), 2.54 (t, J = 7.5 Hz, 2H), 1.80–1.71 (m, 2H), 1.63–1.55 (m, 2H), 1.48–1.37 (m, 4H). [13].C NMR (151 MHz, CD3OD): δ 172.8, 165.2, 159.5, 159.3, 149.8, 121.8 (q, J = 287.3 Hz), 121.2, 112.8, 103.6, 54.6, 39.1, 35.2, 28.7, 28.4, 26.1, 24.7. HRMS-ESI M/z: [M+H]+calcd. 450.1310 for C18H22F3N3O5S, found 450.1312, 98% purity.
N-(5-(4-methoxyphenyl)thiazol-2-yl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (84):
White solid 18 mg (9.4%) mp. 148–153 °C. [1].H NMR (400 MHz, Acetone-d6): δ 7.86 (d, J = 8.9 Hz, 2H), 7.30 (s, 1H), 6.98 (d, J = 8.9 Hz, 2H), 3.85 (s, 3H), 3.34 (q, J = 6.9 Hz, 2H), 2.62 (t, J = 7.4 Hz, 2H), 1.83–1.72 (m, 2H), 1.67–1.56 (m, 2H), 1.51–1.36 (m, 4H). [13].C NMR (151 MHz, Acetone-d6): δ 171.1, 166.4, 159.5, 157.8, 149.4, 127.7, 127.1, 122.6 (q, J = 287.2 Hz), 113.9, 105.2, 90.8 (q, J = 32.5 Hz), 54.7, 39.6, 35.3, 26.2, 24.9. HRMS-ESI M/z: [M+H]+calcd. 476.1361 for C20H24F3N3O5S, found 476.1368, 99% purity.
N-(5-(4-bromophenyl)thiazol-2-yl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido)heptanamide (85):
White solid, 10 mg (4.8%) mp. 145–150 °C. [1].H NMR (600 MHz, Acetone-d6): δ 7.83 (d, J = 8.4 Hz, 2H), 7.54 (d, J = 8.3 Hz, 2H), 7.49 (s, 1H), 3.27 (q, J = 5.4 Hz, 2H), 2.56 (t, J = 7.5 Hz, 2H), 1.75–1.67 (m, 2H), 1.60–1.52 (m, 2H), 1.43–1.34 (m, 4H). [13].C NMR (151 MHz, Acetone-d6): δ 171.3, 166.4, 158.2, 148.2, 134, 131.6, 127.7, 122.6 (q, J = 287.4 Hz), 121, 108, 90.8 (q, J = 32 Hz), 39.6, 35.3, 26.2, 24.9. HRMS-ESI M/z: [M+H]+calcd. 524.0466 for C19H21BrF3N3O4S, found 524.046, 99% purity.
5.3. Synthesis of 86
A mixture of 7-((5-(4-hydroxyphenyl)thiazol-2-yl) amino)-7-oxoheptan-1-aminium chloride 51 (62 mg, 0.174 mmol), trifluoropyruvic acid (27 mg, 0.17 mmol, 0.98 equiv.), HATU (65 mg, 0.17 mmol, 0.98 equiv.), HOBt (23 mg, 0.17 mmol, 0.98 equiv.) and NMM (56 μL, 0.51 mmol, 2.93 equiv.) was stirred in 1 mL dimethylformamide at room temperature overnight. Ethyl acetate was added to the mixture (5 ml), and the mixture was washed several times with ice–water (3 ml). The organic layer was dried (Na2SO4), the solvent was removed under reduced pressure, and the mixture was dissolved in silica and purified by flash chromatography (MeOH/DCM 1.5–2%) to yield N-(5-(4-hydroxyphenyl) thiazol-2-yl)-7-(3,3,3-trifluoro-2,2-dihydroxypropanamido) heptanamide 86 as a white solid (10 mg, 11%, mp. 145–150 °C).
1H NMR (400 MHz, Acetone-d6) δ 7.73 (d, J = 8.8 Hz, 2H), 7.19 (s, 1H), 6.84 (d, J = 8.8 Hz, 2H), 3.30 (q, J = 13.4, 6.7 Hz, 2H), 2.57 (t, J = 7.4 Hz, 2H), 1.77–1.68 (m, 2H), 1.61–1.52 (m, 2H), 1.50–1.33 (m, 4H). [13].C NMR (151 MHz, Acetone-d6): 171.1, 166.4, 157.7, 157.3, 149.7, 127.2, 126.7, 122.6 (q, J = 287.8 Hz), 115.3, 104.6, 39.6, 35.3, 26.2, 24.9. HRMS-ESI M/z: [M+H]+calcd. 462.1310 for C19H22F3N3O5S, found 462.1329, 98% purity.
5.4. Synthesis of 87
A mixture of tert-butyl (7-((5-(4-hydroxyphenyl) thiazol-2-yl) amino)-7-oxoheptyl) carbamate 52 (183 mg, 0.44 mmol), 1,8-Diazabicyclo[5.4.0]undec-7-ene (75 μL, 0.5 mmol, 1.14 equiv) and ethyl iodide (160 μL, 2 mmol, 4.55 equiv) was refluxed in acetone (3 mL) for 6 h, after which the solvent was removed under reduced pressure. The reaction mixture was diluted with ethyl acetate (5 mL) and washed with 1 M HCl (5 mL) and brine (2 × 5 mL) twice. The organic layer was dried (Na2SO4), concentrated, dried under vacuum, and used in the next reaction without further purification. To the crude mixture was added 20% trifluoroacetic acid in dichloromethane (1 ml: 4 ml), and the mixture was stirred at room temperature for 4 h. The solvent was removed under reduced pressure, and the mixture was dried under vacuum, followed by addition of N, N - dimethyl formamide (1 ml), trifluoro pyruvic acid (64 mg, 0.4 mmol, 1 equiv), HATU (152 mg, 0.4 mmol, equiv,), HOBt (54 mg, 0.4 mmol, 1 equiv) and NMM (132 μl, 1.2 mmol, 3 equiv). The mixture was stirred for 4 h, ethyl acetate was added to the mixture (5 ml), and the mixture was washed several times with ice–water (3 ml). The organic layer was dried (Na2SO4), the solvent was removed under reduced pressure, and the mixture was dissolved in silica and purified by flash chromatography (MeOH/DCM 1.5–2%) to yield N-(5-(4-ethoxyphenyl) thiazol-2-yl)-7-(3,3,3-trifluoro-2,2-dihydroxy propanamido)heptanamide 87 as a white solid (10 mg, 4.66%, mp. 138–143 °C, 3 steps).
[1]H NMR (600 MHz, Acetone-d6): 1H NMR (600 MHz, Acetone) δ 7.79 (d, J = 8.8 Hz, 23H), 7.25 (s, 1H), 6.91 (d, J = 8.8 Hz, 2H), 4.04 (q, J = 7.0 Hz, 2H), 3.28 (q, J = 7.1 Hz, 2H), 2.56 (t, J = 7.5 Hz, 2H), 1.76–1.66 (m, 2H), 1.62–1.52 (m, 2H), 1.42–1.36 (m, 4H), 1.34 (t, J = 7.0 Hz, 3H). [13].C NMR (151 MHz, CD3OD): 172.4, 165.2, 158.9, 157.9, 149.7, 129.9, 127.4, 126.9, 121.8 (q, J = 287.3 Hz), 115.1, 105.1, 94.3 (q, J = 32.3 Hz), 63.1, 39.1, 35.1, 28.6, 28.4, 26.1, 24.9, 13.8. HRMS-ESI M/z: [M+H]+calcd. 490.1623 for C21H26F3N3O5S, found 490.1623, 98% purity.
6. Biology
6.1. Cell Culture and cell Viability assays for cancer and normal cell lines
HCT116 human colon cancer cells, NCI–H522 human lung cancer cells, HT1080 fibrosarcoma cells, HeLa cervical cancer cells, U2OS osteosarcoma cells, MDA-MB-231 and MDA-MB-468 breast cancer cells, WI38 diploid human cell line and human retinal pigment epithelial RPE cells were preserved in Dulbecco’s Modified Eagle’s medium (Mediatech, Inc.) supplemented with 10% calf serum (Atlanta Biologicals) or 10% fetal bovine serum (Gemini Bio-Products # 100–106) and 1000 U/ml of both Penicillin and Streptomycin (Mediatech, Inc.) at 37 °C with 5% CO2. Cell feasibility was examined using methylene blue staining: cells were plated at a rate of 5000/well in 96 well plates (n = 3) respectively and treated the next day. Three days after treatment, cells were fixed/stained in methylene blue saturated in 50% ethanol for 30 min at room temperature. Excessive water was used to wash off extra dye from the plates. The retained dye in the plates was dissolved in 0.1 N HCl and absorbance was measured at 668 nm.
6.2. Western blotting
NCI–H522 cell pellets obtained were lysed using lysis buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 0.5% NP-40, (supplemented with 1 μg/ml aprotinin, 2 μg/ml leupeptin, 1 μg/ml pepstatin A, 1 mM DTT, and 0.1 M PMSF) for 30 min on ice and centrifuged at 13*103 g for 25 min at 4 °C. The protein levels of the obtained lysates were normalized using BCA Protein Assay Kit (Pierce) and separated by SDS-polyacrylamide gel electrophoresis (12.5% acrylamide). Transfer to polyvinylidene difluoride membranes (Millipore) was followed by obstruction of membranes with blocking buffer containing 5% (w/v) non-fat dry milk dissolved in PBST [1X PBS containing 0.05% (v/v) Tween 20] for 1 h at room temperature. Membranes were then incubated with corresponding primary antibodies overnight at 4 °C. The membranes were then washed (3 × 15 min each) with PBST and incubated with secondary antibodies conjugated to horse-radish peroxidase, obtained from Biorad, and used at a dilution of 1:10 000. Bound antibodies were detected using enhanced chemiluminescence (Biorad).
6.3. Inhibitor testing with HDAC isoforms
In a half-area 96-well white opaque plate (Corning), recombinant HDAC1 (1 μL of 3 ng/μL, BPS Bioscience), HDAC2 (1 μL of 1 ng/μL, BPS Bioscience), HDAC3 (1 μL of 30 ng/μL, BPS Bioscience), HDAC4 (1 μL of 5 ng/μL, BPS Bioscience), HDAC6 (1 μL of 35 ng/μL, BPS Bioscience), HDAC8 (1 μL of 70 ng/μL, BPS Bioscience), or buffer alone (1 μL) was added to HDAC-Glo™ buffer (43 μL, Promega). Serial dilutions or single concentrations of inhibitors (1 μL in DMSO, concentrations described in the supporting information) or DMSO alone (1 μL) were added to the enzyme solution, followed by 3 h incubation at room temperature. The HDAC-Glo™ reagent (5 μL), prepared per manufacturer recommendations, was added to each reaction. Luminescent signal was measured every 3 min over the course of 30 min using an M−Plex Infinite 200 Pro (Tecan). To determine IC50 values, the luminescent signal at peak reading was first background corrected with the signal from a background control reaction without HDAC enzyme. Percent deacetylase activity values were calculated by dividing the background-corrected signal with inhibitor by the background-corrected signal without inhibitor. IC50 values were calculated by fitting the percent deacetylase activity remaining as a function of inhibitor concentration to a sigmoidal dose–response curve (y = 100/(1+(x/IC50)z), where y = percent deacetylase activity and × = inhibitor concentration) using non-linear regression with KaleidaGraph 5.0 software.
6.4. S9 fraction assay
A protocol similar to the one employed by Rudraraju et al. [41] using Human Liver S9 fractions instead of liver microsomes was used for the metabolism studies. The assay was initiated by incubating a mixture of the substrate (0.02 mM), buffer (0.1 M), and NADPH (1 mM) at 37 °C for 10 min, followed by addition of the S9 fraction (0.5 mg) to the mixture and gentle mixing. The total volume of the reaction mixture was maintained at 1 mL. Experiments were done in duplicate 450 μL aliquots of the mixture were collected at different time intervals (1 h, 2 h) for compound 60 and 200 μL aliquots every 15 min for the TFMK 91, as it has been reported earlier that it had a half-life less than 30 min [11]. The reactions in the aliquots were terminated by adding an equal volume of ethyl acetate, mixing thoroughly, and transferring into micro centrifuge tubes. They were vortexed for 2 min and then subjected to centrifugation at 10 000×g for 15 min to remove the proteins. The top organic layers were extracted, evaporated under reduced pressure. The organic residues from the aliquots were dissolved in methanol first, then diluted 1:1 with water, and analyzed using LC-ESI-MS. A 450 μL aliquot of compound 60 with concentration of 0.02 mM contains 9 nmol, which after evaporation, was dissolved in 500 μL of methanol to give 18 μM in concentration of 60. This was diluted 1:1 with water to give a final concentration of 9 μM, which we used for LC-ESI-MS. Similarly, 200 μL of TFMK 91 at a concentration of 0.02 mM contains 4 nmol, which after removing ethyl acetate, and dissolving in 250 μL of methanol gives 16 μM concentration of 91. This was diluted 1:1 with water to give 8 μM final concentration for LC–ESI-MS.
6.5. LC-ESI-MS protocol
For analysis of the aliquots obtained, the LC–ESI–MS protocol shown in Tables S9 and S10 was used. The solvent flow rate was 0.2 mL/min, and the sample injection volume was 20 μL. Mobile phase A consisted of water containing 0.1% formic acid, while mobile phase B was acetonitrile containing 0.1% formic Acid. The column was equilibrated with 30% of mobile phase A and 70% of mobile phase B. The retention times for compound 60, compound 92 (TFMHA) and compound 91 (TFMK) were 7.5 min (±0.1 min), 8.13 min and 10.5 min (±0.3 min). (Additional technical details are included in the Supporting Information). At the start, the parameters for both starting materials were established. The M+1 for compound 60 was m/z 370 (hydrate) with a small percentage having an M+1 of m/z 352 m/z (ketone). Its MS/MS peak gave only m/z 352. The compound 91 had M+1 peaks of m/z 302 (ketone), m/z 320 (hydrate) and another peak of m/z 338 corresponding to a dihydrate, while the MS/MS corresponded to m/z 302 with a lesser amount of m/z 320. The TFMHA 92 had an M+1 peak of 354 m/z (14C). (Additional mass spectra are displayed in the supporting information).
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health grant R15CA213185 to L.M.V.T, grant R15GM120712 to W.R.T, and grant GM131821 to MKHP. The authors thank the Developmental Therapeutics Program of the National Cancer Institute, Bethesda, MD, USA for cytotoxicity studies in the human tumor cell lines screen.
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix B. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2022.114807.
Appendix A. Supplementary Data
Supplementary data to this article can be found online at.
Data availability
Data will be made available on request.
References
- [1].Siegel RL, Miller KD, Fuchs HE, Jemal A, Cancer statistics, CA A Cancer J. Clin. 72 (2022) 7–33, 2022. [DOI] [PubMed] [Google Scholar]
- [2].Ho TCS, Chan AHY, Ganesan A, Thirty years of HDAC inhibitors: 2020 insight and hindsight, J. Med. Chem. 63 (2020) 12460–12484. [DOI] [PubMed] [Google Scholar]
- [3].Rodd AL, Ververis K, Karagiannis TC, Current and emerging Therapeutics for cutaneous T-cell lymphoma: histone deacetylase inhibitors, Lymphoma 2012. 1–10 (2012). [Google Scholar]
- [4].Witt O, Deubzer HE, Milde T, Oehme I, HDAC family: what are the cancer relevant targets? Cancer Lett. 277 (2009) 8–21. [DOI] [PubMed] [Google Scholar]
- [5].Choi MA, et al. , Design, synthesis and biological evaluation of a series of CNS penetrant HDAC inhibitors structurally derived from amyloid-β probes, Sci. Rep. 9 (2019) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ning Z-Q, et al. , Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity, Cancer Chemother. Pharmacol. 694 69 (2011) 901–909, 2011. [DOI] [PubMed] [Google Scholar]
- [7].Thangapandian S, John S, Lee Y, Arulalapperumal V, Lee KW, Molecular modeling study on tunnel behavior in different histone deacetylase isoforms, PLoS One 7 (2012), 49327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Vaidya GN, et al. , Paradigm shift of “classical” HDAC inhibitors to “hybrid” HDAC inhibitors in therapeutic interventions, Eur. J. Med. Chem. 209 (2021), 112844. [DOI] [PubMed] [Google Scholar]
- [9].Shen S, Kozikowski AP, Why hydroxamates may not Be the best histone deacetylase inhibitors - what some may have forgotten or would rather forget? ChemMedChem 11 (2016) 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].He J, et al. , Synthesis and biological evaluation of HDAC inhibitors with a novel zinc binding group, Front. Chem. 8 (2020) 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Frey RR, et al. , Trifluoromethyl ketones as inhibitors of histone deacetylase, Bioorg. Med. Chem. Lett 12 (2002) 3443–3447. [DOI] [PubMed] [Google Scholar]
- [12].Suzuki T, et al. , Thiol-based SAHA analogues as potent histone deacetylase inhibitors, Bioorg. Med. Chem. Lett. 14 (2004) 3313–3317. [DOI] [PubMed] [Google Scholar]
- [13].Wada CK, et al. , α-Keto amides as inhibitors of histone deacetylase, Bioorg. Med. Chem. Lett 13 (2003) 3331–3335. [DOI] [PubMed] [Google Scholar]
- [14].Lv W, Zhang G, Barinka C, Eubanks JH, Kozikowski AP, Design and synthesis of mercaptoacetamides as potent, selective, and brain permeable histone deacetylase 6 inhibitors, ACS Med. Chem. Lett. 8 (2017) 510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Geurs S, et al. , Identification of mercaptoacetamide-based HDAC6 inhibitors via a lean inhibitor strategy: screening, synthesis, and biological evaluation, Chem. Commun. 58 (2022) 6239–6242. [DOI] [PubMed] [Google Scholar]
- [16].Tavares MT, Kozikowski AP, Mercaptoacetamide Shen S, A promising zinc-binding group for the discovery of selective histone deacetylase 6 inhibitors, Eur. J. Med. Chem. 209 (2021), 112887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].He X, et al. , Medicinal chemistry updates of novel HDACs inhibitors (2020 to present), Eur. J. Med. Chem. 227 (2022), 113946. [DOI] [PubMed] [Google Scholar]
- [18].Chen K, Xu L, Wiest O, Computational exploration of zinc binding groups for HDAC inhibition, J. Org. Chem. 78 (2013) 5051–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Malátková P, Wsól V, Carbonyl reduction pathways in drug metabolism, Drug Metab. Rev. 46 (2014) 96–123. [DOI] [PubMed] [Google Scholar]
- [20].Veale A, et al C Orally Active Trifluoromethyl Ketone Inhibitors of Human Leukocyte Elastase. J. Med. Chem. 40, 3173–3181 (1997). [DOI] [PubMed] [Google Scholar]
- [21].Scarpelli R, et al. , Studies of the metabolic stability in cells of 5-(trifluoroacetyl) thiophene-2-carboxamides and identification of more stable class II histone deacetylase (HDAC) inhibitors, Bioorg. Med. Chem. Lett. 18 (2008) 6078–6082. [DOI] [PubMed] [Google Scholar]
- [22].Yoshida M, Kijima M, Akita M, Beppu T, Potent and Specific Inhibition of Mammalian Histone Deacetylase Both in Vivo and in Vitro by Trichostatin A*, 1990. [PubMed] [Google Scholar]
- [23].Jones P, et al. , 2-Trifluoroacetylthiophenes, a novel series of potent and selective class II histone deacetylase inhibitors, Bioorg. Med. Chem. Lett 18 (2008) 3456–3461. [DOI] [PubMed] [Google Scholar]
- [24].Bottomley MJ, et al. , Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain, J. Biol. Chem. 283 (2008) 26694–26704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gong CJ, et al. , Design, synthesis and biological evaluation of bisthiazole-based trifluoromethyl ketone derivatives as potent HDAC inhibitors with improved cellular efficacy, Eur. J. Med. Chem. 112 (2016) 81–90. [DOI] [PubMed] [Google Scholar]
- [26].Frühauf A, Meyer-Almes FJ, Non-hydroxamate zinc-binding groups as warheads for histone deacetylases, Mol 26 (5151 26) (2021) 5151, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kelly CB, Mercadante MA, Leadbeater NE, Trifluoromethyl ketones: properties, preparation, and application, Chem. Commun. 49 (2013) 11133–11148. [DOI] [PubMed] [Google Scholar]
- [28].Kurohara T, et al. , Identification of novel histone deacetylase 6-selective inhibitors bearing 3,3,3-trifluorolactic amide (TFLAM) motif as a zinc binding group, Chembiochem 22 (2021) 3158–3163. [DOI] [PubMed] [Google Scholar]
- [29].Vanommeslaeghe K, De Proft F, Loverix S, Tourw??, D., P. Geerlings, Theoretical study revealing the functioning of a novel combination of catalytic motifs in histone deacetylase, Bioorg. Med. Chem. 13 (2005) 3987–3992. [DOI] [PubMed] [Google Scholar]
- [30].El Kaïm L, Pinot-Périgord E, Trifluoropyruvamides from isocyanides and trifluoroacetic anhydride, Tetrahedron 54 (1998) 3799–3806. [Google Scholar]
- [31].Suzuki T, et al. , Novel histone deacetylase inhibitors: design, synthesis, enzyme inhibition, and binding mode study of SAHA-Based non-hydroxamates, Bioorg. Med. Chem. Lett. 13 (2003) 4321–4326. [DOI] [PubMed] [Google Scholar]
- [32].Suzuki T, Miyata N, Non-hydroxamate histone deacetylase inhibitors, Curr. Med. Chem. 12 (2005) 2867–2880. [DOI] [PubMed] [Google Scholar]
- [33].Sagheer OM, Mohammed MH, Ibraheem ZO, Wadi JS, Tawfeeq MF, Synthesis of gamma biguanides butyric acid analogues as HDAC inhibitors and studying their cytotoxic activity, Mater. Today Proc. 47 (2021) 5983–5991. [Google Scholar]
- [34].Wang J-J, Yu W, Wang J, Yu W, Anti-markovnikov hydroazidation of alkenes by visible-light photoredox catalysis, Chem. Eur J. 25 (2019) 3510–3514. [DOI] [PubMed] [Google Scholar]
- [35].Sokolenko TM, Davydova YA, Yagupolskii YL, Efficient synthesis of 5′-fluoroalkoxythiazoles via α-bromo-α-fluoroalkoxyacetophenones Hantzsch type cyclization with thioureas or thioamides, J. Fluor. Chem. 136 (2012) 20–25. [Google Scholar]
- [36].Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL, Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation, Proc. Natl. Acad. Sci. U.S.A. 100 (2003) 4389–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Depetter Y, et al. , Selective pharmacological inhibitors of HDAC6 reveal biochemical activity but functional tolerance in cancer models, Int. J. Cancer 145 (2019) 735–747. [DOI] [PubMed] [Google Scholar]
- [38].Al-Hamashi AA, et al. , A new class of cytotoxic agents targets tubulin and disrupts microtubule dynamics, Bioorg. Chem. 116 (2021), 105297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kerr E, et al. , Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis, Cell Death Differ. 19 (2012) 1317–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chaitanya GV, Alexander JS, Babu PP, PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration, Cell Commun. Signal. 8 (2010) 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].V Rudraraju A, Vitro metabolic stability study of new cyclen based antimalarial drug leads using RP-HPLC and LC-MS/MS, Mod. Chem. Appl. 2 (2014). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.