


Abstract
Recently we have found that mitoxantrone, like Adriamycin, can be activated by formaldehyde and subsequently form adducts which stabilise double-stranded DNA in vitro. This activation by formaldehyde may be biologically relevant since formaldehyde levels are elevated in those tumours in which mitoxantrone is most cytotoxic. In vitro transcription analysis revealed that these adducts block the progression of RNA polymerase during transcription and cause truncated RNA transcripts. There was an absolute requirement for both mitoxantrone and formaldehyde in transcriptional blockage formation and the activated complex was found to exhibit site specificity, with blockage occurring prior to CpG and CpA sites in the DNA (non-template strand). The stability of the adduct at 37°C was site dependent. The half-lives ranged from 45 min to ~5 h and this was dependent on both the central 2 bp blockage site as well as flanking sequences. The CpG specificity of mitoxantrone adduct sites was also confirmed independently by a λ exonuclease digestion assay.
INTRODUCTION
Adriamycin is an anthracycline antibiotic which is one of the most versatile anticancer agents used in the clinic today (1). The major dose-limiting factor influencing the use of Adriamycin and other anthracyclines is the associated cardiotoxicity (1–3). The ability of these anticancer drugs to be reduced to semiquinone derivatives results in a number of reactions which produce oxygen radicals and result in cardiotoxicity, at least partly because of the reduced amount of protective enzymes in the heart (and other muscle tissue) (3,4).
Mitoxantrone (Novatrone™) lacks this cardiotoxic side-effect. It is a synthetic anthracenedione compound (1,5) which was originally synthesised based on the anthracycline structure, in an attempt to find anthracycline analogues which lack the cardiotoxic response but retain the potent cytotoxicity of the anthracyclines. Although not a true anthracycline, mitoxantrone is considered a partial analogue of the anthracyclines and has proven to be one of the best drug candidates to arise from extensive studies which have attempted to find cytotoxic but less cardiotoxic analogues. However, unlike Adriamycin, mitoxantrone is not active against a wide spectrum of tumours, being used specifically for the treatment of advanced breast cancer, non-small cell lung cancer, non-Hodgkins lymphoma, non-lymphocytic leukaemia and melanoma (6).
The mechanism of action of mitoxantrone has not yet been fully elucidated. It has been found that mitoxantrone accumulates in cells and concentrates in the nucleus, where it interacts with nucleic acids (5,7,8). Previous studies have shown that mitoxantrone intercalates into DNA (9), specifically at CpG and CpA sites (10), and can be oxidatively activated to bind to DNA (11,12), but the mechanism and binding properties remain unresolved. Mitoxantrone has also been documented to target topoisomerase II, causing DNA strand breaks (13,14). Recently we reported that mitoxantrone can be activated by formaldehyde in vitro to produce adducts which stabilise DNA such that they function as interstrand crosslinks (15). This finding may be biologically relevant, since cells of myeloid origin have increased levels of formaldehyde (16) and mitoxantrone is effective against myeloid tumours. The attraction of neutrophils to sites of inflammation of advanced solid tumours causes release of hydrogen peroxide upon respiratory burst (17). Hydrogen peroxide produces oxygen radicals (e.g. hydroxyl radicals) which oxidise Tris (in vitro) and the biologically available polyamines such as spermine to generate formaldehyde (18,19). This provides an increased level at the site of these solid tumours and may also result in activation of mitoxantrone. Therefore, tumours which do not contain increased levels of formaldehyde may be resistant to mitoxantrone because the drug cannot be activated sufficiently in these cells.
Recent studies have shown that the mitoxantrone–formaldehyde–DNA adduct stabilises DNA sufficiently to prevent strand separation (15). In susceptible cells such prevention of strand separation may impair progression of replicative enzymes and therefore prevent cell division and proliferation. We present here the consequence of these DNA adducts on two other biologically relevant processes (transcription and exonuclease digestion) and show that formaldehyde-activated mitoxantrone–DNA adducts are detrimental to these processes, and occur predominantly at CpG and CpA sequences.
MATERIALS AND METHODS
Materials
Mitoxantrone was kindly provided by Lederle Laboratories (Pearl River, NY). NTPs, 3′-O-methylguanine (MeG), 3′-O-methylcytidine (MeC), [α-32P]dCTP, [α-32P]UTP (3000 Ci/mmol), ribonuclease inhibitor (RNA guard, human placenta) and Escherichia coli RNA polymerase were purchased from Amersham Pharmacia Biotech. Acrylamide and urea were obtained from ICN Biomedicals, bisacrylamide and ammonium persulphate were from Bio-Rad Laboratories and TEMED and nuclease-free BSA were purchased from Promega. Phenol and λ exonuclease were from Gibco BRL and formaldehyde was purchased from BDH. DNA polymerase (Klenow fragment) and the restriction enzymes SmaI, PvuII and HindIII were obtained from New England Biolabs and EcoRI and BamHI were from Promega. Nensorb™ 20 columns were purchased from NEN Research Products and glycogen was from Boehringer Mannheim. Plasmid purification kits and Taq polymerase were purchased from Qiagen.
DNA isolation and purification
The plasmid pCC1 (20) was isolated using a DNA purification kit from Qiagen. For transcription assays the plasmid was restriction digested with PvuII and HindIII to isolate a 512 bp fragment containing the lac UV5 promoter. The 512 bp fragment was purified by electroelution and phenol/chloroform extraction. This fragment was used for all subsequent transcription studies. Two DNA sequences within the 512 bp fragment were amplified by PCR for exonuclease studies. One primer set [5′-TCC CCC GGG CCT ATA TCG CCG ACA TCA CC (+163 to +182) and 5′-CCG GAA TTC GAC TCC TGC ATT AGG AAG C (+342 to +361)] contained a SmaI and EcoRI restriction site, respectively, and generated a 200 bp DNA fragment, while the second set [5′-TCC CCC GGG TGT GGA ATT GTG AGC GGA TA (–3 to +16) and 5′-CGC GGA TCC GGT GAT GTC GGC GAT ATA GG (+162 to +183] contained a SmaI and BamHI cut site, respectively, and generated a 185 bp DNA fragment. These fragments were designed for 3′-end-labelling upon restriction digestion and were isolated as described above for the 512 bp fragment.
Transcription assay
DNA was reacted with mitoxantrone and formaldehyde prior to initiation of transcription in order to enable all unbound formaldehyde to be removed by ethanol precipitation. After precipitation, DNA was resuspended in a transcription mix containing 10 mM DTT, 150 µg/ml BSA, 1 U/µl RNA guard and RNA polymerase in transcription buffer (40 mM Tris pH 8.0, 0.1 M KCl, 3 mM MgCl2 and 0.1 M EDTA). Transcription assays were performed as previously published (21), however, the DNA (90 µM bp) was treated with drug prior to transcription initiation, resulting in a 25 µM bp DNA concentration in the transcription mix. Subsequent to transcription, equal volumes of transcription termination buffer (10 M urea, 10% sucrose, 40 mM EDTA, 0.1% xylene cyanol and 0.1% bromophenol blue in 2× TBE) was added to all samples and denatured at 90°C for 5 min followed by 5 min on ice.
λ exonuclease digestion
The two PCR-derived DNA fragments were cut with SmaI and either EcoRI or BamHI and then 3′-end-labelled at the EcoRI and BamHI sites using the Klenow fragment of E.coli DNA polymerase I and [α-32P]dATP. The DNA fragments were then purified using Nensorb 20 chromatography columns and were resuspended in TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0) containing 200 µM bp sonicated calf thymus DNA. End-labelled DNA (30 µM bp) was then incubated with 30 µM mitoxantrone and up to 25 mM formaldehyde at 37°C for 2 h. The samples were ethanol precipitated and resuspended in buffer containing 67 mM glycine and 2.5 mM MgCl2, pH 9.4, 50 µg/ml BSA and 5 U λ exonuclease. Samples were digested at 37°C for 40 min prior to denaturing in an equal volume of formamide loading dye (90% formamide, 10 mM EDTA, 0.1% xylene cyanol, 0.1% bromophenol blue) at 90°C for 5 min. Maxam–Gilbert G sequencing was also performed as described previously (21).
Electrophoresis
The DNA samples from transcription and exonuclease-treated reactions were separated on a 12% denaturing acrylamide gel which was preheated to 50°C. The samples were subjected to electrophoresis at 2000 V for 1.5–2 h in TBE buffer. The gels were then fixed (10% methanol, 10% acetic acid) and vacuum dried on a Bio-Rad Model 583 gel drier and exposed to a phosphor screen overnight. The gels were analysed using a Model 400B PhosphorImager and ImageQuant software (Molecular Dynamics, CA).
RESULTS
Concentration and time dependence of transcriptional blockages
When E.coli RNA polymerase encounters a bulky DNA adduct, elongation of the nascent RNA is blocked, thus producing a truncated transcript. The length of these RNA transcripts is consequently dependent on the position of the drug–DNA adduct on the DNA template. If no adducts are encountered, RNA polymerase transcribes to the end of the fragment, yielding a 379 base full-length transcript.
Truncated transcripts were produced when the promoter-containing template was reacted with mitoxantrone and formaldehyde, and the dependence on formaldehyde concentration is illustrated in Figure 2. An increase in blockage with a corresponding decrease in the full-length RNA is associated with an increase in formaldehyde concentration (Fig. 2A). Controls lacking formaldehyde, mitoxantrone or both compounds produced only the full-length transcript, with no evidence of any sequence-dependent blockage to the progression of RNA polymerase. This indicates the requirement for both formaldehyde and mitoxantrone for the production of adducts which have the ability to block transcription. The extent of blockage at individual sites was quantitated as a percentage of the total number of transcripts in each lane (Fig. 2B) and revealed a linear increase in lesion frequency with increasing formaldehyde concentration. The percentage of full-length transcript was quantitated (Fig. 2C) and shows a concomitant decrease in the full-length transcript with increasing formaldehyde concentration.
Figure 2.
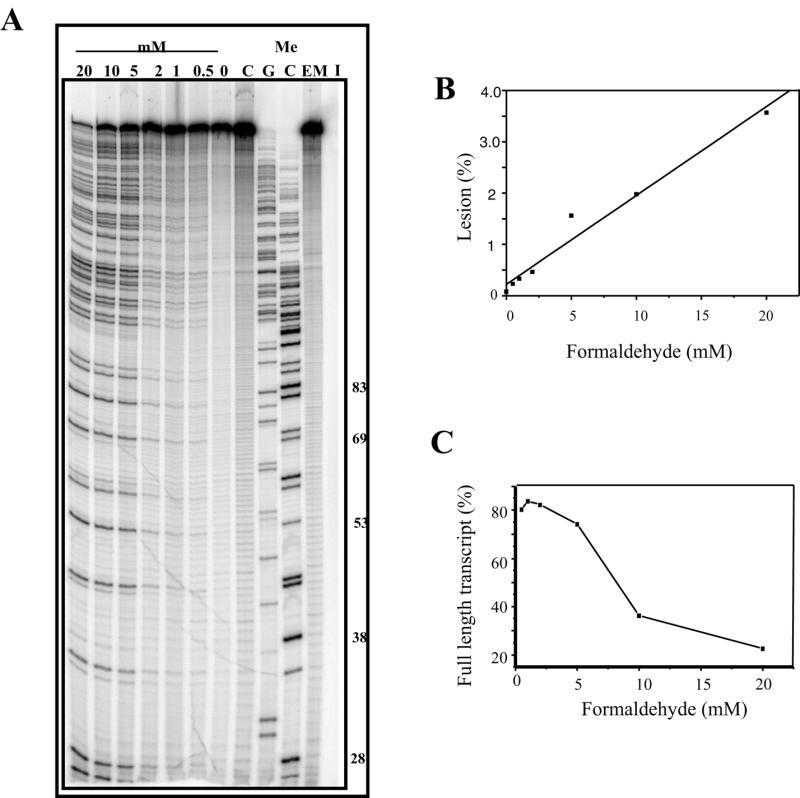
Mitoxantrone-induced transcriptional blockages with increasing formaldehyde. (A) DNA (90 µM bp) was incubated with 20 µM mitoxantrone and formaldehyde (0–20 mM as indicated) in transcription buffer for 2 h. The samples were ethanol precipitated and resuspended in transcription reaction mix. Transcription was initiated at the lac UV5 promoter of the 512 bp fragment prior to transcript elongation for 5 min and denaturation in transcription termination buffer at 90°C for 5 min. The samples were then subjected to electrophoresis at 2000 V for 1.5 h on a 12% acrylamide gel. I is an initiation control which has not been elongated. C is an elongated control lacking mitoxantrone and EM is an elongated control representing undamaged DNA lacking both mitoxantrone and formaldehyde. MeC and MeG are sequencing lanes which were obtained using methylated nucleotides during elongation. The length of a range of MeC-terminated transcripts are shown to the right of the figure. (B) Quantitation of block sites from the gel shown in (A). The mole fraction of block site 4 (44mer) was calculated and plotted as a percentage of the entire lane at each formaldehyde concentration. (C) Quantitation of reduction in full-length transcript. The percentage of full-length transcript (A) is shown as a function of formaldehyde concentration.
Transcriptional blockage was also dependent on mitoxantrone concentration (Fig. 3). Quantitation of the blockage revealed a linear increase in blockage intensity with increasing mitoxantrone concentration (Fig. 3B) and a decrease in the fraction of full-length transcript (Fig. 3C).
Figure 3.

Dependence on mitoxantrone concentration of transcriptional blockage. (A) The 512 bp DNA fragment was reacted with mitoxantrone (0–50 µM) and 5 mM formaldehyde in transcription buffer for 2 h at 37°C. The DNA was then ethanol precipitated, resuspended in transcription reaction mix prior to initiation and transcription elongation at 37°C for 5 min. Lane C is a control lacking formaldehyde and other controls are as stated in Figure 1A. (B) Quantitation of increase in transcriptional blockage from (A). The percentage of block 4 (44mer) as a fraction of the entire lane is shown as a function of mitoxantrone concentration. (C) Reduction in percent of full-length transcript from (A) with increasing mitoxantrone concentration.
The 512 bp DNA fragment was incubated with mitoxantrone and formaldehyde for times up to 30 min, and Figure 4 illustrates the time dependence for production of blockages. Quantitation revealed an increase in blockage frequency with increasing time (Fig. 4B), with a 50% loss of the full-length transcript after 30 min (Fig. 4C).
Figure 4.
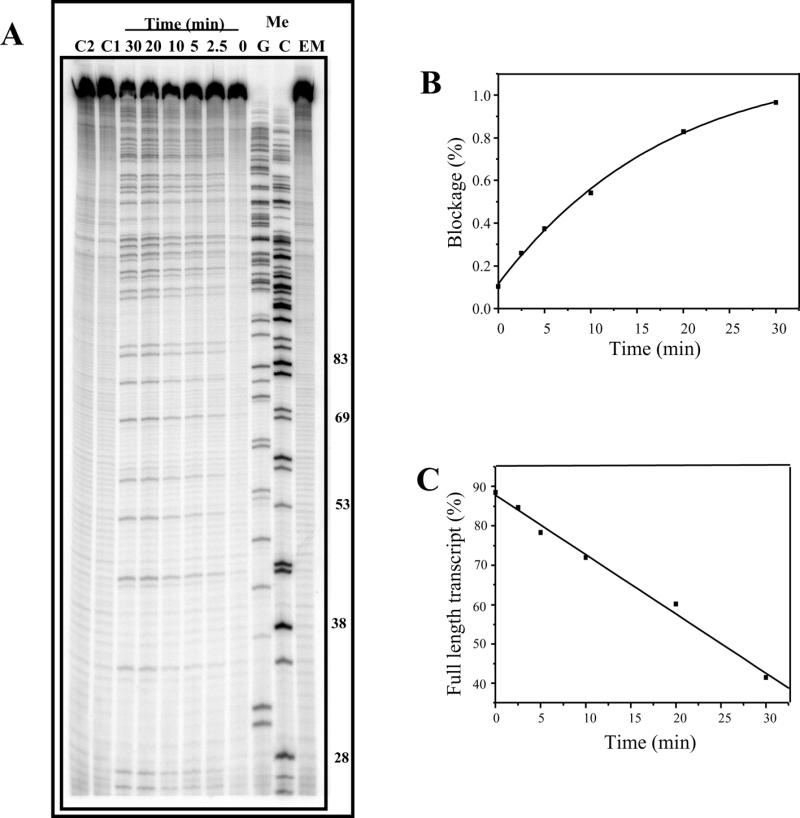
Time dependence of formation of mitoxantrone–DNA adducts. (A) DNA (90 µM bp) was incubated with 20 µM mitoxantrone and 6 mM formaldehyde for 0–30 min at 37°C. Reactions were terminated by ethanol precipitation prior to transcription initiation with subsequent elongation for 5 min. C1 and C2 are controls lacking mitoxantrone and formaldehyde, respectively. (B) The mole fraction of transcriptional block 4 (44mer) is plotted as a percentage of all transcripts in the lane and expressed as a function of drug–DNA reaction time. (C) Loss of full-length transcript (A) with incubation time.
Stability of adducts at 37°C
As mitoxantrone–formaldehyde–DNA adducts have previously been shown to exhibit thermal instability in an in vitro crosslinking assay (15), it was of interest to compare the stability of these adducts to the lesions detected as transcriptional blockages. DNA was reacted initially to yield a high fraction of transcriptional blockages and their stability at 37°C was then assessed at increasing transcription elongation times (Fig. 5A). First order kinetic analysis of the loss of blockages at individual sites revealed that there is great variation in stability at different sites (Fig. 5B). The half-lives for 11 well-resolved blockage sites varied from 45 min to >5 h; these values are summarised in Table 1, together with the DNA sequence flanking each mitoxantrone blockage site.
Figure 5.

Stability of mitoxantrone–DNA adducts at 37°C. (A) The 512 bp fragment was reacted with 25 µM mitoxantrone and 5 mM formaldehyde for 2 h at 37°C. Following ethanol precipitation, transcription was initiated prior to elongation for 5–150 min as shown at 37°C. (B) First order kinetic analysis of the loss of transcriptional blockage at individual adduct sites as indicated. The half-lives of adducts at individual sites are summarised in Table 1.
Table 1. Stability of adducts at individual drug sites.

Site specificity of transcriptional blockages
The mole fraction of each blockage site was quantitated (Fig. 5A, lane 6) and is shown as a function of the DNA sequence (Fig. 6A). Nine blockages were at CG sequences and nine at CA sequences. The most intense blockage was at a CG site (blockage number 6 in Fig. 6A), but overall there was no significant difference between the relative occupancy at CG and CA sites.
Figure 6.

Sequence specificity of mitoxantrone–DNA adducts. (A) The mole fraction of each mitoxantrone transcriptional blockage site (Fig. 4A, lane 5) was determined and is shown as a function of the sequence of the 512 bp DNA sequence. (B) Sequence specificity of λ exonuclease blockage sites on the 185 bp fragment.
Detection of mitoxantrone adducts by λ exonuclease
DNA fragments (which reside within the 512 bp DNA fragment used in the transcription assays) were generated to investigate whether these adducts could block the digestion of DNA by λ exonuclease and to provide an alternative means of identifying the location of mitoxantrone adducts. The digestion studies revealed that reaction of DNA with mitoxantrone and formaldehyde produced adducts which blocked λ exonuclease 5′→3′ DNA digestion (Fig. 7A). The blockage frequency increased with increasing formaldehyde concentration (Fig. 7B) and the majority of block sites were specific to CpG sites (Fig. 7C). To enable a direct comparison of mitoxantrone-induced transcriptional and λ exonuclease blockages, a 185 bp DNA fragment was utilised with a sequence overlapping that assessed in the transcription assay. Fewer blockages were detected by λ exonuclease digestion as compared to the transcription assay, and the majority of these were again at CG sites (Fig. 6B). These correspond to the same sites as detected by the transcription assay, and this suggests that this assay is more sensitive than the λ exonuclease digestion assay, since adducts at CA sequences were detected by RNA polymerase but to a lesser extent by λ exonuclease.
Figure 7.


Blockage of λ exonuclease. (A) 3′-End-labelled DNA (30 µM bp) was incubated with mitoxantrone (30 µM) and formaldehyde (0–25 mM) for 2 h at 37°C. Samples were ethanol precipitated and digested with λ exonuclease for 40 min at 37°C. Reactions were terminated by the addition of formamide loading dye followed by denaturation at 90°C and electrophoresis on a 12% acrylamide denaturing gel. G is a Maxam–Gilbert sequencing lane. Controls lacking exonuclease digestion showed no blockage and yielded ~50-fold more full-length DNA (data not shown). The presence of full-length DNA fragments in the digested, drug-treated samples indicates that the DNA was not completely digested by λ exonuclease. Individual blockage sites are numbered on the left hand side. (B) Quantitation of block sites from the gel in (A). Mole fraction of increasing λ exonuclease blocks are shown relative to the formaldehyde concentration. (C) Sequence specificity of adducts which block λ exonuclease. The direction of exonuclease progression is 5′→3′ and the fragment represents the 200 bp fragment from primer set 1.
DISCUSSION
Formaldehyde activation of mitoxantrone
Our recent studies have found that mitoxantrone can be activated by formaldehyde to form adducts in DNA in vitro (15). These adducts stabilised DNA such that it was resistant to strand separation in interstrand crosslinking assays. Stability studies revealed that the adduct was labile and therefore not a classical covalent interstrand crosslink, like those formed with platinum and mustard anticancer agents (22). The mitoxantrone adduct is presumably structurally similar to that found with Adriamycin, where adducts function as ‘virtual’ interstrand crosslinks (23). To further investigate and characterise the binding of formaldehyde-activated mitoxantrone to DNA, two investigations were undertaken to determine the effect these lesions had on biologically important systems, e.g. transcription and exonuclease digestion. The results revealed that formaldehyde-activated mitoxantrone blocked the progression of both E.coli RNA polymerase and λ exonuclease in a sequence-dependent manner.
In vitro transcription assays confirmed that both formaldehyde and mitoxantrone were required in order to produce transcriptional blockage, since at the concentrations used neither compound alone blocked RNA polymerase progression. Although formaldehyde itself can react with DNA at high concentrations in in vitro cell systems (24–26), it did not produce transcriptional blockage at the concentrations used in this assay.
The time dependence of formation of transcriptional blockage revealed that the block frequency began to plateau at 30 min. This reaction time is consistent with the time required for maximal crosslinking in in vitro crosslinking assays (15). The blockages, however, were less intense than those seen in transcription studies using other drugs such as Adriamycin (27). In those studies, lower drug concentrations resulted in significantly higher levels of transcriptional block and therefore complete inhibition of the full-length transcript (27). In in vitro crosslinking studies (15) lower drug concentrations also resulted in complete crosslinking and this is due to the requirement for only one adduct in the entire 3496 bp DNA duplex to stabilise DNA in order to prevent strand separation.
Sequence specificity
The major transcriptional blockages detected were at CpG and CpA sites on the non-template strand of DNA. This sequence specificity is in striking contrast to that observed for Adriamycin and the structurally related antibiotic barminomycin, both of which exhibit a strong preference for adduct formation at GpC sequences (27,28). Exonuclease studies on the same region of the 512 bp fragment revealed that formaldehyde-activated mitoxantrone blockages were specific to CpG sites and this suggests that adducts at CpG sequences may distort the DNA helix to produce a lesion which can block the progression of exonuclease, whereas the CpA lesion cannot. Since exonuclease generally recognises only drug modifications in the DNA minor groove or those causing a major distortion of the DNA helix (29), it is possible that the two lesions are structurally different. The proposed structure of the mitoxantrone–DNA adduct (15) implies the likelihood of hydrogen bonding to the second DNA strand. The hydrogen bonding at CpA/TpG sequences may therefore be weaker due to the absence of the guanine residue on the second strand and, if so, this may also contribute to the reduced number of blockage sites detected by the λ exonuclease digestion assay.
Although the sequence specificity and occupancy of mitoxantrone adducts (Fig. 6) suggests no significant effect of flanking sequence on adduct formation and frequency, the importance of the flanking sequence is revealed by the stability studies. The two most stable sites detected so far for formaldehyde-activated mitoxantrone were both at ACGC sequences (Table 1). Analysis of a more extensive range of adduct sites will be required to ascertain if flanking sequences contribute significantly to the stability of these mitoxantrone–DNA adducts. The sequence specificity found in transcription and exonuclease studies parallels the sequence specificity for intercalation studies. Mitoxantrone has been found to preferentially intercalate (detected in a transcription assay only at low temperatures) at CG and CA sites in DNA (10). Since mitoxantrone is therefore more abundant at these preferred intercalation sequences, this may result in subsequent activation by formaldehyde and covalent binding to neighbouring nucleotides on the DNA.
Adduct stability
Past work using an in vitro crosslinking assay has revealed the lability of the mitoxantrone–DNA ‘virtual crosslink’. The adducts were found to be heat labile, indicating that the lesion is not a true covalent interstrand crosslink. By comparison with Adriamycin, it is therefore likely that formaldehyde-activated mitoxantrone might also be covalently bound to only one strand of DNA, most likely the N2 amino group of guanine residues at CpG and CpA (i.e. TpG) dinucleotides, with sufficient stabilisation of the local DNA region to resist strand separation arising from intercalation of the drug plus additional hydrogen bonding, similar to that reported recently for Adriamycin–DNA adducts (18,23,30,31).
To further investigate the stability of these adducts and to gain additional insight into the likely structure of the adduct, the stability of block sites in transcription was assessed at 37°C. Recent work with barminomycin found that transcriptional blockages exhibited half-lives ranging up to 2 h, with one site >200 min. The mitoxantrone adducts therefore exhibit stability similar to that found at many of the barminomycin adduct sites. Adriamycin-induced DNA adducts, however, are more stable, with half-lives of 3 and 40 h (32). The stability of mitoxantrone–formaldehyde adducts at CG sites and CA sites indicates that these sites are likely to contain an interstrand (‘virtual’) crosslink, with covalent attachment to guanine on only one DNA strand. It is possible that CA sites contain the same type of lesion that occurs at CG sites, but with a difference in hydrogen bonding to the second strand due to the lack of guanine (on the non-covalently bound strand), resulting in reduced stabilities. The suggested structure of the mitoxantrone–formaldehyde–DNA adduct (15) is consistent with the present transcriptional stability studies, since loss of less stable hydrogen bonds (resulting in separation of double-stranded DNA) could possibly leave the monoadduct attached to one strand of the DNA, sufficient to block transcription. This provides an explanation as to why transcriptional blockage generally appears to be more stable than adducts detected by in vitro crosslinking assays. The mere breakage of hydrogen bonds in this case could result in the loss of DNA stabilisation and therefore lead to strand separation.
Biological/medical implications
The importance of formaldehyde in the activation of mitoxantrone has now been established in several independent in vitro assays. The activity of mitoxantrone against myeloid and solid tumours is also consistent with the proposed mechanism of activation of mitoxantrone by formaldehyde, which is associated with higher levels of formaldehyde in these tumours (16,33). The results of the current study clearly demonstrate the ability of mitoxantrone adducts to perturb critical cellular processes, including transcription and DNA exonuclease activity. As the inhibition of similar enzymatic functions is known to lead to cell death, such DNA damage may be important in the mechanism of action of mitoxantrone. The adducts are also likely to induce apoptosis, as demonstrated for other cytotoxic agents which form DNA adducts (34).
Conclusions
Information from in vitro crosslinking, transcription and exonuclease assays indicates that mitoxantrone reacts rapidly with formaldehyde and DNA to form adducts which stabilise DNA and block the progression of DNA-dependent enzymes. The exact chemical nature of this adduct remains elusive and mass spectrometry experiments are currently in progress to establish the number of mitoxantrone chromophores involved and the contribution of formaldehyde. The majority of these adducts are at CpG and CpA sites in DNA, and the stability of these adducts is dependent on flanking sequences. The most stable lesions detected to date are at ACGC sequences. The preference for CpG sites is of interest as the frequency of these sequences are quite low in the genome and a high proportion are methylated. Studies are currently in progress to determine if mitoxantrone is more active against methylated CpG sites, as has been found with the CpG-specific drug Mitomycin C (35).
Figure 1.

Structure of Adriamycin and mitoxantrone.
REFERENCES
- 1.De Vita V.T., Hellman,S. and Rosenberg,S.A. (1993) In De Vita,V.T. (ed.), Cancer—Principles and Practice of Oncology. J.B. Lippincott, Philadelphia, PA.
- 2.Myers C.E., Mimnaugh,E.G., Yeh,G.C. and Sinha,B.K. (1988) In Lown,J.W. (ed.), Anthracycline and Anthracenedione-based Anti-cancer Agents. Elsevier, Amsterdam, The Netherlands, pp. 527–569.
- 3.Pratt W.B., Ruddon,R.W., Ensminger,W.D. and Maybaum,S. (1994) The Anticancer Drugs, 2nd Edn. Oxford University Press, New York, NY.
- 4.Lown J.W., Chen,H. and Plambeck,J.A. (1982) Biochem. Pharmacol., 31, 575–581. [DOI] [PubMed] [Google Scholar]
- 5.Feofanov A., Sharonov,S., Kudelina,I., Fleury,F. and Nabiev,I. (1997) Biophys. J., 73, 3317–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cornbleet M.A., Stuart-Harris,R.C., Smith,I.E., Coleman,R.E., Rubens,R.D., McDonald,M., Mouridsen,H.T., Rainer,H., Van Oosterom,A.T. and Smyth,J.F. (1984) Eur. J. Cancer Oncol., 20, 1141–1146. [DOI] [PubMed] [Google Scholar]
- 7.Feofanov A., Sharonov,S., Fleury,F., Kudelina,I. and Nabiev,I. (1997) Biophys. J., 73, 3328–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konopa J. (1988) In Borowski,E. and Shugar,D. (eds), Molecular Aspects of Chemotherapy. Pergamon Press, New York, NY, pp. 83–94.
- 9.Lown J.W., Morgan,A.R., Yen,S.F., Wang,Y.H. and Wilson,W.D. (1985) Biochemistry, 24, 4028–4035. [DOI] [PubMed] [Google Scholar]
- 10.Panousis C. and Phillips,D.R. (1994) Nucleic Acids Res., 22, 1342–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mewes K., Blanz,J., Ehninger,G., Gebhardt,R. and Zeller,K.P. (1993) Cancer Res., 53, 5135–5142. [PubMed] [Google Scholar]
- 12.Reszka K., Hartley,J.A., Kolodziejczyk,P. and Lown,J.W. (1989) Biochem. Pharmacol., 113, 173–178. [DOI] [PubMed] [Google Scholar]
- 13.Routier S., Bernier,J.L., Catteau,J.P., Riou,J.F. and Bailly,C. (1998) Anticancer Drug Des., 13, 407–415. [PubMed] [Google Scholar]
- 14.Hande K.R. (1998) Biochim. Biophys. Acta, 1400, 173–184. [DOI] [PubMed] [Google Scholar]
- 15.Parker B.S., Cullinane,C. and Phillips,D.R. (1999) Nucleic Acids Res., 27, 2918–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thorndike J. and Beck,W.S. (1977) Cancer Res., 37, 1125–1132. [PubMed] [Google Scholar]
- 17.Edwards S.W. and Swan,T.F. (1986) Biochem. J., 237, 601–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taatjes D.J., Gaundiano,G., Resing,K. and Koch,T.H. (1997) J. Med. Chem., 40, 1276–1286. [DOI] [PubMed] [Google Scholar]
- 19.Taatjes D.J., Gaudiano,G. and Koch,T.H. (1997) Chem. Res. Toxicol., 10, 953–961. [DOI] [PubMed] [Google Scholar]
- 20.Cullinane C. and Phillips,D.R. (1993) Nucleic Acids Res., 21, 1857–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phillips D.R. and Cullinane,C. (1997) In Fox,K. (ed.), Drug–DNA Interaction Protocols. Humana Press, Totowa, NJ, pp. 127–146.
- 22.Lawley P.D. and Phillips,D.H. (1996) Mutat. Res., 355, 13–40. [DOI] [PubMed] [Google Scholar]
- 23.Zeman S.M., Phillips,D.R. and Crothers,D.M. (1998) Proc. Natl Acad. Sci. USA, 95, 11561–11565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Costa M. and Zhitkovich,A. (1997) J. Toxicol. Environ. Health, 50, 433–449. [DOI] [PubMed] [Google Scholar]
- 25.Craft T.R., Bermudez,E. and Skopek,T.R. (1987) Mutat. Res., 176, 147–155. [DOI] [PubMed] [Google Scholar]
- 26.Chaw Y.F.M., Crane,L.E., Lange,P. and Shapiro,R. (1980) Biochemistry, 19, 5525–5531. [DOI] [PubMed] [Google Scholar]
- 27.Cullinane C. and Phillips,D.R. (1990) Biochemistry, 29, 5638–5646. [DOI] [PubMed] [Google Scholar]
- 28.Perrin L.C., Cullinane,C., Kimura,K. and Phillips,D.R. (1999) Nucleic Acids Res., 27, 1781–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mattes W.B. (1990) Nucleic Acids Res., 18, 2723–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fenick D.J., Taatjes,D.J. and Koch,T.H. (1997) J. Med. Chem., 40, 2452–2461. [DOI] [PubMed] [Google Scholar]
- 31.Taatjes D.J., Gaundiano,G., Resing,K. and Koch,T.H. (1996) J. Med. Chem., 39, 4135–4138. [DOI] [PubMed] [Google Scholar]
- 32.Van Rosmalen A., Cullinane,C., Cutts,S.M. and Phillips,D.R. (1995) Nucleic Acids Res., 23, 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ebeler S.E., Clifford,A.J. and Shibamoto,T. (1997) J. Chromatogr. B. Biomed. Sci. Appl., 702, 211–215. [DOI] [PubMed] [Google Scholar]
- 34.Gieseler F., Nussler,V., Brieden,T., Kunze,J. and Valsamas,S. (1998) Int. J. Clin. Pharmacol. Ther., 36, 25–28. [PubMed] [Google Scholar]
- 35.Millard J.T. and Beachy,T.M. (1993) Biochemistry, 32, 12850–12856. [DOI] [PubMed] [Google Scholar]