

Abstract
Background:
Mast cells (MCs) within the airway epithelium in asthma are closely related to airway dysfunction, but crosstalk between airway epithelial cells (AECs) and MCs in asthma remains incompletely understood. Human rhinovirus (RV) infections are key triggers for asthma progression and AECs from individuals with asthma may have dysregulated anti-viral responses.
Objective:
We utilize primary AECs in an ex vivo coculture model system to examine crosstalk between AECs and MCs following epithelial RV infection.
Methods:
Primary AECs were obtained from children with asthma (n=11) and healthy children (n=10), differentiated at air-liquid interface, and cultured in the presence of laboratory of allergic diseases-2 (LAD2) MCs. AECs were infected with RV serogroup-A 16 (RV16) for 48 hours. RNA isolated from both AECs and MCs underwent RNA-sequencing (RNAseq) analysis. Direct effects of epithelial-derived interferons on LAD2 MCs were examined by qPCR.
Results:
MCs increased expression of pro-inflammatory and anti-viral genes in AECs. AECs demonstrated a robust antiviral response following RV16 infection that resulted in significant changes in MC gene expression, including upregulation of genes involved in anti-viral responses, leukocyte activation, and type-2 (T2) inflammation. Subsequent ex vivo modeling demonstrated that IFN-β induces MC T2 gene expression. The effects of AEC donor phenotype were small relative to the effects of viral infection and the presence of MCs.
Conclusions:
There is significant crosstalk between AECs and MCs, which are present in the epithelium in asthma. Epithelial-derived interferons not only play a role in viral suppression, but further alter MC immune responses including specific T2 genes.
Keywords: asthma, rhinovirus, mast cell, airway epithelium, T2 inflammation, interferon
Capsule Summary:
Murphy et al study the bidirectional communication between primary airway epithelial cells and mast cells in the context of viral infection, identifying that epithelial viral infection induces diverse mast cell immune responses, including type-2 inflammation.
Graphical Abstract
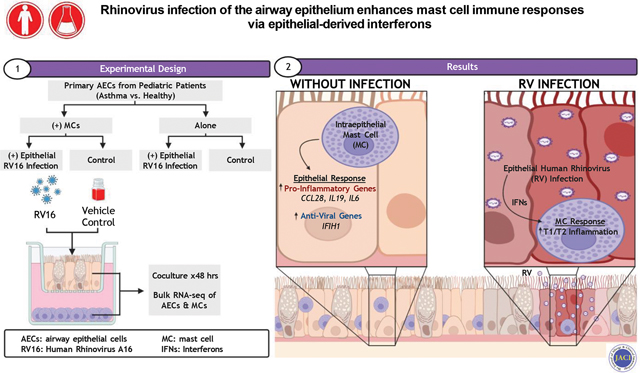
INTRODUCTION
Airway inflammation in asthma is heterogenous and regulated in a time and context dependent manner by multiple cell types and numerous external stimuli, including inhaled allergens, pollutants, and respiratory viruses (1). We recently demonstrated that mast cells (MCs) shift from the subepithelial space to the airway epithelium in individuals with asthma and their presence is tightly correlated with T2 inflammation and airway hyperresponsiveness (AHR) (2). However, MCs have been implicated in both T2 and non-T2 mechanisms of airway inflammation and there is emerging evidence that bidirectional communication between MCs and the epithelium plays a key role in regulating airway inflammation in asthma (1, 3). We developed an ex vivo model with airway epithelial cells (AECs) differentiated at air-liquid interface (ALI) cocultured with laboratory of allergic diseases-2 (LAD2) MCs and found that AECs and MCs engage in a feedforward loop in which MCs stimulated with IL-33 increase T2 gene expression in MCs and concurrently promote epithelial expression of IL33. This feedforward loop was accentuated in asthmatic AECs suggesting that there are additional alterations in the airway epithelium in asthma essential in modulating interactions between these cell types (2).
Human rhinovirus (RV) infection is a common trigger for acute exacerbations and persistence of asthma in both pediatric and adult populations (4, 5). There are several RV serogroups with RV-A serogroup 16 (RV16) being the most common cause of lower respiratory tract infections in adults and a common cause of upper and lower respiratory tract infections in children. RV16 gains cell entry by binding to intercellular adhesion molecule-1 (ICAM-1), which is expressed on multiple cell types, but typically initiates disease through infection of upper and lower AECs. Viral infection of AECs generates increased expression and production of epithelial-derived interferons (IFNs), which serve as important regulators of the host innate immune response. However, there have been conflicting results on differences in primary AEC responses from individuals with asthma with some studies demonstrating increased viral replication and reduced IFN responses (6) while others have found no significant differences (7–9). In vivo studies have demonstrated increased levels of T2 cytokines and eosinophilic inflammation following RV infection and have implicated IL-33 as a central regulator of this response (10, 11). Although CD4+ T cells and type-2 innate lymphoid cells (ILC2s) have been implicated as the key sources of T2 cytokines following RV infection, the role of MCs in regulating the T2 inflammatory response in this context has received less attention.
Here we examine communication between AECs and MCs both at baseline and in the context of acute viral infection. We utilized our coculture model system with primary AECs isolated from atopic children with well-defined asthma and from non-atopic healthy children that were differentiated at ALI, and cultured in the presence or absence of LAD2 MCs in the basolateral compartment. We then infected AECs with RV16 for 48 hours and performed bulk RNA-sequencing (RNAseq) of both AECs and MCs to further examine the effects of asthma and MCs in this system. Based on our analysis of transcriptomics data, we examined the effects of epithelial-derived IFNs on MC gene expression.
METHODS
Pediatric Study Subjects
Children ages 6–18 years undergoing an elective surgical procedure requiring endotracheal intubation and general anesthesia were recruited for this study and characterized for the presence or absence of asthma as previously described (12, 13). Parental informed consent and assent from the child were obtained prior to inclusion in the study, and the Seattle Children’s Hospital Institutional Review Board approved the study protocol.
Pediatric Primary AEC Isolation, Proliferation, and Differentiation
During the elective surgery, AECs were obtained by blind bronchial brushings through an endotracheal tube then established in primary culture and cryopreserved as previously described (12). Using passage 2 or 3 cells, AECs were differentiated to an organotypic state at ALI for 3 weeks using methods previously described (14).
Pediatric Primary AEC-LAD2 MC Coculture Model with Epithelial RV16 Infection
AEC ALI organotypic cultures were differentiated in PneumaCult™-ALI Medium (Stemcell™ Technologies) on 12 mm 0.4 μm permeable polyester membrane transwell inserts (Corning®). LAD2 MCs at a concentration of 0.5 × 106 cells/mL were added to the basolateral chamber. Either 100 μl of RV16 at a MOI of 1:1 suspended in Hank’s Balanced Salt Solution (HBSS) (100 μg/ml) or 100 μl HBSS (vehicle control) were added to the apical surface of AECs for 48 hours. Following infection of AECs with RV16 for 48 hours, RNA was extracted from the AECs and LAD2 MCs using a RNAqueous™-Micro Total RNA Isolation Kit (ThermoFisher/Life Technologies) (Supplemental Figure 1).
AEC and LAD2 RNA Sequencing
Using 1 ug total RNA with a RNA integrity number (RIN) ≥8, polyadenylated RNA was selected and purified using oligo-dT conjugated magnetic beads followed by cDNA library construction using the TruSeq Stranded mRNA Sample Prep Kit (Illumina, #RS-122-2103). Full details regarding RNAseq methods are available in the online supplement. Genes with a raw count of at least 10 in one of the libraries went into further analysis, leaving 16,950 unique genes from AECs and 15,842 unique genes from LAD2 MCs. RNAseq data have been deposited at the Gene Expression Omnibus repository (GSE206680).
Bioinformatics Analysis
Differential gene expression analysis was performed using the DESeq2 program in R (R Foundation for Statistical Computing, Vienna, Austria, www.R-project.org) (15). These analyses were performed independently to allow for direct comparisons between conditions rather than subset analyses within a larger model. The same techniques were used to determine differences based upon AEC phenotype. Details regarding individual analyses are available in the online supplement. Differentially expressed genes (DEGs) were defined as those with a false discovery rate (FDR) <0.05 using the Benjamini-Hochberg procedure. Functional associations and biological significance of DEGs were assessed using WebGestalt 2019 (WEB-based GEne SeT AnaLysis Toolkit) (16). Gene Ontology (GO) biologic process, molecular function, and cellular component, as well as canonical pathways from KEGG and Reactome were used as databases. Ingenuity Pathway Analysis (IPA, QIAGEN Redwood City, www.qiagen.com/ingenuity) was performed on differentially expressed genes with a FDR <0.05 and used to identify enriched canonical pathways, construct regulatory networks, and identify upstream regulators (17).
Measuring RV16 Replication
We measured RV16 replication in AECs and MCs using the Genesig® Human Rhinovirus Subtype 16 PCR Kit (Primerdesign®) as previously described (18).
LAD2 Mast Cell Stimulation
LAD2 MCs were stimulated with IFN-β (Abcam, Cambridge, UK) at 1 ng/mL. Blocking antibody studies were performed using an anti-IFNAR2 antibody at 5 μg/ml (clone MMHAR-2, PBL Assay Science, Piscataway, NJ) or IgG2A isotype control antibody at 5 μg/ml (clone eBM2a, eBioscience, Thermo Fisher Scientific, Waltham, MA). qPCR analysis was conducted using TaqMan primer probe sets with quantification of IL4, IL5, and IL13 genes relative to the endogenous control gene HPRT1 using the delta Ct method.
RV infection time course experiment
We performed a time course experiment to clarify the time course of changes in IL33, IFNB1, and IFNL1 following RV16 infection of human primary AECs at ALI obtained from 4 different pediatric, non-atopic, non-asthmatic donors (2). Either 100 μl of RV16 at a multiplicity of infection (MOI) of 1:1 suspended in HBSS (100 μg/ml) or 100 μl HBSS (vehicle control) were added to the apical surface of AECs. RNA was isolated at 4, 12, 24, 48, and 72 hours following RV16 infection. qPCR analysis was conducted using TaqMan primer probe sets with quantification of IL33, IFNB1, and IFNL1 genes relative to the endogenous control gene HPRT1 using the delta Ct method.
Statistics
Statistical methods are explicitly discussed within individual figure legends and detailed in the online supplement.
RESULTS
Study Population Characteristics
The characteristics of pediatric AEC donors are presented in Table 1. There were no significant differences in the age, sex, baseline FEV1 % predicted, and fractional exhaled nitric oxide (FeNO) between the groups, but atopic children with asthma had more airflow obstruction measured by the FEV1/FVC ratio, significantly higher total serum immunoglobulin E (IgE) levels and higher number of positive allergen-specific IgE concentrations relative to non-atopic healthy children.
Table 1.
Study population characteristics.
| Asthma (n = 11) | Healthy Controls (n = 10) | P value | |
|---|---|---|---|
|
| |||
| Age (years) | 10.5 (6 – 17) | 11.8 (6 – 18) | 0.50 |
| Sex (female) | 7/11 | 5/10 | 0.55 |
| FEV1 (% predicted) | 98.6 (80 – 122) | 101.1 (92 – 117) | 0.66 |
| FEV1/FVC | 0.81 (0.70 – 0.92) | 0.88 (0.81 – 0.98) | 0.02 |
| FeNO (ppb) | 23.5 (6 – 68) | 11.7 (6 – 27) | 0.10 |
| Total IgE (kIU/L) | 380.6 (1 – 1069) | 16.2 (1 – 52) | 0.01 |
| # (+) Allergen-Specific IgE Results | 2.82 (0 – 6) | 0 | 0.002 |
| Controller Treatment | 5/11 | ||
Five of the 11 children with asthma were on controller therapy: 3 children were on low dose fluticasone monotherapy, 1 child was on montelukast monotherapy, and 1 child was on low dose fluticasone and montelukast dual therapy. Mean values and ranges are listed. P values represent the result of unpaired t tests. FEV1, forced exhaled volume in one second; FVC, forced vital capacity; FeNO, fractional exhaled nitric oxide.
MCs upregulate AEC expression of pro-inflammatory genes
Overall, gene expression patterns between AECs obtained from children with asthma versus healthy children were similar when cultured in isolation with identification of only eight differentially expressed genes (Supplemental Table 1). To investigate the effect of MCs on AECs, we directly compared gene expression patterns between all AECs cultured alone or in the presence of LAD2 MCs and identified 111 differentially expressed genes. AECs cultured in the presence of MCs demonstrated increased expression of several pro-inflammatory cytokines and genes associated with anti-viral responses (Fig. 1A). These include IL19 (19–22), CCL28 (23), and IL6 (24, 25) that have been implicated in asthma pathogenesis and IFIH1, which encodes the RIG-I-like receptor and viral RNA sensor MDA5 that restricts replication of respiratory syncytial virus (RSV) and RV (26). These differentially expressed genes were enriched in GO pathways for “cytokine metabolic response”, “interleukin-6 production”, “negative regulation of cell adhesion”, “negative regulation of immune system process”, “angiogenesis”, and “epithelial cell proliferation” (Fig. 1B). We utilized IPA to identify significant upstream regulators of these differentially expressed genes and found that type I IFNs, signaling through TLR3 and TLR9, IL-17A, and TGF-β were predicted to be activated with the changes in epithelial gene expression associated with exposure to MCs (Fig. 1C). We separately compared gene expression between AECs obtained from children with asthma versus healthy controls cultured with LAD2 MCs and found only a single DEG (PIP5K1B) encoding for phosphatidylinositol-4-phosphate 5-kinase type-1β, which has previously been implicated in childhood asthma susceptibility (27).
Figure 1.
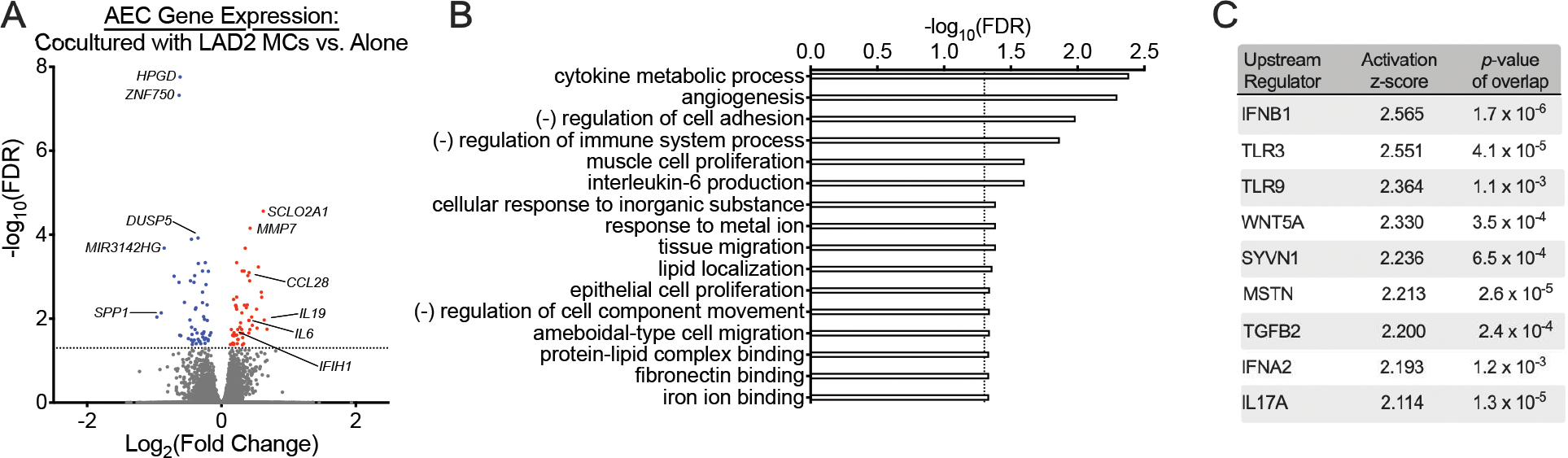
Mast cells (MCs) alter airway epithelial cell (AEC) gene expression patterns. (A) A volcano plot showing fold-change differences in gene expression between AECs cultured in the presence of LAD2 MCs versus AECs cultured alone. Red indicates genes that have significantly higher expression and blue indicates genes that have significantly lower expression in AECs exposed to MCs (FDR <0.05). (B) Gene Ontology (GO) biologic processes overrepresented amongst the 111 differentially expressed genes between AECs cultured in the presence or absence of MCs using WebGestalt 2019 (FDR <0.05). (C) Upstream regulator analysis of the 111 differentially expressed genes between AECs cultured in the presence or absence of MCs using Ingenuity Pathway Analysis.
Epithelial responses to rhinovirus infection are modestly altered by MCs
RV16 infection of AECs cultured alone resulted in 5398 differentially expressed genes (Fig. 2A) that were highly enriched in pathways involved in the anti-viral response and IFN signaling (Supplemental Table 2). There were no differentially expressed genes identified upon comparison of AECs obtained from children with asthma and healthy controls following viral infection. However, we detected non-significantly higher RV copy numbers in AECs obtained from healthy children in comparison with AECs obtained from children with asthma (P = 0.06, Supplemental Fig. 2A). Previous work from our group demonstrated that RSV infection resulted in an exaggerated IFN response in AECs obtained from asthmatic subjects with airflow obstruction (defined as FEV1/FVC <0.85 and FEV1 <100% predicted) (12). Of the 11 asthmatic AEC donors in our analysis, only 5 met these criteria for airflow obstruction. In contrast to previous results, we did not identify significant changes in gene expression patterns either at baseline or following RV infection amongst this subgroup in comparison to AECs obtained from healthy children (data not shown). However, we did identify lower RV copy numbers in AECs obtained from asthmatic subjects with airflow obstruction in comparison to AECs obtained from healthy children (P = 0.02, Supplemental Fig. 2B).
Figure 2.
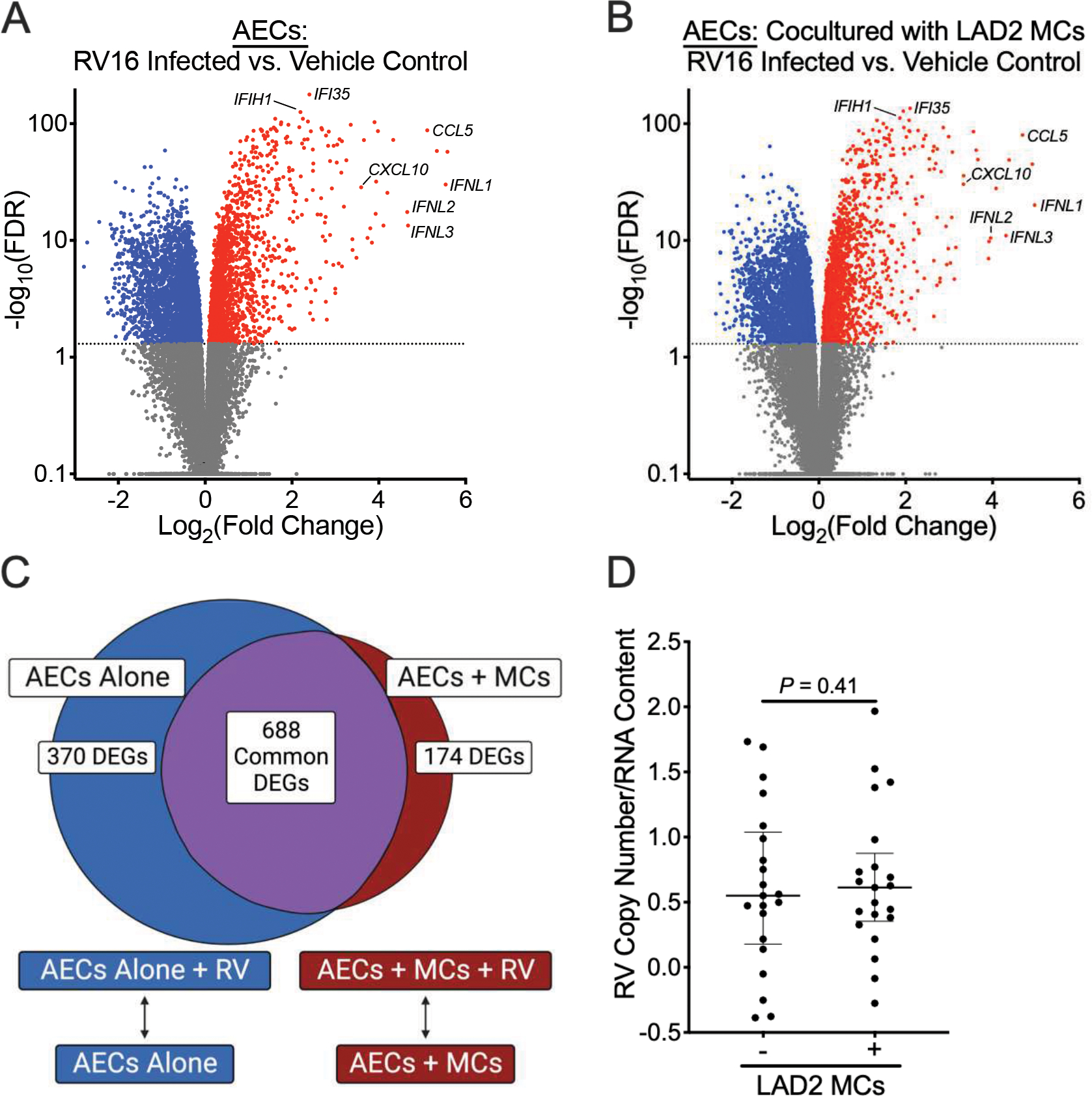
Mast cells (MCs) modestly alter airway epithelial cell (AEC) responses to human rhinovirus A16 (RV16) infection. (A, B) Volcano plots showing fold-change differences between RV16-infected AECs and AECs treated with vehicle control in the absence of LAD2 MCs (A) or presence of LAD2 MCs (B). (C) Venn diagram demonstrating overlapping and non-overlapping differentially expressed genes (FDR <0.05 and a log2 fold change ≥1.0 or ≤−1.0) between AECs at baseline and following RV16 infection in the presence or absence of MCs. (D) RV16 copy numbers relative to RNA concentration between AECs cultured in the presence or absence of MCs. RV copy numbers are log-transformed. P value represents the results from a Wilcoxon matched-pairs signed rank test.
RV16 infection of AECs cultured in the presence of LAD2 MCs resulted in 4731 differentially expressed genes and similarly demonstrated upregulation of IFNs and IFN-stimulated genes in comparison to RV16 infection of AECs cultured alone (Figure 2B). Upon comparison of gene expression patterns between AECs infected with RV16 cultured in the presence of MCs versus AECs infected with RV16 cultured alone, we identified 688 common DEGs that were enriched in genes involved in the anti-viral response and IFN signaling (Fig. 2C; Supplemental Tables 3 and 4). 174 DEGs exclusive to AECs cultured in the presence of MCs were enriched in genes involved in cellular motility and included upregulation of genes encoding the chemokine CXCL9 and the IL-36 receptor (IL1RL2), both of which have been implicated in regulating airway inflammation in asthma (28, 29). 370 DEGs exclusive to AECs cultured alone were enriched in genes involved in cell adhesion but also included downregulation of the prostaglandin E2 receptor (PTGER2) and upregulation of nitric oxide synthase 2 (NOS2). However, despite these differences in gene expression patterns, there were no significant differences in RV16 copy number in AECs following viral infection in the presence or absence of MCs (Fig. 2D).
RV16 infection of AECs alters MC gene expression patterns
To examine the effect of epithelial viral infection on MCs, we compared MC gene expression in conditions with and without RV infection of AECs. We identified differential expression of 1897 genes, which included increased expression of several IFN-stimulated genes and genes implicated in response to viral infection, in MC conditions exposed to RV16-infected AECs (Fig. 3A). These differentially expressed genes were enriched in GO biologic processes involved in response to viral infection, including responses to IFN-α, IFN-β, and IFN-γ, as well as the response to IL-4 (Fig. 3B). An analysis for upstream regulators of these differentially expressed genes identified IFN-α, IFN-β, IFN-γ, and IFN-λ amongst the upstream regulators with the highest activation z-scores, suggesting that epithelial-derived IFNs may play a key role in regulating the alterations in gene expression patterns amongst MCs cocultured with RV-infected AECs (Fig. 3C). Again, we did not identify significant differences in LAD2 MC gene expression between conditions cultured with AECs obtained from children with asthma compared with AECs obtained from healthy controls.
Figure 3.
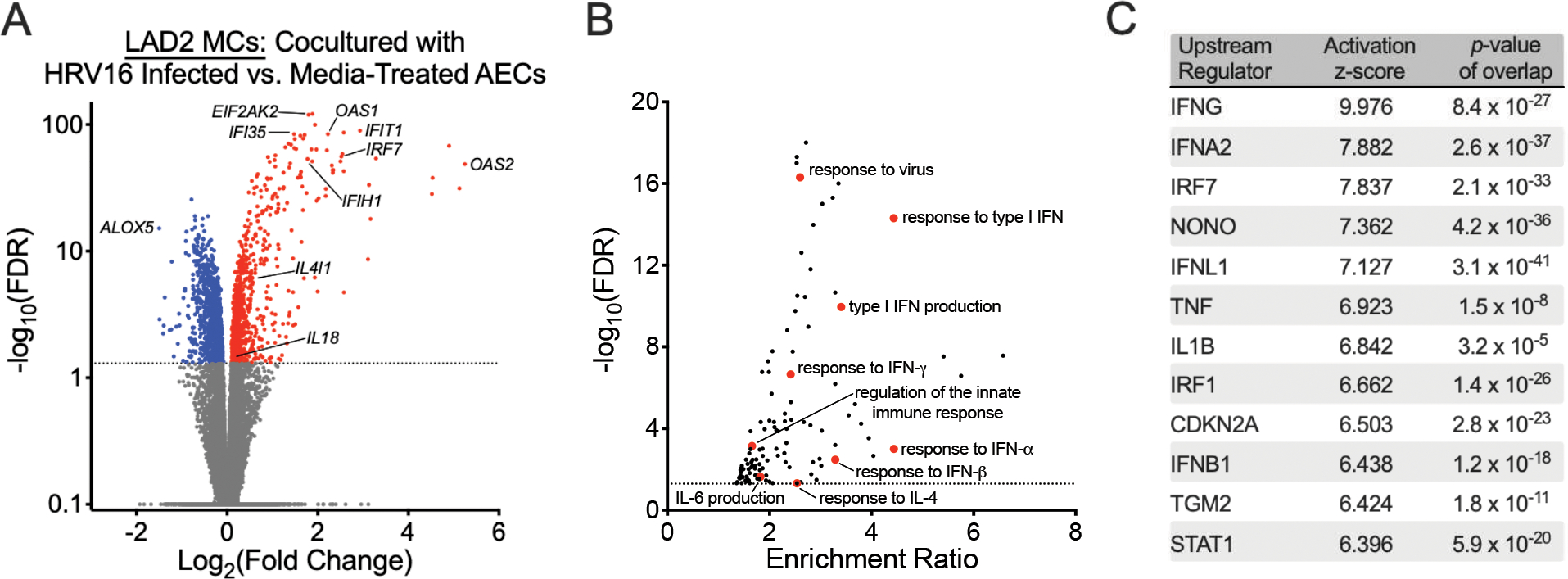
Human rhinovirus A16 (RV16) infection of airway epithelial cells (AECs) alter mast cell (MC) gene expression patterns. (A) A volcano plot showing fold-change differences in gene expression between LAD2 MCs cultured in the presence of RV16-infected AECs versus AECs treated with vehicle control identified using a paired analysis. Red indicates genes that have significantly higher expression and blue indicates genes that have significantly lower expression in the LAD2 MCs cultured in the presence of RV16-infected AEC group (FDR <0.05). (B) Gene Ontology (GO) biologic processes overrepresented amongst the 1897 differentially expressed genes between AECs cultured in the presence of RV16-infected AECs versus AECs treated with vehicle control using WebGestalt 2019 (FDR <0.05). (C) Upstream regulator analysis of the 1897 differentially expressed genes between AECs cultured in the presence of RV16-infected AECs versus AECs treated with vehicle control using Ingenuity Pathway Analysis.
Given that MCs express ICAM-1 and can be directly infected by rhinoviruses, we wanted to confirm that these alterations in gene expression were not related to direct infection with RV16 (30). We detected extremely low RV copy numbers in MCs relative to RV16-infected AECs, suggesting that changes in MC gene expression may have been related to communication with the epithelium, exposure to viral components, or low-level infection with RV (Supplemental Fig. 3). However, we did not directly expose MCs to RV16 in this study, which may have served as a superior positive control condition.
In addition to identifying genes involved in a type-1 (T1) inflammatory response, we identified increased expression of genes implicated in T2 inflammation. IL-4 inducer-1 (IL4I1) has been implicated in M2 polarization of macrophages and T2 inflammation (31). IL18 is a pleotropic pro-inflammatory cytokine that has been implicated in asthma (32–38) and is capable of inducing T2 cytokine production in basophils and MCs (39, 40).
IFN-β Induces MC T2 Gene Expression
Given that we identified type I, II, and III IFNs as upstream regulators of global transcriptional changes in MCs exposed to RV-infected AECs coupled with the known associations between RV infection and T2 inflammation, we hypothesized that epithelial-derived IFNs can directly activate MC T2 inflammatory responses without confounding by exposure to viral products or through direct RV16 infection. LAD2 MCs stimulated with IFN-β directly induced IL4, IL5, and IL13 expression in LAD2 MCs (Fig. 4A–C). The effects of IFN-β on IL4 (Fig. 4D) and IL13 expression (Fig. 4F) were completely attenuated and the effects on IL5 expression (Fig. 4E) were partially attenuated in the presence of an IFNAR blocking antibody, which serves as the common receptor for type I IFNs.
Figure 4.
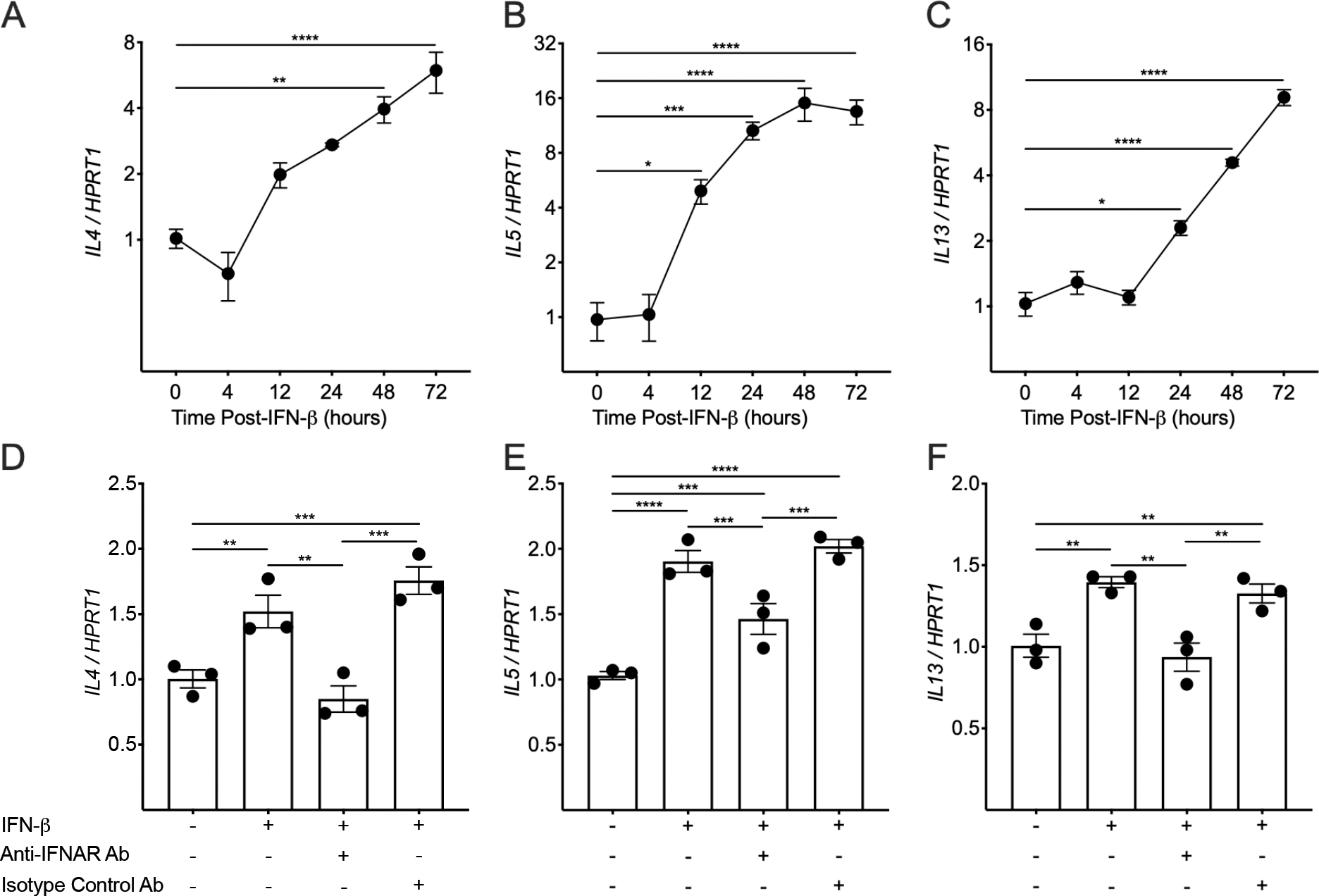
IFN-β induces type-2 gene expression in mast cells (MCs). (A-C) PCR analysis was performed on Laboratory of Allergic Diseases-2 (LAD2) MCs stimulated with IFN-β1 (1 ng/mL) for 4, 12, 24, 48, and 72 hours. IL4 (A), IL5 (B), and IL13 (C) expression levels in comparison to the housekeeping gene HPRT1 (each data point represents the mean of three PCR reactions). Mean values are shown with error bars representing the standard error of the mean. (D-F) LAD2 MCs were stimulated with IFN-β1 (1 ng/mL) for 24 hours in the presence or absence of an anti-IFNAR antibody or isotype control antibody. IL4 (D), IL5 (E), and IL13 (F) expression levels in comparison to the housekeeping gene HPRT1 (each data point represents the mean of three PCR reactions). The P values in (A-F) are the result of a one-way ANOVA with correction for multiple comparisons using the 2-stage step-up method of Benjamini, Krieger, and Yekutieli. * indicates a P values <0.05, ** indicates a P value <0.01, *** indicates a P value <0.001, and **** indicates a P value <0.0001.
RV16 Infection Induces Early Epithelial IL33 Expression
IL-33 has previously been implicated in the T2 inflammatory response following RV infection of individuals with asthma and is also known to stimulate MC T2 gene expression, prompting us to evaluate expression of IL33 and epithelial-derived IFNs at multiple timepoints following RV16 infection of primary AECs obtained from healthy children (Fig. 5). Epithelial IL33 expression peaked at 4 hours while IFNB1 and IFNL1 expression peaked at 24 hours following RV16 infection.
Figure 5.
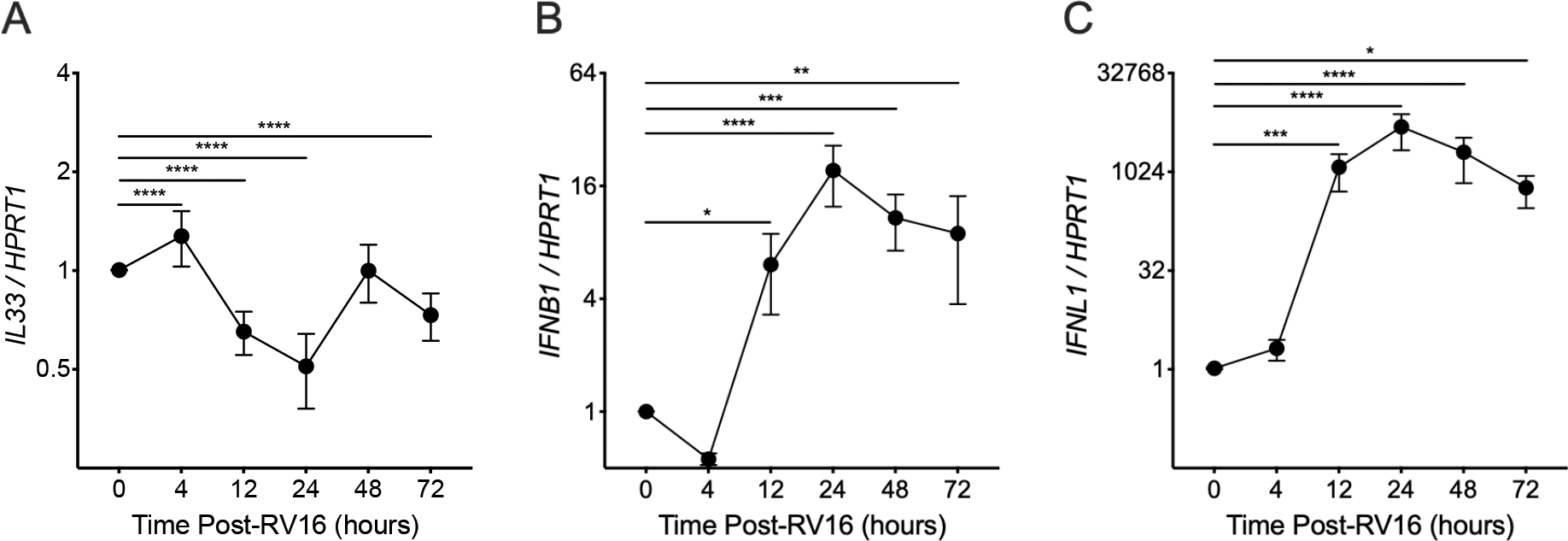
PCR analysis was performed on airway epithelial cells (AECs) obtained from non-atopic healthy children differentiated in air-liquid interface organotypic cultures and exposed to human rhinovirus A16 (RV16). IL33 (A), IFNB1 (B), and IFNL1 (C) expression levels in comparison to the housekeeping gene HPRT1 were assessed at various time points following infection (each data point represents the mean value of 4 individual donors; individual AEC donors had 3 replicates at each timepoint and each replicate was the result of three PCR reactions). Mean values are shown with error bars representing the standard error of the mean. P values represent the result of two-way ANOVA analyses. * indicates a P values <0.05, ** indicates a P value <0.01, *** indicates a P value <0.001, and **** indicates a P value <0.0001.
DISCUSSION
Here we studied the bidirectional communication between the airway epithelium and MCs, focusing on the role of this intercellular signaling in regulating airway inflammation in the context of acute viral infection (Fig. 6). We performed transcriptomic analyses of an ex vivo model using primary AECs from children with asthma and healthy controls infected with RV16 cocultured with LAD2 MCs. First, we observed that MCs promote increased expression of several pro-inflammatory genes in AECs but the magnitude of these effects were modest relative to the effects of RV16 infection on the epithelium. Secondly, we found that epithelial RV16 infection results in significant changes in MC gene expression patterns, including increased expression of genes involved in both T1 and T2 inflammation. We identified extremely low RV counts in MCs cultured with RV16-infected AECs, suggesting that exposure to viral components or low-level infection may have induced some changes in MC transcriptional patterns. However, upstream regulator analyses indicated that epithelial-derived IFNs also may have altered MC gene expression, which prompted additional ex vivo modeling that demonstrated that IFN-β can directly induce MC IL4, IL5, and IL13 expression and is mediated through the primary receptor for type I IFNs (IFNAR2). This represents a novel mechanism through which epithelial viral infection can connect T1 and T2 inflammatory pathways in asthma.
Figure 6.
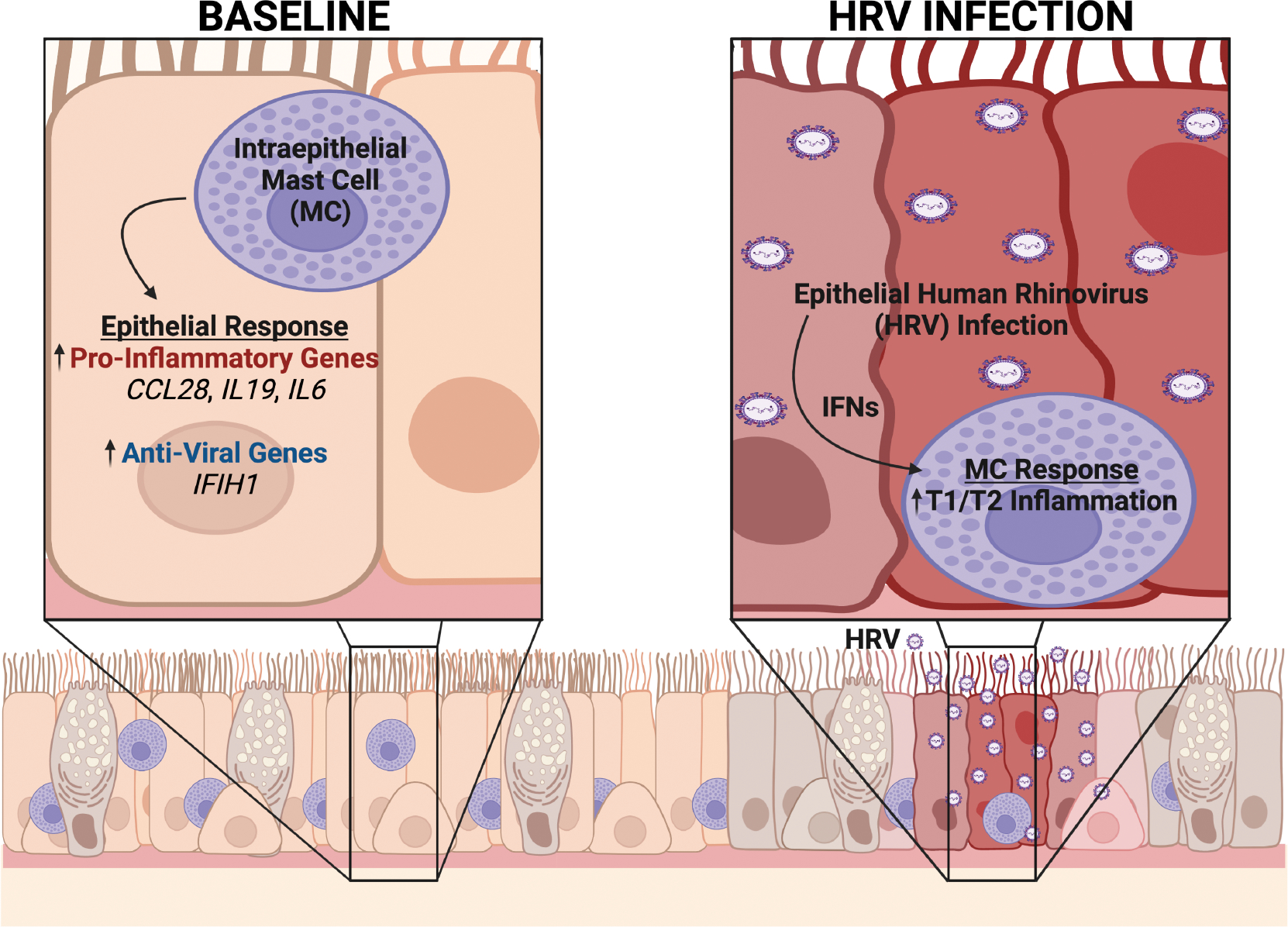
Ex vivo coculture modeling suggests bidirectional communication between airway epithelial cells (AECs) and mast cells (MCs) within the epithelial compartment in asthma. At baseline, MCs reside within the airway epithelium in asthma and our transcriptomic analyses suggest that MCs induce epithelial expression of pro-inflammatory and anti-viral genes. However, following human rhinovirus (RV) infection, epithelial-derived interferons enhance MC type-1 (T1) and type-2 (T2) immune responses. Figure created with biorender.com.
RV infection is known to induce T1 immune responses but can also lead to an enhanced T2 inflammatory response in individuals with asthma, although the key cellular sources and mechanisms responsible for generating this response are unclear (10, 11). CD4+ T cells, ILC2s, and monocytes have been implicated in the response to viral infection in asthma but here we focused on the interaction between the epithelium and MCs due to their proximity in asthma, although the role of MCs in regulating responses to viral infection has received less attention (10, 41, 42). A previous study demonstrated that RSV infection of AECs cocultured with a human mast cell line (HMC-1) promotes increased MC degranulation and multiple investigators have demonstrated that MCs are permissive to RV infection, which causes release of IL-6, IL-8, TNF-α, and IFN-β (30, 43, 44). Here we identify increased expression of genes associated with both T1 and T2 inflammation in MCs cocultured with RV16-infected AECs. One potential mechanism is through generation of IL-33, which has been identified following RV infection in clinical samples and was found by our lab to participate in a feedforward loop regulating T2 responses through MCs. Here we show that IL33 expression is transiently increased at early timepoints following RV16 infection but was not sustained at the 48-hour time point. Based on this early increase in IL33, we postulate that IL-33 may play a role in governing AEC-MC communication in the early stages following viral infection, but this was not identified in our coculture model evaluating gene expression at 48 hours post-epithelial RV16 infection and represents a limitation of performing transcriptomic analyses at a single timepoint. However, we identified epithelial-derived IFNs as important upstream regulators of global MC transcriptional changes following exposure to RV16-infected AECs, which motivated further examination of the direct effects of key epithelial-derived IFNs and revealed that IFN-β induces MC IL4, IL5, and IL13 gene expression. Previous studies have demonstrated that IFN-γ, the type II IFN primarily released from leukocytes, can induce IL-4 and IL-13 production from cord blood-derived MCs but, to our knowledge, this is the first study to directly demonstrate that an epithelial-derived IFN such as IFN-β can promote enhanced MC T2 gene expression (45). These findings suggest that increased levels of IFN-β in the epithelial compartment following epithelial viral infection can promote a T2 inflammatory response in MCs, representing a previously unrecognized mechanism through which respiratory viral infections can drive airway inflammation in a manner that is dependent upon both T1 and T2 signals and may be particularly relevant for individuals with asthma, where MCs reside in the epithelial compartment.
We also found that MCs enhance AEC expression of multiple pro-inflammatory genes and genes involved in the response to viral infection, which is consistent with a previous study demonstrating increased epithelial release of IL-6, CXCL1, and CXCL8 following coculture with HMC-1 cells (46). Here we identified multiple additional genes with increased expression including IL19, which has been implicated in asthma based upon increased expression in asthmatic AECs (19), increased expression in lung tissue as well as higher levels of IL-19 protein in the blood and saliva of individuals with asthma (20). IL19 SNPs have also been associated with recurrent wheezing in infants following RSV infection and in regulating inflammatory responses to viral infection (21, 22). We also observed upregulation of CCL28, IL6, and IFIH1 and downregulation of DUSP5 in AECs cocultured with MCs. CCL28 has been suggested as a therapeutic target in asthma as it is elevated after viral infection and blocking CCL28 in a murine model reduces AHR and mucous cell metaplasia following murine respirovirus infection (23). IL6 is a ubiquitous pro-inflammatory cytokine released following viral infections that is implicated in severe asthma and stimulates CD4+ T cells to secrete IL-4 (24, 25). IFIH1 encodes the RIG-I-like receptor and viral RNA sensor MDA5, which induces a response that restricts RSV and RV replication (26). DUSP5, a phosphatase involved in regulating MAPK signaling, has been identified as a negative regulator of IL-33-dependent function of murine eosinophils but its role in epithelial IL-33 signaling has not been examined (47). It should be noted that the changes in epithelial transcriptional patterns induced by MCs were relatively modest in comparison to the epithelial response to RV16 infection, which was notable for pronounced upregulation of IFNs and IFN-stimulated genes. In this model, the presence of MCs in the basolateral compartment did not markedly alter epithelial transcriptional programs or affect viral replication in AECs. One possible explanation is that while MCs increase expression of pro-inflammatory genes in the absence of viral infection, MCs may also serve to suppress epithelial inflammatory responses following viral infection including through release of tryptase, which was recently found to reduce type I and type III IFN expression and reduced IFN-β release following exposure to poly(I:C) (48). Overall, these results reveal the capacity of MCs to regulate epithelial transcriptional patterns in a manner that is relevant to asthma.
Finally, although we utilized primary AECs from atopic children with asthma and non-atopic healthy children, the differences in expression between the groups were minor relative to the changes in the epithelium induced by MCs and the changes in MCs induced by epithelial viral infection. We suspect that this lack of difference may be driven by heterogeneity in our epithelial donor population. Prior studies of primary AECs from individuals with asthma and healthy controls differentiated at ALI have demonstrated variable results with some studies indicating significant alterations in gene expression patterns (14, 49, 50) and others revealing overall conservation of baseline gene expression (51), potentially due to loss of some phenotype-specific characteristics in ex vivo culture conditions (52). The antiviral response appears similarly heterogeneous as initial studies of AECs in monolayer culture found compromised IFN responses in AECs from individuals with asthma following RV infection (6) but subsequent studies of both RV and RSV infection of AECs differentiated at ALI did not replicate these results (7–9, 12, 53). A recent study has also suggested that the overall IFN and anti-viral response may be conserved between asthmatic and control AECs but there is an overall delayed response in asthmatic AECs, although investigators did not identify any difference in viral copy number or viral replication (54). Here we demonstrate that AECs obtained from children with mild to moderate asthma and atopic disease differentiated at ALI have similar transcriptional responses to RV infection at 48 hours compared with AECs obtained from non-atopic healthy children. Although we did not see an enhanced IFN transcriptional response in AECs from individuals with asthma as we had identified following RSV infection in relation to altered lung function, we did find lower levels of RV replication in AECs derived from asthmatic children with reduced lung function. These results suggest that the epithelial anti-viral response from individuals with asthma may vary based upon specific endotypic differences between asthma subpopulations.
The strengths of this study include our novel experimental design, which for the first time utilizes transcriptomics to examine interactions between AECs obtained from carefully phenotyped children and MCs in the context of RV16 infection. This design allowed enough power to detect differences not only in the overall epithelial response to MC exposure and viral infection but also to detect significant differences based on AEC donor phenotype in response to these same conditions. Our bioinformatics analysis identified epithelial-derived IFNs as key upstream regulators of global MC transcriptional changes, which resulted in confirmation that IFN-β induces T2 gene expression in MCs.
However, our study also has several limitations, which include a lack of data at the protein level confirming the presence of epithelial-derived IFNs and the critical T2 cytokines in the cell culture supernatant of our experimental conditions. Additionally, further exploration of the effects of epithelial RV16 infection in our coculture model system at additional time points and in the presence of blocking antibodies targeted at epithelial-derived IFNs would have further strengthened our conclusions. We also acknowledge several limitations that reduce this study’s generalizability to the in vivo environment. First, ex vivo modeling of primary AECs cannot precisely replicate the airway microenvironment and our coculture system does not allow for the direct cell-cell contact that occurs within the epithelial compartment of asthmatic individuals with intraepithelial MCs, which may significantly alter in vivo epithelial and MC responses. Second, we purposely utilized LAD2 MCs to avoid introducing additional transcriptional heterogeneity from MCs derived from human donors to focus on differences in gene expression patterns between AECs obtained from asthmatic and non-asthmatic donors. However, LAD2 MCs may not fully replicate responses of MCs residing in the lung tissue. Third, the role of intraepithelial MCs in asthma has primarily been characterized in adults but the importance of AEC-MC communication in pediatric populations is not well understood. Thus, examination of pediatric AECs and MCs may not replicate the responses of AECs obtained from adult subjects. Also, it is important to clarify that we used RV16, an RV-A serogroup virus, in our model system and thus our results may not translate directly to other RV serogroups (RV-B or RV-C) or other common viral pathogens causing lower respiratory tract infections in children (such as RSV). Finally, although our pediatric study population was well characterized and asthmatic subjects were determined to have atopic disease, asthma remains a heterogeneous disease and the epithelial responses from our study population may not mimic the epithelial responses of AECs obtained from patients with more severe disease, higher disease activity at the time of collection, or different disease endotypes.
In summary, we utilized a coculture system to model the intraepithelial environment in asthma and assess AEC and MC responses to epithelial RV16 infection. MCs enhanced epithelial expression of pro-inflammatory and anti-viral genes but did not alter RV replication. Importantly, viral infection of the epithelium significantly upregulated MC expression of T1 and T2 inflammatory genes, which our upstream regulator analysis suggested was regulated by epithelial-derived IFNs. We subsequently confirmed that IFN-β promotes MC IL13 expression, which represents a novel mechanism by which the epithelium drives T2 airway inflammation following RV infection and may be critical to regulating airway inflammation in asthma, particularly for individuals with asthma who have intraepithelial MCs.
Supplementary Material
{kind=link}
Key Messages:
Mast cells (MCs) reside within the airway epithelial compartment in asthma where they participate in significant cross-talk with airway epithelial cells (AECs), resulting in activation of pro-inflammatory and anti-viral genes in AECs.
Human rhinovirus (RV) infection of AECs promotes dramatic changes in MC gene expression, including activation of type-1, type-2, and type-3 immune responses.
Interferon-β, an epithelial-derived type I interferon, directly induces MC type 2 gene expression.
Funding Sources:
This work was supported by NIH grants U19AI125378 (SFZ); K24AI150991 (JSD); K24AI130263, R01HL153979 (TSH); F32 HL159889 (RCM).
Abbreviations:
- MCs
mast cells
- AECs
airway epithelial cells
- AHR
airway hyperresponsiveness
- ALI
air-liquid interface
- LAD2
laboratory of allergic diseases-2
- RV16
human rhinovirus-A serogroup 16
- ICAM-1
intercellular adhesion molecule-1
- IFNs
interferons
- ILC2s
type-2 innate lymphoid cells
- RNAseq
RNA-sequencing
- FEV1
forced exhaled volume in one second
- FVC
forced vital capacity
- FeNO
fractional exhaled nitric oxide
- DEGs
differentially expressed genes
- FDR
false discovery rate
Footnotes
Conflict of Interest Disclosure Statement: Dr. Hamerman reports receiving consulting fees from aTyr Pharma. Dr. Altman reports receiving consulting fees from Regeneron. All other authors have no conflicts of interest or disclosures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Murphy RC, Pavord ID, Alam R, Altman MC. Management Strategies to Reduce Exacerbations in non-T2 Asthma. J Allergy Clin Immunol Pract. 2021;9(7):2588–97. [DOI] [PubMed] [Google Scholar]
- 2.Altman MC, Lai Y, Nolin JD, Long S, Chen CC, Piliponsky AM, et al. Airway epithelium-shifted mast cell infiltration regulates asthmatic inflammation via IL-33 signaling. J Clin Invest. 2019;129(11):4979–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murphy RC, Hallstrand TS. Exploring the origin and regulatory role of mast cells in asthma. Curr Opin Allergy Clin Immunol. 2021;21(1):71–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gavala ML, Bertics PJ, Gern JE. Rhinoviruses, allergic inflammation, and asthma. Immunol Rev. 2011;242(1):69–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kusel MM, de Klerk NH, Kebadze T, Vohma V, Holt PG, Johnston SL, et al. Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J Allergy Clin Immunol. 2007;119(5):1105–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wark PA, Johnston SL, Bucchieri F, Powell R, Puddicombe S, Laza-Stanca V, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201(6):937–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez-Souza N, Favoreto S, Wong H, Ward T, Yagi S, Schnurr D, et al. In vitro susceptibility to rhinovirus infection is greater for bronchial than for nasal airway epithelial cells in human subjects. J Allergy Clin Immunol. 2009;123(6):1384–90.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bochkov YA, Hanson KM, Keles S, Brockman-Schneider RA, Jarjour NN, Gern JE. Rhinovirus-induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal Immunol. 2010;3(1):69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sykes A, Macintyre J, Edwards MR, Del Rosario A, Haas J, Gielen V, et al. Rhinovirus-induced interferon production is not deficient in well controlled asthma. Thorax. 2014;69(3):240–6. [DOI] [PubMed] [Google Scholar]
- 10.Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo-Torralbo MB, Footitt J, et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med. 2014;190(12):1373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Message SD, Laza-Stanca V, Mallia P, Parker HL, Zhu J, Kebadze T, et al. Rhinovirus-induced lower respiratory illness is increased in asthma and related to virus load and Th1/2 cytokine and IL-10 production. Proc Natl Acad Sci U S A. 2008;105(36):13562–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altman MC, Reeves SR, Parker AR, Whalen E, Misura KM, Barrow KA, et al. Interferon response to respiratory syncytial virus by bronchial epithelium from children with asthma is inversely correlated with pulmonary function. J Allergy Clin Immunol. 2018;142(2):451–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murphy RC, Lai Y, Barrow KA, Hamerman JA, Lacy-Hulbert A, Piliponsky AM, et al. Effects of Asthma and Human Rhinovirus A16 on the Expression of SARS-CoV-2 Entry Factors in Human Airway Epithelium. Am J Respir Cell Mol Biol. 2020;63(6):859–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopez-Guisa JM, Powers C, File D, Cochrane E, Jimenez N, Debley JS. Airway epithelial cells from asthmatic children differentially express proremodeling factors. J Allergy Clin Immunol. 2012;129(4):990–7.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47(W1):W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krämer A, Green J, Pollard J, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30(4):523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanderwall ER, Barrow KA, Rich LM, Read DF, Trapnell C, Okoloko O, et al. Airway epithelial interferon response to SARS-CoV-2 is inferior to rhinovirus and heterologous rhinovirus infection suppresses SARS-CoV-2 replication. bioRxiv. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang F, Wachi S, Thai P, Loukoianov A, Tan KH, Forteza RM, et al. Potentiation of IL-19 expression in airway epithelia by IL-17A and IL-4/IL-13: important implications in asthma. J Allergy Clin Immunol. 2008;121(6):1415–21, 21.e1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saheb Sharif-Askari F, Saheb Sharif-Askari N, Goel S, Mahboub B, Ansari AW, Temsah MH, et al. Upregulation of interleukin-19 in severe asthma: a potential saliva biomarker for asthma severity. ERJ Open Res. 2021;7(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ermers MJ, Janssen R, Onland-Moret NC, Hodemaekers HM, Rovers MM, Houben ML, et al. IL10 family member genes IL19 and IL20 are associated with recurrent wheeze after respiratory syncytial virus bronchiolitis. Pediatr Res. 2011;70(5):518–23. [DOI] [PubMed] [Google Scholar]
- 22.Azuma YT, Nishiyama K. Interleukin-19 enhances cytokine production induced by lipopolysaccharide and inhibits cytokine production induced by polyI:C in BALB/c mice. J Vet Med Sci. 2020;82(7):891–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas MA, Buelow BJ, Nevins AM, Jones SE, Peterson FC, Gundry RL, et al. Structure-function analysis of CCL28 in the development of post-viral asthma. J Biol Chem. 2015;290(7):4528–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peters MC, McGrath KW, Hawkins GA, Hastie AT, Levy BD, Israel E, et al. Plasma interleukin-6 concentrations, metabolic dysfunction, and asthma severity: a cross-sectional analysis of two cohorts. Lancet Respir Med. 2016;4(7):574–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dienz O, Rincon M. The effects of IL-6 on CD4 T cell responses. Clin Immunol. 2009;130(1):27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asgari S, Schlapbach LJ, Anchisi S, Hammer C, Bartha I, Junier T, et al. Severe viral respiratory infections in children with. Proc Natl Acad Sci U S A. 2017;114(31):8342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kho AT, Sharma S, Qiu W, Gaedigk R, Klanderman B, Niu S, et al. Vitamin D related genes in lung development and asthma pathogenesis. BMC Med Genomics. 2013;6:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hasegawa T, Okazawa T, Uga H, Kurata H, Mori A. Serum CXCL9 as a potential marker of T1 inflammation in the context of eosinophilic asthma. Allergy. 2019;74(12):2515–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong H, Hao Y, Li W, Yang W, Gao P. IL-36 cytokines: their roles in asthma and potential as a therapeutic. Front Immunol. 2022. Jul 12;13:921275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akoto C, Davies DE, Swindle EJ. Mast cells are permissive for rhinovirus replication: potential implications for asthma exacerbations. Clin Exp Allergy. 2017;47(3):351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yue Y, Huang W, Liang J, Guo J, Ji J, Yao Y, et al. IL4I1 Is a Novel Regulator of M2 Macrophage Polarization That Can Inhibit T Cell Activation via L-Tryptophan and Arginine Depletion and IL-10 Production. PLoS One. 2015;10(11):e0142979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong CK, Ho CY, Ko FW, Chan CH, Ho AS, Hui DS, et al. Proinflammatory cytokines (IL-17, IL-6, IL-18 and IL-12) and Th cytokines (IFN-gamma, IL-4, IL-10 and IL-13) in patients with allergic asthma. Clin Exp Immunol. 2001;125(2):177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanaka H, Miyazaki N, Oashi K, Teramoto S, Shiratori M, Hashimoto M, et al. IL-18 might reflect disease activity in mild and moderate asthma exacerbation. J Allergy Clin Immunol. 2001;107(2):331–6. [DOI] [PubMed] [Google Scholar]
- 34.Harada M, Obara K, Hirota T, Yoshimoto T, Hitomi Y, Sakashita M, et al. A functional polymorphism in IL-18 is associated with severity of bronchial asthma. Am J Respir Crit Care Med. 2009;180(11):1048–55. [DOI] [PubMed] [Google Scholar]
- 35.Imaoka H, Gauvreau GM, Watson RM, Smith SG, Dua B, Baatjes AJ, et al. Interleukin-18 and interleukin-18 receptor-α expression in allergic asthma. Eur Respir J. 2011;38(4):981–3. [DOI] [PubMed] [Google Scholar]
- 36.Higa S, Hirano T, Mayumi M, Hiraoka M, Ohshima Y, Nambu M, et al. Association between interleukin-18 gene polymorphism 105A/C and asthma. Clin Exp Allergy. 2003;33(8):1097–102. [DOI] [PubMed] [Google Scholar]
- 37.Lee CC, Lin WY, Wan L, Tsai Y, Tsai CH, Huang CM, et al. Association of interleukin-18 gene polymorphism with asthma in Chinese patients. J Clin Lab Anal. 2008;22(1):39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng D, Hao Y, Zhou W, Ma Y. The relationship between interleukin-18 polymorphisms and allergic disease: a meta-analysis. Biomed Res Int. 2014;2014:290687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol. 2001;19:423–74. [DOI] [PubMed] [Google Scholar]
- 40.Nakanishi K Unique Action of Interleukin-18 on T Cells and Other Immune Cells. Front Immunol. 2018;9:763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jurak LM, Xi Y, Landgraf M, Carroll ML, Murray L, Upham JW. Interleukin 33 Selectively Augments Rhinovirus-Induced Type 2 Immune Responses in Asthmatic but not Healthy People. Front Immunol. 2018;9:1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marshall JS, Portales-Cervantes L, Leong E. Mast Cell Responses to Viruses and Pathogen Products. Int J Mol Sci. 2019;20(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shirato K, Taguchi F. Mast cell degranulation is induced by A549 airway epithelial cell infected with respiratory syncytial virus. Virology. 2009;386(1):88–93. [DOI] [PubMed] [Google Scholar]
- 44.Liu H, Tan J, Liu J, Feng H, Pan D. Altered mast cell activity in response to rhinovirus infection provides novel insight into asthma. J Asthma. 2020;57(5):459–67. [DOI] [PubMed] [Google Scholar]
- 45.Oldford SA, Salsman SP, Portales-Cervantes L, Alyazidi R, Anderson R, Haidl ID, et al. Interferon α2 and interferon γ induce the degranulation independent production of VEGF-A and IL-1 receptor antagonist and other mediators from human mast cells. Immun Inflamm Dis. 2018;6(1):176–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cao J, Ren G, Gong Y, Dong S, Yin Y, Zhang L. Bronchial epithelial cells release IL-6, CXCL1 and CXCL8 upon mast cell interaction. Cytokine. 2011;56(3):823–31. [DOI] [PubMed] [Google Scholar]
- 47.Holmes DA, Yeh JH, Yan D, Xu M, Chan AC. Dusp5 negatively regulates IL-33-mediated eosinophil survival and function. EMBO J. 2015;34(2):218–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramu S, Akbarshahi H, Mogren S, Berlin F, Cerps S, Menzel M, et al. Direct effects of mast cell proteases, tryptase and chymase, on bronchial epithelial integrity proteins and anti-viral responses. BMC Immunol. 2021;22(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kicic A, Sutanto EN, Stevens PT, Knight DA, Stick SM. Intrinsic biochemical and functional differences in bronchial epithelial cells of children with asthma. Am J Respir Crit Care Med. 2006;174(10):1110–8. [DOI] [PubMed] [Google Scholar]
- 50.Kicic A, Hallstrand TS, Sutanto EN, Stevens PT, Kobor MS, Taplin C, et al. Decreased fibronectin production significantly contributes to dysregulated repair of asthmatic epithelium. Am J Respir Crit Care Med. 2010;181(9):889–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reeves SR, Barrow KA, White MP, Rich LM, Naushab M, Debley JS. Stability of gene expression by primary bronchial epithelial cells over increasing passage number. BMC pulmonary medicine. 2018;18(1):91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen P, Edelman JD, Gharib SA. Comparative evaluation of miRNA expression between in vitro and in vivo airway epithelium demonstrates widespread differences. Am J Pathol. 2013;183(5):1405–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patel DA, You Y, Huang G, Byers DE, Kim HJ, Agapov E, et al. Interferon response and respiratory virus control are preserved in bronchial epithelial cells in asthma. J Allergy Clin Immunol. 2014;134(6):1402–12.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Veerati PC, Troy NM, Reid AT, Li NF, Nichol KS, Kaur P, et al. Airway Epithelial Cell Immunity Is Delayed During Rhinovirus Infection in Asthma and COPD. Front Immunol. 2020;11:974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.