

Abstract
Mounting evidence indicates that disruptions in bidirectional communication pathways between the central nervous system (CNS) and peripheral immune system underlie the etiology of pathologic pain conditions. The purpose of this review is to focus on the cross-talk between these two systems in mediating nociceptive circuitry under various conditions, including nervous system disorders. Elevated and prolonged proinflammatory signaling in the CNS is argued to play a role in psychiatric illnesses and chronic pain states. Here we review current research on the dynamic interplay between altered nociceptive mechanisms, both peripheral and central, and physiological and behavioral changes associated with CNS disorders.
Keywords: stress, pain, neuroimmunology
Introduction
In response to actual or potential tissue damage, pain has both unpleasant emotional and sensory components. It is a complex experience involving sensory-discriminative, affective-motivational, and cognitive-evaluative dimensions (Fig. 1). Notably, pain is divided into four major sub-types, each with its own sensory components and clinical features (Woolf 2020). Nociceptive pain is described as pain arising from peripheral nociceptor activation, where a nociceptor is defined as a specialized sensory receptor for painful stimuli. Subtypes of nociceptive pain include visceral pain and musculoskeletal pain, which may result in clinical syndromes such as osteoarthritis. Neuropathic pain is described as pain caused by injury or disease of the somatosensory nervous system, such as pain experienced after spinal cord injury. Inflammatory pain refers to increased sensitivity due to inflammation and associated tissue damage, including pain associated with rheumatoid arthritis (RA). Under these sensitized conditions, pain can be elicited by normally innocuous stimuli, known as allodynia, or pain can be exaggerated and prolonged in response to noxious stimuli, known as hyperalgesia. Primary hyperalgesia is a direct consequence of peripheral sensitization, which results from post-translational changes in ion channels in the peripheral terminals of primary afferents, and results in increased sensitivity to afferent nerve stimuli. Additionally, hypersensitivity can spread even further through the effects of central sensitization, which increases the responsiveness of nociceptive neurons in the central nervous system (CNS) through synaptic plasticity and associated changes in downstream central circuits. Finally, dysfunctional pain is used to describe pain states in which there is no clear evidence of active inflammation or nervous system damage. This type of chronic pain is associated with a broad range of clinical disorders, including fibromyalgia, irritable bowel syndrome, interstitial cystitis, and temporomandibular joint disease. By understanding and appreciating these various types of pain and their associated mechanisms, more effective treatment and management strategies are possible.
Figure 1.
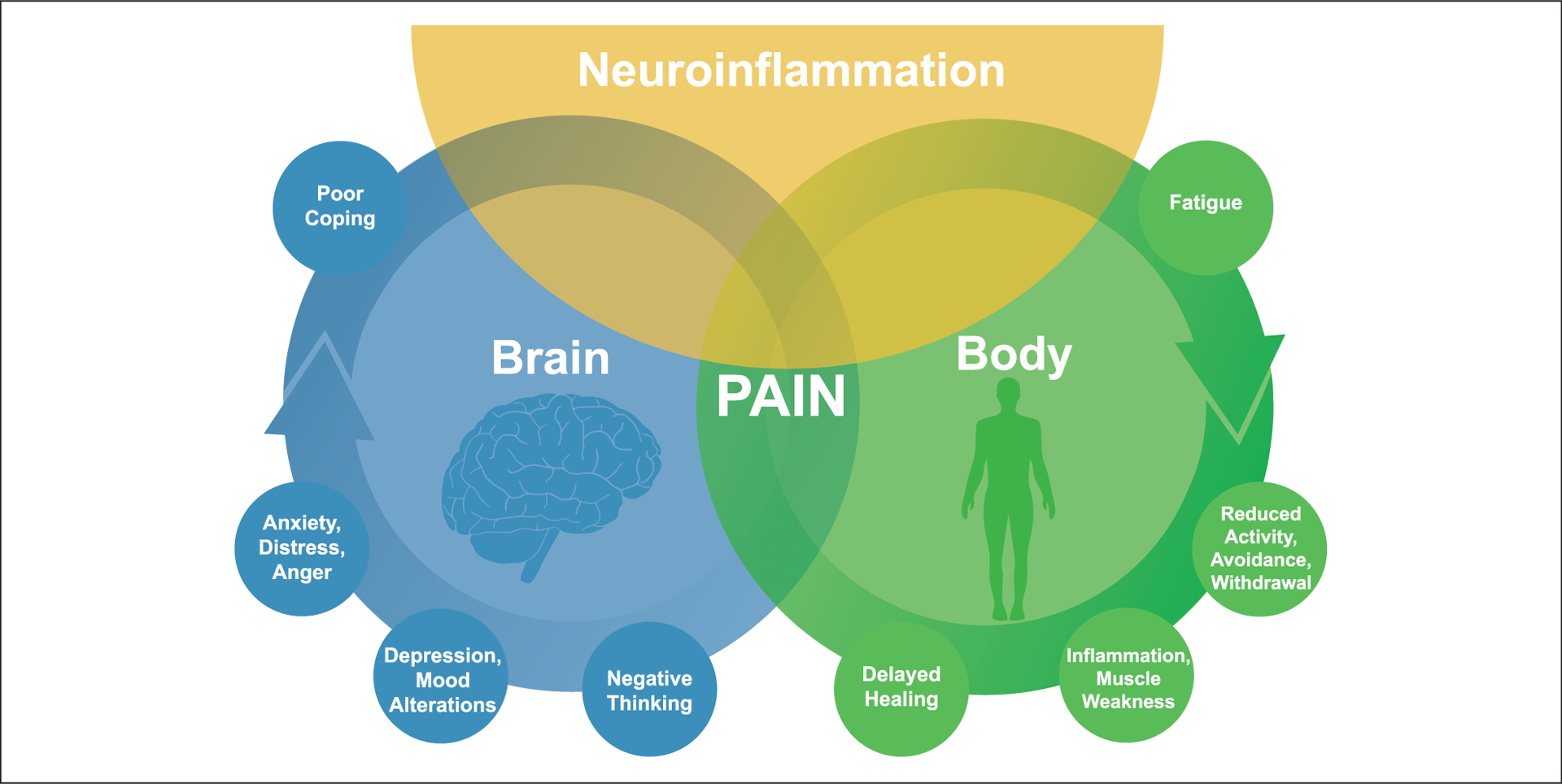
Pain is a complex experience involving sensory-discriminative, affective-motivational, and cognitive-evaluative dimensions. The experience of pain and responses to pain result from interactions of biological, psychological, and social factors. The interactions among these pathways are complex and reciprocal. Emerging evidence suggests that neuroinflammation in the peripheral and central nervous systems plays a critical role in the onset and progression of pain. Inflammation signals the brain to induce sickness responses that include increased pain and negative affect. Therefore, immune activation and inflammation play a critical role in the cross-talk between the central nervous system and peripheral immune system during states of stress, psychiatric illness, and abnormal pain conditions. Understanding neuroimmune mechanisms that underlie pain and comorbid symptoms may yield novel therapeutic strategies that integrate a collaborative and interdisciplinary approach in the treatment of chronic pain.
At the level of the brain and spinal cord, a complex network of regulatory mechanisms actively controls central immune signaling to influence pain perception. Meant to be a protective mechanism, pain processing is affected by the bidirectional communication between the CNS and peripheral immune system in response to various challenges. Emerging evidence indicates that dysregulation of this communication underlies the etiology of persistent and exaggerated pain symptoms (Hore and Denk 2019). Several chronic pain syndromes associated with neuropathic and inflammatory pain components often present with distinct neural and immune features that contribute to the pathophysiology and outcomes of these diseases (Otmishi and others 2008). For example, RA causes activation of proinflammatory signaling pathways resulting in chronic joint and systemic inflammation. Chronic inflammation alters neuropeptide processing, causing nociceptor sensitization to chemical, mechanical, and thermal stimulation. Soluble cytokines and inflammatory mediators can directly sensitize nociceptors, resulting in joint inflammation and bone destruction. In addition to these physical symptoms, RA patients are prone to developing depression (Kojima and others 2009). Several studies have supported the bidirectional association between systemic inflammation and depression involving activation of the immune-brain pathway (Hore and Denk 2019; Marchand and others 2005; Vojvodic and others 2019). Chronic pain and depression have been shown to share common mechanisms of altered neurotransmitter metabolism and neuroplastic change in the peripheral and central nervous systems (Kojima and others 2009). For example, interleukin (IL)-1β levels are elevated in the cerebrospinal fluid of patients with RA (Krock and others 2018) and depression (Miller and others 2009). Accordingly, immune modulators such as steroids and cytokine antagonists have been shown to relieve pain and depressive symptoms in patients suffering from severe RA (Otmishi and others 2008). Therefore, the emerging development of immunotherapies for various pain states with comorbid depression should consider the overlapping mechanisms between inflammation, depression, and chronic pain.
Clinically, over the past several decades, the comorbidity of pain and psychiatric diagnoses has been established (Ashkinazi and Vershinina 1999). However, animal models of pain have provided valuable information on the molecular mechanisms of comorbid anxiety, depression, and cognitive impairments that are frequently associated chronic pain conditions. For example, an inflammatory model of pain, in which complete Freund’s adjuvant is injected into the hindpaw of rodents, has been shown to promote behavioral hyperalgesia in addition to depressive-like symptoms (Ren and others 1992). Similarly, a model of neuropathic pain, induced by spared nerve injury (SNI), has been shown to induce sensory hypersensitivity, anhedonia, and behavioral despair (Wang and others 2011). Notably, psychoemotional stress has been shown to modulate nociceptive circuitry, increase susceptibility to experience pain, and exacerbate existing pain (Ashkinazi and Vershinina 1999). In support of this, repeated social defeat (RSD) is a murine model of psychosocial stress that activates brain regions associated with stress, facilitates neuroinflammatory events including increased cytokine production, and promotes the development of anxiety, social avoidance, and mechanical allodynia (McKim and others 2018; Sawicki and others 2018). Using animal models to characterize the roles of immune activation and inflammation in the bidirectional relations between the CNS and immune system during states of abnormal pain processing and CNS disorders have increased our understanding of the molecular mechanisms that underlie their often-comorbid presence.
Overall, the aim of this review is to discuss new evidence for the interactions between the nervous and immune systems in mediating physiological and behavioral changes associated with adverse pain conditions. We will describe CNS and immune system modulation of pain pathways in a variety of clinical and preclinical models of nervous system disorders. Additionally, we will describe the significant overlap between mental health and pain processing and discuss the need for a multidisciplinary approach to improve treatment of chronic pain and comorbid mental health issues. This review will shed new light on the neuroimmune interactions that may contribute to the comorbidity of pain and psychiatric diagnoses.
Disruptions in Bidirectional CNS to Immune System Communication: Psychiatric Disease, Abnormal Pain, and Stress
As introduced above, pain is a multidimensional experience that is associated with high rates of mental health disorders. Most studies investigating the causal nature of this association have pointed to a bidirectional relationship. For instance, patients may develop depression or anxiety as a result of living with chronic pain, and a history of a mental health diagnosis is considered a risk factor for developing chronic pain (Goesling and others 2018). Among patients being treated for depression, half report physical pain symptoms. Accordingly, more than half of pain patients report depression and cognitive deficits (Goesling and others 2018). Among patients with chronic back or neck pain, nearly a quarter of them had a mental disorder during the past 12 months, with major depression being the most common (Xu and others 2020). Similarly, among study participants with any mood or anxiety disorder, chronic pain disorder was the most common physical condition reported (Askari and others 2017).
Functional imaging studies have begun to outline the complexity of neurobiology that may underlie the connection between pain and mental illness. Chronic back pain is one of the most commonly reported painful conditions and thus represents a significant focus of clinical investigation. Functional magnetic resonance imaging studies have demonstrated that high and sustained levels of back pain engage brain areas involved in the emotional, cognitive, and motivational processing of pain. For instance, patients with chronic back pain exhibited evidence of functional reorganization of the primary somatosensory cortex, and that amount of reorganizational change was correlated with the chronicity of pain (Flor and others 1997). Chronic back pain was also found to be associated with reduced gray matter density in the dorsolateral prefrontal cortex and anterior thalamus (Apkarian and others 2004). Notably, increased activity of the medial prefrontal cortex has been demonstrated in patients with high levels of back pain (Baliki and others 2006) and depressive disorders (Chung and others 2018). Functional imaging studies in patients with complex regional pain syndrome, which is manifested by sensory, motor, and autonomic symptoms, revealed functional cortical changes that reversed with resolution of symptoms (Henry and others 2011). The results of these studies suggest that CNS reorganization likely contributes to abnormal pain processing associated with chronic pain conditions and implicate neural circuitry in the persistence of physical pain and negative moods.
At the molecular level, mounting evidence indicates that dysregulated and proinflammatory signaling between the CNS and immune system underlies the etiology of psychiatric disease and pathologic pain conditions. Recent clinical evidence arguing for neuroimmune interactions in chronic pain was demonstrated in patients suffering from lumbosacral radicular pain (Das and others 2018). Treatment with pulsed radiofrequency, a target selective treatment with posited immunomodulatory effects, was associated with a significant reduction in pain and functional improvement in patients with lumbosacral radicular pain, and this was also associated with altered lymphocyte populations and inflammatory cytokine levels in the cerebrospinal fluid (Das and others 2018). These results suggest a possible immune-mediated mechanism of the analgesic response, and thus immunotherapy may be a promising treatment option for chronic pain patients. Although chronic pain fundamentally results from dysfunction of the nociceptive circuitry at varying levels of the nervous system, immune cells have been shown to mediate this dysfunction (Marchand and others 2005). For instance, positron emission tomography studies in chronic pain patients showed persistent activation of microglia, the resident immune cells of the CNS, in the thalamus (Jeon and others 2017). Similarly, neuroimaging in depressed patients showed that symptom severity correlated with microglial activation within brain regions implicated in mood regulation, including the prefrontal cortex and thalamus (Setiawan and others 2015). Additionally, postmortem studies of depressed suicide victims demonstrated microglial activation and macrophage accumulation within similar brain regions (Jakobsson and others 2015). RNA sequencing on microglia isolated from resected human brain tissue revealed that microglial-specific genes overlapped significantly with genes implicated in several neurodegenerative and psychiatric disorders (Gosselin and others 2017).
In addition to these central mechanisms of action, several studies have identified elevations in circulating levels of proinflammatory cytokines in patients suffering from chronic pain and depression, including IL-1β, tumor necrosis factor (TNF)-α, and IL-6 (Hodes and others 2015). For instance, proinflammatory cytokine levels were elevated in patients suffering from conditions associated with hypersensitivity and hyperalgesia (Sommer 2001). Notably, increased cytokine levels in adolescence have been associated with increased susceptibility to develop depression in adulthood. For instance, children with higher levels of IL-6 at age 9 years were at greater risk of developing major depressive disorder by age 18 years compared to the general population with low levels of IL-6 (Khandaker and others 2014). Additionally, studies have shown that the incidence of depression increases with age and is frequently associated with chronic pain (Zis and others 2017). Based on the extent of evidence for the involvement of cytokines in the development of depression, clinical investigations have focused on the use of cytokine antagonists as a means to improve depressive symptoms. For instance, clinical trials are currently underway for the use of TNF antagonists like adalimumab, etanercept, and infliximab for the treatment of depressive episodes of bipolar disorder (Vojvodic and others 2019). Emerging evidence indicates that inflammatory markers are elevated not only in patients with depression but also in patients with other CNS disorders, including anxiety. For example, elevated levels of circulating C-reactive protein, IL-6, and TNF-α have been identified in patients with anxiety disorders (Costello and others 2019). In support of these clinical findings, repeated social defeat stress in mice increased IL-1β, TNF-α, and IL-6 levels in both the periphery and CNS, which corresponded with the development of anxiety, social avoidance, and mechanical allodynia (McKim and others 2018; Sawicki and others 2019). Similarly, in a mouse model of neuropathic pain, social isolation increased depressive-like behavior and mechanical allodynia in mice exposed to SNI that corresponded with increased IL-1β expression in the prefrontal cortex (Norman and others 2010). These studies suggest that peripheral and central inflammation are critical contributing factors to the comorbidity of pain and CNS disorders.
Stress can provoke and escalate the proinflammatory CNS signaling and immune dysregulation implicated in the etiology of mood disorders (Hodes and others 2015) and hypersensitivity to noxious stimuli (Marchand and others 2005). Exposure to chronic environmental, psychological, or social stressors increases the risk of developing several types of psychiatric disorders (Gilman and others 2013). Chronic exposure to adverse social conditions promotes a “transcriptional fingerprint” on peripheral monocytes that is characterized by increased proinflammatory gene expression (Powell and others 2013). Repeated or chronic stressors activate bidirectional communication pathways between the CNS and peripheral immune system, converging to promote a heightened neuroinflammatory environment (Fig. 2). It is important to note that neural activation of brain regions implicated in stress responses demonstrates stressor specificity, and stressors are interpreted in the brain within specific stress-responsive neurocircuitry (Wohleb and others 2014). Activation of this neurocircuitry stimulates sympathetic nervous system (SNS) and hypothalamic-pituitary-adrenal axis (HPA) activity, thus allowing the CNS to effectively communicate with the immune system (Wohleb and others 2014). For instance, exposure to repeated social defeat stress promotes SNS-dependent monocyte trafficking from the bone marrow (BM) to the brain, leading to dynamic interactions between BM-derived monocytes, endothelial cells, and resident microglia (Weber and others 2017). Notably, stress can prime the neuroinflammatory response to subsequent inflammatory challenges, resulting in chronic neuroinflammation and immune dysregulation, which has been argued to facilitate the etiology of mood disorders (Hodes and others 2015) and hypersensitivity (Marchand and others 2005). Overall, psychological stress initiates a cascade of neuroimmune responses involving brain-to-immune and immune-to-brain signaling that converge to influence mood and behavior.
Figure 2.
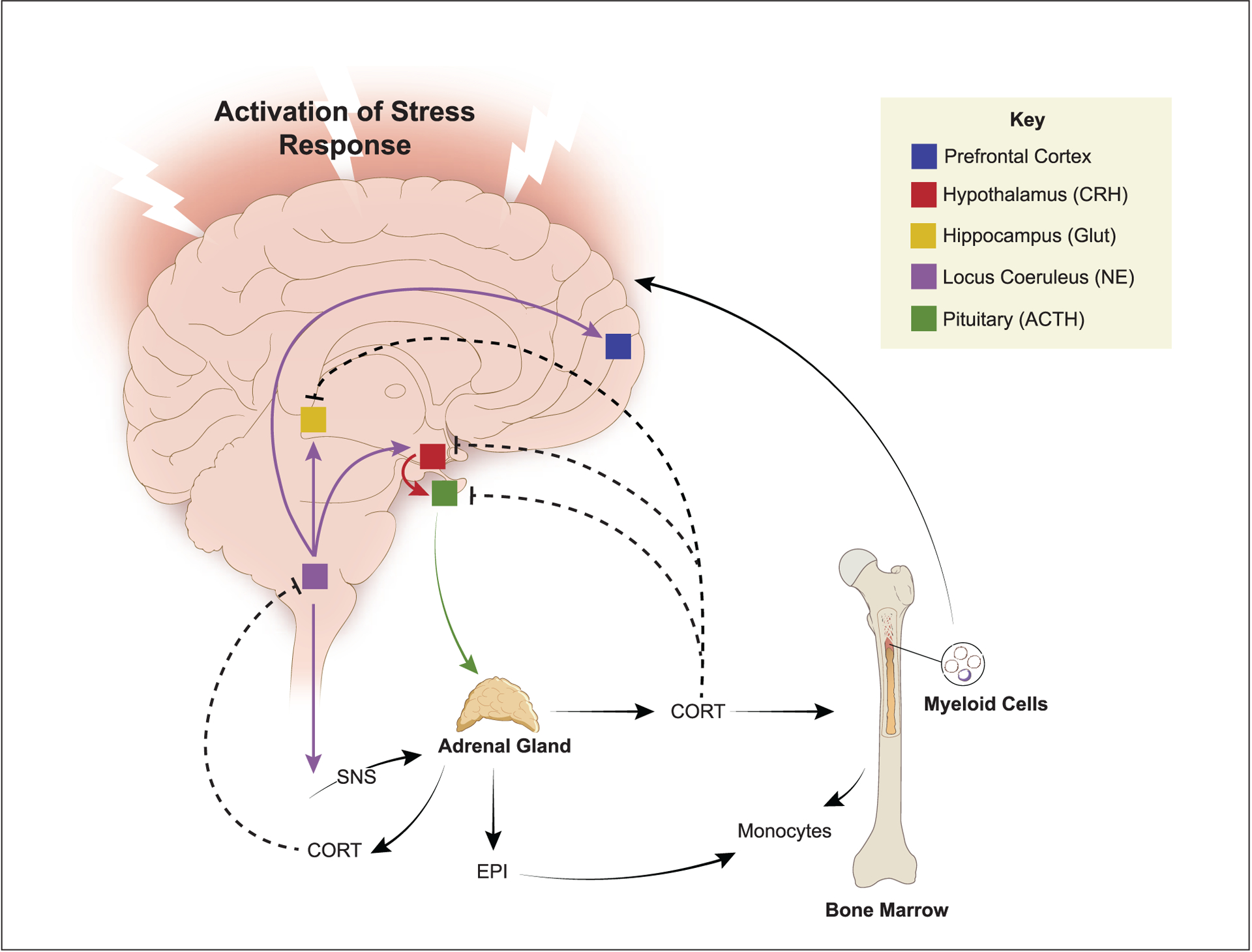
Interpretation of psychological stress in the brain activates neuroendocrine pathways that signal into the periphery, including the hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic nervous system (SNS). HPA activation leads to the release of circulating corticosterone/cortisol (CORT) that promotes trafficking of monocytes from the bone marrow, which feedback to the brain to influence behavior. Activation of the SNS leads to increased release of epinephrine (EPI) in circulation, which primes the monocytic response to future activation. CORT provides negative feedback at multiple levels of HPA regulation and inhibits central noradrenergic release (dashed lines indicate negative feedback). Another critical element of HPA and SNS activation is that these signals are relayed to the immune system. Therefore, a major component of the stress response involves relaying information from the brain to peripheral organs and the immune system via HPA and SNS neuroendocrine pathways.
The Neuroimmune Interface Modulates Nociceptive Pathways and Stress-Related Behaviors
As illustrated in Figure 3, the experience of pain depends on efficient transmission of nociceptive information from peripheral nociceptor neurons to second order interneurons in the spinal cord, and then onto supraspinal structures (Grace and others 2014). First-order primary afferent neurons transmit nociceptive signals from a peripheral stimulus site to the CNS, which is then transmitted at central synapses via release of neurotransmitters involving glutamate and neuropeptides. These neurons synapse with second-order nociceptive projection neurons in the spinal dorsal horn, which then project to supraspinal sites, including cortical and subcortical regions via third-order neurons. Third-order neurons project to the somatosensory cortex and allow for the perception of pain. Activation of descending serotonergic and noradrenergic projections to the spinal cord can additionally regulate the action of second-order nociceptive projection neurons, thus influencing the response and perception of a painful experience. The descending pain modulating system can inhibit or facilitate peripheral nociceptive input. This system receives input from the prefrontal and cingulate cortices, anterior insula, amygdala, and hypothalamus, thus allowing differential processing of nociceptive information during affective or cognitive processes. Notably, sustained activation of descending modulatory pathways that facilitate pain transmission is argued to play a role in the pathogenesis of chronic pain states. Despite the significance of neuronal pain facilitation mechanisms, growing evidence suggests a critical role for central immune signaling in the pathogenesis of abnormal pain processing (Grace and others 2014).
Figure 3.
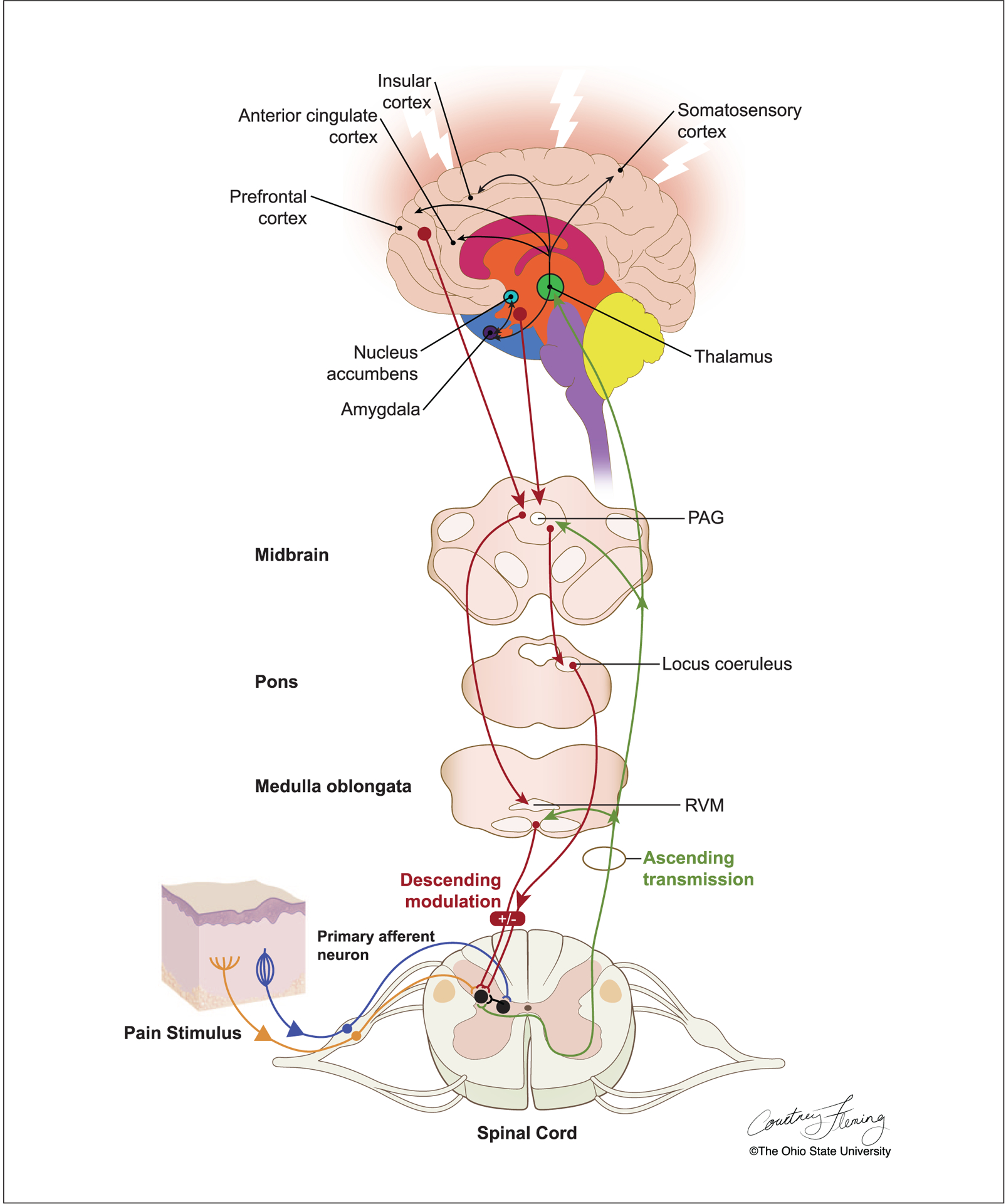
The experience of pain depends on efficient transmission of nociceptive information from peripheral nociceptor neurons to second order interneurons in the spinal cord, and then onto supraspinal structures. First-order primary afferent neurons transmit nociceptive signals from a peripheral stimulus site to the spinal cord via the dorsal root ganglion and synapse with second-order nociceptive projection neurons in the spinal dorsal horn. Secondary order projection neurons ascend in the contralateral spinothalamic and spinoreticular tracts that relay the signal to cortical centers. Descending pathways projecting from the periaqueductal gray (PAG) in the midbrain and the rostral ventromedial medulla (RVM) to the dorsal horn influence pain transmission.
Microglia act as first responders to a host of neuronally derived mediators, and on detection of these signals, transition into a state of reactive gliosis. In their reactive state, microglia release proinflammatory cytokines, including IL-1β, IL-6, and TNF-α. Mounting evidence indicates that these cytokines are critical modulators of nociceptor activity and pain sensitization (Zhang and An 2007). For example, proinflammatory cytokines have been shown to directly activate and sensitize nociceptors, as well as modulate excitatory synaptic transmission at central terminals (Zhou and others 2016). Consistent with their pronociceptive roles, IL-1β produced hyperalgesia following intraperitoneal, intracerebroventricular, or intraplantar injection (Watkins and others 1994), and TNF-α produced mechanical and thermal hyperalgesia following intraplantar injection (Perkins and Kelly 1994). Notably, administration of antagonists to IL-1β (Sweitzer and others 2001) or TNF-α (Schäfers and others 2003) was effective in preventing inflammatory hyperalgesia and nerve-injury induced mechanical allodynia. Additionally, intrathecal infusion of IL-6 induced tactile allodynia and thermal hyperalgesia, and administration of anti-IL-6 neutralizing antibody alleviated these pain behaviors (Zhou and others 2016). Numerous animal models of nerve injury have demonstrated that administration of minocycline, a known inhibitor of microglial activation, decreased inflammatory cytokine expression and reduced pain behaviors (Barcelon and others 2019; Hains and Waxman 2006). Notably, clinical studies have provided strong evidence for the role of cytokines in modulating human chronic pain conditions. For example, postmortem studies of individuals who reported suffering from chronic pain showed elevated levels of TNF-α and IL-1β in the dorsal spinal cord (Shi and others 2012). Additionally, elevated levels of IL-1β levels have been detected in the cerebrospinal fluid of patients with complex regional pain syndrome (Alexander and others 2005). Moreover, patients treated with inhibitors of IL-6 demonstrated reduced pain and improved mood symptoms (Choy and Calabrese 2018). Several studies indicate that cytokines contribute to the facilitatory descending pain pathway by inducing NMDA (N-methyl-d-aspartate) receptor phosphorylation, thus influencing transmission of glutamate. In support of this, antagonist to IL-1 receptor and TNF-blocking antibody decreased enhanced NMDA receptor phosphorylation in the rostral ventromedial medulla (RVM), a major component of brainstem descending pain modulatory circuitry, and also prevented the development of behavioral hypersensitivity (Wei and others 2008). Additionally, administration of an NMDA receptor antagonist blocked the development of allodynia produced by intra-RVM administration of TNF-α and IL-1β (Wei and others 2008). Therefore, cytokines and central glial-neuronal interactions likely contribute to descending pain facilitation.
Microglia have also been shown to enhance and transform the output of pain transmission neurons (Trang and others 2012). A critical modulator of microglial activity is adenosine triphosphate (ATP), an endogenous ligand of the P2-purinoceptor family consisting of P2X ionotropic and P2Y metabotropic receptors. Microglia express numerous subtypes of P2 receptor, and several have been implicated in the pathogenesis of neuropathic pain, including P2X4, P2X7, and P2Y12. Studies have demonstrated that ATP-stimulated microglia can change the output of ascending nociceptive pathways arising from neurons in lamina I of the spinal dorsal horn. The change in output of the lamina I neurons contributes to clinical symptoms of pain, including mechanical allodynia, hyperalgesia, and spontaneous pain (Trang and others 2012; Woolf and Salter 2000). Therefore, microglia-neuron communication is bidirectional and posits ATP as a key molecular substrate allowing these cell types to interact.
Chemokines are other important messenger molecules between cells of the immune system. Released locally from peripheral blood monocytes at sites of inflammation, chemokines play a key role in the inflammatory response by recruiting additional leukocytes to sites of damage (Abbadie and others 2009). Newly differentiated monocytes express high levels of monocyte marker Ly6C (Ly6Chi). Ly6Chi monocytes have a high capacity for proinflammatory processes and express a chemokine receptor profile that allows for trafficking to inflamed tissue. Ly6Chi monocytes are positive for C-C chemokine receptor type 2 (CCR2) that detects chemotactic cytokine ligand 2 (CCL2) to influence cell trafficking. As they shift toward an anti-inflammatory phenotype characterized by increased expression of fractalkine receptor (CX3CR1), monocytes will reduce Ly6C and CCR2 expression (Auffray and others 2007). Similar to cytokines, chemokines are also involved in neuroimmune modulation of pain processing and mood regulation. Perhaps the most well-recognized signaling pathways between sensory neurons and microglia include CCL2/CCR2 and CX3CL1/CX3CR1 (Auffray and others 2007). Notably, CX3CR1 is constitutively expressed in the RVM, which can stimulate activation of microglial cells to produce cytokines and thus modulate the facilitatory descending pain pathway (Zhang and An 2007). The importance of CCL2/CCR2 and CX3CL1/CX3CR1 signaling pathways in peripheral nerve and neuronal hyper-excitability have been well characterized in the context of peripheral nerve injury (Grace and others 2014). However, emerging evidence posits a critical role for CCL2/CCR2 signaling in the maintenance of chronic pain states in the absence of injury. For instance, administration of a CCR2 antagonist in a preclinical model of chronic pain provided analgesia in mice (Rosen and others 2018). Furthermore, mice lacking CCL2 or CCR2 failed to develop pain-related behavior in a model of inflammatory pain (Miotla Zarebska and others 2017). Notably, the repeated social defeat model of stress has provided several lines of evidence for the importance of CCL2/CCR2 and CX3CL1/CX3CR1 signaling pathways in facilitating pain and mood dysregulation in the absence of injury. For example, genetic knockout of CCR2 or CX3CR1 prevented monocyte recruitment to the brain and blocked the development of anxiety-like behavior (Wohleb and others 2013). It is important to note that stress still caused the release of Ly6Chi monocytes into circulation independent of CCR2 or CX3CR1, thus indicating that the release of monocytes into circulation is not sufficient for monocytes to traffic to the brain. Rather, monocyte recruitment to the brain requires signaling from the CNS. Therefore, it is likely that circulating peripheral monocytes are actively recruited to the brain through CNS communication, rather than passive diffusion. Interestingly, mechanical allodynia during repeated social defeat stress occurred independent of peripheral monocyte recruitment to the spinal cord. Despite this observation, stress-induced mechanical allodynia corresponded with enhanced CCL2 and CCR2 gene expression in the spinal cord (Sawicki and others 2019). CCR2 is expressed by sensory neurons and, when ligated by CCL2, can directly excite nociceptive neurons to promote pain behavior (Miotla Zarebska and others 2017). Therefore, it is plausible that CCL2/CCR2 signaling pathways influence stress-induced pain behaviors independent of monocyte recruitment. The role of CCL2/CCR2 signaling in chronic pain conditions is further supported by clinical studies. For example, the CCR2 antagonist AZD2423 (AstraZeneca) showed trends toward reduced paroxysmal pain, paresthesia, and dysesthesia, providing clinical evidence for an important analgesic effect of CCR2 antagonists (Kalliomäki and others 2013). Emerging evidence posits a role for C-X-C motif chemokine ligand 1 (CXCL1)/C-X-C motif chemokine receptor 2 (CXCR2) signaling in the periaqueductal gray in descending pain facilitation (Ni and others 2019). CXCL1 has been shown to play a critical role in the development and maintenance of inflammatory and neuropathic pain through its preferred receptor, CXCR2. The ventrolateral periaqueductal gray (vlPAG) is a critical component of the descending pain modulatory network and exerts excitatory or inhibitory control on pain transmission through the RVM, which in turn projects to the spinal dorsal horn. Most recently, it was demonstrated that micro-administration of CXCL1 neutralizing antibody attenuated mechanical allodynia in rats with established bone cancer pain, and vlPAG application of CXCL1 induced pain hypersensitivity. Notably, a CXCR2 antagonist blocked CXCL1-induced mechanical allodynia and reduced pain hypersensitivity associated with bone cancer (Ni and others 2019). Therefore, the CXCL1-CXCR2 signaling cascade may have a critical role in glial-neuron interactions and in descending facilitation of pain.
Toll-like receptors (TLRs) are a family of pathogen recognition receptors that play a significant role in the communication between neurons and immunocompetent cells within the CNS (Austin and Moalem-Taylor 2010). TLRs are responsible for sensing damage or danger signals, and subsequently translating this into a central immune signal that can be interpreted and responded to by neurons and other immunocompetent cells within the CNS. Once activated, TLRs mediate proinflammatory cytokine production, leading to enhanced central and peripheral immune signaling and subsequent exaggerated pain transmission (Ji 2015). Several preclinical pain models have demonstrated a role for CNS TLR involvement in activation of pain. For example, the development and maintenance of nerve injury–induced allodynia was blocked in animals deficient in TLR2 signaling (Kim and others 2007). Following peripheral nerve injury, mice genetically deficient in functional TLR4 exhibited attenuated behavioral hypersensitivity and decreased expression of spinal microglial markers and proinflammatory cytokines (Tanga and others 2005). Furthermore, pharmacological blockade of TLR4 has been shown to prevent (Bettoni and others 2008) and rapidly reverse preclinical models of neuropathic pain (Hutchinson and others 2010). Notably, emerging clinical data have provided support for the role of TLR4 in human pain states. For example, patients suffering from discogenic pain exhibited increased TLR4 expression that was dependent on the degree of intervertebral disk degeneration (Klawitter and others 2014). Interestingly, both preclinical and clinical data suggest that TLR-related mechanisms can mediate stress-induced adaptations involved in the development of mental health disorders including major depressive disorder (Liu and others 2014). In support of this, deletion of TLR2 and TLR4 mitigated microglial activation and altered neuronal activation associated with repeated social defeat stress, which corresponded with reduced social avoidance and anxiety behaviors (Nie and others 2018). Furthermore, the development of mechanical allodynia during repeated social defeat stress corresponded to increased TLR4 gene expression in the spinal cord (Sawicki and others 2019). It is important to note, however, that these TLR-mediated events occurred in the absence of injury; therefore, TLR2/4 signaling pathways are critical to the central immune signaling process in the maintenance of homeostasis within both the peripheral and central nervous systems. Understanding the role of TLR-mediated neuroinflammatory signaling holds considerable promise for the development of novel therapies for the management of patients suffering from comorbid pain and psychiatric disorders.
Though neuroimmune mechanisms are a common element, the precise relationship between stress, behavior, and pain perception is not clear. As illustrated in Figure 4, pain processing is mediated by the function of several intracellular and extracellular molecular messengers involved in signal transduction in the peripheral and central nervous systems. Following a neuronal insult, first-order neurons initiate alterations in neuronal and biochemical processing at central synapses and descending projections, which manifests as loss of endogenous inhibitory control or enhancement of pain facilitation at these sites (Grace and others 2014). In the spinal dorsal horn, these effects can be manifested through the phosphorylation of various receptors, including NMDA and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors, thus increasing synaptic efficacy. The result of these processes is seen in many clinical syndromes of pain, in which pain is no longer a protective mechanism but rather arises spontaneously, such as in cases of allodynia and hyperalgesia.
Figure 4.
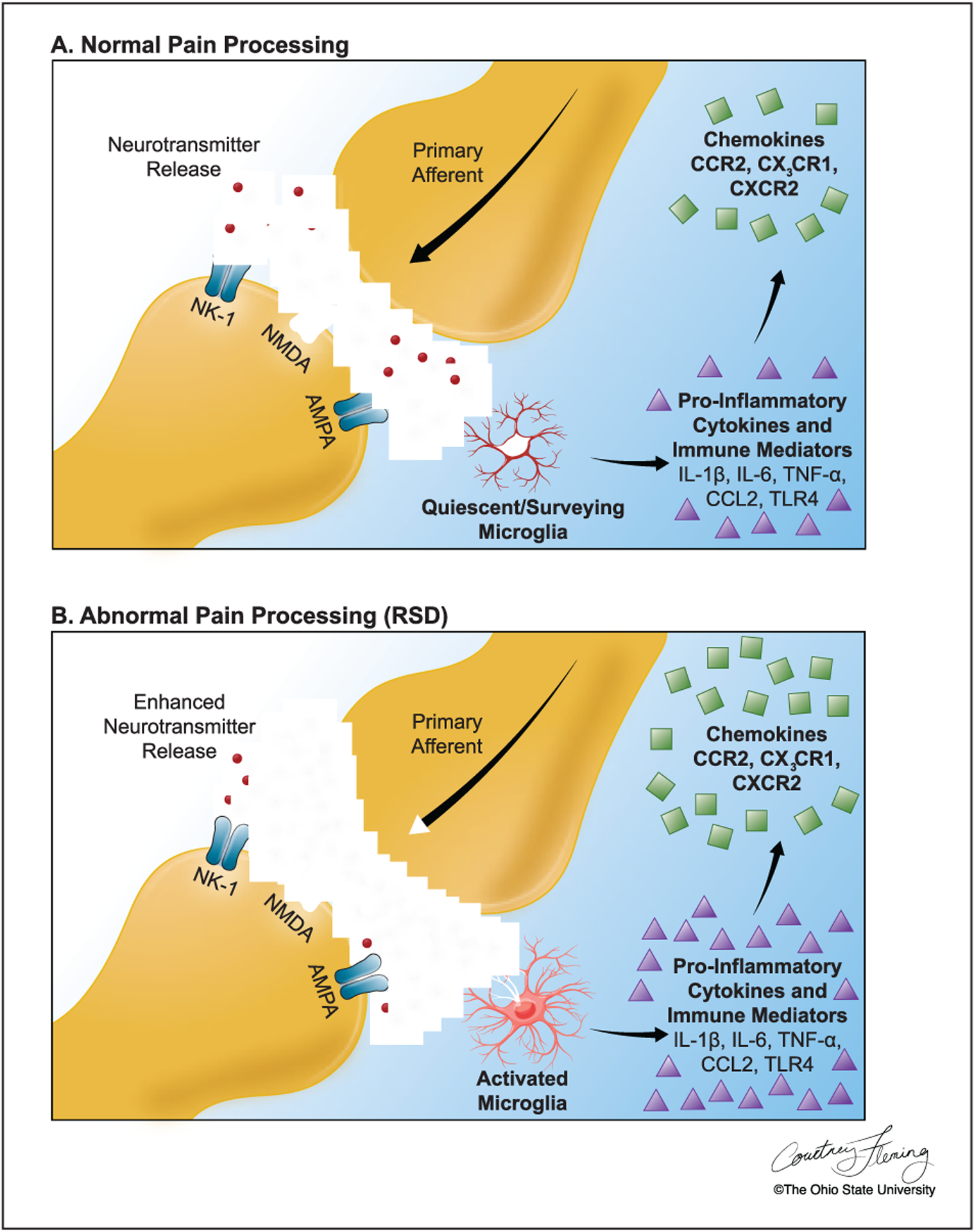
(A) In the dorsal horn, incoming afferent pain signals cause the release of neurotransmitters that bind to and activate postsynaptic receptors on pain transmission neurons. Microglia are present but quiescent, actively surveying the neuronal environment and responding to minute homeostatic disturbances. As a protective mechanism, microglia produce proinflammatory cytokines and chemokines to support endangered neurons. (B) With repeated social defeat (RSD) stress, incoming afferent signals are increased, and presynaptic release of neurotransmitters is enhanced. Microglia become further activated and increase production of proinflammatory cytokines/immune mediators and chemokines that further increase presynaptic release and postsynaptic hyperexcitability.
By stimulating stress-reactive neurocircuitry, repeated social defeat stress promotes development of anxiety, social avoidance, and mechanical allodynia, modeling the coexistence of inflammation, psychiatric conditions, and altered pain processing observed in patients following stress. Repeated social stress induced activation of microglia specifically within the dorsal horn of the spinal cord (Sawicki and others 2019), the first relay for pain transmission in the CNS. Stress-induced microglial activation corresponded with the release of inflammatory and immune mediators, converging to promote a heightened inflammatory environment within the spinal cord (Sawicki and others 2019). Similarly, restraint stress potentiated nerve injury-induced tactile allodynia that corresponded with activation of dorsal horn microglia (Alexander and others 2009). Additionally, a critical role for spinal microglial activation in the development of visceral hyperalgesia was demonstrated in a model of chronic psychological stress in rats (Bradesi and others 2009). Thus, microglial activation and the release of proinflammatory markers in the spinal cord likely contribute to abnormal pain processing associated with psychological stress (Fig. 4). These results suggest that microglial activation during stress may induce a reorganization of the circuitry within the spinal cord dorsal horn that mediates the development of mechanical allodynia. In support of this, several studies demonstrate that functional plasticity changes are accompanied by structural remodeling and reorganization of circuits that may contribute to chronic pain conditions (Kuner and Flor 2017).
Evidence using the repeated social defeat model of stress has begun to reveal a mechanism by which microglia promote the development of allodynia during stress in the absence of injury (Sawicki and others 2019). To determine the role of microglia in abnormal pain processing during RSD, microglia were depleted throughout the spinal cord using colony-stimulating factor 1 receptor (CSF1R) antagonist PLX5622 (Fig. 5). Following 14 days of treatment with PLX5622, microglia were eliminated from the spinal cord based on Iba-1 labeling for microglial activation (Fig. 5A and B). Additionally, the gene expression of microglia-related CX3CR1was reduced by PLX5622 in the cervical, thoracic, and lumbar spinal cord regardless of stress exposure (Fig. 5C–E). Notably, depletion of microglia throughout the CNS prevented the development of mechanical allodynia during RSD and reduced the expression of critical inflammatory markers involved in nociceptive signaling (Fig. 6). Following treatment with PLX5622, mice were either exposed to stress or left undisturbed as controls. Mechanical allodynia was determined by the von Frey behavior test at baseline and 12 hours after the first, third, and final day of RSD. Prior to stress exposure, all four treatment groups exhibited comparable baseline measurements of allodynia. Notably, depletion of microglia with PLX5622 prevented RSD-induced allodynia after three and six days of stress (Fig. 6A). Similarly, in an animal model of neuropathic pain, significant alleviation of both mechanical and cold allodynia was demonstrated in mice treated with PLX5622 (Lee and others 2018). Microglial activation is associated with the release of proinflammatory markers in the spinal cord that may contribute to exaggerated pain states associated with psychological stress and nerve injury. In support of this, PLX5622 treatment reduced IL-1β, TLR4, and CCR2 levels in the spinal cord after exposure to repeated social defeat stress (Fig. 6B–D). Similarly, blocking spinal microglia with PLX5622 reduced IL-1β and TNF-α expression following partial sciatic nerve ligation (Lee and others 2018). Together, these results suggest that disruption in microglial functioning likely influences the neurocircuitry that underlies the development of pain associated with stress and injury.
Figure 5.
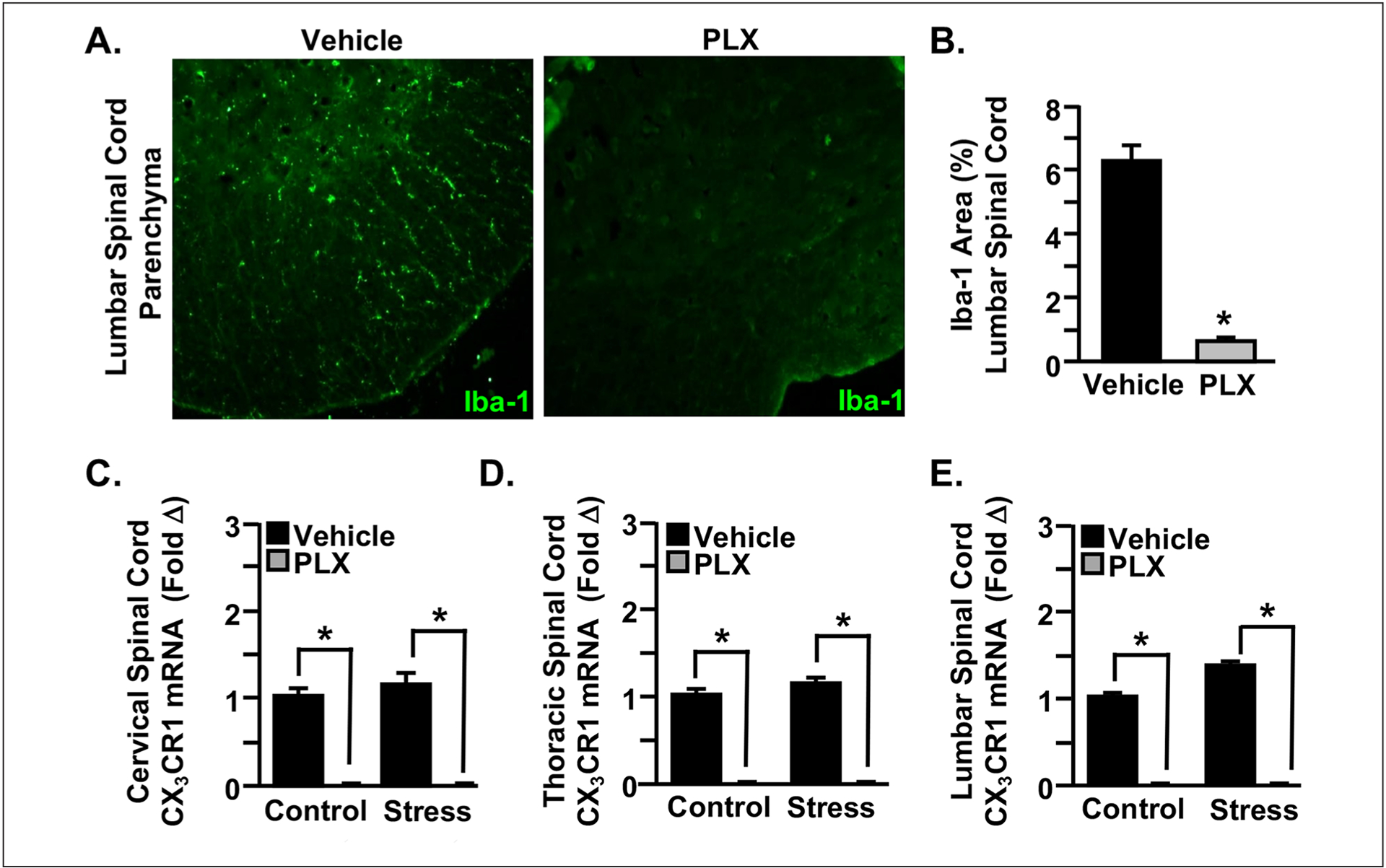
Pharmacological ablation of microglia with colony-stimulating factor 1 receptor antagonist PLX5622 allows for investigation of the role of microglia in the development of pain by stress. Mice were provided ad libitum diets of PLX5622 (PLX) or vehicle (Veh) chow for 14 days. (A, B) Following 14 days of treatment with PLX5622, microglia were eliminated from the spinal cord based on Iba-1 labeling for microglial activation. (C–E) The gene expression of microglia-related CX3CR1 was reduced by PLX5622 throughout the spinal cord in mice exposed to repeated social defeat (RSD; Stress) and those left undisturbed as controls. Modified from Sawicki and others (2019).
Figure 6.
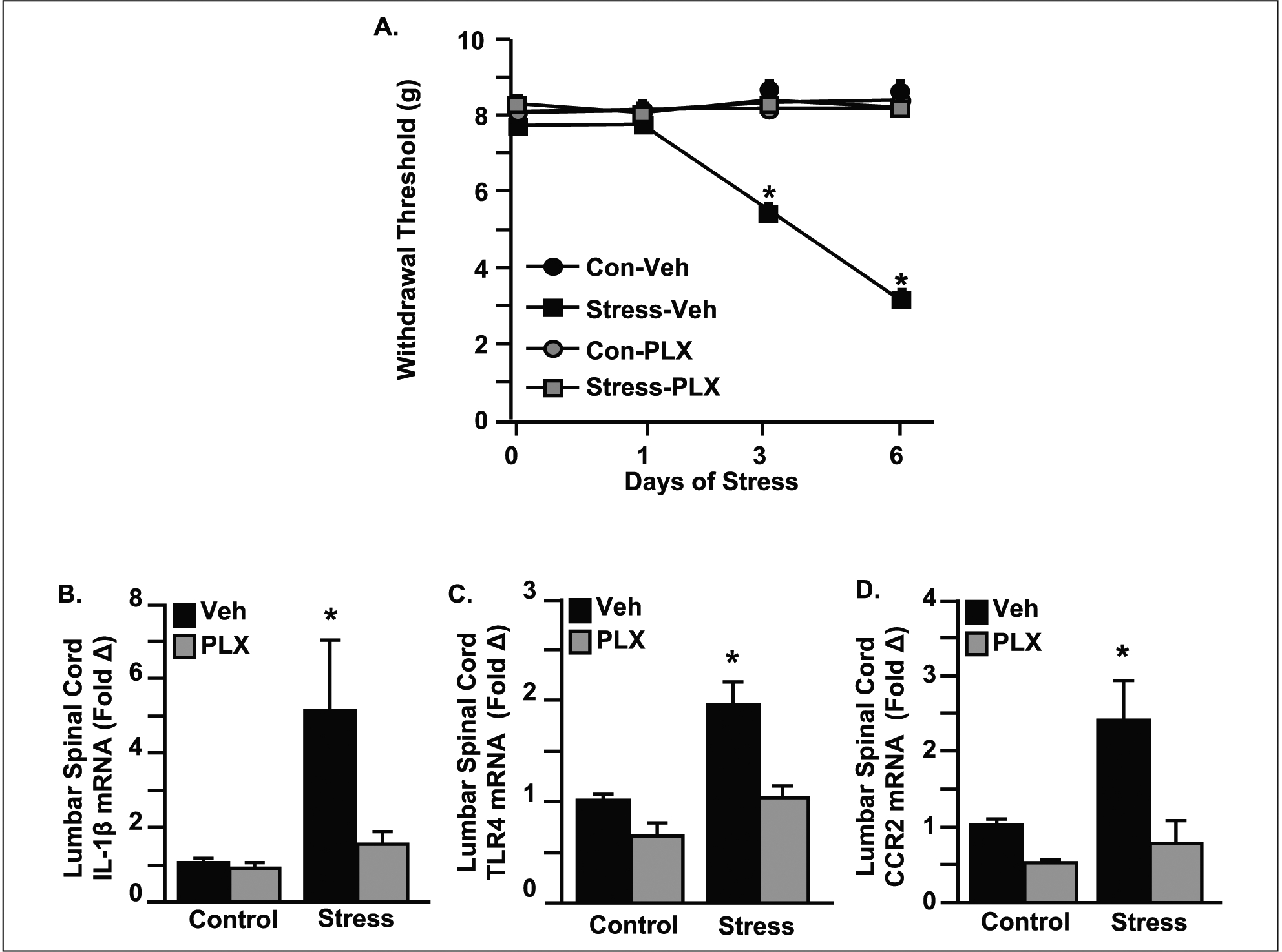
Microglia facilitate the transmission of pain to the brain through an inflammatory-driven mechanism. Mice were provided ad libitum diets of PLX5622 (PLX) or vehicle (Veh) chow for 14 days and then exposed to repeated social defeat (RSD; Stress) or left undisturbed as controls. (A) Mice were tested for mechanical allodynia before exposure to RSD and 12 hours after the first, third, and sixth day of stress. Before stress, each of the four treatment groups had comparable baseline withdrawal thresholds of mechanical stimulation to the hindpaw using the von Frey behavior test. Microglial depletion by PLX5622 prevented RSD-induced allodynia after three and six days of RSD. (B–D) The gene expression of critical inflammatory markers involved in nociceptive signaling were increased in the spinal cord with RSD, and induction of these genes was attenuated by microglial elimination with PLX5622. Modified from Sawicki and others (2019).
A Multidisciplinary Approach to Mental Health and Chronic Pain Treatment
Based on the complexity of neuroimmune dysfunction in chronic pain, clinicians and scientists alike need to approach treatment strategies from an interdisciplinary perspective. Traditional pharmacotherapy, most commonly with opiates, often fails patients afflicted with chronic pain secondary to side effect intolerance and reduced efficacy over time due to receptor down- regulation. Furthermore, targeting aberrant peripheral nerve activity in isolation fails to address CNS aspects of development and maintenance of chronic pain syndromes (Farrell and others 2018).
Despite clear disruption in peripheral and local immune regulation during various conditions of stress or mental illness, there is a lack of concordance between damage/inflammation and pain. For instance, a poor relationship exists between pathological changes evident in functional magnetic resonance imaging or computed tomography scans of the spine and the presence or absence of lumbar pain (Boden and others 1990). Accordingly, no pathologic or radiographic evidence of peripheral nociceptive damage in chronic pain is able to predict who will experience pain, or the severity of pain (Goesling and others 2018). This suggests that the peripheral and central nervous systems both contribute to the perception of pain, by determining which nociceptive input is detected by sensory nerves in the peripheral tissue.
For many individuals with chronic pain conditions, there is no identifiable cause. Pain in idiopathic chronic conditions appears to result from abnormalities in pain processing rather than just from damage or inflammation of peripheral structures (Giesecke and others 2004). For instance, patients with RA suffer from debilitating pain, long considered to be due to the inflammatory processes associated with the autoimmune response (ten Klooster and others 2007). However, patients with RA have reported pain when their disease activity was at a minimum or in remission (ten Klooster and others 2007). Similarly, the majority of patients suffering from temporomandibular joint disorder, a cluster of conditions characterized by persistent pain induced by capsule inflammation or damage, do not experience pain relief from anti-inflammatory drugs or surgical treatment (Dimitroulis 1998). Notably, the clinical course of temporomandibular joint disorders does not reflect a progressive disease but instead a multifaceted disorder that is influenced by several interacting affective conditions, including stress, anxiety, and depression, which serve to sustain the disease. Collectively, all these studies strongly implicate the complex and reciprocal interrelationship between affective disorders and pain.
Advances in our understanding of chronic pain have led to the development of new treatment modalities that target both biological and psychological causes. It is becoming increasingly clear that a combination of pharmacologic therapies and behavioral interventions would most benefit chronic pain patients. Especially considering the fact that patients suffering from mental health disorders tend to experience worse pain and functioning (Goesling and others 2018), it is critical to optimize mental health treatment in these patients. Emerging evidence indicates that certain antidepressants exhibit a moderate analgesic effect and are recommended for treatment of several pain conditions (Mercier and others 2013). It is important to note that pain processing and mood are controlled by similar neurotransmitters including norepinephrine, serotonin, glutamate, and GABA (γ-aminobutyric acid) (Mercier and others 2013). Therefore, patients suffering from abnormal pain processing and depression may respond to similar pharmacological treatments. In support of this approach, tricyclic antidepressants and selective norepinephrine reuptake inhibitors have been used effectively for the treatment of both chronic pain and depression (Goesling and others 2018). Despite this, it is critical to consider the effect of the medication on both pain and mood in chronic pain patients with comorbid psychiatric diagnoses. For instance, the addition of certain classes of drugs may worsen several psychiatric conditions or may interfere with current psychiatric medications. Therefore, psychiatrists serve an important role in chronic pain management through the evaluation of specific treatment modalities on mental health symptoms.
With iatrogenic lesions, electrical stimulation, and local perfusion of analgesics/anesthetics, various parts of the pain pathway (e.g., peripheral nerve, dorsal root, spinal cord, midbrain, thalamus, and cingulate cortex) can be therapeutic targets. Interest in neurostimulation and neuromodulation, both non-invasive and invasive strategies, has renewed in response to the opiate epidemic. Repetitive transcranial magnetic stimulation (rTMS) and transcranial direct current stimulation (tDCS) are believed to alter maladaptive plasticity within the pain circuitry of the thalamus; rTMS by stimulating neuronal firing and tDCS by altering neuronal excitability through changes in resting membrane potential. In a preclinical model of neuropathic pain, motor cortex stimulation in rodents reversed mechanical hyperalgesia and was associated with inhibition of microglial activity and decreased proinflammatory cytokines in the dorsal horn of the spinal cord (Silva and others 2015). Placement of a deep brain stimulator (DBS) electrode into various brain regions including the thalamus (e.g., ventroposterolateral nucleus), posterior limb of the internal capsule, and periventricular and/or periaqueductal gray matter, has been used clinically to treat refractory pain. Much debate surrounds DBS for treatment of pain, including mechanisms of action, ideal location for placement, and effectiveness (Farrell and others 2018). A growing body of clinical pain literature has focused on the endogenous descending pain modulatory systems. In particular, spinal cord stimulation (SCS) is a successful and common treatment strategy for patients with refractory chronic pain conditions. According to the American Association of Neurologic Surgeons, about 50,000 spinal cord stimulators are implanted per year throughout the world, with a clinical effectiveness of 60% to 85% (Sivanesan and others 2019). Although the precise mechanism by which SCS modulates the pain experience is unclear, SCS is intended to stimulate large myelinated nerve fibers in the spinal cord dorsal columns to block nociceptive inputs from small unmyelinated nerve fibers. SCS has proven to be most effective for failed back surgery syndrome (FBSS), multiple sclerosis, and complex regional pain syndrome, but less effective for phantom limb pain and postherpetic neuralgia. Notably, patients suffering from chronic pain and FBSS often report symptoms of anxiety and depression, which can modulate and amplify the pain experience. A recent clinical study demonstrated that SCS not only reduces pain in FBSS but also improves symptoms of anxiety and depression (Robb and others 2017). Similarly, clinical trials using burst stimulation of SCS found pain relief and improvement of mood in patients suffering from chronic pain (Sivanesan and others 2019). These studies and others have led to clinical investigations of spinal cord stimulation for neuropsychiatric disorders, memory, addiction, and other neurologic diseases. The molecular mechanisms of neurostimulation and neuromodulation are not known at this time, but anti-inflammatory processes may be important. Use of vagal nerve stimulators for treatment of inflammatory bowel disease in preclinical rodent models was associated with less disease burden and presence of less inflammatory markers including TNF-α, IL-6, and resident gut immune cell activation (Cheng and others 2019). Although inflammation does not always correlate with pain perception due to complex psychoemotional aspects, the coexistence of proinflammatory states in the periphery and CNS warrants continued investigations of therapies that can impact both inflammatory cascades and neuronal circuitry, including cognitive-behavioral interventions, pharmacotherapy, and neurostimulation/modulation.
Conclusions and Future Directions
In summary, it is becoming increasingly evident that neuroimmune interactions play a critical role in the development and progression of pain. Preclinical animal models have been instrumental in advancing our knowledge of the mechanisms underlying the pathogenesis of pain and its comorbidities. However, to better understand the complex dialogue between the immune and nervous systems during human chronic pain conditions, it is critical to integrate a collaborative and interdisciplinary approach. Considering the overlap between chronic pain and mental health, there is a clear need to facilitate a strategy for integrating psychiatry into chronic pain management. Moreover, the physiological and conceptual overlaps between pain and stress offer an opportunity for the use of interdisciplinary tools to aid in our understanding of peripheral and central nociceptive mechanisms under various conditions. Therefore, to develop a more integrative model of pain management, future treatment strategies should consider the contribution of neuroimmune interactions to chronic pain. Novel methods to manipulate neuroimmune pain transmission hold considerable promise for the millions of individuals living with chronic pain.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by NIMH grants R01-MH-093473 and R01-MH097243 to JFS. CMS was supported by NIDCR Training Grant T32-DE014320 and F30-DE026075.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- Abbadie C, Bhangoo S, De Koninck Y, Malcangio M, Melik-Parsadaniantz S, White FA. 2009. Chemokines and pain mechanisms. Brain Res Rev 60(1):125–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander GM, van Rijn MA, van Hilten JJ, Perreault MJ, Schwartzman RJ. 2005. Changes in cerebrospinal fluid levels of pro-inflammatory cytokines in CRPS. Pain 116(3):213–9. [DOI] [PubMed] [Google Scholar]
- Alexander JK, DeVries AC, Kigerl KA, Dahlman JM, Popovich PG. 2009. Stress exacerbates neuropathic pain via glucocorticoid and NMDA receptor activation. Brain Behav Immun 23(6):851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apkarian AV, Sosa Y, Sonty S, Levy RM, Harden RN, Parrish TB, and others. 2004. Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. J Neurosci 24(46):10410–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkinazi IY, Vershinina EA. 1999. Pain sensitivity in chronic psychoemotional stress in humans. Neurosci Behav Physiol 29(3):333–7. [DOI] [PubMed] [Google Scholar]
- Askari MS, Andrade LH, Filho AC, Silveira CM, Siu E, Wang YP, and others. 2017. Dual burden of chronic physical diseases and anxiety/mood disorders among São Paulo Megacity Mental Health Survey Sample, Brazil. J Affect Disord 220:1–7. [DOI] [PubMed] [Google Scholar]
- Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, and others. 2007. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317(5838):666–70. [DOI] [PubMed] [Google Scholar]
- Austin PJ, Moalem-Taylor G. 2010. The neuro-immune balance in neuropathic pain: involvement of inflammatory immune cells, immune-like glial cells and cytokines. J Neuroimmunol 229(1–2):26–50. [DOI] [PubMed] [Google Scholar]
- Baliki MN, Chialvo DR, Geha PY, Levy RM, Harden RN, Parrish TB, and others. 2006. Chronic pain and the emotional brain: specific brain activity associated with spontaneous fluctuations of intensity of chronic back pain. J Neurosci 26(47):12165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcelon EE, Cho WH, Jun SB, Lee SJ. 2019. Brain microglial activation in chronic pain-associated affective disorder. Front Neurosci 13:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettoni I, Comelli F, Rossini C, Granucci F, Giagnoni G, Peri F, and others. 2008. Glial TLR4 receptor as new target to treat neuropathic pain: efficacy of a new receptor antagonist in a model of peripheral nerve injury in mice. Glia 56(12):1312–9. [DOI] [PubMed] [Google Scholar]
- Boden SD, Davis DO, Dina TS, Patronas NJ, Wiesel SW. 1990. Abnormal magnetic-resonance scans of the lumbar spine in asymptomatic subjects. A prospective investigation. J Bone Joint Surg Am 72(3):403–8. [PubMed] [Google Scholar]
- Bradesi S, Svensson CI, Steinauer J, Pothoulakis C, Yaksh TL, Mayer EA. 2009. Role of spinal microglia in visceral hyperalgesia and NK1R up-regulation in a rat model of chronic stress. Gastroenterology 136(4):1339–48, e1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Shen H, Chowdhury R, Abdi T, Selaru F, Chen JDZ. 2019. Potential of electrical neuromodulation for inflammatory bowel disease. Inflamm Bowel Dis. Epub Nov 29. doi: 10.1093/ibd/izz289 [DOI] [PubMed] [Google Scholar]
- Choy EHS, Calabrese LH. 2018. Neuroendocrine and neuro-physiological effects of interleukin 6 in rheumatoid arthritis. Rheumatology (Oxford) 57(11):1885–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung G, Kim SJ, Kim SK. 2018. Metabotropic glutamate receptor 5 in the medial prefrontal cortex as a molecular determinant of pain and ensuing depression. Front Mol Neurosci 11:376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello H, Gould RL, Abrol E, Howard R. 2019. Systematic review and meta-analysis of the association between peripheral inflammatory cytokines and generalised anxiety disorder. BMJ Open 9(7):e027925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B, Conroy M, Moore D, Lysaght J, McCrory C. 2018. Human dorsal root ganglion pulsed radiofrequency treatment modulates cerebrospinal fluid lymphocytes and neuroinflammatory markers in chronic radicular pain. Brain Behav Immun 70:157–65. [DOI] [PubMed] [Google Scholar]
- Dimitroulis G 1998. Temporomandibular disorders: a clinical update. BMJ 317(7152):190–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell SM, Green A, Aziz T. 2018. The current state of deep brain stimulation for chronic pain and its context in other forms of neuromodulation. Brain Sci 8(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flor H, Braun C, Elbert T, Birbaumer N. 1997. Extensive reorganization of primary somatosensory cortex in chronic back pain patients. Neurosci Lett 224(1):5–8. [DOI] [PubMed] [Google Scholar]
- Giesecke T, Gracely RH, Grant MA, Nachemson A, Petzke F, Williams DA, and others. 2004. Evidence of augmented central pain processing in idiopathic chronic low back pain. Arthritis Rheum 50(2):613–23. [DOI] [PubMed] [Google Scholar]
- Gilman SE, Trinh NH, Smoller JW, Fava M, Murphy JM, Breslau J. 2013. Psychosocial stressors and the prognosis of major depression: a test of Axis IV. Psychol Med 43(2):303–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goesling J, Lin LA, Clauw DJ. 2018. Psychiatry and pain management: at the intersection of chronic pain and mental health. Curr Psychiatry Rep 20(2):12. [DOI] [PubMed] [Google Scholar]
- Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, and others. 2017. An environment-dependent transcriptional network specifies human microglia identity. Science 356(6344). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace PM, Hutchinson MR, Maier SF, Watkins LR. 2014. Pathological pain and the neuroimmune interface. Nat Rev Immunol 14(4):217–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. 2006. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci 26(16):4308–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry DE, Chiodo AE, Yang W. 2011. Central nervous system reorganization in a variety of chronic pain states: a review. PM R 3(12):1116–25. [DOI] [PubMed] [Google Scholar]
- Hodes GE, Kana V, Menard C, Merad M, Russo SJ. 2015. Neuroimmune mechanisms of depression. Nat Neurosci 18(10):1386–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hore Z, Denk F. 2019. Neuroimmune interactions in chronic pain—an interdisciplinary perspective. Brain Behav Immun 79:56–62. [DOI] [PubMed] [Google Scholar]
- Hutchinson MR, Lewis SS, Coats BD, Rezvani N, Zhang Y, Wieseler JL, and others. 2010. Possible involvement of toll-like receptor 4/myeloid differentiation factor-2 activity of opioid inactive isomers causes spinal proinflammation and related behavioral consequences. Neuroscience 167(3):880–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson J, Bjerke M, Sahebi S, Isgren A, Ekman CJ, Sellgren C, and others. 2015. Monocyte and microglial activation in patients with mood-stabilized bipolar disorder. J Psychiatry Neurosci 40(4):250–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SY, Seo S, Lee JS, Choi SH, Lee DH, Jung YH, and others. 2017. [11C]-(R)-PK11195 positron emission tomogra phy in patients with complex regional pain syndrome: a pilot study. Medicine (Baltimore) 96(1):e5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR. 2015. Neuroimmune interactions in itch: Do chronic itch, chronic pain, and chronic cough share similar mechanisms? Pulm Pharmacol Ther 35:81–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalliomäki J, Attal N, Jonzon B, Bach FW, Huizar K, Ratcliffe S, and others. 2013. A randomized, double-blind, placebo-controlled trial of a chemokine receptor 2 (CCR2) antagonist in posttraumatic neuralgia. Pain 154(5):761–7. [DOI] [PubMed] [Google Scholar]
- Khandaker GM, Pearson RM, Zammit S, Lewis G, Jones PB. 2014. Association of serum interleukin 6 and C-reactive protein in childhood with depression and psychosis in young adult life: a population-based longitudinal study. JAMA Psychiatry 71(10):1121–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Kim MA, Cho IH, Kim MS, Lee S, Jo EK, and others. 2007. A critical role of toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J Biol Chem 282(20):14975–83. [DOI] [PubMed] [Google Scholar]
- Klawitter M, Hakozaki M, Kobayashi H, Krupkova O, Quero L, Ospelt C, and others. 2014. Expression and regulation of toll-like receptors (TLRs) in human intervertebral disc cells. Eur Spine J 23(9):1878–91. [DOI] [PubMed] [Google Scholar]
- Kojima M, Kojima T, Suzuki S, Oguchi T, Oba M, Tsuchiya H, and others. 2009. Depression, inflammation, and pain in patients with rheumatoid arthritis. Arthritis Rheum 61(8):1018–24. [DOI] [PubMed] [Google Scholar]
- Krock E, Jurczak A, Svensson CI. 2018. Pain pathogenesis in rheumatoid arthritis-what have we learned from animal models? Pain 159 (suppl 1):S98–109. [DOI] [PubMed] [Google Scholar]
- Kuner R, Flor H. 2017. Structural plasticity and reorganisation in chronic pain. Nat Rev Neurosci 18(2):113. [DOI] [PubMed] [Google Scholar]
- Lee S, Shi XQ, Fan A, West B, Zhang J. 2018. Targeting macrophage and microglia activation with colony stimulating factor 1 receptor inhibitor is an effective strategy to treat injury-triggered neuropathic pain. Mol Pain 14:1744806918764979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Buisman-Pijlman F, Hutchinson MR. 2014. Toll-like receptor 4: innate immune regulator of neuroimmune and neuroendocrine interactions in stress and major depressive disorder. Front Neurosci 8:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand F, Perretti M, McMahon SB. 2005. Role of the immune system in chronic pain. Nat Rev Neurosci 6(7):521–32. [DOI] [PubMed] [Google Scholar]
- McKim DB, Weber MD, Niraula A, Sawicki CM, Liu X, Jarrett BL, and others. 2018. Microglial recruitment of IL-1β-producing monocytes to brain endothelium causes stress-induced anxiety. Mol Psychiatry 23(6):1421–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier A, Auger-Aubin I, Lebeau JP, Schuers M, Boulet P, Hermil JL, and others. 2013. Evidence of prescription of antidepressants for non-psychiatric conditions in primary care: an analysis of guidelines and systematic reviews. BMC Fam Pract 14:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AH, Maletic V, Raison CL. 2009. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65(9):732–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotla Zarebska J, Chanalaris A, Driscoll C, Burleigh A, Miller RE, Malfait AM, and others. 2017. CCL2 and CCR2 regulate pain-related behaviour and early gene expression in post-traumatic murine osteoarthritis but contribute little to chondropathy. Osteoarthritis Cartilage 25(3):406–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni H, Wang Y, An K, Liu Q, Xu L, Zhu C, and others. 2019. Crosstalk between NFκB-dependent astrocytic CXCL1 and neuron CXCR2 plays a role in descending pain facilitation. J Neuroinflammation 16(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie X, Kitaoka S, Tanaka K, Segi-Nishida E, Imoto Y, Ogawa A, and others. 2018. The innate immune receptors TLR2/4 mediate repeated social defeat stress-induced social avoidance through prefrontal microglial activation. Neuron 99(3):464–79.e7. [DOI] [PubMed] [Google Scholar]
- Norman GJ, Karelina K, Morris JS, Zhang N, Cochran M, Courtney DeVries A. 2010. Social interaction prevents the development of depressive-like behavior post nerve injury in mice: a potential role for oxytocin. Psychosom Med 72(6):519–26. [DOI] [PubMed] [Google Scholar]
- Otmishi P, Gordon J, El-Oshar S, Li H, Guardiola J, Saad M, and others. 2008. Neuroimmune interaction in inflammatory diseases. Clin Med Circ Respirat Pulm Med 2:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins MN, Kelly D. 1994. Interleukin-1β induced-desArg-9bradykinin-mediated thermal hyperalgesia in the rat. Neuropharmacology 33(5):657–60. [DOI] [PubMed] [Google Scholar]
- Powell ND, Sloan EK, Bailey MT, Arevalo JM, Miller GE, Chen E, and others. 2013. Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via β-adrenergic induction of myelopoiesis. Proc Natl Acad Sci U S A 110(41):16574–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren K, Hylden JL, Williams GM, Ruda MA, Dubner R. 1992. The effects of a non-competitive NMDA receptor antagonist, MK-801, on behavioral hyperalgesia and dorsal horn neuronal activity in rats with unilateral inflammation. Pain 50(3):331–44. [DOI] [PubMed] [Google Scholar]
- Robb LP, Cooney JM, McCrory CR. 2017. Evaluation of spinal cord stimulation on the symptoms of anxiety and depression and pain intensity in patients with failed back surgery syndrome. Ir J Med Sci 186(3):767–71. [DOI] [PubMed] [Google Scholar]
- Rosen JM, Yaggie RE, Woida PJ, Miller RJ, Schaeffer AJ, Klumpp DJ. 2018. TRPV1 and the MCP-1/CCR2 Axis Modulate Post-UTI Chronic Pain. Sci Rep 8(1):7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicki CM, Kim JK, Weber MD, Faw TD, McKim DB, Madalena KM, and others. 2019. Microglia promote increased pain behavior through enhanced inflammation in the spinal cord during repeated social defeat stress. J Neurosci 39(7):1139–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicki CM, Kim JK, Weber MD, Jarrett BL, Godbout JP, Sheridan JF, and others. 2018. Ropivacaine and bupiva-caine prevent increased pain sensitivity without altering neuroimmune activation following repeated social defeat stress. Brain Behav Immun 69:113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfers M, Svensson CI, Sommer C, Sorkin LS. 2003. Tumor necrosis factor-α induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci 23(7):2517–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G, and others. 2015. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 72(3):268–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Gelman BB, Lisinicchia JG, Tang SJ. 2012. Chronic-pain-associated astrocytic reaction in the spinal cord dorsal horn of human immunodeficiency virus-infected patients. J Neurosci 32(32):10833–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva GD, Lopes PS, Fonoff ET, Pagano RL. 2015. The spinal anti-inflammatory mechanism of motor cortex stimulation: cause of success and refractoriness in neuropathic pain? J Neuroinflammation 12:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivanesan E, Maher DP, Raja SN, Linderoth B, Guan Y. 2019. Supraspinal mechanisms of spinal cord stimulation for modulation of pain: five decades of research and prospects for the future. Anesthesiology 130(4):651–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer C 2001. [Cytokines in neuropathic pain]. Anaesthesist 50(6):416–26. [DOI] [PubMed] [Google Scholar]
- Sweitzer S, Martin D, DeLeo JA. 2001. Intrathecal interleukin-1 receptor antagonist in combination with soluble tumor necrosis factor receptor exhibits an anti-allodynic action in a rat model of neuropathic pain. Neuroscience 103(2):529–39. [DOI] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. 2005. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A 102(16):5856–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Klooster PM, Veehof MM, Taal E, van Riel PL, van de Laar MA. 2007. Changes in priorities for improvement in patients with rheumatoid arthritis during 1 year of anti-tumour necrosis factor treatment. Ann Rheum Dis 66(11):1485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trang T, Beggs S, Salter MW. 2012. ATP receptors gate microglia signaling in neuropathic pain. Exp Neurol 234(2):354–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojvodic J, Mihajlovic G, Vojvodic P, Radomirovic D, Vojvodic A, Vlaskovic-Jovicevic T, and others. 2019. The impact of immunological factors on depression treatment—relation between antidepressants and immunomodulation agents. Open Access Maced J Med Sci 7(18):3064–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Goffer Y, Xu D, Tukey DS, Shamir DB, Eberle SE, and others. 2011. A single subanesthetic dose of ketamine relieves depression-like behaviors induced by neuropathic pain in rats. Anesthesiology 115(4):812–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Wiertelak EP, Goehler LE, Smith KP, Martin D, Maier SF. 1994. Characterization of cytokine-induced hyperalgesia. Brain Res 654(1):15–26. [DOI] [PubMed] [Google Scholar]
- Weber MD, Godbout JP, Sheridan JF. 2017. Repeated social defeat, neuroinflammation, and behavior: monocytes carry the signal. Neuropsychopharmacology 42(1):46–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Guo W, Zou S, Ren K, Dubner R. 2008. Supraspinal glial-neuronal interactions contribute to descending pain facilitation. J Neurosci 28(42):10482–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, McKim DB, Sheridan JF, Godbout JP. 2014. Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-to-brain communication that influences mood and behavior. Front Neurosci 8:447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, Powell ND, Godbout JP, Sheridan JF. 2013. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci 33(34):13820–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ. 2020. Capturing novel non-opioid pain targets. Biol Psychiatry 87(1):74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. 2000. Neuronal plasticity: increasing the gain in pain. Science 288(5472):1765–9. [DOI] [PubMed] [Google Scholar]
- Xu Y, Wang Y, Chen J, He Y, Zeng Q, Huang Y, and others. 2020. The comorbidity of mental and physical disorders with self-reported chronic back or neck pain: results from the China Mental Health Survey. J Affect Disord 260:334–41. [DOI] [PubMed] [Google Scholar]
- Zhang JM, An J. 2007. Cytokines, inflammation, and pain. Int Anesthesiol Clin 45(2):27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YQ, Liu Z, Liu ZH, Chen SP, Li M, Shahveranov A, and others. 2016. Interleukin-6: an emerging regulator of pathological pain. J Neuroinflammation 13(1):141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zis P, Daskalaki A, Bountouni I, Sykioti P, Varrassi G, Paladini A. 2017. Depression and chronic pain in the elderly: links and management challenges. Clin Interv Aging 12:709–20. [DOI] [PMC free article] [PubMed] [Google Scholar]