


Abstract
The hairpin ribozyme is a small catalytic RNA that catalyses reversible sequence-specific RNA hydrolysis in trans. It consists of two domains, which interact with each other by docking in an antiparallel fashion. There is a region between the two domains acting as a flexible hinge for interdomain interactions to occur. Hairpin ribozymes with reverse-joined domains have been constructed by dissecting the domains at the hinge and rejoining them in reverse order. We have used both the conventional and reverse-joined hairpin ribozymes for the design of a hairpin-derived twin ribozyme. We show that this twin ribozyme cleaves a suitable RNA substrate at two specific sites while maintaining the target specificity of the individual monoribozymes. For characterisation of the studied ribozymes we have evaluated a quantitative assay of sequence-specific ribozyme activity using fluorescently labelled RNA substrates in conjunction with an automated DNA sequencer. This assay was found to be applicable with hairpin and hairpin-derived ribozymes. The results demonstrate the potential of hairpin ribozymes for multi-target strategies of RNA cleavage and suggest the possibility for employing hairpin-derived twin ribozymes as powerful tools for RNA manipulation in vitro and in vivo.
INTRODUCTION
Nucleic acids, long viewed as targets for drugs, nowadays attract attention as drugs themselves. Specific interference with gene expression at the level of mRNA is a promising approach to decreasing the level of proteins for which the intracellular disappearance is of biological interest or therapeutic benefit to a given tissue or organism. The demonstration that RNA can be cleaved by cis- or trans-acting ribozymes has potentially important therapeutic implications. Like antisense oligonucleotides, ribozymes can inactivate the target RNA by complementary base pairing, but additionally have the capacity to cleave more than one copy of the target RNA by dissociating from the cleavage products and binding to another target molecule. Most studies performed to date have described the use of ribozymes as therapeutic agents for viral diseases and cancer. Ribozymes have been shown to be active in transfected cells and have been successfully used to inhibit HIV replication (1–3). Nevertheless, when considering ribozymes as a potential tool for HIV therapy, the extended variability of viral sequences has to be taken into account. The problems arising from the high mutability of viruses like HIV can be overcome by applying ribozymes that are capable of cleaving the virus RNA at several conserved sites. In fact, there are examples that targeting several sites simultaneously by antisense DNAs, as well as the use of expression vectors for multi-targeted hammerhead ribozymes, prevented the development of escape mutants (4–8).
Like the hammerhead ribozyme, the hairpin ribozyme is a small catalytic RNA suitable for gene therapeutic application. Hairpin ribozymes work in cis (intramolecularly) in nature, but they have been engineered to work in trans (intermolecularly) and thus catalyse site-specific reversible cleavage on the 5′-side of a GUC triplet (Fig. 1a), generating a 2′,3′-cyclic phosphate and a free 5′-hydroxyl group (for recent reviews see 9–11). It has been shown that the two domains of the hairpin ribozyme, one containing helix 1, helix 2 and loop A and the second containing helix 3, helix 4 and loop B (Fig. 1a), can be joined to each other in a variety of different ways and still maintain ribozyme activity (12–17). Several lines of experimental evidence suggest that the two domains are involved in interdomain interactions during catalysis (17–23). The proposed models of the hairpin ribozyme tertiary structure (22,24) describe the catalytically active conformer as a three-dimensional complex where the two domains closely approach each other by bending via the hinge at the junction between helix 2 and helix 3. The required tertiary interaction can be achieved from different initial positions of both domains. Thus, hairpin ribozymes with reverse-joined domains have been constructed by dissecting the two domains of the parent ribozyme at the hinge between helix 2 and helix 3 and rejoining them in a reverse configuration (12,15).
Figure 1.
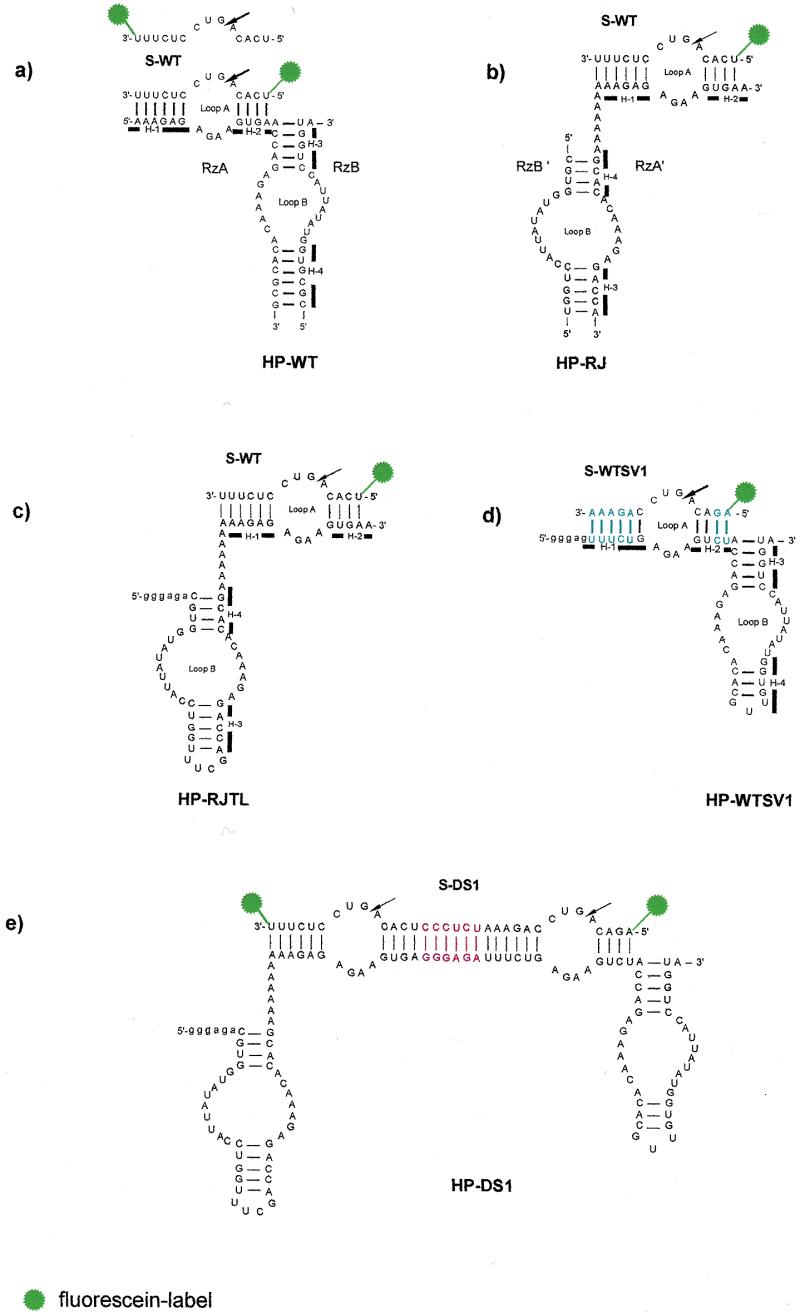
Secondary structures of the studied ribozymes. The four helices (H-1–H-4) in HP-WT, HP-RJ, HP-RJTL and HP-WTSV1 are indicated by bars. The arrows denote the sites of cleavage. Vector nucleotides resulting from in vitro transcription of HP-RJTL, HP-WTSV1 and HP-DS1 with T7 RNA polymerase are indicated in lower case. (a) Conventional hairpin ribozyme HP-WT with wild-type sequence. The fluorescently 5′- and 3′-labelled substrate S-WT is shown. (b) Reverse-joined hairpin ribozyme HP-RJ derived from HP-WT by dissecting the loop A and loop B domains at the hinge and rejoining them via an A6 linker in reverse order. (c) Reverse-joined ribozyme HP-RJTL derived from HP-RJ with helix 3 capped by the UUCG tetraloop. (d) Conventional hairpin ribozyme with sequence variation HP-WTSV1. Base pairs which are reversed in comparison to HP-WT are highlighted in blue. (e) Twin ribozyme HP-DS1 derived from combination of the monoribozymes HP-RJTL and HP-WTSV1. The sequence introduced as spacer between both ribozyme units is highlighted in red.
Our work focuses on the design of hairpin-derived twin ribozymes which are capable of cleaving a RNA substrate at two specific sites. This type of ribozyme would be an initial step towards the construction of multi-targeted hairpin ribozymes for antiviral therapy. Further, the hairpin ribozyme is also an efficient ligase (25,26). If it is possible to combine and to control cleavage and ligation activity, a hairpin-derived twin ribozyme might be a potential tool for RNA manipulation in vitro, with the possibility in future of applying a similar method in vivo. By cleaving a RNA fragment out of a defective RNA followed by ligation of the correct fragment, such types of ribozymes might open the way to ‘revise’ genetic instructions, instead of just to destroy them. In general, the construction of a hairpin ribozyme targeting more than one site within the substrate RNA yields further insight into the structural limits of hairpin ribozyme-catalysed RNA cleavage and can provide information on the structural and mechanistic requirements for catalysis.
MATERIALS AND METHODS
Synthesis and purification of oligonucleotides
Chemical synthesis of RNA was based on 2′-O-t-butyldimethylsilyl (TBDMS) nucleoside protection (27,28) and involved sequential couplings of the β-cyanoethyl-(N,N-diisopropyl)-phosphoramidite of 5′-O-dimethoxytrityl-2′-O-TBDMS nucleosides with phenoxyacetyl amino group protection for A, p-iso-propyl-phenoxyacetyl amino group protection for G and benzoyl protection for C (Amersham Pharmacia Biotech), essentially by the method of Scaringe et al. (29). Oligoribonucleotides S-WT, S-WTSV1, S-DS1, HP-WT-RzA, HP-WT-RzB, HP-RJ-RzA′ and HP-RJ-RzB′ (14–37 bases) were synthesised on a 1 µmol scale. Fluorescein labelling at the 5′-end was achieved using ‘fluoreprime’ fluorescein amidite (Amersham Pharmacia Biotech) as last monomer building block in the nucleotide chain to be coupled during solid phase synthesis. For labelling at the 3′-end controlled pore glass (CPG) was used to which fluorescein was attached via a thiourea functionality as a succinate linkage (ChemGenes Corp.). It contained a dimethoxytrityl-protected hydroxyl group which after deprotection was used for chain elongation. The synthesis was performed trityl-off; removal of the base protecting groups was accomplished using saturated methanolic ammonia at room temperature for 16–24 h. Treatment with triethylamine trihydrofluoride/DMF (3:1) at 55°C for 1.5 h (30) was carried out to remove silyl groups, followed by precipitation with n-butanol. Oligoribonucleotides were purified by strong anion exchange chromatography on a MonoQ HR 5/5 column (Amersham Pharmacia Biotech), flow rate 1 ml/min, using buffer A (0.02 M sodium acetate, 20% acetonitrile, pH 6.5) and buffer B (0.02 M sodium acetate, 1 M sodium chloride, 20% acetonitrile) at 0% buffer B for 2 min, followed by gradients of 0–40% B over 5 min, 40–80% B over 40 min, 80–100% B over 2 min. Desalting was achieved via extensive dialysis against water. The purity of oligoribonucleotides was assayed by ion exchange and reverse phase chromatography as well as by electrophoresis on a 20% polyacrylamide gel in the presence of 7 M urea.
Large RNA molecules HP-WTSV1, HP-RJTL and HP-DS1 (50, 59 and 114 bases) were transcribed in vitro from oligodeoxyribonucleotide templates. Our strategy involved the synthesis of DNA templates containing the ribozyme sequence linked to the T7 RNA polymerase-specific promoter sequence. DNA templates were synthesised as previously described (31) on a 0.2 µmol scale using 3′-O-(2-cyanoethyl-N,N-diisopropyl)-phosphoramidite building blocks having phenoxyacetyl amino group protection for A and G, isobutyryl protection for C (Amersham Pharmacia Biotech). Synthesis was performed trityl-on; deblocking was performed using 32% aqueous ammonia at room temperature overnight. Dimethoxytrityl-containing oligonucleotides were purified by HPLC using an RP 18 column (150 × 13 mm, 5 ml/min; Machery and Nagel) with buffer A (0.1 M triethyl ammonium acetate, 5% acetonitrile, pH 7) and buffer B (0.1 M triethyl ammonium acetate, 30% acetonitrile, pH 7) at 0% B for 2 min, followed by gradients of 0–40% B over 5 min and 40–100% B over 30 min (oligonucleotides <50 bases), 0–40% B over 5 min, 40–70% B over 30 min and 70–100% B over 5 min (oligonucleotides 50–100 bases) or 0–40% B over 5 min, 40–70% B over 40 min and 70–100% B over 5 min (oligonucleotides >100 bases). Product-containing fractions were evaporated to dryness in vacuo, redissolved in 80% acetic acid and allowed to react at room temperature for 40 min. Acetic acid was removed by evaporation in vacuo, followed by co-evaporation with water and ethanol. The resulting product was dissolved in water and extracted twice with ethyl acetate. The aqueous layer was evaporated to dryness and the oligomers stored at –20°C. Purity was checked by electrophoresis on 15 or 20% denaturing polyacrylamide gels and by reverse phase chromatography.
Templates for transcription were generated from two synthetic DNA strands, overlapping by 9–20 complementary bases, dependent on the primary structure of the RNA to be synthesised. After annealing, the DNA templates were completed enzymatically using DNA polymerase I, Klenow fragment exo– (MBI Fermentas) and the four deoxyribonucleoside triphosphates (Amersham Pharmacia Biotech) in the presence of dithiotreitol (DTT) at concentrations of 0.04 U/µl polymerase, 0.4 mM each deoxyribonucleoside triphosphates, 5 mM DTT for 2 h at 37°C. Double-stranded DNA was purified by electrophoresis through 15–20% native polyacrylamide gels. Product-containing bands were eluted from the gel with buffer (0.5 M sodium acetate, 1 mM EDTA, 50 mM Tris–HCl, pH 7.4) followed by extraction with phenol and ethanol precipitation. RNAs were synthesised by transcribing the DNA templates with T7 RNA polymerase (MBI Fermentas) in the presence of the four ribonucleoside triphosphates (Amersham Pharmacia Biotech) using concentrations of 1 µM DNA template, 4 U/µl T7 RNA polymerase, 2 mM each ribonucleoside triphosphates for 1 h at 37°C. Final purification was achieved by electrophoresis on 20% denaturing polyacrylamide gels, elution of the product-containing bands with buffer (0.5 M sodium acetate, 1 mM EDTA, 50 mM Tris–HCl, pH 7.4) and precipitation with ethanol.
Determination of ribozyme kinetic parameters
Kinetic parameters of cleavage reactions were determined under multiple turnover conditions and each measurement was repeated at least once. Individual solutions of ribozymes (2–20 nM, 10 µl) in 10 mM Tris–HCl (pH 7.5) containing 12 mM magnesium chloride and, separately, the corresponding fluorescein-labelled substrate RNAs (20–300 nM, 40 µl) in 10 mM Tris–HCl (pH 7.5) containing 12 mM magnesium chloride were incubated at 90°C for 1 min and then allowed to cool to 37°C over 15 min. Cleavage reactions were started by mixing the ribozyme and substrate solutions to give a final volume of 50 µl. Aliquots (3 µl) were removed at eight suitable time intervals and the reactions quenched by addition to 6 µl of stop mix (7 M urea, 50 mM EDTA). The samples were analysed by polyacrylamide gel electrophoresis on 15% denaturing gels, using an ALF DNA sequencer as described previously (32). Note that addition of bromphenol blue to the stop mix should be avoided, since the dye is photoactive at 488 nm, the wavelength of the argon laser. Thus, the signals resulting from bromphenol blue would disturb quantitative analysis of the cleavage reaction (C.Schmidt and S.Müller, unpublished observations). The data were processed and quantitated using the Software package FM 1.2 A.L.F. (Pharmacia).
Kinetic parameters were obtained from plotting reciprocal initial rates normalised to a fixed ribozyme concentration (1/kobs) against reciprocal substrate concentrations (1/[S]) using Lineweaver–Burk plots.
RESULTS
Quantitative assay of sequence-specific ribozyme activity
Functional characterisation of ribozymes via analysis of reaction kinetics requires large numbers of individual measurements. Current methods rely primarily on the use of radiolabelled RNA substrates and thus require tedious electrophoretic separation and quantitation of reaction products for each data point in any experiment. To make the analysis more efficient we have used an automated DNA sequencer to determine amounts of both reactants and products of the hairpin cleavage reaction by virtue of their fluorescence. As we have shown in previous studies on the quantitation of sequence-specific endonuclease activity (32), progress of the reaction is monitored by the change in relative peak areas (linear response provided).
To use this assay for quantification of ribozyme activity, it was necessary first to investigate whether the rather bulky fluorescein molecule has any influence on formation of the ribozyme–substrate complex and on catalysis. We have used a three-stranded hairpin ribozyme (33), assembled from two catalytic strands (RzA and RzB) and either the 5′- or 3′-labelled substrate strand (Fig. 1a). In this way each strand is sufficiently short to be prepared by RNA solid phase chemical synthesis. The three-stranded ribozyme is reported to have kinetic properties similar to those of the normal two-stranded hairpin ribozyme (34) and has been used in many studies. The hairpin ribozyme cleaves the substrate 5′ to G within loop A, reducing the length of the RNA strand carrying the fluorescent label at the 5′-end from 14 to 5 nt (and from 14 to 9 nt if the fluorescein is attached at the 3′-end). Thus, for both cases (5′- and 3′-labelling), reactants and products of the reaction could be separated by polyacrylamide gel electrophoresis and detected by virtue of their fluorescence. While the fluorescent quantum yield of fluorescein when attached at either the 5′- or 3′-end is different, it remains invariant for uniformly labelled (either 5′- or 3′-end) substrate and corresponding product RNAs. This we have evaluated by producing equimolar mixtures of substrate and product RNAs and comparing the individual peak areas in gel electrophoresis (ALF). Thus, the fluorescence yield for the 5′-labelled substrate and the 5′-labelled cleavage product or the 3′-labelled substrate and 3′-labelled product, respectively, is the same. Since both the reactant and the product of the reaction were measured, combined areas were standardised to 100%. This allows the measurement to be independent of pipetting errors when loading the gel and also of the quite pronounced sensitivity differences between individual detector channels. Using the available software package A.L.F. Manager the output data of the DNA sequencer were analysed by integrating the relative peak areas.
Steady-state parameters of hairpin cleavage were obtained from determination of the initial rates of reaction. In both cases (fluorescein 5′- or 3′-labelling) the reaction velocities, once normalised to a fixed ribozyme concentration, followed a conventional Michaelis–Menten curve. Thus, fluorescein-labelled RNAs are suitable substrates for the hairpin ribozyme, whether the fluorescein label is attached at the 5′- or 3′-end. The obtained catalytic parameters for the 5′-labelled substrate (Km = 50 nM and kcat = 0.2 min–1) are consistent with those (Km = 40 nM and kcat = 0.23 min–1) found using a 32P-end-labelled substrate (35). For the 3′-labelled substrate the Km (19 nM) is smaller by a factor of two, while the kcat (0.18 min–1), within error, falls in the same range as the kcat for the radiolabelled and the fluorescein 5′-labelled substrates. The error is ~30% of the mean for Km and 10% for kcat using either fluorescently labelled substrate.
The 2-fold reduction in Km observed for the fluorescein 3′-labelled substrate in comparison to radiolabelled or 5′-labelled samples can be interpreted, in a simple Michaelis–Menten mechanism, as tighter binding of the ribozyme to the substrate caused by increased stacking energy of the intercalating fluorescein, located at the 3′-terminus, in the ground state. Attachment of the fluorescein at the 5′-terminus seems to influence Km less; presumably intercalation from this position is not as favourable. Furthermore, the different chemical nature of the fluorescein labels used for either 5′- or 3′-labelling (Fig. 3) might affect binding of the ribozyme to the substrate. Although the 2-fold reduction in Km observed for the 3′-labelled substrate is not detrimental to the determination of ribozyme kinetics, we preferred using 5′-labelled substrates since the results obtained are more easily compared with hairpin ribozyme studies using radiolabelled samples.
Figure 3.

Fluorescein building blocks used for RNA labelling by solid phase synthesis. (a) 5,6-Carboxy isomer of the fluorescein phosphoramidite building block used for 5′-labelling. (b) Polymer-bound fluorescein building block used for 3′-labelling.
Ribozyme design
For construction of the twin ribozyme HP-DS1 (Fig. 1e) our strategy involved the combination of a conventional hairpin ribozyme with a second hairpin ribozyme consisting of reverse-joined domains. The kinetic properties of both ribozyme units were studied separately before joining them in the twin ribozyme. Therefore, both three-stranded hairpin ribozymes were assembled from a substrate strand and two catalytic strands, thus allowing the preparation of each strand by RNA solid phase chemical synthesis, or two-stranded hairpin ribozymes consisting of a substrate strand and a one-piece catalytic strand were prepared by a combination of chemical synthesis and in vitro transcription techniques. The 5 or 6 nt overhang at the 5′-end of the transcribed ribozyme strands HP-RJTL, HP-WTSV1 and HP-DS1 (Fig. 1c–e) result from the required start sequence for T7 RNA polymerase (36) and are essential for successful transcription of the hairpin sequences used.
Design of the reverse-joined hairpin ribozyme unit
Reverse-joined hairpin ribozymes were first described by Komatsu et al. (12,15). This type of ribozyme is derived from the conventional hairpin ribozyme by dissecting the two domains at the hinge between helices 2 and 3 and rejoining helix 4 to helix 1 via a linker of six unpaired bases. Following this construction principle we have synthesised the reverse-joined hairpin ribozyme HP-RJ (Fig. 1b) and studied the kinetic properties in comparison to the conventional hairpin ribozyme HP-WT (Fig. 1a). In HP-RJ, the A6 linker acts as the flexible hinge between helices 1 and 4 and was found to be of optimal length for catalytic activity (C.Schmidt and S.Müller, unpublished results). For kinetic analysis, HP-RJ as well as HP-WT were assembled from the two catalytic strands (RzA and RzB or RzA′ and RzB′, respectively) and the corresponding substrate RNA S-WT, carrying a fluorescein label at the 5′-end (Fig. 1a and b). Kinetic parameters were determined under multiple turnover conditions with 10 mM Tris–HCl (pH 7.5) and 12 mM magnesium chloride at a temperature of 37°C. The Tris concentration was decreased from the conventional 40 to 10 mM, because we have observed an inhibitory effect of Tris–HCl on the cleavage reaction, which is rather strong for HP-RJ and relatively weak for HP-WT (data not shown). Therefore, we kept the Tris concentration rather low.
Comparing the kinetic parameters found for HP-RJ with those for HP-WT reveals a difference which is reflected in kcat rather than in Km (Table 1). Whereas the Km (145 nM) for HP-RJ is only three times higher than the Km (50 nM) for HP-WT, the kcat is 40-fold lower (kcat, HP-RJ = 0.005 min–1, kcat, HP-WT = 0.2 min–1). Among other reasons, this result can possibly be attributed to reduced stability of helices 3 and 4 of HP-RJ. Both helices are each 1 bp shorter than the corresponding helices in HP-WT (Fig. 1a and b). This is very likely to cause destabilisation of the loop B domain structure. Independent folding of the two domains of the hairpin ribozyme is rapid and precedes docking into the closed complex (20). If the loop B domain is unable to fold into a stable structure, docking of the two domains will be substantially effected. In addition, the A6 linker introduced to allow interdomain interactions to occur causes enhanced flexibility of the three-dimensional active conformation and subsequently decreases activity. Thus, the sub-optimal secondary and tertiary structure of the loop B domain together with the high hinge flexibility are a reasonable explanation for the reduced rate constant of HP-RJ.
Table 1. Kinetic data for cleavage reactions catalysed by HP-WT, HP-RJ, HP-RJTL and HP-WTSV1 (fluorescein 5′-labelled substrates were used).
| Km (nM) | kcat (min–1) | kcat/Km (min–1 µM–1) | |
|---|---|---|---|
| HP-WT | 50 ± 18 | 0.2 ± 0.02 | 4 |
| HP-RJ | 145 ± 41 | 0.005 ± 0.001 | 0.03 |
| HP-RJTL | 26 ± 11 | 0.06 ± 0.01 | 2.31 |
| HP-WTSV1 | 50 ± 15 | 0.05 ± 0.006 | 1 |
To reconcile these data we have synthesised another reverse-joined hairpin ribozyme HP-RJTL (Fig. 1c). The sequence of HP-RJTL is analogous to HP-RJ, but helix 3 is capped by the RNA tetraloop sequence 5′-UUCG-3′. The tetraloop forms a very stable structure (37) and thus enhances stability of the ribozyme HP-RJTL (Fig. 1c). The new ribozyme HP-RJTL showed much higher cleavage efficiency than HP-RJ (Table 1). The individual kinetic parameters were Km = 26 nM and kcat = 0.06 min–1. While the effect on Km was marginal (5-fold reduced), the kcat of HP-RJTL was increased 12-fold compared to HP-RJ, suggesting that introduction of the UUCG tetraloop assists proper folding of the loop B domain and, subsequently, tertiary folding into a three-dimensional ribozyme–substrate complex. The catalytic efficiency (kcat/Km = 2.31 min–1 µM–1) of the reverse-joined hairpin ribozyme with a tetraloop, HP-RJTL, approaches kcat/Km (4 min–1 µM–1) of the conventional hairpin ribozyme HP-WT. Therefore, HP-RJTL can be considered as a practical tool for site-specific RNA cleavage. In addition to the stabilising effect, closing helix 3 in HP-RJTL is advantageous for our further strategy to construct a twin ribozyme with a one-piece catalytic strand (Fig. 1e).
Design of the conventional hairpin ribozyme unit (sequence variation)
The substrate binding sequence of the reverse-joined ribozyme HP-RJTL is identical to that of the conventional hairpin ribozyme. In order to later distinguish between both predicted cleavage sites in the twin ribozyme, it was necessary to design a different substrate-binding sequence of the conventional hairpin ribozyme unit. Thus, the ribozyme HP-WTSV1 used here (Fig. 1d) has been modified from the original hairpin motif by minor changes within the sequence needed for specific binding of the substrate. To minimise interference with stability of the ribozyme–substrate complex, we used a partially reversed sequence. The 5′-terminal bases AAAGA of helix 1 of the original hairpin motif were changed to UUUCU and the 3′-terminal bases GA of helix 2 to CU, targeting a corresponding substrate with a 5′-AG and a 3′-AAAGA terminus (Fig. 1a and d). In view of our strategy to construct a one-piece catalytic twin ribozyme strand we immediately closed helix 4 by the standard GUU loop as it appears in the normal hairpin ribozyme (34).
Using the fluorescence assay described above for quantification of the ribozyme reaction a double peak was observed for the 5mer cleavage product (Fig. 2, lane 2). There is also a double peak for the 14mer substrate, suggesting that this effect results from the fluorescein label itself, rather than from non-specific cleavage. The fluorescein phosphoramidite used for 5′-labelling was a mixture of the 5-carboxy and 6-carboxy fluorescein isomers (Fig. 3a). Oligonucleotides labelled with either isomer can usually be separated by reverse phase chromatography. One could expect the same for gel electrophoresis, considering the fact that not only the mass but also the overall structure is responsible for migration of a sample through the gel. The difference in retention times between the two signals corresponding to the 5mer cleavage product was ~1 min (Fig. 2, lane 2). The retention time difference between n and n – 1 fragments was ~3 min under the given conditions. Therefore it is not very likely that the second peak results from non-specific cleavage to give a 4mer or 6mer. The hairpin ribozyme-catalysed cleavage reaction delivers 5′-fragments with 2′,3′-cyclic phosphate termini. Thus another explanation for the observed two product signals might be subsequent hydrolysis of the cyclic phosphate to a phosphate. To test this hypothesis we carried out large scale cleavage of S-WTSV1 and separated both cleavage products by HPLC on a MonoQ column. We chose this strategy because the difference between two RNA fragments carrying either isomer is more pronounced in shorter sequences and therefore separation of the 5mer cleavage products was easier than separation of the two 14mer substrate RNA species. In separate experiments the two products were tested as substrates for hairpin-catalysed ligation. Both RNA fragments could be ligated to the corresponding 5′-OH fragment in the presence of HP-WTSV1, resulting in the two peaks of the 14mer substrate RNA (data not shown). This suggests that both cleavage products were terminated by a 2′,3′-cyclic phosphate and that the appearance of the double peak in the 5mer cleavage product as well as in the 14mer substrate indeed results from the two fluorescein isomers. This, however, is surprising, since the wild-type substrate S-WT labelled with the same fluorescein isomer mixture did not show this behaviour (Fig. 2, lane 1). S-WT is different from S-WTSV1 in two bases of the 5′-terminal sequence and five bases of the 3′-terminal sequence (Fig. 1a and d). It seems likely that the different sequences of S-WT and S-WTSV1 cause the two species labelled with the two fluorescein isomer to separate to differing extents in the two RNAs. To further prove this hypothesis we synthesised the fluorescein 5′-labelled five base sequence 5′-AGACA-3′ corresponding to the sequence of the cleavage product of HP-WTSV1. This 5mer sequence has no phosphate or cyclic phosphate at the 3′-end but, nevertheless, we observed a double peak after electrophoresis through a 15% denaturing polyacrylamide gel (Fig. 2, lane 3). Since it lacks the terminal phosphate, it migrates through the gel more slowly than the corresponding fragment that results from cleavage of S-WTSV1. From these results we conclude that the double peak clearly observed for the 5mer fragment indeed results from identical oligomers labelled with either 5-carboxy or 6-carboxy fluorescein. The appearance of double peaks for some oligonucleotides can be avoided by also using isomeric pure fluorescein phosphoramidite building blocks, as shown for example in Figure 3b, for 5′-labelling. Preliminarily, for kinetic analysis both individual peak areas corresponding to the two isomers were considered.
Figure 2.

Primary data on the cleavage of fluorescein 5′-labelled S-WT by the wild-type hairpin ribozyme HP-WT (lane 1) and of fluorescein 5′-labelled S-WTSV1 by the hairpin ribozyme with varied sequence HP-WTSV1 (lane 2). Cleavage conditions: 20 nM ribozyme, 200 nM substrate, 10 mM Tris–HCl pH 7.5, 12 mM MgCl2, 37°C. Lane 3 shows the migration of a separately synthesised fluorescein 5′-labelled RNA (5′-AGACA-3′) corresponding to the sequence of the 5mer fragment resulting from cleavage of S-WTSV1 by HP-WTSV1. cp, 2′,3′-cyclic phosphate. All lanes show autoscaled data (ordinate, fluorescence intensity in arbitrary units).
Characterisation of ribozyme HP-WTSV1 showed a cleavage efficiency that was 4-fold lower than for the parent conventional ribozyme (Table 1). The catalytic efficiency (kcat/Km) was determined as 1 min–1 µM–1. The individual kinetic parameters were Km = 50 nM and kcat = 0.05 min–1 (Table 1). For the hairpin ribozyme in its natural four-way junction conformation, Murchie et al. (38) have shown that the conformation of the junction is dependent on the sequence of the central base pairs on each arm and that this influences the activity of the ribozyme. A modified species with the same sequence at the hinge between the loop A and loop B domains as HP-WTSV1 exhibited 10-fold lower activity than the natural form (38). Therefore, we attribute the lower activity of HP-WTSV1 compared to the wild-type hairpin ribozyme to a hinge sequence that is detrimental for efficient tertiary folding.
RNA double cleavage by the twin ribozyme
Following our strategy to combine two catalytic units in one molecule to produce a twin ribozyme, we synthesised the ribozyme HP-DS1 (Fig. 1e). The reverse-joined hairpin ribozyme HP-RJTL (Fig. 1c) and the conventional hairpin with modified substrate-binding sequence HP-WTSV1 (Fig. 1d) seem to be particularly well-suited for combination as a twin ribozyme because both units showed nearly equivalent catalytic efficiency in individual activity tests. Furthermore, the resulting structure of the twin ribozyme is advantageous since the flexible hinge regions required for bending are located at the outside of the complex, such that both hairpin motifs enjoy sufficient conformational freedom to bend into the active three-dimensional structure. We additionally incorporated a six base sequence (5′-GGGAGA-3′) in the centre of the substrate binding domain of HP-DS1. This should increase the distance between the two ribozyme units and thus exclude possible steric hindrance for tertiary folding.
The twin ribozyme was assembled from the catalytic strand HP-DS1 and the substrate strand S-DS1 (Fig. 1e). The 34mer substrate RNA has a fluorescein label at both the 3′- and 5′-ends, allowing cleavage at both predicted sites to be followed. Specific cleavage by the reverse-joined ribozyme unit within the twin ribozyme would produce a 9mer and a 25mer RNA, cleavage by the conventional hairpin ribozyme unit would produce a 5mer and a 29mer RNA. Both the 29mer and the 25mer fragments are still substrates for single cleavage at the second site. Therefore the 5mer and 9mer cleavage products are expected to accumulate during the reaction and should be found as main products. Since the substrate is labelled at the 3′- and 5′-ends, both short product RNAs as well as the longer product strands are detectable and the cleavage reaction can be followed.
First, cleavage was carried out under standard conditions as described above. In the presence of 10 mM MgCl2 cleavage at the predicted sites resulting in the characteristic 5mer and 9mer fragments could be clearly detected (Fig. 4a). However, after 70 min only a small fraction of substrate S-DS1 was cleaved. It has been shown by Earnshaw and Gait (39) that the presence of polyamines like spermine or spermidine in the reaction mixture boosts cleavage rates considerably. To make the twin ribozyme more efficient we made use of this property of polyamines and carried out the cleavage reaction catalysed by HP-DS1 in the presence of different ratios of magnesium ions/spermine. Furthermore, the reaction was allowed to proceed for a longer time in order to produce clearly detectable products. Best results were obtained with 7 mM MgCl2, 5 mM spermine. There is clear conversion of a slow moving species (210 min) into three faster moving species (85, 86 and 105 min) with reaction time. Further, two smaller peaks appear at 162 and 180 min, which after an initial increase later decreased again (Fig. 4b). In comparison to authentic samples the individual signals could be assigned as follows: 85/86 min, 5′-labelled 5mer product; 105 min, 3′-labelled 9mer product; 162 min, 5′-labelled 25mer product; 180 min, 3′-labelled 29mer product; 210 min, 3′,5′-labelled 34mer substrate. For the 5mer cleavage product again two peaks with a 1 min retention time difference (85 and 86 min) were detected. This is consistent with the predicted cleavage by the HP-WTSV1 unit resulting in the 5mer fragment labelled by 5-carboxy or 6-carboxy fluorescein. As mentioned above, the migration difference for isomer labelled fragments is more pronounced for short sequences. Even the 14mer substrate RNA in the individual cleavage experiment with HP-WTSV1 showed a peak with a shoulder rather than two separated signals after electrophoretic separation. Therefore, it is not surprising that the characteristic double peak for the 5′-labelled 25mer at 162 min was not observed, although it contains the fluorescein isomers attached at the same site as in the 5mer cleavage product. Also, the signal of the double labelled 34mer substrate S-DS1 at 210 min is not divided into two peaks, because the length of this RNA levels off the effect brought about by the fluorescein isomers at the 5′-terminus. For 3′-labelling a distinct fluorescein derivative attached to a polymer support was used (Fig. 3b). This material was homogeneous and thus no double peaks were detected at 180 min for the 3′-labelled 29mer and at 105 min for the 3′-labelled 9mer cleavage product.
Figure 4.
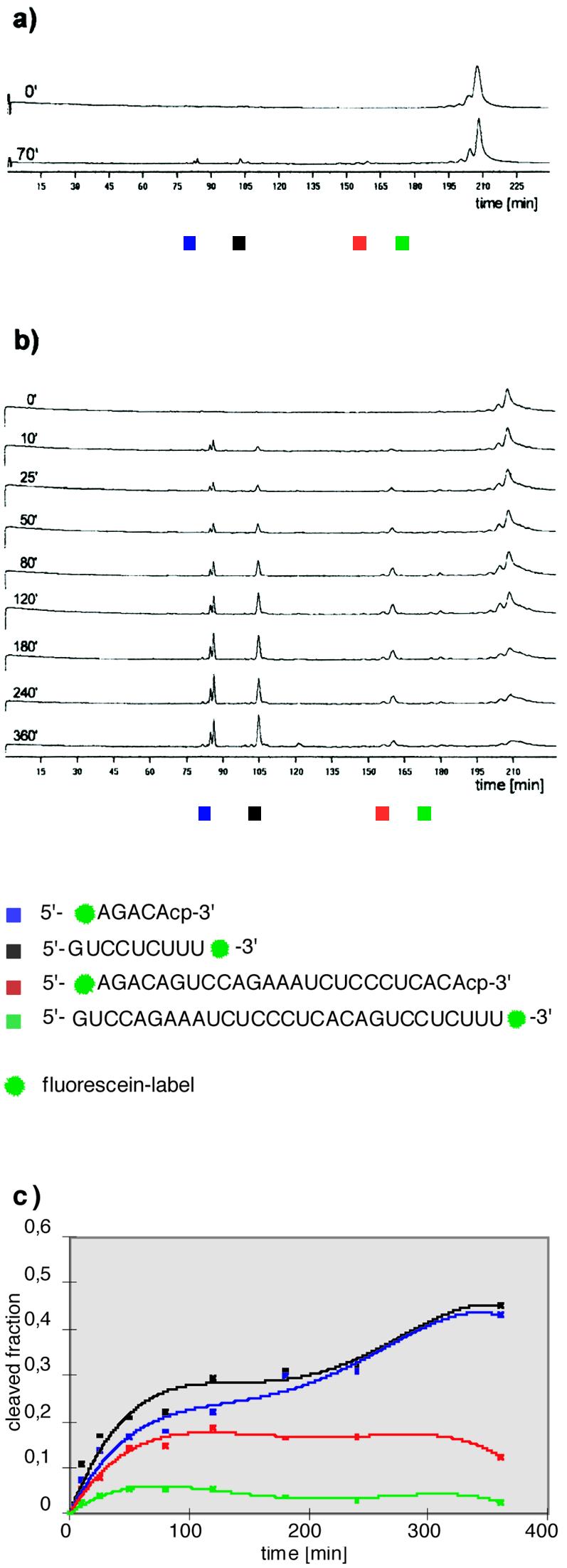
RNA double cleavage by the twin ribozyme HP-DS1. The coloured squares refer to the sequences of the cleavage products. (a) Primary data on the cleavage of S-DS1 by HP-DS1 in the presence of Mg2+ after gel electrophoresis on the ALF DNA sequencer. Cleavage conditions: 20 nM HP-DS1, 200 nM S-DS1, 10 mM Tris–HCl pH 7.5, 12 mM MgCl2, 37°C. (b) Primary data on the cleavage of S-DS1 by HP-DS1 in the presence of Mg2+ and spermine after gel electrophoresis on the ALF DNA sequencer. Cleavage conditions: 20 nM HP-DS1, 200 nM S-DS1, 10 mM Tris–HCl pH 7.5, 7 mM MgCl2, 5 mM spermine, 37°C. All lanes in (a) and (b) show autoscaled data (ordinate, fluorescence intensity in arbitrary units). (c) Formation of 5mer (blue), 9mer (black), 25mer (red) and 29mer (green) fragments as a function of time. In a separate experiment, which involved cleavage of a double labelled substrate RNA at one specific position, the contribution of the fluorescein label at the 5′-end and the fluorescein label at the 3′-end to the fluorescence intensity of the substrate RNA was determined to be 3:2. This factor was used for standardising peak areas in quantification of the twin ribozyme cleavage reaction.
Analysis of the cleavage reaction shows that the substrate RNA is cleaved at two specific sites following incubation with the twin ribozyme, leading to the characteristic 5mer and 9mer fragments and to a 20mer, which cannot be detected by laser-induced fluorescence, as main products. Whereas the fractions of the 5mer and 9mer produced increase with time, the fractions of the 25mer and the 29mer decrease after an initial increase, based on cleavage occurring at the second specific site (Fig. 4c). The 29mer intermediate does not accumulate as significantly as the 25mer, indicating that cleavage by the reverse-joined ribozyme unit within the twin ribozyme occurs faster than cleavage by the conventional ribozyme unit.
DISCUSSION
The systematic search for high efficiency ribozymes began only a few years ago and has focused mainly on different ribozyme motifs, target sites and flanking sequences. The multi-targeting strategy introduced by Taira et al. (5) is directed towards the treatment of viral diseases. Multi-target hammerhead ribozymes maintain the target specificity of the individual ribozymes and also significantly raise the overall cleavage efficiency per catalytic RNA molecule (7). The importance of this strategy has been demonstrated by several groups (4–8), however, only the hammerhead motif was used in those studies.
In this work we describe the rational design of a hairpin-derived twin ribozyme which is capable of cleaving a suitable RNA substrate at two specific sites. Such a ribozyme is not only a first step towards multi-targeted hairpin ribozymes. Since the hairpin ribozyme has cleavage as well as ligation activity (25,26), a hairpin-derived twin ribozyme could possibly, under appropriate conditions, be used to introduce sequence alterations into RNA in vitro. In addition, it might serve as a tool for RNA repair in vivo, related to the strategy first described by Sullenger and Cech (40), who used trans-splicing group I introns for repair of a defective mRNA in vivo. Targeted trans-splicing has been demonstrated by several groups since then (41–47) and although there are still major problems to be overcome, this method offers an interesting new way of correcting monogenic diseases. In view of problems of specificity and cell delivery it might be advantageous to replace the rather large group I intron by smaller structures such as the twin ribozyme described here. However, the possible ability of the twin ribozyme to support RNA repair in vivo is beyond the scope of the present discussion and first has to be evaluated carefully.
A quantitative assay, which rests on fluorescently labelled substrates in conjunction with a DNA sequencer, was used to evaluate the cleavage reaction catalysed by the hairpin and the designed hairpin-derived ribozymes. This assay achieves high sample throughput by parallel measurement of multiple samples. Because of its capacity to produce and to process large sets of experimental data rapidly and conveniently, the system is particularly well suited to the determination of reaction kinetics. Compared to radioactive assay of ribozyme activity, the procedure is easier to handle and less time consuming with respect to both acquisition and processing of data. The substrates are non-radioactive and can be stored for long periods of time. There are neither health hazards nor waste disposal problems. Furthermore, the fluorescently labelled samples can be used for further studies of hairpin ribozyme mechanism and structure by fluorescence resonance energy transfer or base-specific quenching, as previously described by Walter et al. (20,21).
The combination of a conventional hairpin ribozyme unit together with a reverse-joined hairpin ribozyme allowed us to produce a twin ribozyme that cleaved a RNA substrate at two selected sites. To our knowledge, this is the first example of a hairpin-derived ribozyme combining two catalytic units in one molecule. Both the conventional and the reverse-joined hairpin motif combined in the twin ribozyme still have sufficient conformational freedom to fold into the catalytically active complex. Analysis of the cleavage reaction revealed that the RNA substrate was cleaved at two specific sites and that the twin ribozyme maintained the target specificity of the individual ribozymes HP-RJTL and HP-WTSV1. There is even the same clear double peak for the 5′-labelled 5mer cleavage product as observed in the cleavage reaction catalysed by the monoribozyme HP-WTSV1 (Figs 2 and 4). The cleavage activity of the twin ribozyme is boosted in the presence of the polyamine spermine. This corresponds very well to higher cleavage rates obtained for the wild-type hairpin ribozyme as well as for reverse-joined hairpin ribozymes in the presence of spermine (39; C.Schmidt and S.Müller, manuscript in preparation). This result demonstrates that mechanistic details of the monoribozymes are conserved in the twin ribozyme.
Polyamines are found in all cells and are essential for eukaryotic proliferation. Although the quantities of free intracellular polyamines are unknown, it seems likely that twin ribozyme-supported RNA manipulation inside cells could be significantly enhanced by spermine.
The results presented here show that hairpin-derived twin ribozymes can be successfully applied to specific RNA fragmentation and suggest that such a type of ribozyme may have utility in a direction besides that previously envisioned for the hairpin ribozyme. Further characterisation of the twin ribozyme will be required before second generation twin ribozymes with optimised properties can be designed. As an initial effort towards this objective we aim to investigate whether cleavage at both sites occurs independently of each other or if there is some sort of cooperative stimulation. Our future studies include the characterisation of ligation activity as well as the cleavage/ligation equilibrium of the twin ribozyme and, based on the results, optimisation of the structure for functional design.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Christina Schönherr for valuable technical assistance, particularly for synthesis of oligonucleotides. We are grateful to U. Koert, H.-J. Fritz and E.S. Gromova for helpful discussions and critical reading of this manuscript. This work was supported by Deutsche Forschungsgemeinschaft Grant Mu 1396/2-1 and Fonds der Chemischen Industrie.
REFERENCES
- 1.Steinecke P., Herget,T. and Schreier,P.H. (1992) EMBO J., 11, 1525–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lo K.M.S., Biasolo,M.A., Dehni,G., Palu,G. and Haseltine,W.A. (1992) Virology, 190, 176–183. [DOI] [PubMed] [Google Scholar]
- 3.Ojwang J.O., Hampel,A., Looney,D.J., Wong-Stahl,F. and Rappoport,J. (1992) Proc. Natl Acad. Sci. USA, 89, 10802–10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lisziewicz J., Sun,D., Kliotman,M., Agrawal,S., Zamecnik,P. and Gallo,R. (1992) Proc. Natl Acad. Sci. USA, 89, 11209–11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taira K., Nakagawa,K., Nishikawa,S. and Furukawa,K. (1991) Nucleic Acids Res., 19, 5125–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohkawa J., Yuyama,N., Takebe,Y., Nishikawa,S. and Taira,K. (1993) Proc. Natl Acad. Sci. USA, 90, 11302–11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen C.-J., Banerjea,C.B., Harmison,G.G., Hagelund,K. and Schubert,M. (1992) Nucleic Acids Res., 20, 4581–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weizacker F.-V., Blum,H.E. and Wands,J.R. (1992) Biochem. Biophys. Res. Commun., 189, 743–748. [DOI] [PubMed] [Google Scholar]
- 9.Walter N.G. and Burke,J.M. (1998) Curr. Opin. Chem. Biol., 2, 24–30. [DOI] [PubMed] [Google Scholar]
- 10.Hampel A. (1998) Prog. Nucleic Acids Res. Mol. Biol., 58, 1–39. [DOI] [PubMed] [Google Scholar]
- 11.Lilley D.M.J. (1999) FEBS Lett., 452, 26–30. [DOI] [PubMed] [Google Scholar]
- 12.Komatsu Y., Kanzaki,I., Koizumi,M. and Ohtsuka,E. (1995) J. Mol. Biol., 252, 296–304. [DOI] [PubMed] [Google Scholar]
- 13.Butcher S.E., Heckman,J.E. and Burke,J.M. (1995) J. Biol. Chem., 270, 29648–29651. [DOI] [PubMed] [Google Scholar]
- 14.Shin C., Choi,J.N., Song,S.I., Song,J.T., Ahn,J.H., Lee,J.S. and Choi,Y.D. (1996) Nucleic Acids Res., 24, 2685–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Komatsu Y., Kanzaki,I. and Ohtsuka,E. (1996) Biochemistry, 35, 9815–9820. [DOI] [PubMed] [Google Scholar]
- 16.Esteban J.A., Walter,N.G., Kotzorek,G., Heckman,J.E. and Burke,J.M. (1998) Proc. Natl Acad. Sci. USA, 95, 6091–6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pinard R., Heckman,J.E. and Burke,J.M. (1999) J. Mol. Biol., 287, 239–251. [DOI] [PubMed] [Google Scholar]
- 18.Feldstein P.A. and Bruening,G. (1993) Nucleic Acids Res., 21, 1991–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Komatsu Y., Koizumi,M., Nakamura,H. and Ohtsuka,E. (1994) J. Am. Chem. Soc., 116, 3692–3696. [Google Scholar]
- 20.Walter N.G., Hampel,K.J., Brown,K.M.N. and Burke,J.M. (1998) EMBO J., 17, 2378–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walter N.G. and Burke,J.M. (1997) RNA, 3, 392–404. [PMC free article] [PubMed] [Google Scholar]
- 22.Earnshaw D.J., Masquida,B., Müller,S., Sigurdsson,S.Th., Eckstein,F., Westhof,E. and Gait,M.J. (1997) J. Mol. Biol., 274, 197–212. [DOI] [PubMed] [Google Scholar]
- 23.Hampel K.J., Walter,N.G. and Burke,J.M. (1998) Biochemistry, 37, 14672–14682. [DOI] [PubMed] [Google Scholar]
- 24.Butcher S.E., Allain,F.H.-T. and Feigon,J. (1999) Nature Struct. Biol., 6, 212–216. [DOI] [PubMed] [Google Scholar]
- 25.Hegg L.A. and Fedor,M.J. (1995) Biochemistry, 34, 15813–15828. [DOI] [PubMed] [Google Scholar]
- 26.Nesbitt S.-M., Erlacher,H.A. and Fedor,M.J. (1999) J. Mol. Biol., 286, 1009–1024. [DOI] [PubMed] [Google Scholar]
- 27.Ogilvie K.K., Thompson,E.A., Quilliam,M.A. and Westmore,J.B. (1974) Tetrahedron Lett., 2865–2868. [Google Scholar]
- 28.Damha M.J. and Ogilvie,K.K. (1993) In Agrawal,S. (ed.), Methods in Molecular Biology. Humana Press, Totowa, NJ, pp. 81–114.
- 29.Scaringe S.A., Francklyn,C. and Usman,N. (1990) Nucleic Acids Res., 18, 5433–5441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wincott F., DiRenzo,A., Shaffer,C., Grimm,S., Tracz,D., Workman,C., Sweedler,D., Gonzalez,C., Scaringe,S. and Usman,N. (1995) Nucleic Acids Res., 23, 2677–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmidt S., Pein,C.D., Fritz,H.-J. and Cech,D. (1992) Nucleic Acids Res., 20, 2421–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gläsner W., Merkl,R., Schmidt,S., Cech,D. and Fritz,H.-J. (1992) Biol. Chem. Hoppe-Seyler, 373, 1223–1225. [DOI] [PubMed] [Google Scholar]
- 33.Chowrira B.M., Berzal-Herranz,A., Keller,C.F. and Burke,J.M. (1993) J. Biol. Chem., 268, 19458–19462. [PubMed] [Google Scholar]
- 34.Chowrira B.M. and Burke,J.M. (1992) Nucleic Acids Res., 20, 2835–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmidt S., Beigelman,L., Karpeisky,A., Usman,N., Sorensen,U.S. and Gait,M.J. (1996) Nucleic Acids Res., 24, 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milligan J.F. and Uhlenbeck,O.C. (1989) Methods Enzymol., 180, 51–62. [DOI] [PubMed] [Google Scholar]
- 37.Cheong C., Varani,G. and Tinoco,I. (1990) Nature, 346, 680–682. [DOI] [PubMed] [Google Scholar]
- 38.Murchie A.I.H., Thomson,J.B., Walter,F. and Lilley,D.M.J. (1998) Mol. Cell, 1, 873–881. [DOI] [PubMed] [Google Scholar]
- 39.Earnshaw D.J. and Gait,M.J. (1998) Nucleic Acids Res., 26, 5551–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sullenger B.A. and Cech,T.R. (1994) Nature, 371, 619–622. [DOI] [PubMed] [Google Scholar]
- 41.Sullenger B.A. (1995) Chem. Biol., 2, 249–253. [DOI] [PubMed] [Google Scholar]
- 42.Jones J.T., Lee,S.-W. and Sullenger,B.A. (1996) Nature Med., 2, 643–648. [DOI] [PubMed] [Google Scholar]
- 43.Sullenger B.A. (1996) Cytokines Mol. Ther., 2, 201–206. [PubMed] [Google Scholar]
- 44.Phylactou L.A., Darrah,C. and Wood,M.J.A. (1998) Nature Genet., 18, 378–381. [DOI] [PubMed] [Google Scholar]
- 45.Lan N., Howrey,R.P., Lee,S.-W., Smith,C.A. and Sullenger,B.A. (1998) Science, 280, 1593–1595. [DOI] [PubMed] [Google Scholar]
- 46.Köhler U., Ayre,B.G., Goodman,H.M. and Haseloff,J. (1999) J. Mol. Biol., 285, 1935–1950. [DOI] [PubMed] [Google Scholar]
- 47.Ayre B.G., Köhler,U., Goodman,H.M. and Haseloff,J. (1999) Proc. Natl Acad. Sci. USA, 96, 3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]